
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hydrogen peroxide/dimethyl carbonate: a green system for epoxidation of N-alkylimines and N-sulfonylimines. One-pot synthesis of N-alkyloxaziridines from N-alkylamines and (hetero)aromatic aldehydes†
Jamil
Kraïem
 ab,
Donia
Ghedira
b and
Thierry
Ollevier
*a
ab,
Donia
Ghedira
b and
Thierry
Ollevier
*a
aDépartement de chimie, Université Laval, 1045 avenue de la Médecine, Québec (Québec) G1V 0A6, Canada. E-mail: thierry.ollevier@chm.ulaval.ca
bLaboratoire de Développement Chimique, Galénique et Pharmacologique des Médicaments, Faculté de Pharmacie de Monastir, Université de Monastir, Rue Avicenne, 5000 Monastir, Tunisia
First published on 25th July 2016
Abstract
A green method for epoxidation of imines using an environmentally benign oxidant system, H2O2/dimethyl carbonate, was developed. N-Alkyloxaziridines were prepared in high yields from N-alkylamines and (hetero)aromatic aldehydes in one-pot fashion, whereas N-sulfonyloxaziridines have been prepared by using the same oxidant system and 5 mol% of Zn(OAc)2·2H2O as catalyst.
Oxaziridines, first reported by Emmons in 1957,1 exhibit unusual and distinctive reactivity, which is linked to the nature of their N-substituent. For example, N-sulfonyloxaziridines have been used as electrophilic oxygenating agents for enolates,2 alkenes3 and sulfides.4N-Alkyloxaziridines undergo cycloaddition reactions with a variety of heterocumulenes,5,6 alkenes,7 alkynes,8 nitriles9 and arynes10 leading to various five-membered ring heterocycles. In addition, oxaziridines are useful as precursors of nitrones,1 amides,11 secondary amines12 and N,N-disubstituted hydroxylamines.1,13 Within the past decade, the chemistry of oxaziridines has been significantly expanded and now encompasses some new interesting reaction types, among which the enantioselective oxyamination of alkenes with N-sulfonyloxaziridines, catalyzed by Cu(II)14 and Fe(II)15 complexes, and the enantioselective Lewis-base catalyzed cycloaddition of N-sulfonyloxaziridines with ketenes can be mentioned.16
Oxaziridines can be readily prepared from imines, through a number of methods including oxidation with peracids,17 particularly m-chloroperbenzoic acid (MCPBA),18,19 oxidation with O2 in the presence of transition metals,20 oxidation with H2O2 in combination with acetic anhydride,1 nitriles,21,22 and urea23 and oxidation with buffered oxone.24 Perfluoro-oxaziridines have also been prepared by the oxidation of the corresponding perfluoro-imines with H2O2 in the presence of a base.25 These methods usually use toxic solvents26 such as CH3CN![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 11,17 and CH2Cl2.19b Furthermore, most of them generate stoichiometric amounts of undesirable waste, and require tedious work-up and purification.
11,17 and CH2Cl2.19b Furthermore, most of them generate stoichiometric amounts of undesirable waste, and require tedious work-up and purification.
Recently, such synthetic routes utilizing hazardous solvents and reagents and generating toxic waste have become discouraged and there have been much efforts to develop safer and environmentally-benign alternatives. This green chemistry approach was introduced in 1990 with the aim at developing cleaner processes through the design of innovative and environmentally benign chemical reactions. In recent years, increasingly stringent environmental requirements have led to a great interest in the application of sustainable and green oxidations. The concept of green chemistry has been rapidly expanding and important advances have been achieved in this field.27 Interest in new synthetic methods has also been extended to many relevant classes of chemical compounds, such as oxaziridines. In this case, two attempts of eco-friendly synthesis of N-alkyloxaziridines were described in the literature. The first one consisted in the oxidation of imines with H2O2 in acetonitrile, catalyzed by 2 mol% of sodium tungstate,11,27b and the other one described a chemo-enzymatic synthesis of N-alkyloxaziridines using H2O2/octanoic acid system as oxidant of N-alkylimines.17 In both cases, the conversions of imines to oxaziridines did not reach completion and tedious work-up and further purification were needed. Furthermore, these methods did not avoid the use of toxic solvents, such as CH3CN, and are limited to the synthesis of N-alkyloxaziridines rather than other types of oxaziridines.
Among environmentally-friendly oxidation reagents, hydrogen peroxide is particularly attractive, both for its high oxygen content and the nature of its by-products (water and O2). Moreover, diluted aqueous solutions of hydrogen peroxide are stable and they can be easily handled and stored. On the other hand, dimethyl carbonate (DMC) is an interesting green solvent and reagent alternative in organic synthesis due to its high biodegradability and low toxicity defined as the absence of associated irritating or mutagenic effects.28 Furthermore, its industrial production does not require phosgene but only methanol and CO.29 As reported by Rüsch gen. Klaas using DMC with large excess of hydrogen peroxide solution (60%) under chemo-enzymatic catalysis, alkenes were epoxidized with moderate to high yields.30 These authors assume that the active oxidant is the monoperoxy carbonic acid methyl ester (MeO–CO–OOH) generated by the reaction of hydrogen peroxide with DMC, and decomposes to CO2 and methanol after epoxidation of alkenes. In general, O-alkylmonoperoxycarbonic acids (RO–CO–OOH) are known to be effective epoxidizing reagents of olefins.31–34 They are generated in situ by the reaction of ethyl chloroformiate,31 alkoxycarbonyl-1,2,4-triazoles,32 alkoxycarbonylimidazoles33 or dialkyl carbonates using hydrogen peroxide solution,30,34 and they could be utilized without isolation because of their instability. In some cases, O-alkylmonoperoxycarbonic acids have been isolated34 or detected.32 We think that the H2O2/DMC system is still under investigated in synthetic chemistry. Indeed, since the report of Rüsch gen. Klaas in 1999, no other reactions using this green oxidant system have been described in the literature.
In this paper we describe a simple, efficient and eco-friendly synthesis of oxaziridines by use of the green 30% H2O2/DMC system. N-Alkyloxaziridines have been prepared quantitatively in one-pot synthetic route from the corresponding alkylamines and aldehydes, under mild and neutral conditions, whereas N-sulfonyloxaziridines have been prepared with high yields from the corresponding N-sulfonylimines under basic conditions, in the presence of Zn(OAc)2·2H2O as a catalyst.
With the aim of synthesizing N-alkyloxaziridines in a one-pot fashion, we investigated the reaction of t-butylamine 1a (1.0 and 1.5 mmol) with benzaldehyde 2a (1 mmol) and 30% hydrogen peroxide solution, in the presence of a variety of solvents, affording 2-t-butyl-3-phenyloxaziridine 3a (Table 1). We found that, when the one-pot reaction was run in H2O, CH2Cl2, toluene or DMPU, no transformation into the oxaziridine was observed (Table 1, entries 8–11). These solvents are indeed known to be inert towards H2O2. Accordingly, these results indicate that hydrogen peroxide itself is not sufficiently reactive to epoxidize the intermediate imine, whereas, when the reaction was run in DMC, acetonitrile, ethyl acetate, methanol or propylene carbonate (Table 1, entries 1–7), a moderate to high conversion into the oxaziridine was observed. Solvents such as acetonitrile, dialkyl carbonates or ethyl acetate can react with H2O2. Indeed, peroxyimidic acids, generated in situ by perhydrolysis of a nitrile, are known to be effective epoxidizing reagents of olefins and imines (Payne oxidation).22,35 Perhydrolysis of ethyl acetate may also generate peracetic acid as the oxidant of the imine.17,36 In this case, oxaziridine 3a was formed with a moderate yield after 24 h (Table 1, entry 5). The use of methanol as solvent provided the oxaziridine 3a in 50% yield after 24 h (Table 1, entry 6). As shown in Table 1, the best result for the one-pot synthesis of oxaziridine 3a was achieved when the reaction was performed by stirring a mixture of t-butylamine (1.5 mmol), benzaldehyde (1 mmol) and aqueous solution of H2O2 (30%, 5 mmol) in 1 mL of DMC for 15 h, furnishing quantitatively oxaziridine 3a (Table 1, entry 3). We assume that the active oxidant is the monoperoxycarbonic acid methyl ester (MeO–CO–OOH) generated by the reaction of H2O2 with DMC for the following reasons: (i) O-alkylperoxycarbonic acids have already been reported in the literature,31–34 among which O-benzylperoxycarbonic acid, prepared from dibenzyl dicarbonate and H2O2, was isolated and characterized;34 (ii) H2O2 itself is not sufficiently reactive for epoxidation; this hypothesis is supported by the results given in Table 1 (entries 8–11).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Equiv. H2O2 | Equiv. t-BuNH2 | Solvent | Time (h) | Conversion 3ab (%) |
| a Conditions: 1a, 1 mmol of 1b, H2O2 (30%), solvent (1 mL), rt. b Determined by 1H NMR analysis of crude product. c Propylene carbonate. d The oxaziridine was not detected (TLC, 1H NMR). e 1,3-Dimethyltetrahydropyrimidin-2(1H)-one. | |||||
| 1 | 5 | 1 | DMC | 24 | 88 |
| 2 | 3 | 1.5 | DMC | 24 | 85 |
| 3 | 5 | 1.5 | DMC | 15 | 100 |
| 4 | 5 | 1.5 | CH3CN | 20 | 98 |
| 5 | 5 | 1.5 | EtOAc | 24 | 61 |
| 6 | 5 | 1.5 | CH3OH | 24 | 50 |
| 7 | 5 | 1.5 | PCc | 18 | 92 |
| 8 | 5 | 1.5 | H2O | 10 | 0d |
| 9 | 5 | 1.5 | CH2Cl2 | 10 | 0d |
| 10 | 5 | 1.5 | Toluene | 10 | 0d |
| 11 | 5 | 1.5 | DMPUe | 10 | 0d |

The optimized conditions found for the one-pot synthesis of oxaziridine 3a (Table 1, entry 3) were chosen as model conditions for our continuing studies. The substrate scope was then investigated, and a variety of N-alkyloxaziridines were prepared from the corresponding aldehydes and alkylamines, using the 30% H2O2/DMC system (Scheme 1). The condensation of alkylamine 1 with aldehyde 2 provided the imine intermediate, which underwent epoxidation by the oxidizing agent to afford oxaziridine 3. In general, the synthesis of an imine requires removal of water by use of dehydrating agents, such as molecular sieves,37,38 to shift the equilibrium in favor of the imine formation. In our case, the total conversion of amine and aldehyde into the imine is assured by the consumption of the latter through its oxidation. 1H NMR analysis showed total conversion of alkylamines 1 and aldehydes 2 into oxaziridines 3. Pure products were obtained in excellent yields after washing with a saturated solution of sodium sulfite and extraction with ethyl acetate (Table 2). Consequently, additional purification methods such as column chromatography or recrystallization were not needed. Noteworthy, the present oxidation is scalable as the reaction with 1 g of benzaldehyde (10 mmol) with 15 mmol of t-butylamine in 10 mL of DMC, in the presence of 5 mL of 30% H2O2, reached completion after 18 h and afforded oxaziridine 3a in 97% yield.
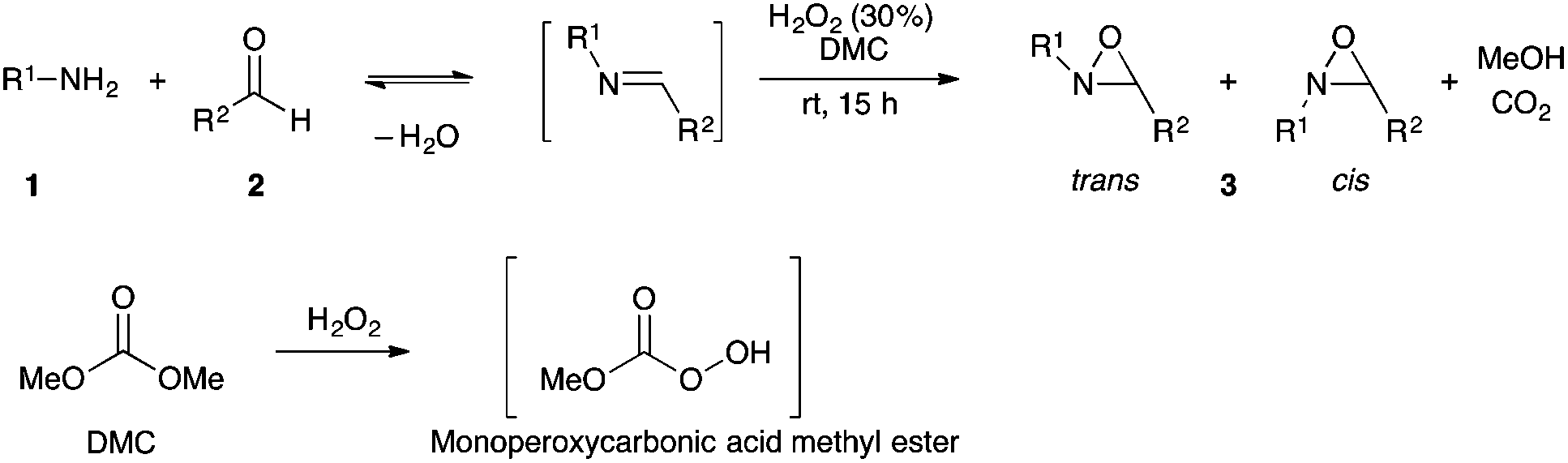 | ||
| Scheme 1 One-pot synthesis of N-alkyloxaziridines. | ||

We found that excess of N-alkylamines and H2O2 is necessary to ensure complete consumption of the starting aldehydes in a reasonable time. The reaction time is attractive if we consider that the one-pot route to N-alkyloxaziridines allows avoiding prior imine synthesis and related work-up. As described in the literature, N-alkylimines were prepared in 18 h at room temperature using CH2Cl2 as a solvent, activated molecular sieves and 2 equiv. of alkylamines with regard to aldehydes,37 and in 48 h by heating an equimolecular mixture of amine and aldehyde in dry toluene, in the presence of activated molecular sieves.38
It was demonstrated that commercial aqueous H2O2 solution in moderate concentration (30%) can be used successfully instead of more concentrated solutions.30 Moreover, this oxidation does not require a catalyst to take place. No significant amount of by-products, resulting from the oxidation of aldehydes and amines with the DMC/H2O2 system, were formed in this reaction. Indeed, the oxidation of aldehydes with hydrogen peroxide requires the presence of selenium oxychloride as catalyst.39 As for alkylamines, they can be easily oxidized by peracids40 or by the H2O2/Na2WO4 system.27b,41 Consequently, the one-pot route from aldehydes and amines to N-alkyloxaziridines probably cannot be achieved by the use of peracids or H2O2/Na2WO4. Thus, the present work represents the first example of a one-pot route to N-alkyloxaziridines from N-alkylamines and aldehydes.42
In this work, N-t-butyloxaziridines were formed exclusively in the trans-isomer form (Table 2). The less stable cis-isomer is less favoured because of the bulkiness of the t-butyl group. Oxaziridines with smaller N-substituents were produced as mixtures of trans and cis-isomers though. These stereoselectivity results are in agreement with the other methods described in the literature.17,22,43,44 The one-pot synthesis of N-alkyloxaziridines, described in this work, provided a high stereoselectivity, affording the trans-isomer as a major product (100:0–90:10, Table 2). The cis/trans ratio was determined by 1H NMR analysis. Indeed, cis and trans-oxaziridines isomers are known to give different 1H and 13C NMR spectra.17,22,43,44 NMR data of cis and trans-oxaziridines isomers is provided in ESI.†
Encouraged by the excellent results obtained using the H2O2/DMC system for the synthesis of N-alkyloxaziridines, we investigated the preparation of other synthetically important oxaziridines, such as N-sulfonyloxaziridines. Our initial attempts to perform the oxidation of N-sulfonylimines into N-sulfonyloxaziridines under the same conditions previously described (H2O2/DMC system, neutral conditions and without catalysis) were unsuccessful (no conversion of the N-sulfonylimine was observed). Therefore, a systematic study was undertaken to define the best reaction conditions for the synthesis of these oxaziridines (Table 3). We investigated the influence of basic medium and that of catalysis with Zn(OAc)2·2H2O and TBAB (tetra-n-butyl ammonium bromide) on the model reaction, based on the following precedents: (i) the epoxidation of N-sulfonylimines with MCPBA,18a,19 CCl3CN/H2O222 and Oxone®24 requires basic medium; (ii) the epoxidation of N-sulfonylimines with CCl3CN/H2O2 requires the use of TBAB as a phase transfer catalyst;22 (iii) Zn(OAc)2 is a non-toxic compound (food additive E650) that showed high catalytic activity in the methoxycarbonylation of aromatic diamines with DMC.45
|
|
|||||
|---|---|---|---|---|---|
| Entry | Equiv. H2O2 | Catalyst | Base | Time (h) | Conversion 5ab (%) |
| a Conditions: 0.5 mmol of 4a, catalyst (5 mol%), base (1.2 equiv.), H2O2 (30%), DMC (1.5 mL), rt. b Determined by 1H NMR analysis of crude product. c The oxaziridine was not detected by TLC. d Reaction carried out in MeOH or in CH2Cl2 instead of DMC. e 0.5 equiv. of Na2CO3 was used instead of 1.2 equiv. | |||||
| 1 | 5 | — | — | 10 | 0c |
| 2 | 10 | — | Na2CO3 | 24 | 62 |
| 3 | 5 | Zn(OAc)2·2H2O | — | 10 | 0c |
| 4 | 5 | TBAB | Na2CO3 | 24 | 23 |
| 5 | 5 | Zn(OAc)2·2H2O | Na2CO3 | 24 | 86 |
| 6d | 5 | Zn(OAc)2·2H2O | Na2CO3 | 10 | 0c |
| 7e | 10 | Zn(OAc)2·2H2O | Na2CO3 | 24 | 86 |
| 8 | 5 | Zn(OAc)2·2H2O | AcONa | 10 | 0c |
| 9 | 5 | Zn(OAc)2·2H2O | NaOH | 24 | 66 |
| 10 | 5 | Zn(OAc)2·2H2O | Imidazole | 10 | 0c |
| 11 | 10 | Zn(OAc)2·2H2O | Na2CO3 | 14 | 99 |
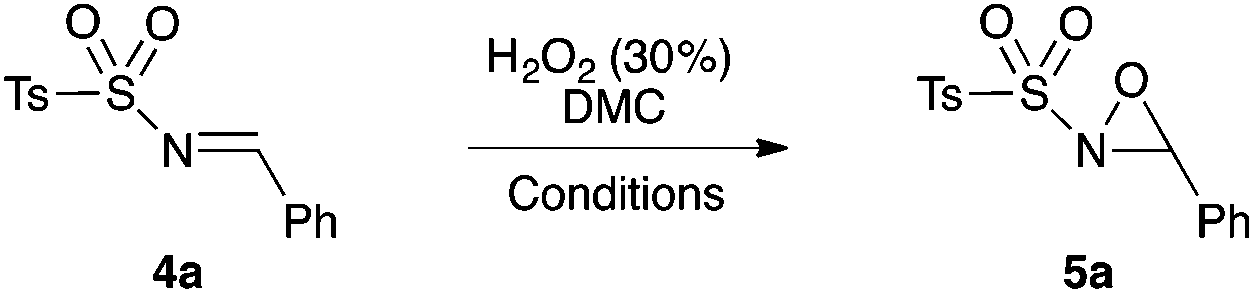
Optimization studies led us to conclude that the basic medium is essential for the epoxidation of N-sulfonylimine 4a (Table 3, entries 1 and 2), but it is not sufficient for completion of the reaction in less than 24 h. The reaction did not work when using bases that are soluble in DMC, such as sodium acetate and imidazole (Table 3, entries 8 and 10). To improve the efficiency of this reaction, we tried two catalysts, i.e. TBAB as a phase transfer catalyst and Zn(OAc)2·2H2O as an activating reactant of DMC. The use 5 mol% of TBAB did not lead to any improvement of the reaction (Table 3, entry 4), whereas 5 mol% of Zn(OAc)2·2H2O would work best for this reaction (Table 3, entries 5 and 11). Thereafter, the reaction reached completion in 14 h when 10 equiv. of H2O2 were used (Table 3, entry 11).
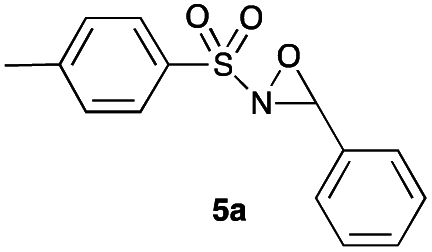
Hence, the optimized conditions found for the synthesis of N-sulfonyloxaziridines starting from N-sulfonylimines (1.2 equiv. of Na2CO3, 5 mol% of Zn(OAc)2·2H2O, 10 equiv. of 30% H2O2, DMC, rt) were chosen as model conditions for our continuing studies. The substrate scope was then investigated. A variety of N-sulfonylimines 4a–k were tested under the optimized conditions, and representative results are summarized in Table 4. N-Sulfonyloxaziridines 5a–k were obtained in high yields after washing with sodium sulfite, extraction with ethyl acetate and filtration through a short pad of silica gel. There was not much difference in reaction time and yields with various functional groups on the benzene ring of the imine system. In all cases, the reaction produced only one N-sulfonyloxaziridine isomer, namely, the thermodynamically favored trans-oxaziridine, as for the existing methods.18a,19,22
|
|
|||
|---|---|---|---|
| Entry | Time (h) | Oxaziridine 5 | Yield 5 (%) |
| a Reaction conditions: N-sulfonylimine (0.5 mmol),46 Zn(OAc)2·2H2O (5 mol%), Na2CO3 (1.2 equiv.), H2O2 (30%, 10 equiv.), DMC (1.5 mL), rt. b Reaction carried out in 3 mL of DMC instead of 1.5 mL to solubilize the imine. | |||
| 1 | 14 |
|
91 |
| 2 | 13 |
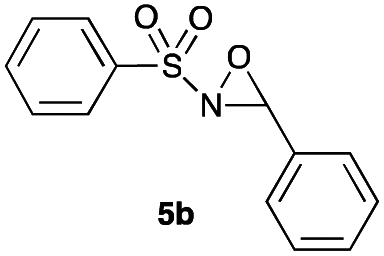
|
86 |
| 3 | 16 |
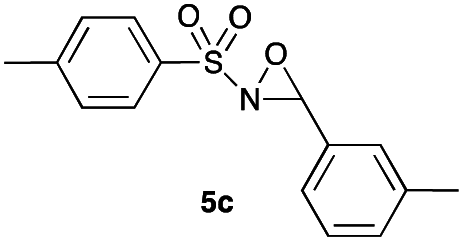
|
88 |
| 4 | 14 |

|
94 |
| 5b | 17 |
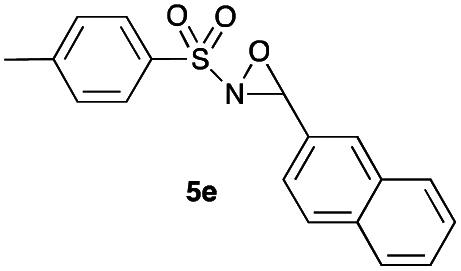
|
87 |
| 6 | 18 |
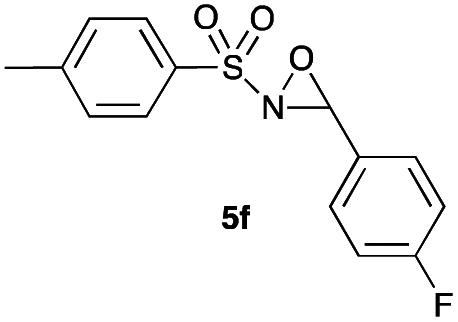
|
89 |
| 7b | 15 |
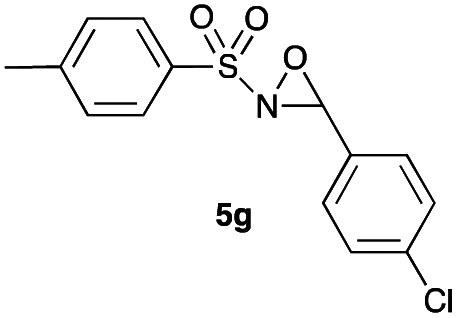
|
88 |
| 8b | 18 |
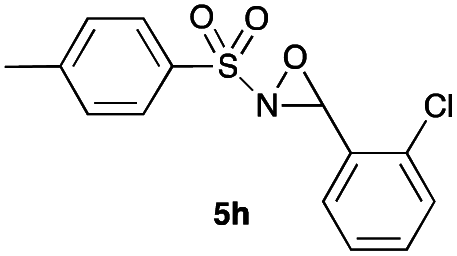
|
90 |
| 9b | 15 |
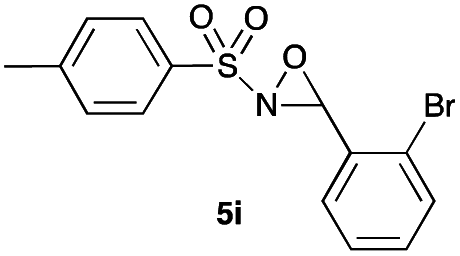
|
85 |
| 10 | 14 |

|
83 |
| 11 | 14 |
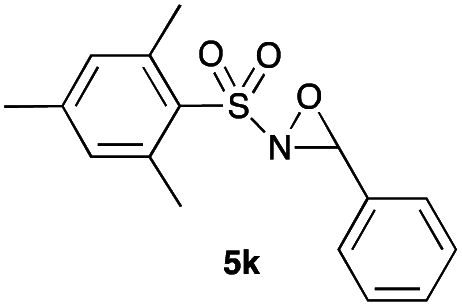
|
93 |

In order to demonstrate the applicability of the present method, a gram scale oxidation of imine 4f was carried out, using 2 g of 4f (7.2 mmol), 10 mL of DMC, 0.9 g of Na2CO3, 7 mL of H2O2 (30%) and 80 mg of Zn(OAc)2·2H2O (5 mol%). The reaction reached completion in 12 h and afforded oxaziridine 5f in 92% yield.
As for epoxidation of imines with peracids,18a we postulated that the reaction proceeds in two steps according to a Baeyer–Villiger type mechanism, rather than the concerted oxygen transfer (epoxidation of olefins). A plausible mechanism illustration is proposed in Fig. 1. The addition of H2O2 to DMC, in the presence of Na2CO3, would generate the methoxy carbonyl peroxydate 6 as an active oxidant, which would react with the electrophilic N-sulfonylimine to give intermediate 7. The latter undergoes intramolecular cyclisation by attack of the sulfonamide anion on the electrophilic oxygen of the methoxy carbonyl peroxide moiety. The role of Na2CO3 may be explained by the generation of the sulfonylamide anion instead of the N–H sulfonamide in 7, which would not be nucleophilic enough because of the adjacent electron-withdrawing sulfonyl group. On the other hand, the role of Zn(OAc)2·2H2O may be explained by its ability to activate the carbonyl group of the DMC and intermediates 6 and 7 to increase the rate of the oxidation.
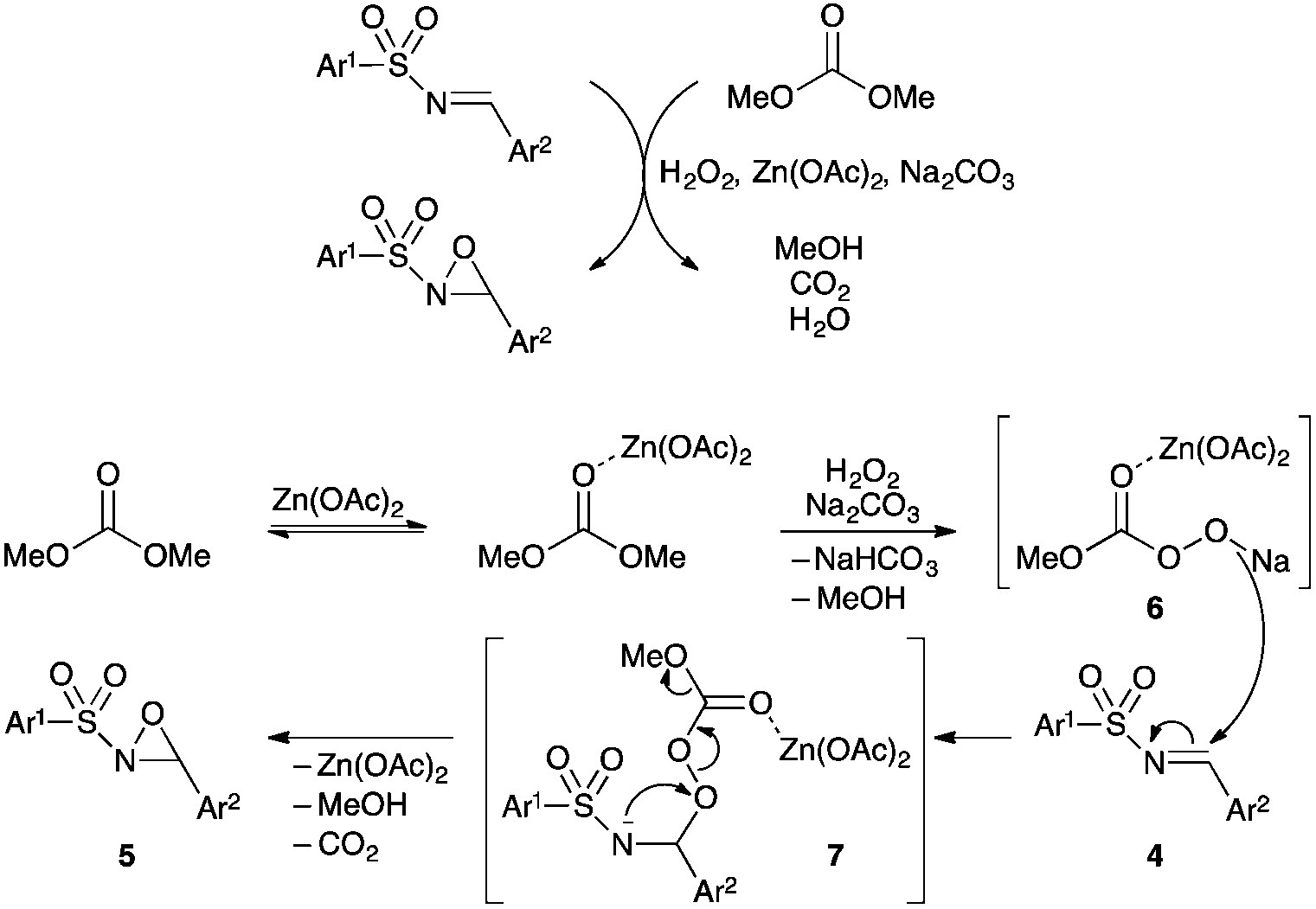 | ||
| Fig. 1 Postulated reaction mechanism for the Baeyer–Villiger-type oxidation of N-sulfonylimines in basic medium under Zn(OAc)2·2H2O catalysis. | ||
The influence of the pH of the reaction media was studied in the synthesis of N-alkyloxaziridines, in order to get more insight into the mechanism of the oxidation of the alkylimine. The oxidation of isolated N-alkylimines was investigated under different pH conditions (Table 5). When the pH was adjusted to 8.5 by adding 1.2 equiv. of Na2CO3, or to 7.0 by adding 0.5 equiv. of t-BuNH2, the obtained results were quite similar to those observed without addition of a base, in both reaction time and conversion (Table 5, entries 1–3). A similar trend was reported for the epoxidation of olefins with the O-ethylperoxycarbonic acid in acidic and basic media.31
|
|
|||||
|---|---|---|---|---|---|
| Entry | R | Additive | Time (h) | Conversion (%) 3b | By-productb (%) |
| a Conditions: imine (1 mmol), H2O2 (30%, 5 mmol), DMC (1 mL), additive, rt. b Determined by 1H NMR analysis of crude product. | |||||
| 1 | t-Bu | — | 12 | 96 | PhCHO (4%) |
| 2 | t-Bu | Na2CO3 (1.2 equiv.) | 12 | >99 | PhCHO (<1%) |
| 3 | t-Bu | t-Bu-NH2 (0.5 equiv.) | 10 | 100 | — |
| 4 | n-Pr | t-Bu-NH2 (1 equiv.) | 10 | 64 (3l) | 3a (33%), PhCHO (3%) |
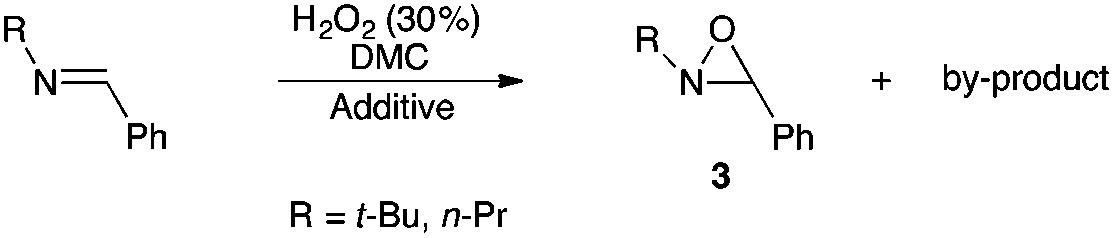
Based on the previous control experiments, we assume that the basic media is not crucial for the one-pot synthesis of N-alkyloxaziridines because the N–H group in the intermediate 8 may be sufficiently nucleophilic for the cyclisation step (Fig. 2). The excess of N-alkylamine used in the optimized conditions (1.5 equiv.) is believed to be necessary to ensure the complete consumption of the aldehyde by shifting the equilibrium towards the imine formation and ensure, accordingly, the total conversion of the imine into the corresponding oxaziridine. This hypothesis is in agreement with the following results: (i) in the presence of 1 equiv. of t-BuNH2, the conversion of benzaldehyde into the oxaziridine was not complete after 24 h of reaction (Table 1, entry 1), (ii) the oxidation of N-propylimine, in the presence of t-BuNH2 (1 equiv.), furnished a mixture of N-propyloxaziridine 3l (64%) and N-t-butyloxaziridine 3a (33%) (Table 5, entry 4), demonstrating the equilibrium taking place in the reaction conditions (Fig. 2).
 | ||
| Fig. 2 Proposed reaction mechanism for the one-pot Baeyer–Villiger-type oxidation of N-alkylimines in neutral medium without catalyst. | ||
For the first time, an eco-friendly one-pot synthesis of N-alkyloxaziridines from the corresponding N-alkylamines and aldehydes has been developed. In this approach, only green DMC is used as solvent and reagent in the presence of an aqueous solution of hydrogen peroxide (30%). Monoperoxy carbonic acid methyl ester, generated in situ by perhydrolysis of DMC, is the active oxidant, which decomposes, after epoxidation of the imine intermediate, to carbon dioxide and methanol. The method has been expanded to the preparation of N-sulfonyloxaziridines from the corresponding N-sulfonylimines. In this case, the reaction was carried out in basic medium under catalysis with 5 mol% of Zn(OAc)2·2H2O. All oxaziridines were obtained in high yields and high purity under simple and minimum manipulation. This procedure appears to be better than the existing methods on cost, ease of manipulation and greenness, and will provide a novel approach to an environmentally benign process for the preparation of oxaziridines.
Acknowledgements
This work was financially supported by the Natural Sciences and Engineering Research Council of Canada (NSERC), the Centre in Green Chemistry and Catalysis (CGCC) and the Ministère de l'enseignement supérieur et de la recherche scientifique tunisien. We thank Marie Gonay for her assistance in performing control experiments.Notes and references
- W. D. Emmons, J. Am. Chem. Soc., 1957, 79, 5739 CrossRef CAS.
- D. A. Evans, M. M. Morrissey and R. L. Dorow, J. Am. Chem. Soc., 1985, 107, 4346 CrossRef CAS.
- D. Prieur, A. El Kazzi, T. Kato, H. Gornitzka and A. Baceiredo, Org. Lett., 2008, 10, 2291 CrossRef CAS PubMed.
- F. A. Davis, S. G. Lal and H. Dupont Durst, J. Org. Chem., 1988, 53, 5004 CrossRef CAS.
- M. Komatsu, Y. Ohshiro, H. Hotta, M. Sato and T. Agawa, J. Org. Chem., 1974, 39, 948 CrossRef CAS.
- J. Kraïem, L. Grosvalet, M. Perrin and B. Ben Hassine, Tetrahedron Lett., 2001, 42, 9131 CrossRef.
- M. Fabio, L. Ronzini and L. Troisi, Tetrahedron, 2007, 63, 12896 CrossRef CAS.
- M. Fabio, L. Ronzini and L. Troisi, Tetrahedron, 2008, 64, 4979 CrossRef CAS.
- L. Troisi, L. Ronzini, F. Rosato and V. Videtta, Synlett, 2009, 1806 CrossRef CAS.
- A. Kivrak and R. C. Larock, J. Org. Chem., 2010, 75, 7381 CrossRef CAS PubMed.
- C. H. Leung, A. M. Voutchkova, R. H. Crabtree, D. Balcells and O. Eisenstein, Green Chem., 2007, 9, 976 RSC.
- F. A. Davis, P. A. Mancinelli, K. Balasubramanian and U. K. Nadir, J. Am. Chem. Soc., 1979, 101, 1044 CrossRef CAS.
- M. L. Di Gioia, A. Leggio, A. Le Pera, A. Liguori and C. Siciliano, J. Org. Chem., 2005, 70, 10494 CrossRef CAS PubMed.
- D. J. Michaelis, K. S. Williamson and T. P. Yoon, Tetrahedron, 2009, 65, 5118 CrossRef CAS PubMed.
- K. S. Williamson and T. P. Yoon, J. Am. Chem. Soc., 2012, 134, 12370 CrossRef CAS PubMed.
- P.-L. Shao, X.-Y. Chen and S. Ye, Angew. Chem., Int. Ed., 2010, 49, 8412 CrossRef CAS PubMed.
- T. B. Bitencourt and M. da Graça Nascimento, Green Chem., 2009, 11, 209 RSC.
- (a) F. A. Davis, J. Lamendola Jr., U. Nadir, E. W. Kluger, T. C. Sedergran, T. W. Panunto, R. Billmers, R. Jenkins Jr., I. J. Turchi, W. H. Watson, J. S. Chen and M. Kimura, J. Am. Chem. Soc., 1980, 102, 2000 CrossRef CAS; (b) F. A. Davis and O. D. Stringer, J. Org. Chem., 1982, 7, 1774 CrossRef.
- (a) Y. Usuki, Y. Wang and J. Aubé, J. Org. Chem., 1995, 60, 8028 CrossRef CAS; (b) J. L. García Ruano, J. Alemá, C. Fajardo and A. Parra, Org. Lett., 2005, 7, 5493 CrossRef PubMed.
- L. Martiny and K. A. Jørgensen, J. Chem. Soc., Perkin Trans. 1, 1995, 699 RSC.
- J. Kraïem, Y. Kacem, J. Khiari and B. Ben Hassine, Synth. Commun., 2001, 31, 263 CrossRef.
- J. Kraïem, R. Othman and B. Ben Hassine, C. R. Chim., 2004, 7, 1119 CrossRef.
- J. A. Damvandi, B. Karami and M. A. Zolfigol, Synlett, 2002, 933 CrossRef.
- F. A. Davis, S. Chattopadhyay, J. C. Towson, S. Lal and T. Reddy, J. Org. Chem., 1988, 53, 2087 CrossRef CAS.
- (a) W. Navarrini and D. D. DesMarteau, Process for the preparation of perfluoro-amino oxaziridines, United States Patent, US5017709-A, 1991 Search PubMed; (b) V. A. Petrov and D. D. DesMarteau, J. Fluorine Chem., 1996, 77, 175 CrossRef CAS.
- D. Prat, J. Hayler and A. Wells, Green Chem., 2014, 16, 4546 RSC.
- (a) Green Catalysis, Homogeneous Catalysis, in Handbook of Green Chemistry, ed. P. T. Anastas and R. H. Crabtree, Wiley-VCH, 2013, vol. 1 Search PubMed; (b) R. Noyori, M. Aoki and K. Sato, Chem. Commun., 2003, 1977 RSC.
- (a) F. Aricò and P. Tundo, Russ. Chem. Rev., 2010, 79, 479 CrossRef; (b) P. Tundo and M. Selva, Acc. Chem. Res., 2002, 35, 706 CrossRef CAS PubMed; (c) P. Tundo, Pure Appl. Chem., 2001, 73, 1117 CrossRef CAS.
- M. M. Mauri, U. Romano and F. Rivetti, Ing. Chim. Ital., 1985, 21, 1 Search PubMed.
- M. Rüsch gen. Klaas and S. Warwel, Org. Lett., 1999, 1, 1025 CrossRef.
- R. D. Bach, M. W. Klein, R. A. Ryntz and J. W. Holubka, J. Org. Chem., 1979, 44, 2569 CrossRef CAS.
- Y. Tsunokawa, S. Iwasaki and S. Okuda, Chem. Pharm. Bull., 1983, 31, 4578 CrossRef CAS.
- Y. Tsunokawa, S. Iwasaki and S. Okuda, Tetrahedron Lett., 1982, 23, 2113 CrossRef CAS.
- R. M. Coates and J. W. Williams, J. Org. Chem., 1974, 39, 3054 CrossRef CAS.
- G. B. Payne and P. H. Williams, J. Org. Chem., 1961, 26, 651 CrossRef CAS.
- E. G. Ankudey, H. F. Olivo and T. L. Peeples, Green Chem., 2006, 8, 923 RSC.
- (a) M. Blümel, P. Chauhan, R. Hahn, G. Raabe and D. Enders, Org. Lett., 2014, 16, 6012 CrossRef PubMed; (b) I. Hachiya, Y. Minami, T. Aramaki and M. Shimizu, Eur. J. Org. Chem., 2008, 1411 CrossRef CAS.
- C. P. Casey and J. B. Johnson, J. Am. Chem. Soc., 2005, 127, 1883 CrossRef CAS PubMed.
- J. B. Firth and H. H. Gething, J. Chem. Soc., 1936, 633 RSC.
- E. J. Corey and A. W. Gross, Tetrahedron Lett., 1984, 25, 491 CrossRef CAS.
- D. R. Boyd, D. C. Neill, C. G. Watson and W. B. Jennings, J. Chem. Soc., Perkin Trans. 2, 1975, 1813 RSC.
- The oxaziridines derived from anilines are known to be unstable and lead to quick decomposition: (a) K. Shinzawa and I. Tanaka, J. Phys. Chem., 1964, 68, 1205 CrossRef CAS; (b) J. S. Splitter and M. Calvin, J. Org. Chem., 1965, 30, 3427 CrossRef CAS; (c) H. Ono, J. S. Splitter and M. Calvin, Tetrahedron Lett., 1973, 4107 CrossRef CAS.
- D. Mohajer, N. Iranpoor and A. Rezaeifard, Tetrahedron Lett., 2004, 45, 631 CrossRef CAS.
- M. Čudić and R. Hermann, Magn. Reson. Chem., 1993, 31, 461 CrossRef.
- (a) T. Baba, A. Kobayashi, T. Yamauchi, H. Tanaka, S. Aso, M. Inomata and Y. Kawanami, Catal. Lett., 2002, 82, 193 CrossRef CAS; (b) T. Baba, A. Kobayashi, Y. Kawanami, K. Inazu, A. Ishikawa, T. Echizenn, K. Murai, S. Aso and M. Inomata, Green Chem., 2005, 7, 159 RSC.
- S. Morales, F. G. Guijarro, J. L. García Ruano and M. B. Cid, J. Am. Chem. Soc., 2014, 136, 1082 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and copies of 1H NMR and 13C NMR. See DOI: 10.1039/c6gc01394e |
| This journal is © The Royal Society of Chemistry 2016 |