
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Heterogeneous palladium-catalysed Catellani reaction in biomass-derived γ-valerolactone†
Dace
Rasina
a,
Arianna
Kahler-Quesada
a,
Simone
Ziarelli
a,
Svenja
Warratz
b,
Hui
Cao
b,
Stefano
Santoro
a,
Lutz
Ackermann
*b and
Luigi
Vaccaro
 *a
*a
aLaboratory of Green Synthetic Organic Chemistry, CEMIN – Dipartimento di Chimica, Biologia e Biotecnologie, Università di Perugia Via Elce di Sotto, 8, 06123 Perugia, Italy. E-mail: luigi.vaccaro@unipg.it
bInstitut für Organische und Biomolekulare Chemie, Georg-August-Universität Göttingen, Tammannstrasse 2, 37077 Göttingen, Germany. E-mail: lutz.ackermann@chemie.uni-goettingen.de
First published on 9th June 2016
Abstract
Herein, we report the unprecedented use of a heterogeneous palladium catalyst for the step-economical Catellani reaction. The substrate scope with encapsulated Pd(OAc)2 (Pd EnCat™ 30) or Pd/Al2O3 proved to be broad, while the renewable biomass-derived γ-valerolactone (GVL) was identified as an effective reaction medium. Mechanistic studies highlighted the possible heterogeneous nature of the Pd/Al2O3 catalyst, while showing that the reaction performed in the presence of Pd EnCat™ 30 is most likely catalysed by leached homogeneous palladium species. The heterogeneous Pd/Al2O3 catalyst can be easily recovered at the end of the reaction and efficiently reused in consecutive reaction runs.
Introduction
In recent years, the direct C–H functionalization of arenes has emerged as a particularly attractive alternative to classical cross-coupling approaches, allowing for late-stage diversifications without the need of prefunctionalization steps.1 Typically, directing groups are required for achieving site-selectivity in intermolecular transformations, mostly occurring at the ortho-position. The Catellani reaction represents an alternative strategy in which the palladium-catalysed C–H ortho-functionalization of aryl iodides is mediated by norbornene, which allows for subsequent ipso-substitution. In the original report by Catellani and coworkers the dialkylation of the aryl iodide in the two ortho-positions was followed by an ipso-vinylation through a Mizoroki–Heck-type process.2 Several modifications to this reaction have subsequently been devised, allowing for different functionalizations to occur both on the ortho- as well as on the ipso-position. The most synthetically useful approaches towards the ipso-functionalization, apart from the Mizoroki–Heck-type alkenylation, include arylations through a Suzuki–Miyaura-type reaction, the alkynylation through a Sonogashira-type process and the hydrogenolysis of the aryl-palladium intermediate.3Thus far, the vast majority of C–H functionalization reactions have relied on homogeneous transition metal catalysis. Only recently the attention has shifted to the use of versatile heterogeneous catalysts for C–H activation chemistry.4 However, to the best of our knowledge, examples of heterogeneous metal-catalysed Catellani reactions have as of yet proven elusive. It is particularly noteworthy that the use of heterogeneous catalysts offers great advantages as compared to homogeneous catalysts, especially in terms of recoverability of the catalysts, ease of purification and minimization of metal contamination.
Thus far, the Catellani reaction has largely been conducted in polar aprotic solvents, such as DMF, DMA or acetonitrile. These solvents, particularly DMF and DMA, present major issues related to their toxicity, and their substitution is highly desirable, especially for large-scale applications.5 Acetonitrile is typically considered the “greenest” option among polar aprotic solvents.6 However, this actually represents a rather poor alternative due to its volatility and toxicity.7
γ-Valerolactone (GVL), is a renewable biomass-derived chemical featuring high polarity, high boiling point, and good stability towards bases and acids. Only few applications employing GVL as reaction media have been disclosed.8 Recently, we have reported for the first time that GVL can be used as an environmentally-benign alternative to common toxic polar aprotic solvents.8i–k In some cases we have also observed that the use of GVL in combination with a heterogeneous catalyst lead to lower product contamination by the metal, compared to the same reaction performed in common polar aprotic solvents.8i–k This is particularly relevant since metal contamination represent a major issue in the synthesis of pharmaceutical compounds.9,10
In this article we report the first example of a Catellani reaction catalysed by a heterogeneous palladium species. The reaction is performed in a sustainable reaction media (GVL) and the optimal catalyst can be easily recovered and reused for consecutive reaction runs. Experimental mechanistic investigations were also conducted in order to clarify the nature of the catalytically active species.
Results and discussion
At the outset of our studies, we tested representative heterogeneous palladium catalysts in the Catellani reaction with methyl 2-iodobenzoate (1a) and methyl acrylate (2a), using K2CO3 as the base and GVL as the medium (Table 1).11 Commercially available encapsulated Pd(OAc)2,12 either with or without PPh3 (Pd EnCat™ 30 and Pd EnCat™ TPP30, respectively) performed with high efficacy, affording the expected product 3a in more than 90% yield (Table 1, entries 1 and 2). The simple Pd/C catalyst gave comparable results, with an isolated yield of 94% (Table 1, entry 3). The addition of 2.5 equivalents of PPh3, either soluble or immobilized on a polystyrene support, led to a slight decrease in catalytic activity, with the product being obtained in 82–87% yield (Table 1, entries 4 and 5). Also commercially available Pd/Al2O3 gave excellent results, providing the expected product in 95% yield (Table 1, entry 6). DMF, NMP, and MeCN were also tested as representative aprotic reaction media (Table 1, entries 7–9). While in the case of DMF satisfactory results were obtained, the use of NMP and MeCN led to poor conversion of the substrate 1a.|
|
|||
|---|---|---|---|
| Entry | Palladium catalyst | Reaction medium | Yieldb (%) |
| a Reaction conditions: 1a (0.330 mmol), 2a (0.195 mmol), norbornene (0.127 mmol), K2CO3 (0.340 mmol), [Pd] (5 mol%), reaction medium (3.75 mL). b Isolated yields. c PPh3 (15 mol%). | |||
| 1 | Pd EnCat™ 30 | GVL | 93 |
| 2 | Pd EnCat™ TPP30 | GVL | 92 |
| 3 | Pd/C | GVL | 94 |
| 4 | Pd/C, TPPc | GVL | 82 |
| 5 | Pd/C, PS-TPPc | GVL | 87 |
| 6 | Pd/Al2O3 | GVL | 95 |
| 7 | Pd/Al2O3 | DMF | 82 |
| 8 | Pd/Al2O3 | NMP | 58 |
| 9 | Pd/Al2O3 | MeCN | 20 |

The effect of different bases – both organic and inorganic – on the reaction was also investigated, with K2CO3 giving the best results (see ESI†).
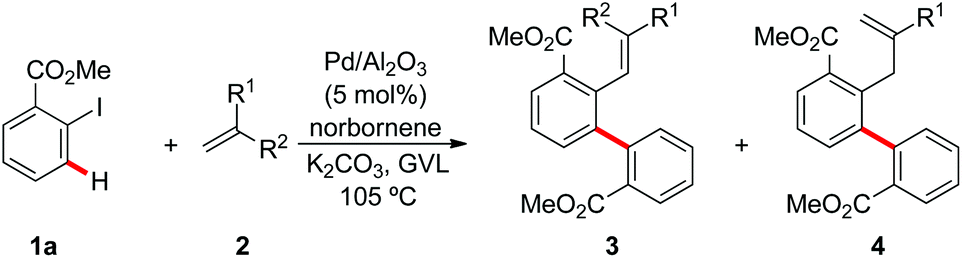
Next, we explored the scope of the reaction between methyl 2-iodobenzoate (1a) and several olefins 2. We chose to conduct all reactions with Pd EnCat™ 30 as well as Pd/Al2O3, in order to investigate possible differences in reactivity between these two rather diverse heterogeneous catalysts. Generally, both catalysts performed well, providing the expected products in moderate to very good yields. In several cases Pd EnCat™ 30 gave slightly better results in terms of yields than Pd/Al2O3. α,β-Unsaturated esters and ketones (2a–c) were efficiently converted to the product by either of the two catalytic systems in yields ranging from 60% to 95% (Tables 2 and 3, entries 1–3). Acrylonitrile (2d) furnished good results in the reaction catalysed by Pd EnCat™ 30 (Table 2, entry 4), while much lower yield was obtained using Pd/Al2O3, which also needed longer reaction times (Table 3, entry 4).
|
|
|||||
|---|---|---|---|---|---|
| Entry | 2 | R1 | R2 | Time | Yieldb (%) |
a Reaction conditions: 1a (0.330 mmol), 2a (0.195 mmol), norbornene (0.127 mmol), K2CO3 (0.340 mmol), [Pd] (5 mol%,), GVL (3.75 mL).
b Isolated yields.
c Product deriving from the ether hydrolysis and subsequent keto/enol tautomerization.
d 1.6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of the acetylated product and the phenol from ester hydrolysis, see ESI for details. :1 mixture of the acetylated product and the phenol from ester hydrolysis, see ESI for details.
|
|||||
| 1 | 2a | CO2Me | H | 24 h | 93 (3a) |
| 2 | 2b | CO2nBu | H | 24 h | 73 (3b) |
| 3 | 2c | COMe | H | 24 h | 77 (3c) |
| 4 | 2d | CN | H | 24 h | 68 (3d) |
| 5 | 2e | OnBu | H | — | — |
| 6 | 2f | OEt | H | 24 h | 48c (3f) |
| 7 | 2g | Ph | H | 24 h | 63 (3g) |
| 8 | 2h | p-AcO-C6H4 | H | 24 h | 79d (3h + 3′h) |
| 9 | 2i | p-Cl-C6H4 | H | 24 h | 68 (3i) |
| 10 | 2j | CO2Me | Me | 2 d | 50 (3j:4j 1:1.9) |
| 11 | 2k | CHO | Me | 24 h | 68 (3k:4k 1:1.6) |

|
|
|||||
|---|---|---|---|---|---|
| Entry | 2 | R1 | R2 | Time | Yieldb (%) |
|
a Reaction conditions: 1a (0.330 mmol), 2a (0.195 mmol), norbornene (0.127 mmol), K2CO3 (0.340 mmol), [Pd] (5 mol%,), GVL (3.75 mL).
b Isolated yields.
c As a 1:4 mixture of the expected product and the phenol deriving from the ester hydrolysis.
|
|||||
| 1 | 2a | CO2Me | H | 24 h | 95 (3a) |
| 2 | 2b | CO2nBu | H | 3 d | 60 (3b) |
| 3 | 2c | COMe | H | 24 h | 68 (3c) |
| 4 | 2d | CN | H | 3 d | 32 (3d) |
| 5 | 2e | OnBu | H | 2 d | 40 (3e) |
| 6 | 2f | OEt | H | — | — |
| 7 | 2g | Ph | H | 24 h | 74 (3g) |
| 8 | 2h | p-AcO-C6H4 | H | 24 h | 54c (3h + 3′h) |
| 9 | 2i | p-Cl-C6H4 | H | 24 h | 86 (3i) |
| 10 | 2j | CO2Me | Me | 7 d | 41 (3j:4j 1:3.2) |
| 11 | 2k | CHO | Me | 3 d | 57 (3k:4k 1:1.5) |
Vinyl alkyl ethers (2e–f) proved to be more challenging, and the efficacy was strongly dependent on the nature of the catalyst (Tables 2 and 3, entries 5 and 6).
Moreover, the reaction conducted on vinyl ethyl ether (2f) with Pd EnCat™ 30 delivered the formyl-substituted product through a Domino Catellani reaction along with hydrolysis and keto/enol tautomerization. Styrene (2g) and para-substituted styrenes (2h–i) reacted efficiently with both catalysts (Tables 2 and 3, entries 7–9).
Interestingly, in both the reactions performed with the two catalysts, the chlorine functionality was well tolerated (Tables 2 and 3, entry 9), which should prove instrumental for further late-stage diversifications.
1,1-Disubstituted olefins were also subjected to the optimized reaction conditions with either Pd EnCat™ 30 or Pd/Al2O3 as the catalysts (Tables 2 and 3, entries 10 and 11). Their reactivity was in line with that observed for the monosubstituted olefins, with yields ranging from 41% to 68%. However, due to the presence of the methyl group on the substrate, the β-elimination step of the final Mizoroki–Heck-type reaction could occur in two positions, thus generating two isomeric products (3 and 4). As to the substitution pattern on the aryl iodides 1, the optimized palladium catalyst also enabled the synthesis of products 3l–3o (Scheme 1).
 | ||
| Scheme 1 Further examples of Pd/Al2O3 catalysed Catellani reactions. | ||
The coupling of the Catellani reaction with a terminal hydrogenolysis step provides step-economical access to meta-difunctionalized aromatic species.3,13 We tested the applicability of this strategy under the optimized reaction conditions with Pd/Al2O3 as the catalyst and benzyl alcohol as hydride donor (Scheme 2). The reaction proceeded efficiently, affording the expected coupling product 5 in high yield.
 | ||
| Scheme 2 Catellani reaction with terminal hydrogenolysis. | ||
A major issue associated with transition metal catalysis, and in particular with palladium chemistry, is the nature of the catalytic active species. Indeed, it frequently was noted that operationally heterogeneous reactions actually occur through the catalysis of soluble leached metal species or, vice versa, that operationally homogeneous reactions were catalysed by in situ formed insoluble metal particles.14 This problem is also of key relevance for C–H functionalization reactions.4 It is worth considering that the actual nature of the catalyst is not only important for mechanistic reasons, but also has practical implications, since it finally controls the possibility to reuse the recovered catalyst and the metal contamination of the products. Unfortunately, assessing the nature of the catalytic active species (homogeneous vs. heterogeneous) is not a simple task. Several experiments have been proposed but none of them is able to give conclusive evidence about the mechanism. Instead, it is necessary to rely on multiple experiments in order to possibly obtain sufficient support to one of the mechanistic scenarios.14
For these reasons, we decided to investigate the nature of the catalytically active species through various approaches, for both the reactions catalysed by Pd EnCat™ 30 and by Pd/Al2O3. First, we performed a hot-filtration test by removing the solid catalyst one hour after the beginning of the representative reaction between 1a and 2a, and allowing the filtrate to stir for 3 days at 105 °C. Interestingly, the two catalysts led to quite different results. Thus, while Pd/Al2O3 gave a very low conversion (7%), Pd EnCat™ 30 furnished 3a in 37% conversion. Second, for the same reaction we performed a mercury-poisoning test for both catalysts. Again, the two catalysts behaved differently. In fact, while the addition of Hg(0) completely hampered the catalytic activity of Pd/Al2O3 with only 4% conversion after 20 hours, the reaction with Pd EnCat™ 30 gave 46% conversion under otherwise identical reaction conditions (see ESI†). These results suggest that leached palladium species might play a key role in the reaction catalysed by Pd EnCat™ 30, but not in the reaction with Pd/Al2O3.15
Third, we measured the amount of palladium leached into the crude product by inductively coupled plasma optical emission spectrometry (ICP-OES), for both catalysts, before and after aqueous work-up (Table 4). The amount of palladium leached from Pd/Al2O3 was very low, even before aqueous work-up (∼2 ppm). However, the extent of metal leaching from Pd EnCat™ 30 is relatively high (52.4 and 10.5 ppm before and after aqueous work-up, respectively), and in line with previous findings15 and with our hot-filtration and Hg-poisoning tests.
| Catalyst | Palladium leaching (ppm) | |
|---|---|---|
| Pd EnCat™ 30 | no work-up | 52.4 |
| aq. work-up | 10.5 | |
| Pd/Al2O3 | no work-up | 2.2 |
| aq. work-up | 0.5 | |
Overall, these results suggest that the two tested catalysts exert their catalytic activity in different ways. While Pd EnCat™ 30 is proposed to catalyse the reaction by releasing homogeneous soluble palladium species, the C–H activation catalysed by Pd/Al2O3 is either genuinely heterogeneous or may proceed through a “release and catch” mechanism.16
From sustainability and chemical efficiency point of views, it is mostly important that the amount of palladium leached into the product is very low and that the solid catalyst can be efficiently reused. Therefore we tested the recyclability of Pd/Al2O3 in the reaction between 1a and 2a in GVL, by recovering the catalyst by simple filtration. The catalyst fully retained its activity for at least three reaction runs, constantly giving full conversion to the products. Thus, the reaction of substrates 1a and 2a was repeated for four consecutive runs giving the desired product 3a constantly in 93–95% yield (Fig. 1).
 | ||
| Fig. 1 Reuse of Pd/Al2O3 in the reaction of 1a and 2a. | ||
Conclusions
In conclusion, we have developed the first Catellani reaction catalysed by a solid palladium catalyst. The reaction proceeded efficiently on a variety of alkenes in the renewable biomass-derived medium GVL. The process could also be merged with a terminal hydrogenolysis, which gave step-economical access to valuable meta-difunctionalized17 arenes. The nature of the catalytically active species was investigated and provided support for the versatile Pd/Al2O3 catalyst to operate in a heterogeneous fashion. Importantly, the catalyst could be efficiently recovered and reused, featuring a very low metal leaching. Studies on the use of other economically attractive organic electrophiles are currently ongoing in our laboratories and will be reported in due course.Experimental
Typical procedure for the Catellani reaction
To a 4 mL vial containing the palladium catalyst (0.00825 mmol, 5 mol%), the base (0.340 mmol), and 2-norbornene (12 mg, 0.127 mmol) was added a GVL solution (3.75 mL) of the aryl iodide 1 (0.33 mmol) and the terminal olefin 2 (0.195 mmol). The resulting mixture was stirred at 105 °C.The solids were then separated from the solution by vacuum filtration and rinsed with cyclopentyl methyl ether. The organic layer was washed with water and dried over anhydrous Na2SO4. The products were isolated by flash column chromatography using a 4:1 mixture of petroleum ether and ethyl acetate as the eluent.
Procedure for catalyst recovery and recycling
After stirring at 105 °C for 24 h, the reaction mixture was centrifuged and the solvent was decanted. The catalyst was washed, first with water (0.2 mL) and GVL (3.0 mL), then with GVL (3.8 mL). New substrates (1a and 2a), reagents and solvent were added and left for 24 h at 105 °C for the next run. In four consecutive reaction runs Pd/Al2O3 consistently led to full conversion to the expected product in 24 h.Acknowledgements
Generous support by the European Research Council under the European Community's Seventh Framework Program (FP7 2007-2013)/ERC Grant agreement no. 307535 is acknowledged. We gratefully acknowledge the Università degli Studi di Perugia and the “Fondazione Cassa di Risparmio di Terni e Narni” for financial support. S. S. gratefully acknowledges the MIUR for financial support through the fellowship PGR123GHQY, funded within “Programma Giovani Ricercatori, Rita Levi Montalcini”. This work was also supported by the REU program of the National Science Foundation under award number DMR-1262908. D. R. was supported by EC 7th Framework Program project REGPOT-CT-2013-316149 Innovabalt.References
- Representative recent reviews on C–H activation: (a) B. Ye and N. Cramer, Acc. Chem. Res., 2015, 48, 1308–1318 CrossRef CAS PubMed; (b) K. Shin, H. Kim and S. Chang, Acc. Chem. Res., 2015, 48, 1040–1052 CrossRef CAS PubMed; (c) O. Daugulis, J. Roane and L. D. Tran, Acc. Chem. Res., 2015, 48, 1053–1064 CrossRef CAS PubMed; (d) Y. Segawa, T. Maekawa and K. Itami, Angew. Chem., Int. Ed., 2015, 54, 66–81 CrossRef CAS PubMed; (e) L. Ackermann, Org. Process Res. Dev., 2015, 18, 260–269 CrossRef; (f) M. Zhang, Y. Zhang, X. Jie, H. Zhao, G. Li and W. Su, Org. Chem. Front., 2014, 1, 843–895 RSC; (g) N. Kuhl, N. Schroeder and F. Glorius, Adv. Synth. Catal., 2014, 356, 1443–1460 CrossRef CAS; (h) T. Mesganaw and J. A. Ellman, Org. Process Res. Dev., 2014, 18, 1097–1104 CrossRef CAS PubMed; (i) S. A. Girard, T. Knauber and C.-J. Li, Angew. Chem., Int. Ed., 2014, 53, 74–100 CrossRef CAS PubMed; (j) G. Rouquet and N. Chatani, Angew. Chem., Int. Ed., 2013, 52, 11726–11743 CrossRef CAS PubMed; (k) S. R. Neufeldt and M. S. Sanford, Acc. Chem. Res., 2012, 45, 936–946 CrossRef CAS PubMed; (l) L. Ackermann, Chem. Rev., 2011, 111, 1315–1345 CrossRef CAS PubMed; (m) J. Wencel-Delord, T. Dröge, F. Liu and F. Glorius, Chem. Soc. Rev., 2011, 40, 4740–4761 RSC; (n) D. J. Schipper and K. Fagnou, Chem. Mater., 2011, 23, 1594–1600 CrossRef CAS; (o) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215–1292 CrossRef CAS PubMed; (p) T. Satoh and M. Miura, Chem. – Eur. J., 2010, 16, 11212–11222 CrossRef CAS PubMed; (q) O. Daugulis, Top. Curr. Chem., 2010, 292, 57–84 CrossRef CAS PubMed; (r) R. Giri, B.-F. Shi, K. M. Engle, N. Maugel and J.-Q. Yu, Chem. Soc. Rev., 2009, 38, 3242–3272 RSC; (s) L. Ackermann, R. Vicente and A. Kapdi, Angew. Chem., Int. Ed., 2009, 48, 9792–9826 CrossRef CAS PubMed , and references cited therein.
- M. Catellani, F. Frignani and A. Rangoni, Angew. Chem., Int. Ed. Engl., 1997, 36, 119–122 CrossRef CAS.
- (a) J. Ye and M. Lautens, Nat. Chem., 2015, 7, 863–870 CrossRef CAS PubMed; (b) A. Martins, B. Mariampillai and M. Lautens, Top. Curr. Chem., 2010, 292, 1–33 CrossRef CAS PubMed; (c) M. Catellani, E. Motti and N. Della Ca’, Acc. Chem. Res., 2008, 41, 1512–1522 CrossRef CAS PubMed; (d) M. Catellani, Top. Organomet. Chem., 2005, 14, 21–53 CAS. For recent examples of Catellani–type reactions, see: (e) X.-X. Wu, Y. Shen, W.-L. Chen, S. Chen, P.-F. Xu and Y.-M. Liang, Chem. Commun., 2015, 51, 16798–16801 RSC; (f) Y. Huang, R. Zhu, K. Zhao and Z. Gu, Angew. Chem., Int. Ed., 2015, 54, 12669–12672 CrossRef CAS PubMed; (g) Z. Dong, J. Wang, Z. Ren and G. Dong, Angew. Chem., Int. Ed., 2015, 54, 12664–12668 CrossRef CAS PubMed; (h) P.-X. Shen, X.-C. Wang, P. Wang, R.-Y. Zhu and J.-Q. Yu, J. Am. Chem. Soc., 2015, 137, 11574–11577 CrossRef CAS PubMed; (i) F. Sun and Z. Gu, Org. Lett., 2015, 17, 2222–2225 CrossRef CAS PubMed; (j) Z. Dong, J. Wang and G. Dong, J. Am. Chem. Soc., 2015, 137, 5887–5890 CrossRef CAS PubMed; (k) X.-C. Wang, W. Gong, L.-Z. Fang, R.-Y. Zhu, S. Li, K. M. Engle and J.-Q. Yu, Nature, 2015, 519, 334–338 CrossRef CAS PubMed; (l) P.-X. Zhou, Y.-Y. Ye, C. Liu, L.-B. Zhao, J.-Y. Hou, D.-Q. Chen, Q. Tang, A.-Q. Wang, J.-Y. Zhang, Q.-X. Huang, P.-F. Xu and Y.-M. Liang, ACS Catal., 2015, 5, 4927–4931 CrossRef CAS; (m) Z. Dong and G. Dong, J. Am. Chem. Soc., 2013, 135, 18350–18353 CrossRef CAS PubMed; (n) X. Sui, R. Zhu, G. Li, X. Ma and Z. Gu, J. Am. Chem. Soc., 2013, 135, 9318–9321 CrossRef CAS PubMed; (o) H. Weinstabl, M. Suhartono, Z. Qureshi and M. Lautens, Angew. Chem., Int. Ed., 2013, 52, 5305–5308 CrossRef CAS PubMed; (p) L. Jiao, E. Herdtweck and T. Bach, J. Am. Chem. Soc., 2012, 134, 14563–14572 CrossRef CAS PubMed; (q) L. Jiao and T. Bach, J. Am. Chem. Soc., 2011, 133, 12990–12993 CrossRef CAS PubMed; (r) A. Rudolph, N. Rackelmann and M. Lautens, Angew. Chem., Int. Ed., 2007, 46, 1485–1488 CrossRef CAS PubMed; (s) T. Wilhelm and M. Lautens, Org. Lett., 2005, 7, 4053–4056 CrossRef CAS PubMed; (t) C. Bressy, D. Alberico and M. Lautens, J. Am. Chem. Soc., 2005, 127, 13148–13149 CrossRef CAS PubMed , and references cited therein.
- For reviews, see: (a) S. Santoro, S. Kozhushkov, L. Ackermann and L. Vaccaro, Green Chem., 2016, 18, 3471–3493 RSC; (b) A. J. Reay and I. J. S. Fairlamb, Chem. Commun., 2015, 51, 16289–16307 RSC; (c) C. Cano, A. F. Schmidt and G. P. McGlacken, Chem. Sci., 2015, 6, 5338–5346 RSC; (d) L. Djakovitch and F.-X. Felpin, ChemCatChem, 2014, 6, 2175–2187 CrossRef CAS. For recent examples, see: (e) K. D. Collins, R. Honeker, S. Vásquez-Céspedes, D.-T. D. Tang and F. Glorius, Chem. Sci., 2015, 6, 1816–1824 RSC; (f) D.-T. D. Tang, K. D. Collins, J. B. Ernst and F. Glorius, Angew. Chem., Int. Ed., 2014, 53, 1809–1813 CrossRef CAS PubMed; (g) D.-T. D. Tang, K. D. Collins and F. Glorius, J. Am. Chem. Soc., 2013, 135, 7450–7453 CrossRef CAS PubMed; (h) T.-H. Park, A. J. Hickman, K. Koh, S. Martin, A. G. Wong-Foy, M. S. Sanford and A. J. Matzger, J. Am. Chem. Soc., 2011, 133, 20138–20141 CrossRef CAS PubMed; (i) S. Kawamorita, T. Miyazaki, H. Ohmiya, T. Iwai and M. Sawamura, J. Am. Chem. Soc., 2011, 133, 19310–19313 CrossRef CAS PubMed; (j) K. Yamazaki, S. Kawamorita, H. Ohmiya and M. Sawamura, Org. Lett., 2010, 12, 3978–3981 CrossRef CAS PubMed; see also: (k) L. Ackermann and R. Vicente, Org. Lett., 2009, 11, 4922–4925 CrossRef CAS PubMed.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2016, 18, 288–296 RSC.
- M. C. Bryan, B. Dillon, L. G. Hamann, G. J. Hughes, M. E. Kopach, E. A. Peterson, M. Pourashraf, I. Raheem, P. Richardson, D. Richter and H. F. Sneddon, J. Med. Chem., 2013, 56, 6007–6021 CrossRef CAS PubMed.
- K. Alfonsi, J. Colberg, P. J. Dunn, T. Fevig, S. Jennings, T. A. Johnson, H. P. Kleine, C. Knight, M. A. Nagy, D. A. Perry and M. Stefaniak, Green Chem., 2008, 10, 31–36 RSC.
- For some selected contributions see: (a) P. Pongrácz, L. Kollár and L. T. Mika, Green Chem., 2016, 18, 842–847 RSC; (b) Z. Zhang, ChemSusChem, 2016, 9, 156–171 CrossRef CAS PubMed; (c) L. Qi, Y. F. Mui, S. W. Lo, M. Y. Lui, G. R. Akien and I. T. Horváth, ACS Catal., 2014, 4, 1470–1477 CrossRef CAS; (d) E. I. Gürbüz, J. M. R. Gallo, D. M. Alonso, S. G. Wettstein, W. Y. Lim and J. A. Dumesic, Angew. Chem., Int. Ed., 2013, 52, 1270–1274 CrossRef PubMed; (e) S. G. Wettstein, D. M. Alonso, Y. Chong and J. A. Dumesic, Energy Environ. Sci., 2012, 5, 81998203 RSC; (f) Z.-Q. Duan and F. Hu, Green Chem., 2012, 14, 1581–1583 RSC; (g) L. Qi and I. T. Horváth, ACS Catal., 2012, 2, 2247–2249 CrossRef CAS; (h) I. T. Horváth, H. Mehdi, V. Fábos, L. Boda and L. T. Mika, Green Chem., 2008, 10, 238–242 RSC. For recent examples from our group, see: (i) G. Strappaveccia, L. Luciani, E. Bartollini, A. Marrocchi, F. Pizzo and L. Vaccaro, Green Chem., 2015, 17, 1071–1076 RSC; (j) G. Strappaveccia, E. Ismalaj, C. Petrucci, D. Lanari, A. Marrocchi, M. Drees, A. Facchetti and L. Vaccaro, Green Chem., 2015, 17, 365–372 RSC; (k) E. Ismalaj, G. Strappaveccia, E. Ballerini, F. Elisei, O. Piermatti, D. Gelman and L. Vaccaro, ACS Sustainable Chem. Eng., 2014, 2, 2461–2464 CrossRef CAS.
- C. E. Garrett and K. Prasad, Adv. Synth. Catal., 2004, 346, 889–900 CrossRef CAS.
- Note for Guidance on Specification Limits for Residues of Metal Catalysts, The European Agency for the Evaluation of Medicinal Products, Evaluation of Medicines for Human Use; London, 17 December 2002; http://www.emea.eu.int.
- E. Motti, G. Ippomei, S. Deledda and M. Catellani, Synthesis, 2003, 2671–2678 CAS.
- (a) S. V. Ley, C. Ramarao, R. S. Gordon, A. B. Holmes, A. J. Morrison, I. F. McConvey, I. M. Shirley, S. C. Smith and M. D. Smith, Chem. Commun., 2002, 1134–1135 RSC; (b) C. Ramarao, D. J. Tapolczay, I. M. Shirley, S. C. Smith and S. V. Ley, Microencapsulated Catalyst Methods of Preparation and Method of Use Thereof, U.S. Patent, 8828902B2, September 9, 2014 Search PubMed.
- S. Deledda, E. Motti and M. Catellani, Can. J. Chem., 2005, 83, 741–747 CrossRef CAS.
- (a) R. H. Crabtree, Chem. Rev., 2012, 112, 1536–1554 CrossRef CAS PubMed; (b) M. Pagliaro, V. Pandarus, R. Ciriminna, F. Béland and P. Demma Carà, ChemCatChem, 2012, 4, 432–445 CrossRef CAS; (c) N. T. S. Phan, M. Van Der Sluys and C. W. Jones, Adv. Synth. Catal., 2006, 348, 609–679 CrossRef CAS; (d) J. A. Widegren and R. G. Finke, J. Mol. Catal. A: Chem., 2003, 198, 317–341 CrossRef CAS.
- (a) S. J. Broadwater and D. T. McQuade, J. Org. Chem., 2006, 71, 2131–2134 CrossRef CAS PubMed; (b) J. M. Richardson and C. W. Jones, Adv. Synth. Catal., 2006, 348, 1207–1216 CrossRef CAS.
- M. Gruttadauria, F. Giacalone and R. Noto, Green Chem., 2013, 15, 2608–2618 RSC.
- A recent review: J. Li, S. De Sarkar and L. Ackermann, Top. Organomet. Chem., 2016, 55, 217–257 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, characterization data, and copies of NMR spectra of known (3a and 5) and new compounds (3b–3o, 3′h, 4j, 4k). See DOI: 10.1039/c6gc01393g |
| This journal is © The Royal Society of Chemistry 2016 |