
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Intensification and deactivation of Sn-beta investigated in the continuous regime†
Daniele
Padovan
,
Charlie
Parsons
,
Manuel
Simplicio Grasina
and
Ceri
Hammond
*
Cardiff Catalysis Institute, Cardiff University, Main Building, Park Place, CF10 3AT, UK. E-mail: hammondc4@cardiff.ac.uk; Web: http://blogs.cardiff.ac.uk/hammond Tel: +44 (0) 29 2087 4082
First published on 13th June 2016
Abstract
Despite a proliferation of research focusing on the synthesis and catalytic chemistry of Sn-containing zeolite beta, research focusing on its intensification lacks behind, prohibiting its further exploitation. In this manuscript, we investigate and optimise the continuous flow activity of Sn-β for a range of sustainable chemical transformations, including the transfer hydrogenation of model and bio-renewable substrates (furfural), and the isomerisation of glucose to fructose. Extended time-on-stream studies reveal Sn-β to be a very stable catalyst during continuous operation in an organic solvent. Spectroscopic methodologies reveal that deactivation in these cases is related to fouling of the micropores with the product and higher molecular weight carbonaceous residue. Periodic regeneration by heat treatment is found to restore full activity, allowing Sn-β to be used for over 700 h continuously with no greater than 20% loss in activity. In contrast, operation in an aqueous medium is extremely disadvantageous, as it causes total destruction of the catalyst and permanent deactivation. In these cases, however, long term activity can still be achieved by modifying the solvent chosen for reaction, with methanol appearing to be a suitable alternative. The promising results presented herein conclusively demonstrate the potential of Sn-β to operate as an industrial heterogeneous catalyst.
Introduction
Given increasing environmental awareness and legislation, and the diminishing supply of traditional feedstock, there is an urgent need for the chemical community to develop alternative methods for chemical and fuel production.1 Indeed, developing processes that are inherently more efficient is one of the most important challenges of contemporary chemistry. Catalysts are able to improve the overall efficiency of any given process. As such, the development of new catalytic materials is one of the cornerstones of green chemistry research.2Zeolites are microporous, crystalline materials, regularly arranged into frameworks of 3–14 Å diameter. With large surface areas, high thermal stability and tuneable structures and composition, it is unsurprising that zeolites have found widespread application as catalysts for petrochemical processes for several decades.3 Traditionally employed as strong Brønsted acid catalysts in their aluminosilicate form, their potential as exceptionally active Lewis acids is realized when their structure is doped with suitable Lewis acidic heteroatoms, such as Ti, Sn and Zr.4 These crystalline, porous, inorganic Lewis acids are subsequently able to pair both the advantages of homogeneous analogues, that is to say high activity and selectivity, with the practical benefits of solid catalysts, which simplify elements of process intensification. Moreover, encapsulation of the Lewis acidic element into the hydrophobic framework is thought to minimises its interaction with H2O, and thereby opens up opportunities to perform aqueous phase Lewis acid catalysis, which is critically important for biomass valorisation.5
One such example, Sn-zeolite β, has rapidly emerged as one of the most promising heterogeneous catalysts in the area of green chemistry. Some notable examples of green chemical processes catalysed by Sn-β include (i) the peracid-free Baeyer–Villiger oxidation of ketones with the green oxidant, H2O2;6 (ii) the production of bio-renewable furanic platform molecules via the isomerisation of glucose to fructose;7 (iii) the conversion of renewable hexoses,8 pentoses9 and trioses10 to bio-renewable and bio-degradable polymer precursors; (iv) the solvent-free hydration of epoxides to 1,2-diols;11 (v) various C–C bond forming reactions,12 (vi) the H2-free, reduction of carbonyl compounds via transfer hydrogenation (TH),13 amongst others.14 We note that TH is an increasingly important method for the reductive valorisation of bio-renewables.
Despite a proliferation of research focusing on its synthesis15 and catalytic chemistry,6–14 research focusing on the intensification of Sn-β (i.e. scale up, deactivation studies) lacks behind, despite its significant green chemistry potential. In the absence of this knowledge, further exploitation of Sn-β on a commercial level is hindered.
Operational lifetime is a critical, but often overlooked, key performance indicator of a heterogeneous catalyst. Several potential processes, such as (i) the strong chemisorption of various adsorbates (poisoning), (ii) textural changes to the catalyst through thermal or mechanical treatment, (iii) the deposition of residue that blocks access to the active sites (fouling), (iv) sintering of catalyst particles, leading to an undesirable decrease in surface area, and (v) active site restructuring, can all negatively impact catalytic activity. Furthermore, for heterogeneous catalysts that operate in the liquid phase, (vi) leaching of the active sites, and (vii) (hydro)thermal dissolution of the catalyst, may also occur.16 Whilst many of these phenomena may be reversible, the length of time a catalyst may operate without requiring regeneration is critical, as regeneration leads to unwanted downtime, a decrease in space-time-yield, and may impact the specific design of the catalytic reactor. Identifying the cause(s) of deactivation, and devising strategies to minimise (or avoid) its impact, are therefore critical areas of catalysis research, as recently described by Sádaba et al. and Lange.17
In this manuscript, we extend our research of stannosilicate catalysis to investigate the stability of Sn-β in the continuous regime. Steady state catalytic testing for TH reactions of cyclohexanone and furfural, in addition to spectroscopic measurements, reveal that Sn-β is an exceptionally stable catalyst for TH chemistry, exhibiting less than 20% loss of activity over 700 h on stream. Partial, non-permanent deactivation during TH is attributed to fouling of the microporous structure with both the reaction product and carbonaceous residue, and this can be overcome through periodic regeneration. In contrast, aqueous phase reactions, such as the isomerisation of glucose to fructose, appear to cause extensive dissolution of the catalyst and its permanent deactivation. Despite this, stability for this class of reactions can still be achieved by modifying the reaction solvent. These promising stability measurements further demonstrate the potential of Sn-β for future industrial exploitation.
Results and discussion
Theoretical background
To optimise our microreactors and gain an initial understanding of continuous flow TH chemistry, we first chose to study the TH of cyclic ketones with an alcohol solvent (Scheme 1). As described above, TH technology provides a mild, selective and safe method for reducing carbonyl groups in the absence of high pressure H2 and inorganic hydride donors, and is therefore a key emerging methodology for the reductive valorisation of biomass.13,18 In addition, several other factors make the TH reaction a useful model reaction for Sn-β catalysis; (i) its intermolecular hydride-shift mechanism is comparable to those mechanisms observed for other important Sn-β catalysed isomerisation;19 (ii) we have recently experimentally demonstrated it to be a suitable model reaction for stannosilicate catalysis, with the TOF values obtained for Sn-β during TH matching those obtained for other reactions, such as glucose isomerisation;20 (iii) it is a single feed reaction, simplifying both the kinetic models and the reactor design; and (iv) unlike other Sn-β catalysed isomerisations, the reaction is – to all intents and purposes – non-equilibrium limited. The thermodynamics for the reaction in study are favourable (ΔH0 = −11.7 kJ mol−1 and ΔS° = −1.5 J mol−1 K−1), and at 25 °C the process is spontaneous with ΔG° = −11.2 kJ mol−1 and an equilibrium constant (Keq) of approximately 94. Furthermore, as 2-butanol is used as a solvent its concentration approximately 50-fold higher than cyclohexanone (0.2 M), and from this two assumptions can be made: (i) 2-butanol concentration is essentially constant i.e. the reaction is pseudo-first order; and (ii) the position of equilibrium is shifted even further in the direction of products causing the expected conversion to be very close to 100%. This allows deactivation processes to be measured on a more detailed level.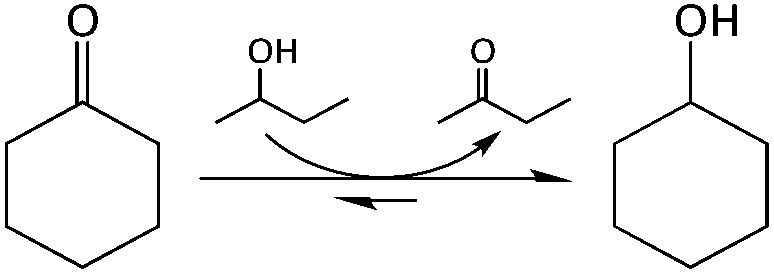 | ||
| Scheme 1 Schematic of the TH of cyclohexanone to cyclohexanol. | ||
Kinetic studies in batch mode
To provide a benchmark for the continuous flow studies, we first evaluated the batch kinetics of the TH reaction. An Arrhenius expression for the TH of cyclohexanone over 10 wt% Sn-β (10Sn-β) was therefore obtained. We chose this catalyst due to its high levels of intrinsic activity, coupled with its very high metal loading, which ensures that high levels of space-time-yield (the key industrial benchmark)1 can be obtained. The synthesis and characterisation of this material was recently extensively described.20 A linear Arrhenius plot for 10Sn-β was observed between 54 and 98 °C, yielding an activation barrier of 39.3 kJ mol−1 (Fig. 1). The relatively low activation energy prompted us to explore potential mass transfer limitations. However, doubling the catalyst (1.0 to 2.0 mol%) led to a linear increase in conversion, confirming the absence of internal mass transfer limitations at these conditions. | ||
| Fig. 1 Kinetic data for the transfer hydrogenation of cyclohexanone in batch mode. (A) Arrhenius plot, obtained through kinetic analysis between 54 and 98 °C, and (B) time-dependent conversion of cyclohexanone in the presence of various amounts of 10Sn-β. | ||
Kinetic and mass transfer studies in continuous flow
Having benchmarked the batch kinetics for 10Sn-β, we subsequently set about exploring its activity in the continuous regime. A continuous Plug Flow Reactor (PFR, Scheme 2) provides several major advantages compared to a stirred tank (slurry) reactor. These advantages include (i) improved control over reaction conditions and safety, (ii) excellent mass- and heat-transfer, (iii) shorter reaction times, (iv) smaller reactor volumes, (iv) scalability and (v) increased space-time-yields.21 More critically given the context of this work, a PFR also permits operation under steady state conditions, allowing catalyst deactivation to be probed. | ||
| Scheme 2 Schematic of PFR utilised for continuous flow studies. | ||
To rule out potential external mass transfer limitations, we first investigated the effect of linear velocity by preparing a range of columns of various length, and monitoring the activity obtained at a fixed contact time (i.e. flow rate was also adjusted so that the overall contact time was kept constant).22 In all these tests, the effective contact time (τeff, eqn (1)) was held at 15 s.
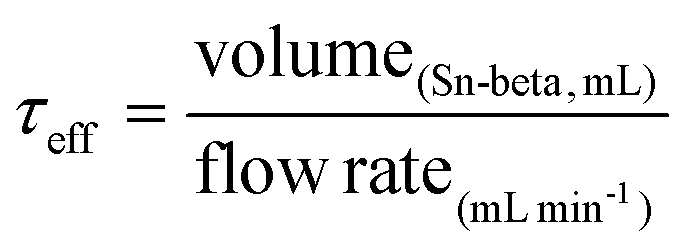 | (1) |
As the reactant flow rate is initially increased, conversion rapidly improved from 63% (at a flow rate of 0.08 mL min−1) to 79% (at 0.10 mL min−1). However, above this particular flow rate, interphase limitations no longer play a role, as the observed conversion is independent of linear velocity. This is a clear indication of film diffusion limitations hindering activity at low flow rates, but the reactor being kinetically relevant at higher flow rates i.e. greater than 0.10 mL min−1. Accordingly all future experiments were conducted at a column length consistent with being in the kinetic regime. Subsequent time online experiments between 80 and 100 °C (Fig. 2B) yielded a satisfactory Arrhenius expression of y = −5014x + 11.2, corresponding to an activation barrier of 41.7 kJ mol−1, in excellent agreement to our batch results. The similar activation barriers found in both batch and continuous mode further confirm the kinetic relevance of the PFR.
 | ||
| Fig. 2 (A) Influence of linear velocity on catalytic activity, and (B) time online data for the MPV transfer hydrogenation of cyclohexanone in continuous mode between 80 and 100 °C. The relevant Arrhenius plot is displayed in the inset. | ||
Initial time on stream studies
Having optimised the PFR to obtain accurate kinetic data, we subsequently focused upon determining the stability of 10Sn-β during extended time on stream TH experiments. τeff (34 s) was optimised so that the yield of CyOH was at or below 98% i.e. some of the reactant could be quantified in the effluent, to ensure that accurate lifetime data could be obtained and that the reactor was not operating in an ‘excess catalyst’ regime. The reaction was repeated at 100 °C several times in order to (i) check reproducibility, and (ii) obtain a variety of ex-reactor samples for ex situ characterisation.Fig. 3 demonstrates the steady state activity of 10Sn-β for the TH of cyclohexanone over a 700 h period. Under the conditions employed, activity is found to decrease from an initial conversion value of ±98% to approximately 70% following 500 h on stream. We note that this is the first time for Sn-β to be tested over such a prolonged reaction period. Both the selectivity to CyOH and the carbon balance remained >95% throughout. On a day-to-day basis approximately a 1–2% decrease in conversion was observed, which suggests 10Sn-β possess exceptional stability for the reaction.
 | ||
| Fig. 3 Time on stream data for 10Sn-β during CyO TH at 100 °C. The regenerated sample data is illustrated by open triangles. | ||
Ex-reactor characterisation
To rationalise the decrease in activity observed, the reaction was terminated after various times on stream, and the entire catalyst bed removed for spectroscopic analysis. Due to the difficulties in separating the catalyst from the diluent, each measurement was performed on the diluted bed, and the data compared to a control sample of fresh catalyst and diluent.XRD analysis and EDXS measurements (ESI Fig. S1 and Table S1†) revealed that the crystalline structure of the zeolite remained intact during TH, and that the Sn concentration of the solid catalyst also remained constant. Clearly, deactivation through dissolution of the zeolite and/or leaching of its active component does not occur.
Despite possessing the 118 isotope, and the dilution of the catalytic powder with SiC, acceptable MAS NMR data was obtained from the ex reactor sample after 200 h on stream (Fig. 4). Given that the precise relaxation times for both framework (−688 ppm) and extra-framework (−602 ppm) Sn have not been fully evaluated, deconvolution and integration of the two major resonances is not feasible, and this unfortunately prohibits fully quantitative interpretation of this data. Nevertheless, semi-quantitative analysis of the NMR spectra indicates that some migration i.e. restructuring, of Sn may have occurred during the 200 h reaction period. This is based on the relative increase of the −602 ppm resonance of extra-framework Sn, compared to the −688 ppm resonance of isomorphously substituted Sn sites. Clearly, the amount of extra-framework Sn increases throughout the reaction, albeit to an unknown extent.
 | ||
| Fig. 4 MAS NMR spectra of (a) fresh i.e. unreacted 10Sn-β, (b) 10Sn-β after 200 h on stream, and (c) bulk SnO2. | ||
In light of this, we also examined the ex reactor series with X-ray Absorption spectroscopy.24 Despite dilution, the spectra are of good quality up to and beyond a distance of 12 Å−1, further indicating that the long-range order (crystalline structure) remains intact throughout the reaction. Information on Sn–Sn scattering interactions, such as those found in extra-framework and oxidic Sn species can be gained from the second shell region. Despite its low intensity, it is clear that the Sn–Sn scattering intensity increases during time on stream (Fig. 5B), further indicating some extraction of Sn from the zeolite framework occurs during operation. However, the intensity of Sn–Sn scattering remains extremely low even after 200 h on stream. Given that the intensity of any XAS signal is directly related to its concentration, this indicates that the extraction of (active) framework Sn atoms is not a major process, and is unlikely to account for the loss in activity.
 | ||
| Fig. 5 (A) X-ray absorption spectra of 10Sn-β at various times on stream, and (B) magnification of the 2.5–4 Å−1 region of the spectra. | ||
TGA analysis (Fig. 6) of the ex reactor sample indicates that significant amounts of the product (CyOH) and some higher-molecular weight carbonaceous residue accumulate on or within the catalyst throughout its operation. The presence of carbonaceous residue was also confirmed by the presence of a “G” peak in the UV-Raman spectrum of the ex reactor sample between 1500 and 1700 cm−1 (Fig. S2†).25 Confirmation of the identity of the adsorbed products was obtained by 1H NMR analysis. After reaction, the used catalyst bed was removed from the reactor, washed in CD3Cl, and the supernatant solution analysed by 1H NMR. As can be seen (ESI Fig. S3†), only CyOH and 2-butanol remained on the catalyst after reaction, confirming the affinity of Sn-β to retain CyOH and not the corresponding ketone.
 | ||
| Fig. 6 TGA data for 10Sn-β. The sample removed after ±500 h on stream (b) is compared to a fresh catalyst sample (a), and two reference materials of 10Sn-β doped with CyO (c) and CyOH (d). | ||
We note here that since the catalyst is being examined in plug flow mode, the deactivation of each catalyst particle is not only related to time on stream, but also to its position in the bed, given that each particle is exposed to a different ratio of reactants and products.21 Thus, the retention of CyOH, and the absence of retained CyO, is not simply a consequence of the reactor operating at high conversion, but indicates that the catalyst has a particular affinity towards retaining CyOH.
The presence of such species within/on the catalyst is notable; given the microporous structure of Sn-β, residual carbonaceous material in the form of CyOH and higher molecular products is likely to lead to a (partial) blocking of the micropores, and hence a loss of activity through decreased active site accessibility. To confirm this, porosimetry measurements of the fresh and used samples were performed. A substantial decrease in micropore volume from 0.23 cm3 g−1 to 0.11 cm3 g−1 was observed following 150 h on stream (ESI Table S1†). Such pronounced decrease in pore volume would clearly inhibit the activity of the catalyst, even if some external enrichment has been observed for these materials.26
In light of these results, we explored the possibility of regenerating the catalyst bed through a calcination procedure, with the aim of removing the carbonaceous material that blocks access of the reactants to the active Sn sites. We employed a conventional heat treatment protocol for zeolite powders (550 °C, 3 h in 30 mL min−1 flowing air), and found that complete regeneration of catalytic activity could be achieved (Fig. 4). This further confirms that major structural degradation to the catalyst throughout operation does not occur, and confirms the role that fouling plays during extended operation. Following regeneration, the reactor was tested for another 200 h, thus taking the total time on stream to over 700 h in total. A comparable deactivation rate was observed during the second extended time on stream period.
Optimisation of continuous flow activity
Due to the retention of products that occurs during extended operation, we subsequently focused upon (re)optimisation of the reaction conditions. We hypothesised that if fouling was the key reason for deactivation, that increased reaction temperatures may improve long-term stability by aiding desorption of the products during the reaction cycle. We consequently performed two addition reactions at 120 and 140 °C for 250 h, in order to elucidate the impact of reaction temperature. The 5 bar overpressure on the reactor column allowed us to work above the boiling point of the solvent used in this study (2-butanol).It is clear from Fig. 7 that 10Sn-β retains excellent levels of stability even at significantly higher reaction temperatures. At both 120 and 140 °C, a slow rate of deactivation over 150 h is observed, with activity decreasing by ±10–20% over this time period. In both cases, a quasi-steady state is achieved after approximately 24 h, which is somewhat earlier than observed for reactions performed at 100 °C (75 h). In both cases, full activity after 170 h can be restored following the regeneration procedure, which demonstrates that the same fouling mechanism likely predominates in these cases.
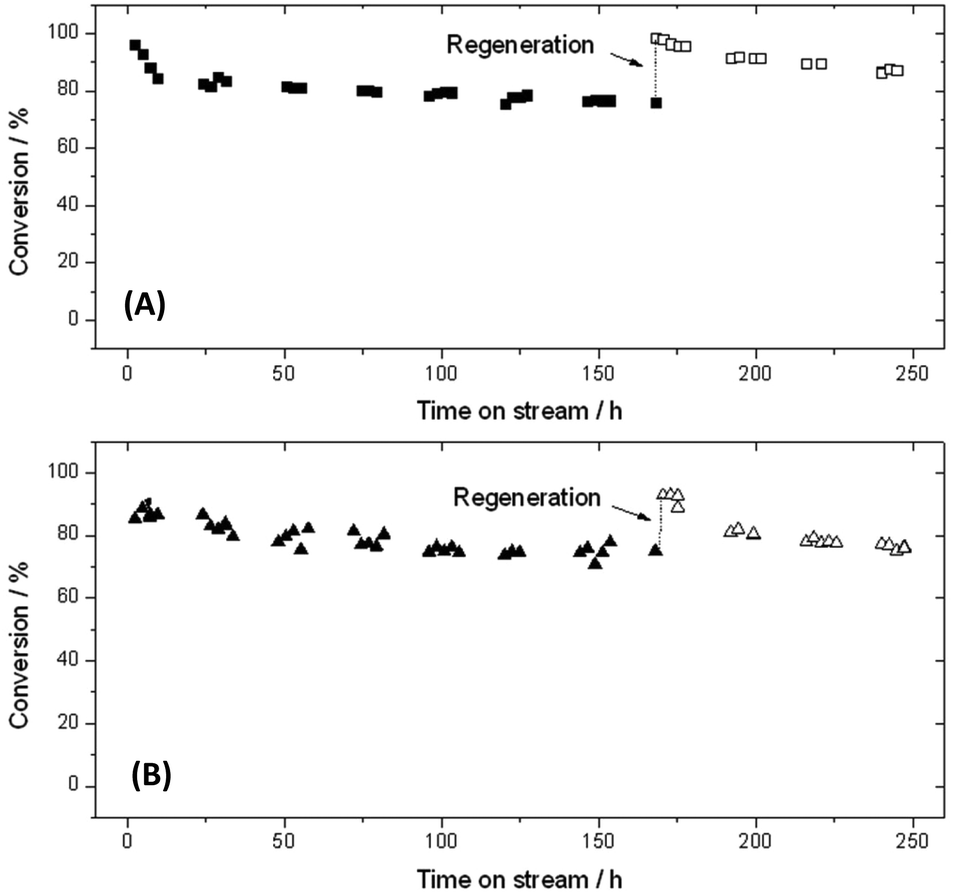 | ||
| Fig. 7 Time on stream data for 10Sn-β at 120 °C (A) and 140 °C (B). | ||
To further probe the effect of reaction temperature on the kinetics of deactivation, we compared the relative activity of each reactor column as a function of (a) relative conversion versus time on stream (Fig. 8A), and (b) relative instantaneous rate of each reaction constant as a function of the quantity of substrate that has passed over the reactor column (Fig. 8B).27 The effective rate of reaction at any given time was calculated by the pseudo-first order rate expression (eqn (2)), and the ‘turnover’ of the system (ρ) was determined by calculating the number of moles of substrate passed over the reactor in a given unit of time, divided by the total of moles of Sn in the reactor column (eqn (4)).
 | (2) |
 | (3) |
 | (4) |
 | ||
| Fig. 8 (A) Relative catalytic activity of 10Sn-β over 170 h on stream between 100 and 140 °C. (B) Relative effective rate constant of 10Sn-β with respect to reactor turnover (ρ) and temperature. | ||
Sn-β clearly performs exceptionally well under each set of reaction conditions, with no greater than 20% loss in relative activity observed during these reactions. Despite the conversion data indicating that increased temperatures do not significantly aid long term stability, the decrease in relative stability is minor compared to the improved productivity of the reactor; at 140 °C, the reactor column is over three times more productive on a space-time-yield scale (i.e. kg of product produced per unit time, per mass of catalyst and per reactor volume) than the 100 °C reactor, given the temperature dependence of the reaction, thus significantly improving the performance of the system on the whole. Furthermore, for a similar number of substrate turnovers, the decrease in effective rate constant is less pronounced at higher temperature (Fig. 8B). Accordingly, operation at elevated temperatures is clearly beneficial. We draw attention to the dimensionless number of substrate turnovers exhibited by the catalyst at 140 °C, which is the equivalent of over 100 batch reactions under standard testing conditions, and the relative performance of the optimised continuous system at 140 °C relative to the batch (Table 1).
| Reactor type | Space-time-yielda | Relative space-time-yield |
|---|---|---|
| a Space-time-yields were calculated at maximum conversion as kg of product produced per cm3 reactor volume, per hour and per kg of catalyst. b Only the liquid volume was used as reactor volume. c Volume of catalyst bed (including diluent) used as reactor volume.28 | ||
| Batchb | 0.76 | 1 |
| Flowc | 31.8 | 41.8 |
Continuous TH of furfural
Having optimised the TH system with our model system, we subsequently focused on the continuous TH of substrates more representative of bio-renewable building blocks. In full agreement to our model system, 10Sn-β displays excellent stability during the catalytic TH of furfural, despite the more complex nature of the highly functionalised substrate. Although there is a gradual decrease in catalytic activity over the first 150 h on stream (from an initial conversion of 84% to 45%), full regeneration of the catalyst can be achieved by re-calcination of the reactor bed in air. This demonstrates that even with highly-functionalised bio-renewable substrates, permanent deactivation during catalytic TH is not observed (Fig. 9). | ||
| Fig. 9 (A) Relative performance of 10Sn-β over 300 h on stream during furfural TH. (B) Selectivity trends observed during the TH of furfural. | ||
Interestingly, the catalyst is much more stable following its first regeneration. Although the origin of this effect is not yet evident, ‘regenerating’ the catalyst bed prior to performing continuous catalysis resulted in exactly the same kinetic behaviour being observed (ESI Fig. S4†). Clearly, positive changes to the catalyst are observed during the first 150 h on stream that cannot be induced by simple calcination procedures alone. Although unexpected, the almost-perfect stability demonstrated by the catalyst during its second and third periods of operation is extremely promising for long-term continuous operation.
In contrast to the model system, where CyOH is the only observed product, some unanticipated selectivity modifications were observed during continuous TH of furfural. Although the anticipated TH product is furfuryl alcohol (Scheme 3), at the initial stages of operation the reaction is almost exclusively selective to the ether product, 2-(butoxymethyl)furan. Although Lewis acidic zeolites have been shown to generate renewable 2,5-bis(alkoxymethyl)furans from 5-HMF,29 the ability to generate such renewable fuel additives from furfural without external hydrogen sources and with Sn-β has not yet been shown. In full agreement to previous studies focused on 2,5-bis(alkoxymethyl)furans, the selectivity observed towards the ether product rapidly diminishes, and the anticipated TH product, furfuryl alcohol, is subsequently observed. At all times, the carbon balance equals ±100%, thus indicating that the catalyst simply loses its ability to mediate the second step of the reaction. This ability to mediate multistep and multifunctional catalytic transformations with a single catalytic material may be of considerable interest in the future when multistep reaction processes are of interest.
 | ||
| Scheme 3 Combined TH and etherification of furfural catalysed by Sn-β. | ||
Continuous isomerisation of glucose in the aqueous phase
Whilst Sn-β is clearly an active, selective and impressively stable catalyst for TH catalysis, we were keen to understand the role that water may play during extended operation. Indeed, a large number of catalytic reactions involving Sn-β are performed in the aqueous phase, or at least in the presence of water, and water-tolerance is often reported to be one of the favourable characteristics of Sn-β. Accordingly, we turned our attention to the isomerisation of glucose to fructose, one of the most important discoveries in Sn-β catalysis, given that it opens a route to furanic platform molecules from highly abundant glucose (Scheme 4). | ||
| Scheme 4 Catalytic routes to furanic platform molecules opened by glucose-fructose isomerisation. | ||
The conventional glucose isomerisation reaction, as pioneered by Davis, is performed in the aqueous phase.7τeff was optimised (1 min 50 s in water) so that the initial conversion of glucose was at or just below 35%, as studies have shown that above this level of conversion a significant decrease in carbon balance is observed. Unfortunately, our results show that under these conditions, 10Sn-β displays poor levels of stability. Over the first 30 h on stream (Fig. 10), Sn-β loses over 90% of its initial activity when the reaction is performed in an aqueous medium. More critically, regeneration of the sample could not be achieved by any methods, indicating deactivation to be permanent. Several methodologies, including XRD (Fig. 11) clearly demonstrated that total destruction of the catalyst was observed after operation in the aqueous phase.
 | ||
| Fig. 10 (A) Relative catalytic activity of 10Sn-β over 170 h on stream between 100 and 140 °C. (B) Relative effective rate constant of 10Sn-β with respect to reactor turnover (ρ) and temperature. | ||
 | ||
| Fig. 11 XRD reflections of fresh and used 10Sn-β samples before and after glucose isomerisation. | ||
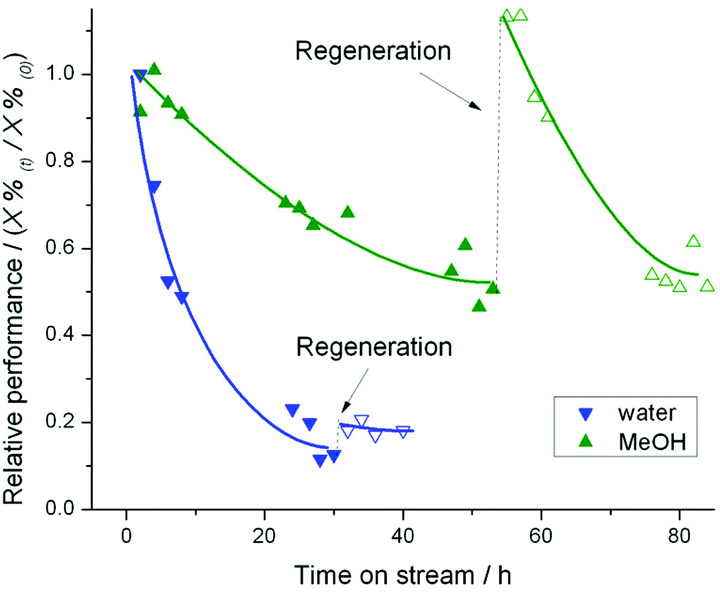
To determine whether the presence of water, glucose, or both were the cause of this extensive destruction, we subsequently modified the isomerisation conditions so that MeOH, instead of water, may be employed as reaction solvent. As can be seen (Fig. 10), performing the isomerisation experiments in an organic media is much more promising. In this case, non-permanent deactivation is observed, and only ±50% deactivation is observed over 50 h on stream. Whilst these results indicate that glucose itself is not detrimental to steady-state operation, it is clear that Sn-β is an unsuitable catalyst for extended steady state operation in the aqueous phase, in contrast to the generally held opinion of its water tolerance.
Conclusions
In this manuscript, the stability of Sn-β under steady state, continuous flow conditions has been evaluated. Time-on-stream studies reveal that Sn-β is a very stable TH catalyst during continuous operation in an organic media. Under optimised conditions, the catalyst is able to operate continuously for over 700 h with at least 80% of its original activity. Evaluation of the reactor system reveals reactions performed at higher temperatures (140 °C) to be optimal in terms of activity and stability. Various spectroscopic methodologies reveal that deactivation during TH is related to fouling of the micropores through continuous deposition of the product and higher molecular weight carbonaceous residue, with micropore volume decreasing by approximately 50% during extended operation. However, periodic regeneration by heat treatment is found to restore full activity. In contrast, the ability of Sn-β to mediate continuous aqueous-phase chemistry is considerably poorer. During glucose isomerisation in the continuous regime, complete loss of catalytic activity is observed in only 30 h on stream, and this loss of activity is related to destruction of the catalytic material under the conditions employed. Whilst this prohibits the use of water as a solvent during the valorisation of bio-renewable feedstock with Sn-β, stable activity can still be achieved by modifying the solvent of the reaction.To the best of our knowledge, this is the most detailed study of continuous flow catalysis with Sn-β. Although the continuous activity of Sn-β is clearly dependent on the precise reaction under going study, and more particularly on the solvent employed during operation, these results conclusively demonstrate the potential of Sn-β for continuous operation, albeit in a non-aqueous medium. The focus of our future studies will be to understand the negative role exhibited by water, and to understand why the regenerated catalyst appears to be more stable than the fresh catalyst during furfural TH.
Experimental
Catalyst synthesis
Commercial zeolite Al-β (Zeolyst, NH4-form) was dealuminated by treatment in HNO3 solution (13 M HNO3, 100 °C, 20 h, 20 mL g−1 zeolite). Solid-state stannation of dealuminated zeolite β was performed the procedure reported in ref. 20, by grinding the appropriate amount of tin(II) acetate with the necessary amount of dealuminated zeolite for 10 minutes in a pestle and mortar. Following this procedure, the sample was heated in a combustion furnace (Carbolite MTF12/38/400) to 550 °C (10 °C min−1 ramp rate) first in a flow of N2 (3 h) and subsequently air (3 h) for a total of 6 h. Gas flow rates of 60 mL min−1 were employed at all times.Catalyst characterisation
A PANalytical X'PertPRO X-ray diffractometer was employed for powder XRD analysis. A CuKα radiation source (40 kV and 30 mA) was utilised. Diffraction patterns were recorded between 6–55° 2θ (step size 0.0167°, time/step = 150 s, total time = 1 h). Sn contents were determined by SEM-EDX. Porosimetry measurements were performed on a Quantachrome Autosorb, and samples were degassed prior to use (90 °C, 5 h, nitrogen flow). Adsorption isotherms were obtained at 77 K. Pore volumes were calculated by the t-plot method. Raman spectra were recorded on a Renishaw inVia Ramanscope, equipped with a 266 nm excitation laser. Samples were measured at <5 mW power to minimise disruption to the sensitive ex reactor samples. TGA analysis was performed on a Labsys TGA-DTA system. Samples were held isothermally at 30 °C for 30 minutes, before being heated to 550 °C (10 °C min−1 ramp rate). MAS NMR experiments were performed at Durham University through the EPSRC UK National solid-state NMR Service. Samples were measured under conditions identical to those reported by Bermejo-Deval et al.,23 and Hammond and co-workers.20 Sn K-edge XAS data was collected by the methods described in detail elsewhere.20Kinetic evaluation and analytical methods
Batch TH reactions with cyclohexanone were performed in a 100 mL round bottom flask equipped with a reflux condenser, which was thermostatically controlled by immersion in a silicon oil bath. The vessel was charged with a 10 mL solution of cyclohexanone in 2-butanol (0.2 M), which also contained an internal standard (biphenyl, 0.01 M). The solution was subsequently heated to the desired temperature (98 °C internal temperature). The reaction was initiated by addition of an appropriate amount of catalyst, typically corresponding to 1 mol% Sn relative to cyclohexanone. The solution was stirred at ±750 rpm with an oval magnetic stirrer bar.Continuous TH reactions were performed in a plug flow, stainless steel, tubular reactor. Our reactor (Scheme 2) was connected to an HPLC pump in order to regulate the reactant flow and allow operation at elevated pressures. The catalyst was mixed with a diluent material (SiC (particle size of 63–75 μm)) in order to avoid back mixing and to minimise the pressure drop, and the bed placed in between two plugs of quartz wool. The diluted sample was densely packed into a ¼′′ stainless steel tube (4.1 mm internal diameter), and a frit of 0.5 μm was placed at the end of the bed in order to avoid any loss of material. The reactor was subsequently immersed in a thermostatted oil bath at the desired reaction temperature. Pressure in the system was controlled by means of a backpressure regulator, and the pressure drop was determined by comparison of the HPLC pump pressure to the outlet pressure measured by a pressure gauge. An overpressure of 5–10 bar was typically employed, depending on reactant flow rate and column length, and this allowed operation above the boiling temperature of the solvent (2-butanol, 98 °C). The reaction feed was identical to that used for batch reactions. Aliquots of the TH reaction solutions were taken periodically from a sampling valve placed after the reactor. Periodic catalyst regeneration was performed heating the whole reactor in a combustion furnace (Carbolite MTF12/38/400) to 550 °C (10 °C min−1) in air (3 h). TH of furfural was performed under identical reaction conditions. All reactants and products during TH were analysed by GC (Agilent 7820, 25 m CP-Wax 52 CB column), and quantified against a biphenyl internal standard. Glucose isomerisation experiments were performed under similar conditions, although all reaction lines were heated to improve the solubility of glucose throughout the system. Reaction feeds of 1 wt% glucose in MeOH and H2O were utilised, and glucose, fructose and mannose were quantified by means of HPLC analysis with sorbitol as external standard. A reaction temperature of 110 °C was employed for all glucose experiments.
Acknowledgements
CH gratefully appreciates the support of The Royal Society for the provision of both a University Research Fellowship (UF140207) and further research funding (RG140754). CH also thanks Dr David Apperley, Dr Fraser Markwell and Dr Eric Hughes of the ESPRC UK National Solid-state NMR Service at Durham University for performing the MAS NMR experiments. Dr Peter Wells (University College London/UK Catalysis Hub) is thanked for measurement of the XAS spectra. Diamond Light Source and the Research Complex at Harwell are thanked for the provision of beamtime (SP8071). Dr Nikolaos Dimitratos is thanked for continued support.References
- F. Cavani and J. H. Teles, ChemSusChem, 2009, 2, 508 CrossRef CAS PubMed; C. Hammond, S. Conrad and I. Hermans, ChemSusChem, 2012, 5, 1668 CrossRef PubMed.
- R. A. Sheldon, Green Chem., 2008, 10, 359 RSC; G. J. Hutchings, J. Mater. Chem., 2009, 19, 1222–1235 RSC.
- A. Corma, Chem. Rev., 1997, 97, 2373 CrossRef CAS PubMed.
- P. Y. Dapsens, C. Mondelli and J. P. Ramirez, Chem. Soc. Rev., 2015, 44, 7025 RSC; M. Moliner, Dalton Trans., 2013, 43, 4197 RSC.
- R. Gounder, Catal. Sci. Technol., 2014, 4, 2877 Search PubMed; Y. R. Leshkov and M. E. Davis, ACS Catal., 2011, 1, 1566 CrossRef CAS.
- A. Corma, L. T. Nemeth, M. Renz and S. Valencia, Nature, 2001, 412, 423 CrossRef CAS PubMed.
- Y. R. Leshkov, M. Moliner, J. A. Labinger and M. E. Davis, Angew. Chem., Int. Ed., 2010, 49, 8954 CrossRef CAS PubMed; M. Moliner, Y. R. Leshkov and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6164 CrossRef PubMed; E. Nikolla, Y. R. Leshkov, M. Moliner and M. E. Davis, ACS Catal., 2011, 1, 408 CrossRef; C. M. Lew;, N. Rajabbeigi and M. Tsapatsis, Ind. Eng. Chem. Res., 2012, 51, 5364 CrossRef; J. D. Lewis, S. Van de Vyver, A. J. Crisci, W. R. Gunther, V. K. Michaelis, R. G. Griffin and Y. R. Leshkov, ChemSusChem, 2014, 7, 2255 CrossRef PubMed.
- M. S. Holm, S. Saravanamurugan and E. Taarning, Science, 2010, 328, 602 CrossRef CAS PubMed.
- M. S. Holm, Y. J. P. Torres, S. Saravanamurugan, A. Riisager, J. A. Dumesic and E. Taarning, Green Chem., 2012, 14, 702 RSC.
- E. Taarning, S. Saravanamurugan, M. S. Holm, J. Xiong, R. M. West and C. H. Cristensen, ChemSusChem, 2009, 2, 625–527 CrossRef CAS PubMed.
- B. Tang, W. Dai, G. Wu, N. Guan, L. Li and M. Hunger, ACS Catal., 2014, 4, 2801 CrossRef CAS.
- S. Van de Vyver, C. Odermatt, K. Romero, T. Prasomsri and Y. R. Leshkov, ACS Catal., 2015, 5, 972–977 CrossRef CAS; S. Van de Vyver and Y. R. Leshkov, Angew. Chem., Int. Ed., 2015, 54, 12554 CrossRef PubMed.
- A. Corma, M. E. Domine, L. Nemeth and S. Valencia, J. Am. Chem. Soc., 2002, 124, 3194 CrossRef CAS PubMed; M. J. Gilkey and B. Xu, ACS Catal., 2016, 6, 1420 CrossRef.
- W. R. Gunther, Y. Wang, Y. Ji, V. K. Michaelis, S. T. Hunt, R. G. Gridffin and Y. R. Leshkov, Nat. Commun., 2012, 3, 1 Search PubMed.
- C. Hammond, S. Conrad and I. Hermans, Angew. Chem., Int. Ed., 2012, 51, 11736 CrossRef CAS PubMed; P. Wolf, C. Hammond, S. Conrad and I. Hermans, Dalton Trans., 2014, 43, 4513 RSC; W. N. P. van der Graaff, G. Li, B. Mezari, E. A. Pidko and E. J. M. Hensen, ChemCatChem, 2015, 7, 1152 CrossRef; J. Dijkmans, D. Gabriels, M. Dusselier, F. de Clippel, P. Vanelderen, K. Houthoofd, A. Malfliet, Y. Pontikes and B. F. Sels, Green Chem., 2013, 5, 2777 RSC; C. C. Chang, H. J. Cho, Z. Wang, X. Wang and W. Fan, Green Chem., 2015, 17, 2943 RSC; P. Y. Dapsens, C. Mondelli, J. Jagielski, R. Hauert and J. P. Ramirez, Catal. Sci. Technol., 2014, 4, 2302 Search PubMed.
- I. W. C. E. Arends and R. A. Sheldon, Appl. Catal., A, 2001, 212, 175 CrossRef CAS.
- I. Sádaba, M. L. Granados, A. Riisager and E. Taarning, Green Chem., 2015, 17, 4133 RSC; J.-P. Lange, Angew. Chem., Int. Ed., 2015, 54, 13186 CrossRef CAS PubMed.
- D. Scholz, C. Aellig, C. Mondelli and J. P. Ramirez, ChemCatChem, 2015, 7, 1551 CrossRef CAS; L. Bui, H. Buo, W. R. Gunther and Y. R. Leshkov, Angew. Chem., Int. Ed., 2013, 52, 1 CrossRef.
- R. Bermejo-Deval, R. S. Assary, E. Nikolla, M. Moliner, Y. Roman-Leshkov, S.-J. Hwang, A. Palsdottir, D. Silverman, R. F. Lobo, L. A. Curtiss and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 9727 CrossRef CAS PubMed.
- C. Hammond, D. Padovan, A. Al-Nayili, P. P. Wells, E. K. Gibson and N. Dimitratos, ChemCatChem, 2015, 7, 3322 CrossRef CAS PubMed.
- R. L. Hartman, J. P. McMullen and K. F. Jensen, Angew. Chem., Int. Ed., 2011, 50, 7502 CrossRef CAS PubMed.
- C. Perego and S. Peratello, Catal. Today, 1999, 52, 133 CrossRef CAS.
- R. Bermejo-Deval, R. Gounder and M. E. Davis, ACS Catal., 2012, 2, 2705 CrossRef CAS; R. Bermejo-Deval, M. Orazov, R. Gounder, S.-J. Hwang and M. E. Davis, ACS Catal., 2014, 4, 2288 CrossRef.
- S. Bordiga, E. Groppo, G. Agostini, J. A. van Bokhoven and C. Lamberti, Chem. Rev., 2013, 113, 1736 CrossRef CAS PubMed.
- P. Beato, E. Schachtl, K. Barbera, F. Bonino and S. Bordiga, Catal. Today, 2013, 205, 128 CrossRef CAS.
- S. Tolberg, A. Katerinopolou, D. D. Falcone, I. Sadaba, C. M. Osmundsen, R. J. Davis, E. Taarning, P. Fristrup and M. S. Holm, J. Mater. Chem. A, 2014, 2, 20252 Search PubMed.
- D. S. Mannel, S. S. Stahl and T. W. Root, Org. Process Res. Dev., 2014, 18, 1503 CrossRef CAS PubMed.
- Flow: 0.15 mL min−1; Sn-beta: 0.0352 g; volume: 0.14 cm3; max conversion: 87%.
- J. D. Lewis, S. V. de Vyver, A. J. Crisci, W. R. Gunther, V. K. Michaelis, R. G. Griffin and Y. R. Leshkov, ChemSusChem, 2014, 7, 2255–2265 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6gc01288d |
| This journal is © The Royal Society of Chemistry 2016 |