
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hydrogenation of oxalic acid using light-assisted water electrolysis for the production of an alcoholic compound†
Sho
Kitano
,
Miho
Yamauchi
*,
Shinichi
Hata
,
Ryota
Watanabe
and
Masaaki
Sadakiyo
International Institute for Carbon-Neutral Energy Research (WPI-I2CNER), Kyushu University, Motooka 744, Nishi-ku, Fukuoka 819-0395, Japan. E-mail: yamauchi@i2cner.kyushu-u.ac.jp; Tel: +81-92-802-6874
First published on 12th May 2016
Abstract
We demonstrate the production of glycolic acid, an industrially important alcoholic compound, via the electrochemical reduction of oxalic acid, which is procurable from biomass, and electro-oxidation of water with the help of renewable light energy for the first time. In principle, this new synthesis system is achievable while minimizing the consumption of fossil resources. We built a precious-metal free electrosynthesis system by employing a TiO2 cathode for oxalic acid reduction and a WO3 photoanode for water oxidation. The alcohol production proceeds during the application of electric power above 2.1 V in the dark. Notably, UV-visible light irradiation of the WO3 photoanode enables glycolic acid electrosynthesis above 0.5 V, which is lower (by 0.6 V) than the theoretical bias, i.e., 1.1 V. Glycolic acid electrosynthesis with an 80% high Faradaic efficiency was achieved on applying a bias of 1.5 V under UV-visible irradiation (λ > 300 nm).
Currently, the chemical industry largely depends on fossil fuels, which are used not only to generate power but also as hydrogen and carbon sources; fossil fuels are also believed to be a contributing factor to global warming due to the increase of CO2 concentrations. Efficient utilization of biomass resources derived from atmospheric CO2 could be a useful strategy to curb global warming by suppressing the consumption of fossil resources as a raw material.1–4 Recently, bio-alcohols, such as ethanol5 and ethylene glycol,6 have been commercialized and utilized as a fuel7 and feedstock.8 However, carbon resources included in biomass are not efficiently converted into alcohols, i.e. the carbon yield in the bio-alcohol production through fermentation is not beyond 50% and the residual carbon is released back into the air.9 Besides, the low production rate is due to the slow enzyme reaction.10
Catalytic hydrogenation of carboxylic acids has attracted much attention as a novel synthetic route for the production of alcoholic compounds from bio-derived materials.11 Several groups have succeeded in the catalytic hydrogenation of organic acids, such as acetic acid,12 stearic acid13 and aromatic carboxylic acids,14,15 to produce the corresponding alcoholic compounds. However, severe reaction conditions are required for the catalytic hydrogenation of organic acids, i.e. high pressure (2–6 MPa) and temperature (100–380 °C)15 with the use of H2 gas. The other drawback of the organic hydrogenation process is the utilization of highly reactive metal hydrides derived from fossil fuels as a hydrogen source, e.g., LiAlH4,16 (BH3)2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 17,18 and HSiEt3,19 to activate highly stable carboxyl groups due to their low electrophilicty20 in organic solvents, resulting in the formation of large amounts of waste.21
17,18 and HSiEt3,19 to activate highly stable carboxyl groups due to their low electrophilicty20 in organic solvents, resulting in the formation of large amounts of waste.21
Alternatively, light-energy-driven electrochemical water splitting using a photoelectrode, such as oxide,22–27 (oxy)nitride28–31 and sulfide,32 is regarded as a clean process for producing hydrogen from water using renewable solar energy and for providing an effective means of energy storage. In this study, we focus on the utilization of the hydrogen generated photocatalytically from water in chemical syntheses and photovoltaic effects to accelerate electrochemical reactions. Thus, the electrochemical reduction of carboxylic acids using water as a hydrogen source with the assistance of light energy would achieve the highly efficient hydrogenation of carboxylic acids with fossil-free hydrogen, i.e., light-energy-driven alcohol synthesis from fossil-free carboxylic acid and water, as shown in Fig. 1. This process is expected to become an excellent, environmentally friendly alcohol production route, if it progresses under largely milder conditions (0.1 MPa, <100 °C) than those of hydrogenation reactions.
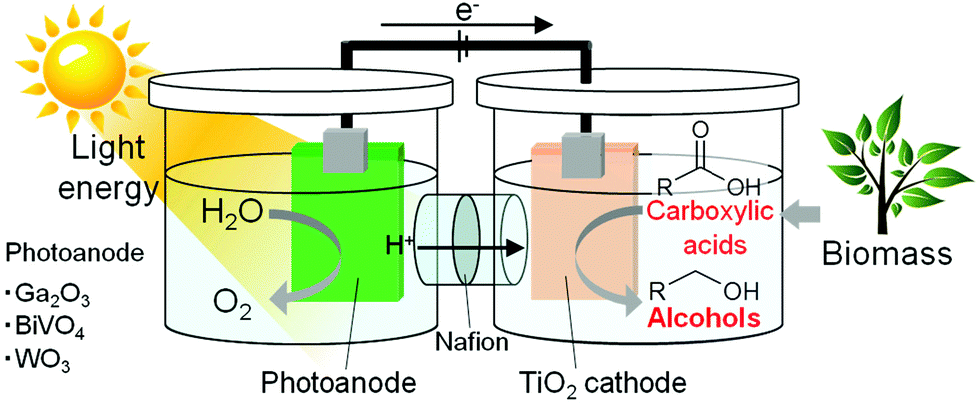 | ||
| Fig. 1 Schematic illustration of light-assisted alcohol electrosynthesis from a fossil-free carboxylic acid and water. | ||
Previously, we have demonstrated the electrochemical reduction of oxalic acid, (COOH)2, OX, a divalent carboxylic acid, to produce glycolic acid, HOOC-CH2OH, GC, an alcoholic compound (α-hydroxy carboxylic acid)33 through the 4-electron reduction of OX with oxidation of water as described in eqn (1)–(3).
Cathode
| (COOH)2 + 4 H+ + 4 e− → HOOC–CH2OH + H2O + 0.13 V vs. RHE | (1) |
Anode
| 2H2O → 4 H+ + 4 e− + O2 + 1.23 V vs. RHE | (2) |
Overall
| (COOH)2 + H2O → HOOCCH2OH + O2 1.1 V | (3) |
The OX is first reduced to glyoxylic acid (HOOC-COH, GO) through two-electron reduction, and then GC is produced from GO through further two-electron reduction34,35 (see Scheme S1 in the ESI†). The GC formation was found to proceed with high selectivity (>98%) and Faradaic efficiency (>95%) in an electrochemical cell equipped with a titanium(IV) dioxide (TiO2) cathode for OX reduction and a Pt wire anode for water oxidation, which corresponds to a direct electric power charge into an alcoholic compound.
Furthermore, in this study, a light-assisted electrochemical alcohol production system was fabricated by adapting oxide semiconductor photoelectrodes as the anode for water oxidation to supply protons and electrons for the hydrogenation of a carboxylic acid, which enables the direct conversion of light energy into low-carbon alcoholic chemicals. Here, we applied Ga2O3,36,37 BiVO438 and WO324,39 particles, which are known as highly active photocatalysts for water oxidation, as the photoanode to oxidize water and to provide electrons and protons for the electro-reduction of OX on an anatase-type TiO2 cathode under UV-visible light irradiation. We successfully produced GC from OX through an electrochemical reaction in the remarkably low bias potential range, i.e., ∼1.5 V, compared with the potentials without light irradiation, ∼2.5 V.
Commercially supplied Ga2O3 and WO3 powders were used in the electrochemical experiments. BiVO4 powder was prepared by using the homogeneous-precipitation method according to a previous report (see the Experimental section in the ESI†).40 All the samples were characterized by X-ray powder diffraction (XRD) and UV-visible diffuse reflection measurements. The Ga2O3, BiVO4 and WO3 powders showed XRD patterns attributable to monoclinic structures and photoabsorption spectra, which coincide with the structure and spectrum of materials reported as highly active photocatalysts for O2 evolution by water splitting (Fig. S1 and S2†).24,36–40 Anodes were prepared by applying a suspended mixture of water, acetylacetone (disperser), Toriton-X (thickener) and the oxide powders on conductive glass using a squeegee method,41 followed by calcination at 450 °C for 4 h. The TiO2 cathode was prepared by drying a methanolic suspension of TiO2 nanoparticles (Japan Reference Catalyst TiO2: JRC-TIO-8, an anatase-type TiO2) dropped onto Ti foil, followed by calcination at 450 °C for 0.5 h. Two-electrode systems were equipped using two-compartment cells, where the cathode and anode were separately mounted in each cell to evaluate an applied bias for OX reduction without re-oxidation of the reduced product at the anode as shown in Fig. 1. All electro-reduction experiments were conducted by introducing a 0.16 M OX cathode solution containing 0.16 M Na2SO4 (pH 1.2) and 0.16 M Na2SO4 anode solution (pH 5.8) at 25 °C. Fig. 2 and S3† show the current–voltage curves for the electro-reduction of OX on Ga2O3, BiVO4 and WO3 photoanodes in the dark, and under UV-visible light (λ > 200 nm) irradiation. We could observe reductive currents on all three anodes by applying external bias above 2.1 V in the dark, as shown in Fig. 2, indicating that the application of 1.0 V as the total overpotential, in addition to 1.1 V of the theoretical cell voltage (eqn (3)), is a requisite for initiating the GC production through OX reduction and water oxidation without light irradiation. Under light irradiation, minimal biases applied to flow reductive current in the system were drastically decreased compared with those observed in the dark. These results clearly indicate that the external applied bias can be enhanced by photoirradiation, causing photoexcitation over photoanodes absorbing UV-visible light energy. The chemical bias in the two-electrode system employing cathode (pH = 1.2) and anode (pH = 5.8) solutions is calculated to be 0.27 V based on the following equation:
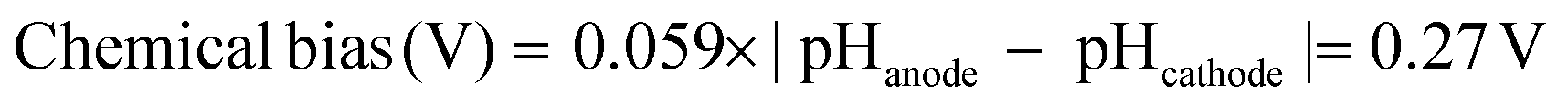
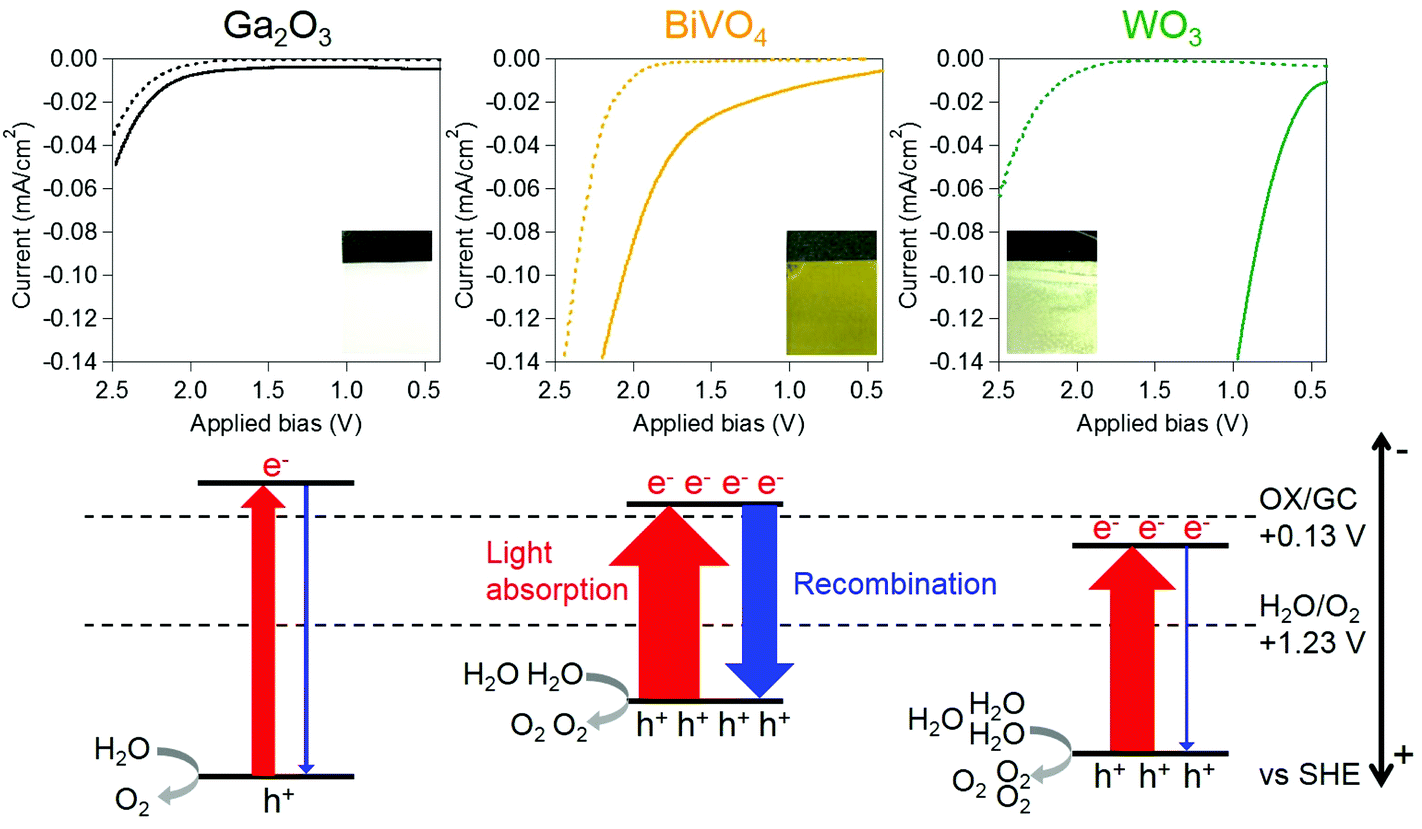 | ||
| Fig. 2 (Upper) Current–voltage curves of electro-reduction of OX using a two-electrode system employing a TiO2 (JRC-TIO-8) cathode in a solution containing OX (0.16 M) and a Na2SO4 solution (pH 1.2, 40 ml, 0.16 M) and Ga2O3, BiVO4 and WO3 photoanodes in Na2SO4 solution (pH 5.8, 40 ml, 0.16 M) at 25 °C under UV-visible light (λ > 200 nm) irradiation (solid line) or in the dark (broken line). The photographs show the photoanodes used for the experiment. (Lower) Schematic illustrations of energy diagrams and photovoltaic performances for the photoanodes under UV-visible light irradiation. | ||
Thus, we can beneficially utilize the chemical bias to lower the applied bias required for the reactions. On the other hand, we conducted OX reduction experiments both under light irradiation and in the dark to investigate the effects of light irradiation on the applied bias under the same experimental conditions. In acidic aqueous solutions having pH < 2, the photoanode composed of Ga2O3 or BiVO4 dissolves and photocatalytic oxidation of co-existing electrolyte anions, SO42−, proceeds on the WO3. Dissolution of the WO3 also occurs in strong alkaline solutions. Thus, in this study, we utilize an aqueous solution of Na2SO4 without pH control, which exhibits neutral pH conditions, for the efficient water oxidation reaction.
The onset potential for the reductive current flow under UV-visible irradiation light depended on the photoanodes; the order of onset bias was as follows: WO3 < BiVO4 < Ga2O3. This order coincides with the photovoltaic performance of photoanodes, i.e., WO3 > BiVO4 > Ga2O3. An observed decrease of the onset biases for the electro-reduction of OX is therefore associated with the photovoltaic performance, which depends on the amount of optically excited electrons and holes and recombination probabilities between such excited species, and corresponds to the activity of photoanodes for water oxidation. Photoexcitation probabilities for the generation of active species strongly depend on the range of photoabsorption wavelengths of photoanodes.42 A wider absorption range of the photoanodes achieves more efficient photoexcitation within the entire irradiation range (λ > 200 nm), generating a larger amount of excited electrons and holes. All the excited electrons and holes, however, are not available because the recombination between some parts of electrons and holes occurs before the water oxidation proceeds.43 Lower recombination probability enables higher reaction probability of holes and water molecules, resulting in higher activity of water oxidation and a larger amount of available excited electrons, i.e., higher photovoltaic performance.44 Therefore, the photoanode, which shows a wide photoabsorption spectrum and low recombination probability, is expected to exhibit a large bias decrease due to the high activity for water oxidation and photovoltaic performance.
The Ga2O3 photoanode exhibited the smallest bias decrease under light irradiation (0.15 V, as shown in Fig. 2) because the narrowest range of photoabsorption wavelengths of the Ga2O3 photoanode (up to 270 nm, as shown in Fig. S2†) corresponds to the smallest amount of excited electrons and holes (as illustrated in Fig. 2), thus resulting in the low activity for water oxidation and photovoltaic performance. Although the BiVO4 photoanode showed the widest absorption spectrum (up to 550 nm) of all the photoanodes, OX reduction over the photoanode required the application of a relatively high bias potential, i.e., 1.7 V, compared with 0.7 V, which was observed over the WO3 anode. This observation clearly shows that the recombination probability is more crucial for water oxidation over the BiVO4 anode. It is known that the BiVO4 photoanode shows a high recombination probability due to the poor mobility of excited electrons (Fig. 2).45–48 By contrast, the WO3 photoanode, absorbing photons at wavelengths less than 480 nm, exhibited the largest bias decrease of all the anodes, as shown in Fig. 2 and S2.† Therefore, the largest decrease of applied bias for OX reduction observed on the WO3 photoanode can be attributed to the relatively wide absorption spectrum and low recombination probability for efficient water oxidation and high photovoltaic performance.24
Reaction conditions of the most active WO3 catalyst were optimized to minimize the applied bias by changing the pH values over the anode and cathode to achieve efficient alcohol electrosynthesis. For this purpose, a three-electrode system, as described in Fig. S4,† was applied to determine an applied potential on the TiO2 cathode by comparing the cathode potential with a reference electrode potential, whereas we could measure the bias applied between a cathode and an anode in the two-electrode system. Electrochemical cells were filled with Na2SO4 aqueous solution (40 ml, 0.2 M) and the pH value in the cathode cell including OX (0.03 M) was controlled to be either 1.0 or 11 by adding H2SO4 or NaOH solution. TiO2 is known as a sufficiently stable oxide compound in both acidic and alkaline conditions and the TiO2 cathode stably worked during the catalytic experiments conducted in this study.
Fig. S5† shows the influence of the pH value on the activity for OX reduction and a product distribution in chronoamperometric electro-reduction with application of a constant cathode potential, i.e., −0.7 V vs. the reversible hydrogen electrode (RHE) at 50 °C for 2 h. At pH values higher than or equal to 7, we could not observe any reduction products, i.e., 0% OX conversion. Decreasing the pH value to less than or equal to 4 led to the generation of both 2- and 4-electron reduction products, i.e., GO and GC, respectively; yields of both the reduction products and the percentage of the GC yield increased with decreasing pH value. Considering that the production of GC and GO are attained via the hydrogenation of OX, a higher proton concentration under lower pH conditions confers a favorable condition for the hydrogenation reaction.49
The influence of the wavelength of irradiation light on the WO3 anode was examined by applying a two-electrode system, which comprised a TiO2 cathode in a solution containing OX (0.03 M) and Na2SO4 solution (pH 1.0, 40 ml, 0.2 M) and a WO3 photoanode in a Na2SO4 solution (pH 5.6, 40 ml, 0.2 M) at 50 °C. Fig. 3 shows the current–voltage curves both in the dark and under the irradiation of light of various wavelengths, such as UV-visible light with λ > 200 and 300 nm and visible light with λ > 400 nm. We observed onsets of approximately 0.5 V under the irradiation of light at all the tested wavelengths, which are 0.6 V smaller than the theoretical bias required for GC production viaOX reduction and water oxidation, i.e., 1.1 V, whereas the onset potentials were observed at approximately 2.1 V in the dark, suggesting that the remarkable bias decrease of 1.6 V was attained by light irradiation. Irradiation of light at λ > 200 nm and 300 nm covers wide energy regions of the absorption spectrum of WO3. Therefore, the rate determining-step for OX reduction under UV-visible light irradiation is probably not the water oxidation over the WO3 anode, where holes with sufficient positive potentials50 are generated, but the electro-reduction of OX on the cathode. Thus, we conclude that the irradiation of UV-visible light with λ > 300 nm efficiently works in the system. Furthermore, almost the same onset potential of reductive current was observed under visible light irradiation, indicating that the WO3 photoanode could efficiently work for the bias decrease under visible light irradiation. These results indicate that the system has applicability under the irradiation of both solar light and only visible light.
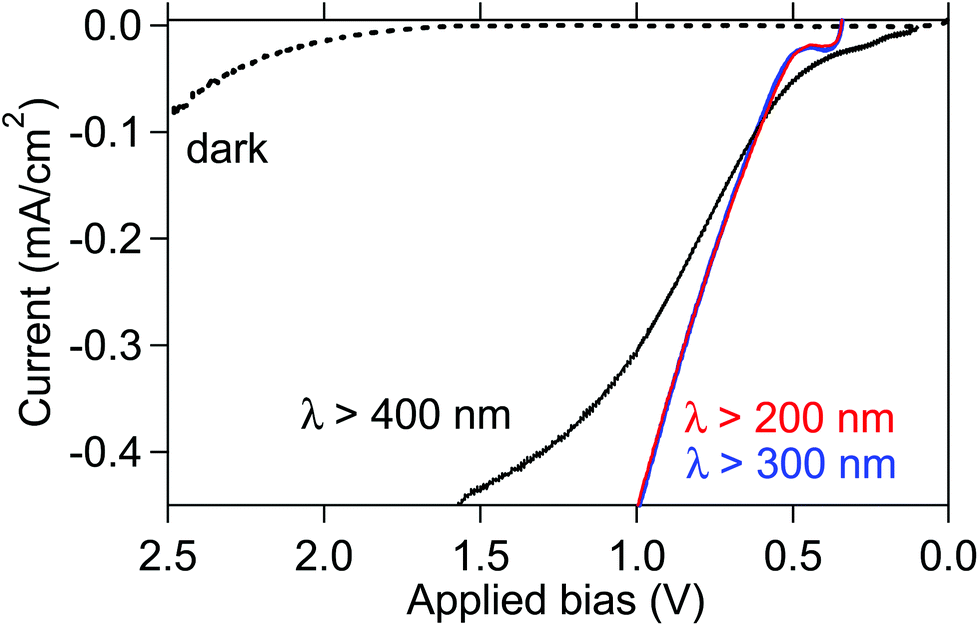 | ||
| Fig. 3 Current–voltage curves of electro-reduction of OX using a two-electrode system comprising a TiO2 (JRC-TIO-7) cathode in solution containing OX (0.03 M) and Na2SO4 solution (pH 1.0, 40 ml, 0.2 M) and a WO3 photoanode in Na2SO4 solution (pH 5.6, 40 ml, 0.2 M) under light irradiation, with wavelength >200 nm (red), >300 nm (blue) or >400 nm (black), and in the dark (broken line). | ||
Chronoamperometric electro-reduction of OX was conducted while applying an external bias of 1.0 or 1.5 V with a two-electrode system employing the TiO2 cathode in OX solution (pH 1.0, 40 ml, 0.03 M OX, 0.2 M Na2SO4) and the WO3 photoanode in Na2SO4 solution (pH 5.6, 40 ml, 0.2 M) at 50 °C for 2 h under irradiation of UV-visible light with λ > 300 nm, visible light with λ > 400 nm or in the dark. Fig. 4 shows the Faradaic efficiencies calculated based on the amount of GO and GC produced under various conditions. No products were detected in the dark even for an external bias below 1.5 V, which is in accordance with the result that no reductive current was observed in the current–voltage curve below 1.5 V, as shown in Fig. 3, and indicates that the system does not work below 1.5 V without irradiation. By contrast, GO and GC production was achieved under irradiations of both UV-visible and visible light, indicating that OX can be reduced with the assistance of light energy absorbed by the WO3 photoanode through an electrochemical reaction system. Higher Faradaic efficiencies for GO and GC production were achieved through the irradiation of UV-visible light and the application of a bias larger than 1.5 V, compared with efficiencies under the irradiation of visible light at 1.0 V. Note that GO and GC were produced with an 80% Faradaic efficiency in total on applying a bias of 1.5 V under the irradiation of UV-visible light. The anode potential on applying a bias of 1.5 V under the irradiation of UV-visible light was observed to be 1.05 V vs. RHE, which is 0.18 V more negative than the theoretical potential of water oxidation, 1.23 V vs. RHE, indicating that the hole generated by photoabsorption could oxidize water molecules.51,52 Furthermore, a constant reductive-current flow at approximately 0.5 mA cm−1 was observed on applying a bias of 1.5 V under the irradiation of UV-visible light for 2 h, except for the initial period, as shown in Fig. S7,† indicating that the system can stably work and reductive products are continuously obtained under the examined conditions. Gaseous products generated in both cathode and anode cells were also analyzed to clarify the entire Faradaic efficiency of the system. Fig. S8† shows the Faradaic efficiencies determined based on the amount of GO, GC and H2 that formed in the cathode cell in OX reduction and O2 that formed in water oxidation in the anode cell on applying a bias of −1.5 V under the irradiation of UV-visible light. Faradaic efficiencies for products in each cell reached 100%, indicating that the reaction substrates were only OX and water and all the electrons formed in water oxidation were consumed for the electro-reduction of OX or water to produce GO, GC and H2 through the circuit. H2 generation occurred with a low evolution rate (7.9 μmol h−1), as a side reaction of hydrogenation of OX on applying a bias of 1.5 V under irradiation of UV-vis light. In our electrochemical hydrogenation system, protons and electrons are provided from electrolyte and electrode separately and react with OX as shown in Scheme S1 (ESI†), resulting in GC production without H2 gas evolution. Therefore, the suppression of hydrogen evolution reaction (HER) over a TiO2 electrode is critically important to achieve a high Faradaic efficiency for OX reduction, which has been discussed in our previous report.33 The TiO2 electrode used in this work also exhibited high activities for OX reduction but low HER activities under the acidic conditions, as reported in Nature Materials.53 Based on these results, we succeeded in the first production of an alcoholic compound, GC, from a carboxylic acid, OX, via water oxidation using light energy through an electrochemical reaction system.
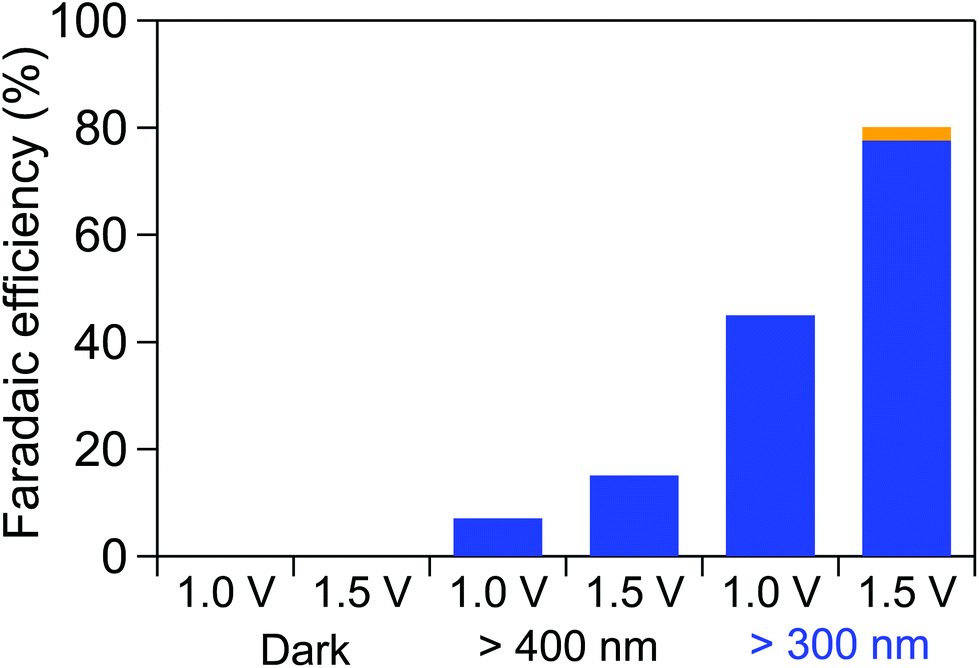 | ||
| Fig. 4 Faradaic efficiencies calculated based on the amount of GO (orange) and GC (blue) formed by applying an external bias of 1.0 or 1.5 V in a two-electrode system comprising a TiO2 (JRC-TIO-7) cathode in OX solution (pH 1, 40 ml, 0.03 M of OX, 0.2 M Na2SO4) and a WO3 photoanode in Na2SO4 solution (pH 5.6, 40 ml, 0.2 M) at 50 °C for 2 h under irradiation of UV visible light with λ > 300 nm, visible light with λ > 400 nm or in the dark. | ||
To improve the product selectivity, we provide an illustration of the relationship between the applied potentials on the TiO2 cathode and the Faradaic efficiencies for the electro-reduction of OX to GO and GC as shown in Fig. 5. The Faradaic efficiencies for GO and GC production in the dark were determined using the three-electrode system, which comprised a TiO2 cathode and Ag/AgCl reference electrode in solution containing OX (0.03 M) and Na2SO4 solution (pH 1.0, 40 ml, 0.2 M) and a Pt anode in Na2SO4 solution (pH 1.0, 40 ml, 0.2 M). Fig. 5 also shows the Faradaic efficiencies obtained using the two-electrode system under the irradiation of UV-visible light with λ > 300 nm and visible light with λ > 400 nm, which are displayed in Fig. 4. The Faradaic efficiencies depended on the TiO2 cathode potential and exhibited a volcano-like tendency regardless of the type of electrode system, indicating that the TiO2 cathode potential was the determining factor in the OX reduction system. The highest Faradaic efficiency, i.e., 99%, was achieved at approximately −0.6 V vs. RHE in the three-electrode system. Fig. S9† shows cyclic voltammogram curves measured using a three-electrode system employing a TiO2 cathode, Pt anode and Ag/AgCl reference electrode in the dark recorded in electrolyte solutions with and without the OX substrate, respectively. The OX reduction current steeply increased below −0.45 V vs. RHE, which implicates the larger current density of OX reduction at −0.6 V vs. RHE of the cathode potential compared to that at −0.45 V vs. RHE in the two-electrode system. On the basis of the results, higher Faradaic efficiencies are also expected to be realized by the negative shift of the cathode potential in the two-electrode system employing the WO3 photoanode.
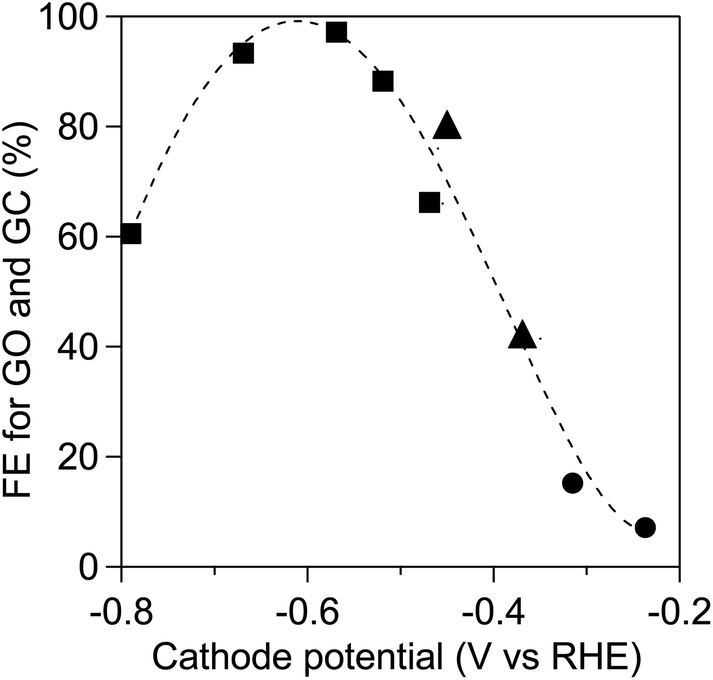 | ||
| Fig. 5 Faradaic efficiencies (FEs) of GO and GC production versus the TiO2 (JRC-TIO-7) cathode potential at 50 °C for 2 h in both the three-electrode system in the dark (square) comprising a TiO2 cathode and the Ag/AgCl reference electrode in a solution containing OX (0.03 M) and Na2SO4 solution (pH 1.0, 40 ml, 0.2 M) and a Pt anode in a Na2SO4 solution (pH 1.0, 40 ml, 0.2 M) and in the two-electrode system comprising a TiO2 (JRC-TIO-7) cathode in a solution containing OX (0.03 M) and Na2SO4 solution (pH 1.0, 40 ml, 0.2 M) and a WO3 photoanode in a Na2SO4 solution (pH 1.0, 40 ml, 0.2 M) under irradiation of UV-visible light (triangle) and visible light (circle). | ||
The performances of OX reduction in the light-assisted system were compared with those of our previous work and other catalytic hydrogenation research studies. The total yields and reaction rates for GO and GC productions in this and previous studies and results in ref. 12–15 are summarized in Table S1.† The total yields obtained using the two-electrode system employing a standard TiO2 catalyst in this work are relatively lower than those in the references because of the smaller active sites on the surface of the standard catalyst. On the other hand, the reaction rate using the three-electrode system employing the highly active porous TiO2 catalyst in our previous work is comparable to those in the references, indicating that the utilization of the highly active TiO2 catalyst having the sufficiently large reactive surface will probably achieve more efficient OX reductions.
In conclusion, we demonstrated GC production through OX electro-reduction and water oxidation using a light-assisted electrochemical reaction system that applies a TiO2 cathode and semiconductor oxide photoanodes. The reaction proceeds by applying a bias potential of more than 2.1 V without light irradiation. Irradiation of UV-visible light on the WO3 photoanode enables a drastic decrease of minimal bias, i.e., 0.5 V, which is 0.6 V smaller than the theoretical bias potential, i.e., 1.1 V, required for GC production viaOX reduction and water oxidation. GC electrosynthesis with an 80% Faradaic efficiency was achieved on applying a bias of 1.5 V under UV-visible irradiation (λ > 300 nm). These results are the first demonstration of a green synthetic process for the production of an alcoholic compound from an organic acid procurable from biomass via electro-oxidation of water with the assistance of light energy.
Acknowledgements
This work was supported by JST-CREST and JSPS KAKENHI grant numbers 25288030 and 24655040.References
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS PubMed.
- M. H. Haider, N. F. Dummer, D. W. Knight, R. L. Jenkins, M. Howard, J. Moulijn, S. H. Taylor and G. J. Hutchings, Nat. Chem., 2015, 7, 1028–1032 CrossRef CAS PubMed.
- T. Matsumoto, M. Sadakiyo, M. L. Ooi, S. Kitano, T. Yamamoto, S. Matsumura, K. Kato, T. Takeguchi and M. Yamauchi, Sci. Rep., 2014, 4, 5620 CAS.
- T. Matsumoto, M. Sadakiyo, M. L. Ooi, T. Yamamoto, S. Matsumura, K. Kato, T. Takeguchi, N. Ozawa, M. Kubo and M. Yamauchi, Phys. Chem. Chem. Phys., 2015, 17, 11359–11366 RSC.
- A. Demirbas, Prog. Energy Combust. Sci., 2007, 33, 1–18 CrossRef CAS.
- N. Ji, T. Zhang, M. Y. Zheng, A. Q. Wang, H. Wang, X. D. Wang and J. G. G. Chen, Angew. Chem., Int. Ed., 2008, 47, 8510–8513 CrossRef CAS PubMed.
- E. de Jong, Avantium, cochair IEA bioenergy Task 42, 2014, vol. 2, pp. 11–12 Search PubMed.
- P. Harmsen and M. Hackmann, Wageningen UR Food & Biobased Research, 2013 Search PubMed.
- H. Kuriyama, Microbe Engineering Handbook, 1990 Search PubMed.
- P. Alvira, E. Tomas-Pejo, M. Ballesteros and M. J. Negro, Bioresour. Technol., 2010, 101, 4851–4861 CrossRef CAS PubMed.
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539–554 RSC.
- T. P. Brewster, A. J. M. Miller, D. M. Heinekey and K. I. Goldberg, J. Am. Chem. Soc., 2013, 135, 16022–16025 CrossRef CAS PubMed.
- H. G. Manyar, C. Paun, R. Pilus, D. W. Rooney, J. M. Thompson and C. Hardacre, Chem. Commun., 2010, 46, 6279–6281 RSC.
- X. J. Cui, Y. H. Li, C. Topf, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2015, 54, 10596–10599 CrossRef CAS PubMed.
- M. Naruto and S. Saito, Nat. Commun., 2015, 6, 8140 CrossRef PubMed.
- J. Seyden-Penne, Reductions by the Alumino- and Borohydrides in Organic Synthesis, 2nd edn, 1997 Search PubMed.
- N. M. Yoon, C. S. Pak, H. C. Brown, S. Krishnam and T. P. Stocky, J. Org. Chem., 1973, 38, 2786–2792 CrossRef CAS.
- A. Pelter, M. G. Hutching, T. E. Levitt and K. Smith, J. Chem. Soc., Chem. Commun., 1970, 347–348 RSC.
- V. Gevorgyan, M. Rubin, J. X. Liu and Y. Yamamoto, J. Org. Chem., 2001, 66, 1672–1675 CrossRef CAS PubMed.
- P. A. Dub and T. Ikariya, ACS Catal., 2012, 2, 1718–1741 CrossRef CAS.
- N. Sakai, K. Kawana, R. Ikeda, Y. Nakaike and T. Konakahara, Eur. J. Org. Chem., 2011, 3178–3183 CrossRef CAS.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- F. Le Formal, N. Tetreault, M. Cornuz, T. Moehl, M. Gratzel and K. Sivula, Chem. Sci., 2011, 2, 737–743 RSC.
- F. Amano, D. Li and B. Ohtani, Chem. Commun., 2010, 46, 2769–2771 RSC.
- K. Iwashina and A. Kudo, J. Am. Chem. Soc., 2011, 133, 13272–13275 CrossRef CAS PubMed.
- J. Gu, Y. Yan, J. W. Krizan, Q. D. Gibson, Z. M. Detweiler, R. J. Cava and A. B. Bocarsly, J. Am. Chem. Soc., 2014, 136, 830–833 CrossRef CAS PubMed.
- D.-D. Qin, Y.-L. Li, T. Wang, Y. Li, X.-Q. Lu, J. Gu, Y.-X. Zhao, Y.-M. Song and C.-L. Tao, J. Mater. Chem. A, 2015, 3, 6751–6755 CAS.
- T. Minegishi, N. Nishimura, J. Kubota and K. Domen, Chem. Sci., 2013, 4, 1120–1124 RSC.
- K. Maeda, M. Higashi, B. Siritanaratkul, R. Abe and K. Domen, J. Am. Chem. Soc., 2011, 133, 12334–12337 CrossRef CAS PubMed.
- M. Higashi, K. Domen and R. Abe, J. Am. Chem. Soc., 2012, 134, 6968–6971 CrossRef CAS PubMed.
- M. G. Kibria, S. Zhao, F. A. Chowdhury, Q. Wang, H. P. T. Nguyen, M. L. Trudeau, H. Guo and Z. Mi, Nat. Commun., 2014, 5, 3825 CAS.
- T. Kato, Y. Hakari, S. Ikeda, Q. X. Jia, A. Iwase and A. Kudo, J. Phys. Chem. Lett., 2015, 6, 1042–1047 CrossRef CAS PubMed.
- R. Watanabe, M. Yamauchi, M. Sadakiyo, R. Abe and T. Takeguchi, Energy Environ. Sci., 2015, 8, 1456–1462 CAS.
- F. M. Zhao, F. Yan, Y. Qian, Y. H. Xu and C. Ma, J. Electroanal. Chem., 2013, 698, 31–38 CrossRef CAS.
- D. J. Pickett and K. S. Yap, J. Appl. Electrochem., 1974, 4, 17–23 CrossRef CAS.
- T. Hisatomi, K. Miyazaki, K. Takanabe, K. Maeda, J. Kubota, Y. Sakata and K. Domen, Chem. Phys. Lett., 2010, 486, 144–146 CrossRef CAS.
- Y. Sakata, Y. Matsuda, T. Yanagida, K. Hirata, H. Imamura and K. Teramura, Catal. Lett., 2008, 125, 22–26 CrossRef CAS.
- A. Kudo, K. Omori and H. Kato, J. Am. Chem. Soc., 1999, 121, 11459–11467 CrossRef CAS.
- K. Sayama, K. Mukasa, R. Abe, Y. Abe and H. Arakawa, Chem. Commun., 2001, 2416–2417 RSC.
- J. Q. Yu and A. Kudo, Adv. Funct. Mater., 2006, 16, 2163–2169 CrossRef CAS.
- S. Kitano, N. Murakami, T. Ohno, Y. Mitani, Y. Nosaka, H. Asakura, K. Teramura, T. Tanaka, H. Tada, K. Hashimoto and H. Kominami, J. Phys. Chem. C, 2013, 117, 11008–11016 CAS.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- S. Murakami, H. Kominami, Y. Kera, S. Ikeda, H. Noguchi, K. Uosaki and B. Ohtani, Res. Chem. Intermed., 2007, 33, 285–296 CrossRef CAS.
- X. B. Chen, S. H. Shen, L. J. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS PubMed.
- Y. Q. Liang, T. Tsubota, L. P. A. Mooij and R. Van de Krol, J. Phys. Chem. C, 2011, 115, 17594–17598 CAS.
- F. F. Abdi, T. J. Savenije, M. M. May, B. Dam and R. van de Krol, J. Phys. Chem. Lett., 2013, 4, 2752–2757 CrossRef CAS.
- M. Zhong, T. Hisatomi, Y. B. Kuang, J. Zhao, M. Liu, A. Iwase, Q. X. Jia, H. Nishiyama, T. Minegishi, M. Nakabayashi, N. Shibata, R. Niishiro, C. Katayama, H. Shibano, M. Katayama, A. Kudo, T. Yamada and K. Domen, J. Am. Chem. Soc., 2015, 137, 5053–5060 CrossRef CAS PubMed.
- T. W. Kim and K. S. Choi, Science, 2014, 343, 990–994 CrossRef CAS PubMed.
- N. R. De Tacconi, R. O. Lezna, R. Konduri, F. Ongeri, K. Rajeshwar and F. M. MacDonnell, Chem. – Eur. J., 2005, 11, 4327–4339 CrossRef CAS PubMed.
- D. E. Scaife, Sol. Energy, 1980, 25, 41–54 CrossRef CAS.
- K. Hashimoto, H. Irie and A. Fujishima, Jpn. J. Appl. Phys., Part 1, 2005, 44, 8269–8285 CrossRef CAS.
- Y. Nosaka, Comprehensive Nanoscience and Technology, in Nanomaterials, 2011, vol 1, pp. 571–605 Search PubMed.
- J. Gu, Y. Yan, J. L. Young, K. X. Steirer, N. R. Neale and J. A. Turner, Nat. Mater., 2016, 15, 456–460 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6gc01135g |
| This journal is © The Royal Society of Chemistry 2016 |