
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNon-isocyanate poly(amide-hydroxyurethane)s from sustainable resources†
Keren
Zhang
,
Ashley M.
Nelson
,
Samantha J.
Talley
,
Mingtao
Chen
,
Evan
Margaretta
,
Amanda G.
Hudson
,
Robert B.
Moore
and
Timothy E.
Long
*
Department of Chemistry, Macromolecules Innovation Institute, Virginia Tech, Blacksburg, VA 24061, USA. E-mail: telong@vt.edu
First published on 19th May 2016
Abstract
A two-step synthesis of epoxidation and carbonation afforded a hetero-functional AB monomer with cyclic carbonate and methyl ester (CC-ME) using plant oil-based methyl 9-decenoate and CO2. A unprecedented one-pot synthetic platform of CC-ME with 1,12-diaminododecane and poly(tetramethylene oxide) (PTMO)-based polyether diamine allowed synthesis of both nonsegmented poly(amide-hydroxyurethane) (PA12HU) and segmented PA12HU-PTMOs with varying polyether contents. 1H NMR spectroscopy confirmed complete conversion of cyclic carbonates and methyl esters to hydroxyurethanes and amides, respectively. Thermal analysis revealed distinctive thermal stability and transitions of PA12HU and PA12HU-PTMOs compared to their precursors and model oligomers. PA12HU and PA12HU-PTMOs were melt compression molded into semicrystalline, free-standing films, except for PA12HU-PTMO100 with 100% polyether diamine. PA12HU-PTMO100 was a viscous liquid with a glass transition temperature (Tg) of −64 °C and zero-shear melt viscosity of 449 Pa s. PA12HU formed a semicrystalline, rigid film with Tg of 11 °C. Polyether incorporation afforded creasable PA12HU-PTMO films with broad glass transitions near −50 °C. Thermal and thermomechanical analysis revealed significant phase-mixing of the hard and soft segments from annealed PA12HU-PTMO films. Polyether soft segments mixed with the amorphous hard segments, forming a miscible soft phase; crystallizable hard segments with ordered hydrogen bonding formed a hard phase. Surface morphological analysis of each PA12HU-PTMO film displayed ribbon-like, hard domains with composition-dependent aspect ratios. PA12HU-PTMOs exhibited higher moisture uptake than traditional thermoplastic polyurethane (TPU) due to resultant hydroxyls. Variable temperature FTIR spectroscopy demonstrated that ordered hydrogen bonding in the crystalline domains was disrupted or dissociated as the crystallites melted. Although tensile strength of segmented PA12HU-PTMOs proved lower than traditional polyurethanes due to phase-mixing, these compositions represent the first examples of film-forming, linear isocyanate-free polyurethanes with mechanical integrity and processability.
Introduction
Sustainable and environmentally friendly materials attract attention due to decreasing petroleum and arising environmental concerns. Polyurethanes provide thermoplastics and thermosets for many applications such as adhesives, sealants, coatings, commodities, automotive, packaging, and insulations materials. However, isocyanates, key starting materials for polyurethane synthesis, exhibit high reactivity and toxicity. Side reactions of isocyanates with water affect cost and demand many safety precautions during storage, transportation, and production of polyurethanes. Furthermore, isocyanate production commonly utilizes phosgene, a more reactive and toxic gas that causes acute safety and environmental hazards. Therefore, developing a more environmentally benign synthetic strategy for polyurethanes to avoid isocyanates remains a priority for many researchers. Literature from the past two decades highlights three primary phosgene-free synthetic routes for preparing non-isocyanate polyurethanes (NIPUs): polyaddition reactions of five-membered cyclic carbonates with amines,1,2 polycondensation of carbonates with amines,3,4 and polycondensation of AB type monomer containing acyl-azides with hydroxyls.5 Endo et al. demonstrated quantitative reactions of a cyclic carbonate with a primary amine after 24 h at room temperature, yielding an isomeric mixture of hydroxyurethanes with primary and secondary hydroxyls.6 This ring opening reaction has emerged as the most promising method to substitute the urethane-forming reaction of isocyanate and alcohol. Cyclic carbonates led to a synthetic route for creating polymer backbones or chemical crosslinks, allowing the synthesis of linear or crosslinked NIPUs for thermoplastics and thermosets, respectively. Webster first reviewed the progress of cyclic carbonate-functionalized monomers and NIPU synthesis, and others comprehensively summarized the most recent efforts in developing NIPUs.7–11 The earliest studies synthesized cyclic carbonate-containing acrylic, methacrylic, and vinyl monomers that allowed post-functionalization of free radically polymerized polymers, mostly used for thermoset materials.12–14 Several studies utilized sustainable resources to produce bio-based NIPUs, and the synthesis generally involves three steps: epoxidation of the carbon–carbon double bonds on an unsaturated fatty acid produces multi-oxiranes; carbon dioxide insertion converts oxiranes to cyclic carbonates; reaction of cyclic carbonates with amines affords crosslinked polyhydroxyurethane networks. Wilkes et al. synthesized a crosslinked NIPU with cyclic carbonate-functionalized soybean oil and amines, and measured their thermomechanical and tensile properties.2,15 Javni et al. also prepared soybean oil-derived, crosslinked NIPU thermosets with the highest stress at break near 6–7 MPa.16,17 Hybrid systems such as carbonated soybean oil-containing epoxy resins have shown comparable toughness with unmodified resins when cured with amines.18 Although further optimization is necessary, crosslinked NIPUs have demonstrated potential to match the mechanical performance of conventional polyurethane thermosets.However, progress lags on developing linear or branched NIPUs to compete with conventional thermoplastic polyurethanes (TPUs). Endo and many others ultilized polyaddition of bis(cyclic carbonate)s and diamine to synthesize linear NIPU.1,19–23 Cramail et al. recently reported the synthesis of aliphatic bis(6-membered cyclic carbonate)s using castor oil-based methyl undecenoate. Polymerization of the cyclic carbonates and diamine produced NIPUs with relative Mn values as high as 23 kDa.22 Another study from the same group also used methyl undecenoate to synthesize amide-containing bis(cyclic carbonate), and melt polymerization yielded amide-containing NIPUs. However, the products were blends due to heterogeneous mixtures of amines and cyclic carbonates. Despite many efforts devoted to developing linear NIPUs for thermoplastics, most studies were limited to synthesis, spectroscopic validations, and thermal properties. Only Nanclares et al. very recently reported stress–strain profiles of segmented bisphenol A-based NIPUs with elastomeric properties.24 To our knowledge, a report does not exist for bio-based NIPU thermoplastics with mechanical ductility or detailed morphological analysis.
This manuscript details the synthesis and characterization of nonsegmented and segmented NIPUs with various physical properties, ranging from film-forming solids to viscous liquids. A commercialized biochemical refinery produced methyl 9-decenoate using metathesis reactions of unsaturated fatty acids. Sequential epoxidation and carbonation yielded a cyclic carbonate-methyl ester (CC-ME) monomer, which is a stable solid at room temperature. An one-pot synthetic platform afforded a series of novel bio-based poly(amide hydroxyurethane)s (PAHUs) using CC-ME, an aliphatic diamine, and a polyether diamine. Polymerization proceeded through ring opening of five-membered cyclic carbonates and amidation of the methyl esters, forming both hydroxyurethanes and amides on the polymer backbone. The reported PAHU series spans both nonsegmented and segmented polyurethanes with tunable thermomechanical properties and crystallinity. The segmented PAHUs with aliphatic diamine incorporation were homogeneous melts during polymerization and compression molded into free-standing films. In addition, atomic force microscopy (AFM) and X-ray scattering experiments elucidated the influence of hard segment percentage on the morphology of annealed PAHU films. The PAHU with the highest soft segment percentage was a clear, viscous liquid that displayed shear thinning during rheological shear-sweep experiment. This manuscript represents the first report of bio-based, semicrystalline NIPUs with mechanical integrity, microphase-separated morphology, and processability. Various physical characterization techniques assisted in establishing the structure–morphology–property relationships of PAHUs. This versatile synthetic platform also enables the preparation of a wide variety of NIPUs with tunable properties using many diamines and cyclic carbonate-ester monomers.
Experimental section
Materials
Methyl 9-decenoate (9-DAME) was provided from Elevance Renewable Sciences, Inc. and used as received. meta-Chloroperoxybenzoic acid (m-CPBA, ≤77%), 1,12-diaminododecane (98%), lithium bromide (LiBr, ≥99%), 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD, 98%), sodium hydroxide solution (NaOH, 1.0 M in water), magnesium sulfate (MgSO4, 99%) sodium sulfite (NaSO3, ≥98%), sodium bicarbonate (NaHCO3, ≥99%), N-methyl-2-pyrrolidone (NMP, ACS grade), and sodium chloride (NaCl, ≥99%) were obtained from Sigma-Aldrich and used as received. Jeffamine® THF-100 is a poly(tetramethylene oxide)/poly(propylene glycol) copolymer-based diamine with 1 kDa molecular weight, obtained from Huntsman Corporation and used as received. Dry CO2 was obtained from Airgas and used as received. Ethyl acetate (EtOAc, ACS grade) and dichloromethane (DCM, ACS grade) were purchased from Spectrum Chemicals and used as received.Analytical methods
1H NMR and 13C NMR spectra were collected in CDCl3, deuterated trifluoroacetic acid (TFA), or deuterated hexafluoroisopropanol (HFIP) on an Agilent U4-DD2 spectrometer operating at 400 MHz, 23 °C. High resolution TOF mass spectroscopy (HRMS) was conducted on an Agilent 6220 mass spectrometer with a TOF analyzer in positive ion mode. Melting points were measured on an Electrothermal IA9000 melting point apparatus with a heating rate of 10 °C min−1. TA Instruments Q50 provided thermogravimetric analysis (TGA) results using a heating ramp at 10 °C min−1 under nitrogen purge. Thermal degradation temperatures (Td,5 wt%) corresponded to the temperature at 5% weight loss. Differential scanning calorimetry (DSC) experiments of all PAHU samples were conducted on a TA instruments Q1000 DSC utilizing a heat/cool/heat procedure under nitrogen flush of 50 mL min−1. Both heating and cooling rates were 10 °C min−1. Cryo-DSC characterized thermal transitions below −80 °C using a TA instruments Q100 DSC. Cryo-DSC employed a heat/cool/heat procedure with heating rate of 10 °C min−1 and cooling rate of 100 °C min−1. The midpoint of the transition in the second heating ramp determined glass transition temperatures (Tg).Dynamic mechanical analysis (DMA) utilized a TA Instruments Q800 dynamic mechanical analyzer in tension mode at an oscillatory amplitude of 10 μm, a frequency of 1 Hz, and a static force of 0.01 N. Rectangular film samples were cooled to −100 °C, equilibrated for 2 min, length measured, and heated at a rate of 3 °C min−1. The peaks of tan delta curves determined Tg values. A Veeco MultiMode scanning probe microscope provided AFM phase and height images using the tapping mode. Samples were imaged with Veeco's Nanosensor silicon tips with spring constants of 10–100 N at set-point ratios near 0.7. A TA Instruments Q5000 thermogravimetric sorption analyzer (TGA-SA) measured room-temperature water sorption of compression molded PAHU samples at relative humidity (RH) steps from 0–95% RH with 5% increments. Each RH step proceeded until the sample weight equilibrated with less than 0.01% weight change in 10 min. An instrumental pre-drying method at 25 °C and 0% RH was applied to each sample until the weight equilibrated. Water sorption was calculated based on the percentage weight gain of a dried sample. An Instron 5500R universal testing instrument measured tensile properties of several segmented PA12HU-PTMO rectangular film samples at a crosshead speed of 10 mm min−1. Tensile analysis data represented an average of five specimens with calculated standard deviations.
Small angle X-ray scattering (SAXS) and wide-angle X-ray diffraction (WAXD) experiments were performed using a Rigaku S-Max 3000 3 pinhole SAXS system, equipped with a rotating anode emitting X-ray with a wavelength of 0.154 nm (Cu Kα). The sample-to-detector distance was 1603 mm for SAXS and 109 mm for WAXD, and the q-range was calibrated using a silver behenate standard. Two-dimensional SAXS patterns were obtained using a 2D multiwire, proportional counting, gas-filled detector, with an exposure time of 1 h. Two-dimensional WAXD diffraction patterns were obtained using an image plate with an exposure time of 1 h. The SAXS data were corrected for sample thickness, and the SAXS/WAXD profiles were vertically shifted to facilitate a comparison of peak positions. All the SAXS and WAXD data were analyzed using the SAXSGUI software package to obtain radially integrated SAXS and WAXD intensity versus the scattering vector q (SAXS) or 2θ (WAXD) respectively, where q = (4π/λ)sin(θ), θ is one half of the scattering angle and λ is the X-ray wavelength.
Variable temperature FTIR (VT-FTIR) experiments were performed on a Varian 670-IR spectrometer (DTGS detector) with Pike Technologies variable temperature GladiATR™ attachment (diamond crystal). The spectra were collected at 4 cm−1 resolution and as an average of 32 scans. The samples were subjected to a temperature ramp of 1 °C min−1, starting from 30 °C to 180 °C, and FTIR spectra were collected every 10 °C beginning from 30 °C. Rheology experiments were conducted on a TA instrument Discovery AR-G2 rheometer with a cone and plate geometry (20 mm in diameter, 2° cone angle, truncation gap 60 μm). The samples were subjected to a shear sweep ramping from 1 s−1 to 200 s−1. All samples were maintained in a desiccator prior to an experiment.
Synthesis of 9,10-cyclic carbonate-methyl decanoate (CC-ME) monomer (Scheme 1)
In the first step, m-CPBA (33 g, 0.14 mol) and DCM (200 mL) were added to a 500 mL, round-bottomed flask equipped with a magnetic stir bar. The flask was placed into an ice bath with constant stirring until m-CPBA dissolved. 9-DAME (20 g, 0.11 mol) was then added into the solution and allowed to stir overnight. White precipitate was removed from the solution using vacuum filtration. DCM was then removed using a rotary evaporator. Additional white precipitate was removed again with vacuum filtration. The filtrate was washed with saturated NaSO3 solution two times, aqueous NaOH (1.0 M) three times, NaHCO3 two times, and brine two times. The resulting light yellow oil was placed at −20 °C overnight, filtered through a 5 μm membrane filter, and dried in vacuo (15 g, 69% yield). The structure and purity of obtained methyl 9,10-epoxydecanoate intermediate were confirmed using NMR spectroscopy (Fig. S1†). 1H NMR (400 MHz, CDCl3): 3.64 (s, 3H, Ha), 2.87 (m, 1H, Hb), 2.72 (dd, 1H, J1 = 3.9 Hz, J2 = 5.0 Hz, Hc), 2.44 (dd, 1H, J1 = 2.8 Hz, J2 = 5.0 Hz, Hd), 2.28 (t, 2H, J = 7.5 Hz, He), 1.59 (m, 2H, Hf), 1.50 (m, 2H, Hg), 1.46–1.19 (m, 8H, Hh). 13C NMR (100 MHz, CDCl3): 174.16, 52.25, 51.36, 47.01, 34.00, 32.39, 29.16, 29.08, 28.95, 25.84, 24.84. HRMS (ES+): m/z calculated for [M + H]+ 201.1485 g mol−1; found 201.1481 g mol−1.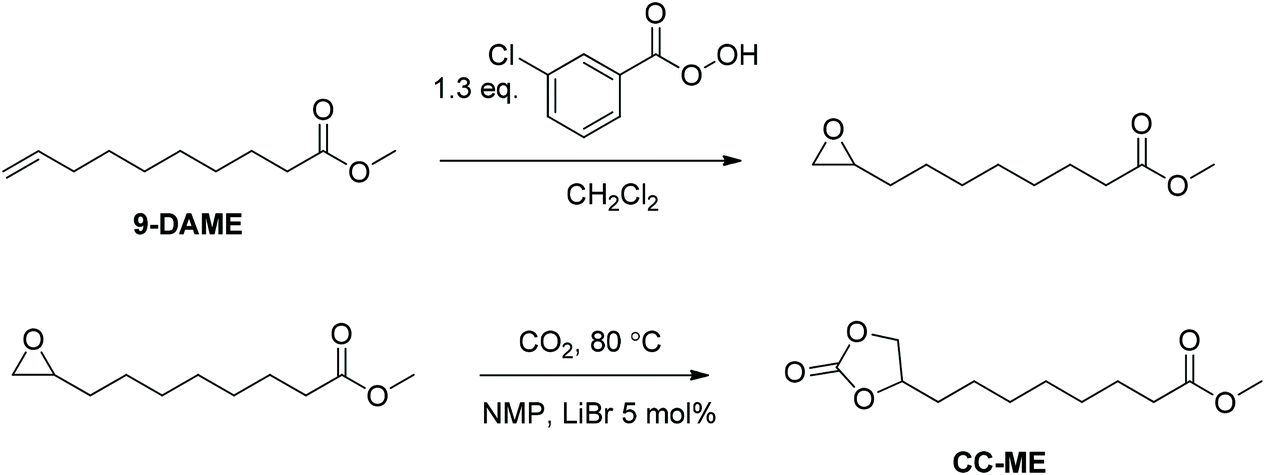 | ||
| Scheme 1 Synthesis of cyclic carbonate-methyl ester (CC-ME) monomers. | ||
In the second step, methyl 9,10-epoxydecanoate (4.0 g, 20 mmol), NMP (10 mL), and LiBr (0.087 g, 1.0 mmol) were added into a 100 mL, round-bottomed flask equipped with a magnetic stir bar. The reaction mixture was stirred at 80 °C with constant CO2 bubbling for 24 h. After cooling back to room temperature, the reaction mixture was dissolved in 100 mL EtOAc and washed with 150 mL brine. The organic phase was dried with MgSO4 and rotatory evaporated to remove EtOAc. Resulting light brown oil was dried in vacuo at 50 °C for 24 h and cooled to room temperature to obtain a salmon color solid (4.9 g, 89% yield) (Fig. S2†). 1H NMR (400 MHz, CDCl3, Fig. S2†): 4.67 (m, 1H, Ha), 4.50 (dd, 1H, J1 = 8.2 Hz, J2 = 8.4 Hz, Hb), 4.05 (dd, 1H, J1 = 7.2 Hz, J2 = 8.4 Hz, Hc), 3.65 (s, 3H, Hd), 2.29 (t, 2H, Hf), 1.82–1.55 (m, 4H, Hg), 1.46–1.28 (m, 8H, Hh). 13C NMR (100 MHz, CDCl3): 174.14, 154.98, 76.93, 69.32, 51.44, 33.95, 33.84, 28.92, 28.88, 28.85, 24.77, 24.28. HRMS (ES+): m/z calculated for [M + H]+ 245.1384 g mol−1; found 245.1384 g mol−1. Melting point 36–37 °C.
One-pot melt polymerization of nonsegmented poly(amide-hydroxyurethane) (PA12HU) (Scheme 2)
9,10-Cyclic carbonate-methyl decanoate (CC-ME) (2.0 g, 8.2 mmol), 1,12-diaminododecane (2.5 g, 8.9 mmol), and TBD (0.057 g, 0.41 mmol) were charged to a dry, 100 mL, round-bottomed flask. The flask was equipped with an overhead stir rod, argon inlet, and connection to vacuum through a dry ice-isopropanol trap. After argon purge for 30 min, the flask was heated to 70 °C to allow melting of the solids. The flask was then heated from 70 °C to 190 °C with constant stirring over 24 h, and the pressure was subsequently reduced to 0.06 mbar at 190 °C for additional 2 h. The resulting polymer was collected without further purification. The structure and purity of PA12HU were confirmed using 1H NMR spectroscopy (Fig. S11†).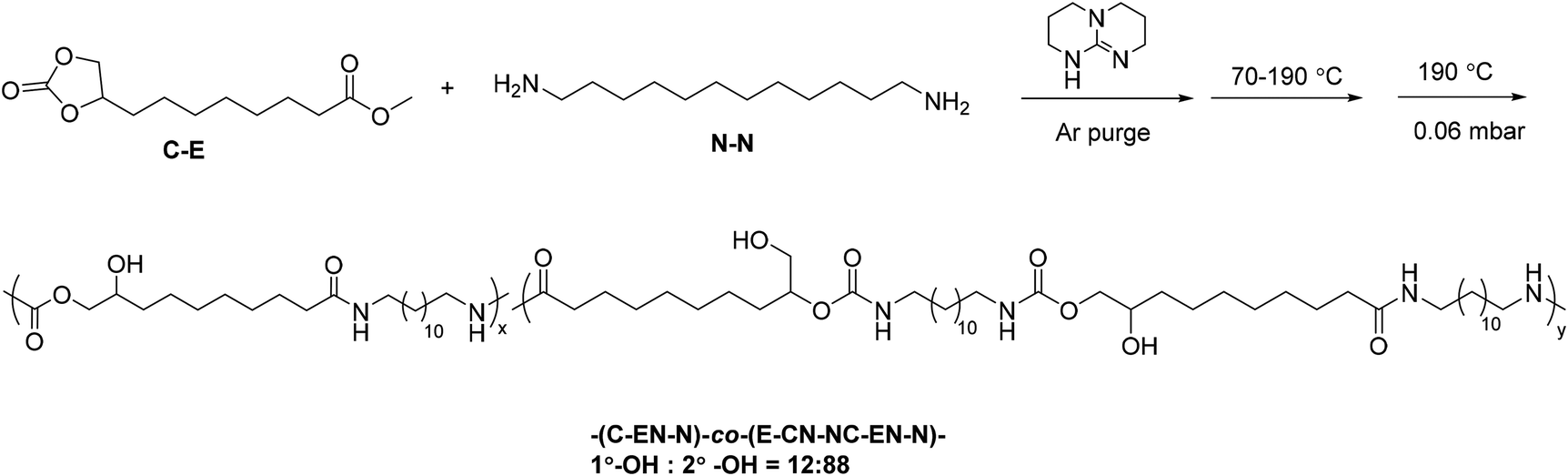 | ||
| Scheme 2 Synthesis of nonsegmented poly(amide-hydroxyurethane) (PA12HU) copolymers. | ||
One-pot melt polymerization of segmented poly(amide-hydroxyurethane) with PTMO soft segment (PA12HU-PTMO) (Scheme 3)
A typical polymerization of segmented PAHU-PTMO was conducted as following: 9,10-cyclic carbonate-methyl decanoate (CC-ME) (2.0 g, 8.2 mmol), 1,12-diaminododecane (0.84 g, 4.2 mmol), PTMO-diamine (THF-100 Jeffamine®) (4.2 g, 4.1 mmol), and TBD (0.057 g, 0.41 mmol) were charged to a dry, 100 mL, round-bottomed flask. The flask was equipped with an overhead stir rod, argon inlet, and connection to vacuum through a dry ice-IPA trap. After argon purge for 30 min, the flask was heated to 70 °C to allow melting of the solids. The flask was then heated from 70 °C to 180 °C with constant stirring over 3 d, and the pressure was subsequently reduced to 0.06 mbar at 180 °C for additional 3 h. The resulting polymer was collected without further purification. The structure and purity of segmented PAHU-PTMO were confirmed using 1H NMR spectroscopy (Fig. S12†).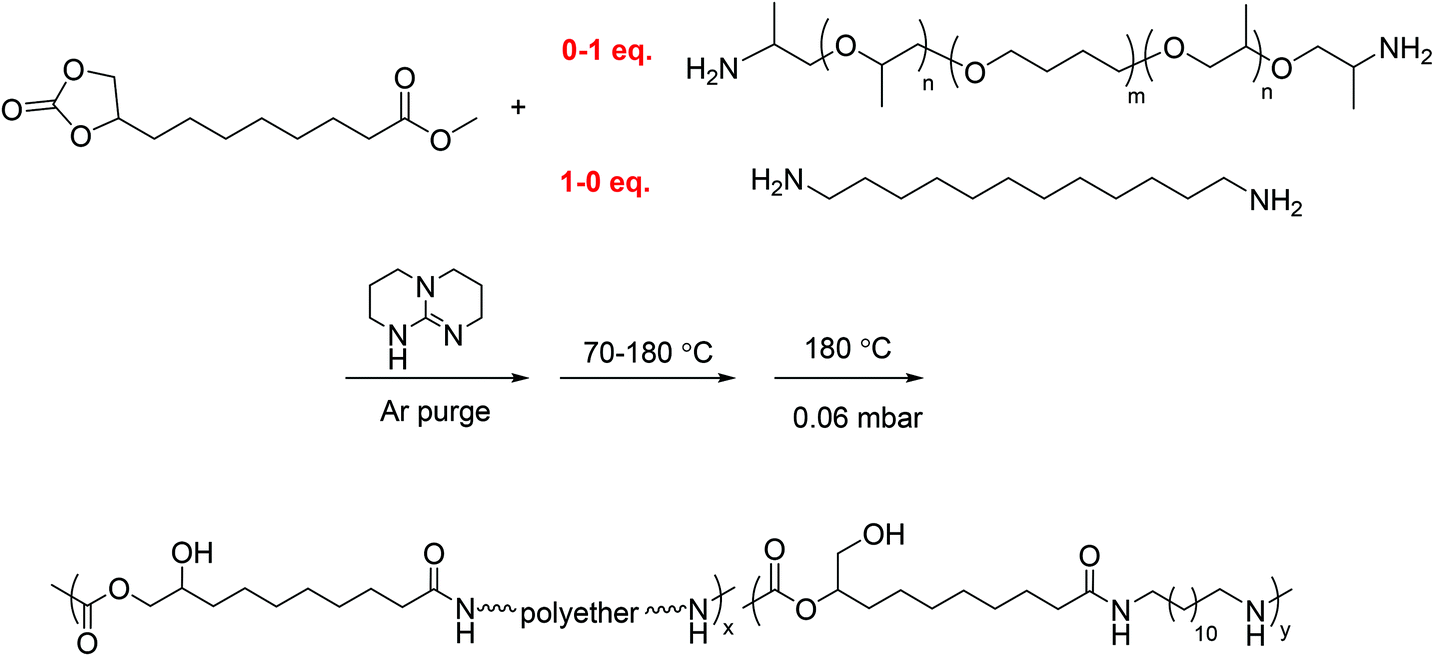 | ||
| Scheme 3 One-pot melt polymerization of segmented poly(amide-hydroxyurethane) with PTMO-based polyether 1 kDa soft segment (PA12HU-PTMO). | ||
Processing and annealing conditions
Nonsegmented PA12HU was melt compression molded at 160 °C for 5 min under 3 ton of compression force and cooled to room temperature, sandwiched between two Kapton® sheets and two steel plates. All segmented PA12HU-PTMO, except for PAHU-PTMO100, were melt compression molded at 130 °C for 5 min under 3 ton of compression force and cooled to room temperature, sandwiched between two Kapton® sheets and two steel plates. Compression molded films were annealed in vacuo for 12 h at 130 °C. Films were cooled using a step-wise procedure wherein the oven temperature was reduced 30 °C and allowed to equilibrate for 3 hours, with the process repeated until room temperature was reached.Results and discussion
Synthesis of CC-ME hetero-difunctional AB monomer
This manuscript reports a synthetic route for a difunctional AB monomer with cyclic carbonate and methyl ester from renewable resources. A proprietary olefin metathesis technology of Elevance Renewable Sciences, Inc. afforded multifunctional small molecules from plant oils. An epoxidation reaction converted methyl 9-decenoate (9-DAME), one of the produced difunctional small molecules, to methyl 9,10-epoxydecanoate (Scheme 1). TLC and NMR spectroscopy confirmed complete removal of the byproduct meta-chlorobenzoic acid to prevent contamination of mono-functional small molecules. In the second step, insertion of a CO2 molecule to the epoxy ring generated a five-membered cyclic carbonate (Scheme 1). The CO2 insertion was quantitative after 24 h. Another one-pot method from Davies et al. also proved effective in converting olefins to cyclic carbonates but less efficient.25 CC-ME monomer synthesis provided near 20 g of pure product with potential scalability. Fig. 1a summarizes the entire process from plant oil to polymerizable, multi-functional monomer. This synthetic route provided an efficient way in using a greenhouse gas as a reagent in functional monomer and polymer synthesis.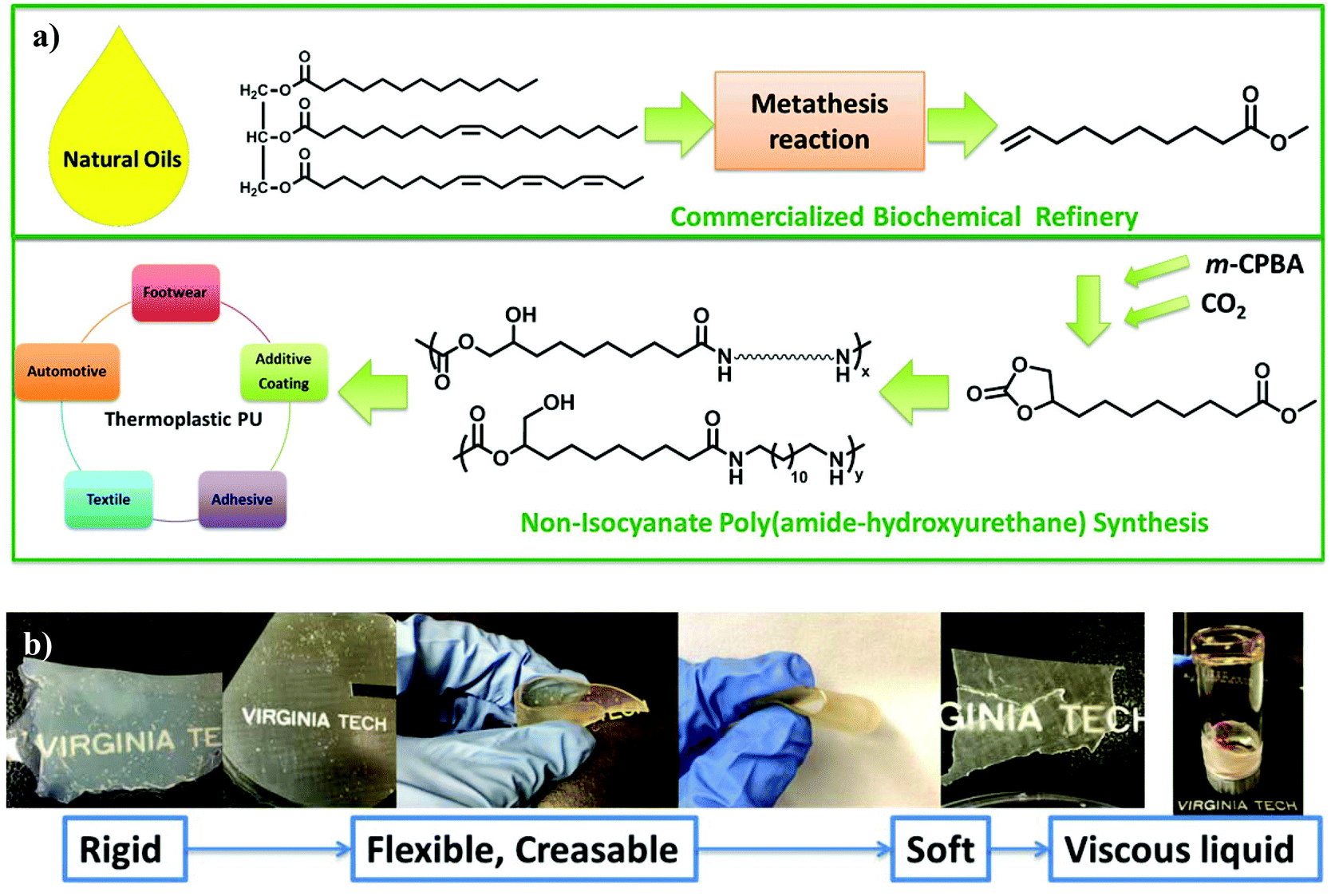 | ||
| Fig. 1 (a) Process of linear segmented PA12HU-PTMO synthesis from unsaturated fatty acid for thermoplastic applications. (b) Melt compression molded films of nonsegmented PA12HU and segmented PA12HU-PTMOs and a liquid of PAHU-PTMO100, with increasing PTMO mol% from left to right. | ||
Nonsegmented PA12HU synthesis
High monomer conversion proves crucial for step growth polymerization.26 Successful polymerization of CC-ME monomer requires quantitative reactions for both the methyl ester and cyclic carbonate functional groups. Investigation of model compound reactions assisted the optimization of reaction conditions for hydroxyurethane formation and amidation (Schemes S1–S3 and Fig. S3–S8†). Scheme 2 illustrates the synthesis of a nonsegmented PAHU using one-pot melt polymerization of CC-ME and 1,12-diaminododecane. The melt polymerization proceeded under an inert atmosphere in one pot. The initial temperature at 70 °C melted the starting materials and minimized sublimation. The final vacuum step aimed to completely remove the methanol byproduct and reach high conversion. Selection of 1,12-diaminododecane rather than 1,6-diaminohexane mitigated the sublimation. The nonsegmented PAHU with dodecyl spacers (PA12HU) existed as a melt-processable, hard solid (Fig. 1b) that dissolved in TFA and HFIP, but not in common organic solvents for conventional TPUs. The challenging solubility in common solvents prohibited size exclusion chromatography (SEC) analysis of PA12HU. Overlaying NMR spectra of PA12HU and CC-ME in deuterated TFA verified the complete conversion of cyclic carbonates and methyl esters (Fig. S9 and S10†). 1H NMR spectroscopy in deuterated HFIP confirmed the structure of PA12HU using a model compound hydroxyurethane methyl ester as the reference (Fig. S11†). PA12HU contained two repeating units as shown in Scheme 2: (C–EN–N) and (E–CN–NC–EN–N) (C: cyclic carbonate, E: methyl ester, N: primary amine). Each repeating unit consisted of isomers with primary and secondary hydroxyls in ratio of 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :88, respectively. Separation of polymers with primary or secondary hydroxyls proved challenging due to the limited solubility in common solvents. The ratio of primary to secondary hydroxyls is tunable through variation of catalyst, reaction temperature, and amine according to Endo et al.19,20
:88, respectively. Separation of polymers with primary or secondary hydroxyls proved challenging due to the limited solubility in common solvents. The ratio of primary to secondary hydroxyls is tunable through variation of catalyst, reaction temperature, and amine according to Endo et al.19,20
Segmented PA12HU-PTMO synthesis
The synthesis of segmented PA12HUs with polyether soft segments proceeded using a one-pot melt polymerization (Scheme 3). Telechelic polyether oligomers with low glass transitions often serve as soft segments for synthesizing segmented polyurethanes, polyureas, and polyamides.27,28 This study utilized commercially available Jeffamine® THF-100, a PTMO-based polyether diamine with approximately 1 kDa molecular weight. Differing the ratio of PTMO-diamine to 1,12-diaminododecane yielded a series of segmented PA12HU-PTMOs with varying extent of soft segment incorporation (Table 1). The synthetic strategy maintained a stoichiometric balance of total diamines and CC-ME to yield segmented PA12HU-PTMOx, where x represents the mol% of PTMO-diamine relative to the total amines. Synthesis of PA12HU-PTMO followed a similar procedure as nonsegmented PA12HU synthesis. Higher polyether diamine mol% led to longer overall reaction time for PA12HU-PTMO synthesis, due to lower concentration of reactive groups in the mixture. THF-100 Jeffamine® features primary amines bonded to secondary carbons rather than primary carbons of 1,12-diaminododecane, which also affected reactivity. 1H NMR confirmed the structure of PAHU-PTMO polymers (Fig. S12†), however, the ratio of the primary to secondary hydroxyls remained undetermined due to the overlapping resonances of the protons near hydroxyls and the protons on polyether carbons. Compression molding of PA12HU-PTMO10 to PA12HU-PTMO67 yielded homogenous, free-standing films (Fig. 1b). PA12HU-PTMO10 film was rigid, similar to nonsegmented PA12HU, while PA12HU-PTMO25–67 films were creasable. The nonsegmented PAHU-PTMO100 existed as a transparent, viscous liquid. Fig. 1b lists the physical appearance of various products with different PTMO-diamine content. Fig. 1a summarizes the entire isocyanate-free synthetic route of linear poly(amide-urethane)s using unsaturated fatty acid and CO2.| PA12HU | PA12HU-PTMO10 | PA12HU-PTMO25 | PA12HU-PTMO33 | PA12HU-PTMO50 | PA12HU-PTMO67 | PAHU-PTMO100 | PTMO-diamine | |
|---|---|---|---|---|---|---|---|---|
| PTMO mol% in total amines | 0% | 10% | 25% | 33% | 50% | 67% | 100% | NA |
| Soft segment wt% | 0% | 19% | 39% | 47% | 59% | 68% | 80% | NA |
| T d (°C, 5 wt%) | 272 | 273 | 302 | 296 | 305 | 316 | 319 | 273 |
| T g (°C) | 11 | NA | −44 | −47 | −53 | −59 | −64 | −85 |
| T m (°C) | 113, 128, 152 | 103, 143 | 61, 96, 127 | 54, 90, 139 | 46, 76, 103 | 39, 68, 93 | NA | 9 |
| ΔHm (J g−1) | 98 | 67 | 52 | 41 | 25 | 15 | 0 | 29 |
| Percent of crystallinity (WAXD) | 25 | 19 | 12 | 13 | 9 | 9 | 0 | NA |
A rheological shear-sweep experiment measured melt viscosities of PAHU-PTMO100 and 1 kDa PTMO diamine to be 449 Pa s and 0.2 Pa s at 25 °C, respectively (Fig. S16†). PAHU-PTMO100 started to shear thin near a shear rate of 25 s−1, while 1 kDa PTMO diamine remained in the linear viscoelastic region up to 200 s−1. The higher viscosity and shear thinning reflected the higher molecular weight of PAHU-PTMO100 compared to the 1 kDa precursor. Hydrogen bonding of the urethanes and amides also contributed to higher viscosity and longer relaxation time for PAHU-PTMO. Direct calculation of molecular weight for PAHUs based on melt viscosity proved challenging. PAHU-PTMO100 exhibited miscibility with DMF, but column interactions prohibited the elution of PAHU-PTMO100 from the SEC with a mobile phase of DMF + 0.05 M LiBr. PA12HU-PTMO solids only dissolved in HFIP and TFA, which limited the availability of molecular weight characterization through SEC.
Thermal analysis
Fig. 2 shows representative TGA weight loss profiles of PA12HU and PA12HU-PTMOs, along with CC-ME monomer and PTMO-diamine controls. Both PA12HU and PA12HU-PTMO demonstrated enhanced thermal stability compared to CC-ME due to polymer formation. Td,5 wt% of PA12HU matched conventional polyurethanes, with the urethane bonds degrading above 210 °C.29 PA12HU clearly exhibited a two-step degradation profile. The second step started near 400 °C, consistent with polyamide degradation.30 Weight loss of PTMO-diamine started near 300 °C and progressed in a single step. PA12HU-PTMOs displayed higher onsets of weight loss with increasing polyether incorporations compared to PA12HU, due to decreasing amount of urethane bonds. PA12HU-PTMOs each followed a single weight loss step due to polyamide content decreasing with increasing polyether incorporation. PA12HU and PA12HU-PTMOs also displayed higher thermal stability than trimers in the model compound analyses, indicative of polymer formation (Fig. S13 and S14†).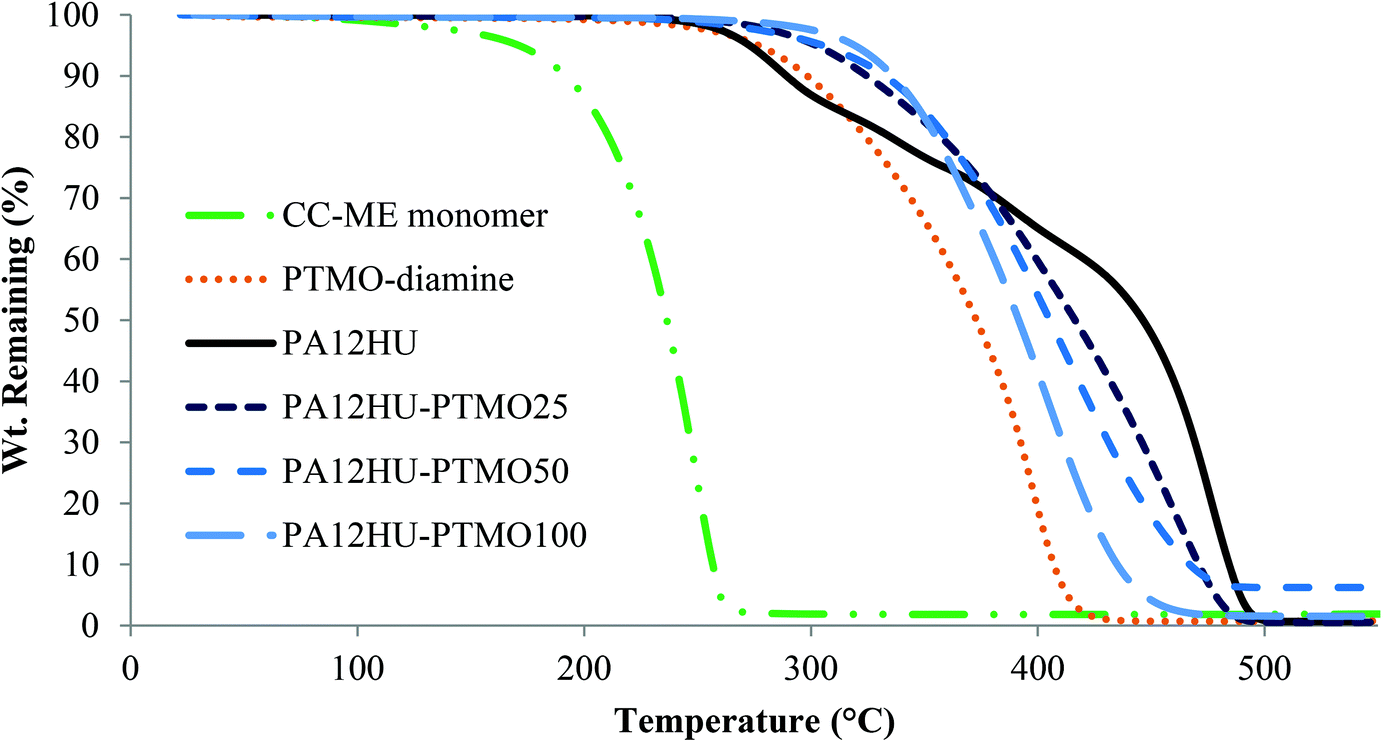 | ||
| Fig. 2 Representative TGA thermograms of nonsegmented PA12HU and segmented PA12HU-PTMOs, along with PTMO-diamine precursor and CC-ME monomer under nitrogen purge. | ||
DSC revealed thermal transitions of PA12HU and PA12HU-PTMOs, comparing to PTMO diamine (Fig. 3), CC-ME monomer (melting point 36–37 °C), and trimers (Fig. S14†). The differences between their thermal transitions again confirmed polymer formation. Table 1 summarizes compositions and thermal properties of PA12HU and PA12HU-PTMOs. Heat of fusion (ΔHm) was normalized against weight percent of the hard segments. PA12HU showed a Tg of 11 °C and multiple melting exothermic peaks, indicative of a semicrystalline polymer. The complex melting exotherms presumably resulted from different crystalline structures, due to possible differences in packing of the dodecane units flanked when by amides or hydroxyurethanes. However, other explanations such as recrystallization of imperfect crystals and re-melting lamellae with different thicknesses should not be ruled out without further investigation.31 Bidentate hydrogen bonding of amide and urethane units generally assists crystallization in polyamides and polyurethanes, respectively.28 The highest melting point of PA12HU was significantly lower than typical polyamide melting points (180–270 °C),32 due to hydroxyurethane units disrupting the long range periodic packing of polyamide segments. Shorter amide segment length generally results in thinner lamellae with lower melting points. The primary hydroxyls and secondary hydroxyls on the polymer chains also possibly interfered with chain packing, as most reported poly(hydroxyurethane)s are amorphous.10
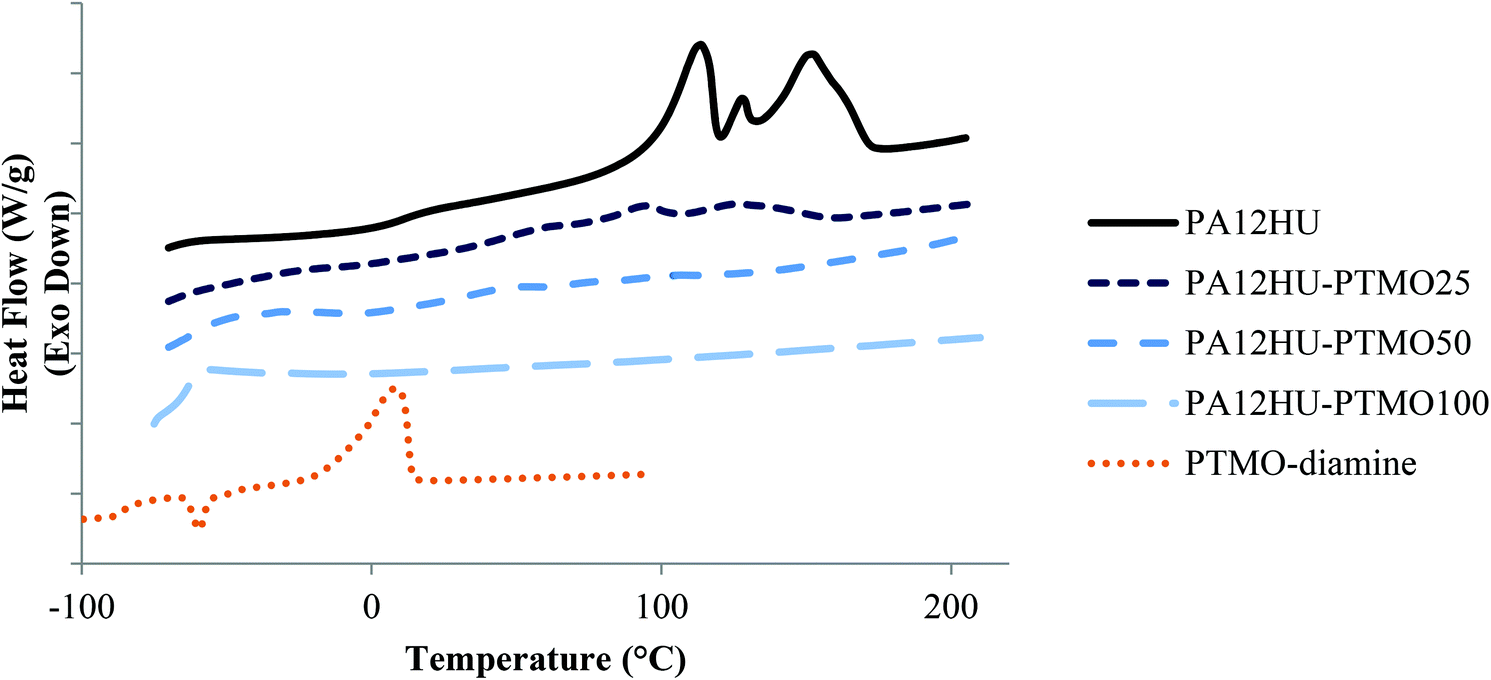 | ||
| Fig. 3 Representative DSC thermograms of nonsegmented PA12HU and segmented PA12HU-PTMOs, compared with PTMO-diamine 1 kDa. | ||
DSC thermograms of segmented PA12HU-PTMOs contained melting exotherms except for amorphous PAHU-PTMO100 (Fig. 3). However, the ΔHm values were significantly lower than the nonsegmented PA12HU (Table 1). Furthermore, ΔHm decreased with increasing soft segment content. This discussion only involves qualitative comparisons due to unknown ΔHm for fully crystalline PAHU. Although the DSC profile of PTMO-diamine showed an exothermic melting peak near 0 °C, PA12HU-PTMOs did not display any thermal transition indicative of crystallinity of the soft segments. Polymer formation and relatively low polyether molecular weight (1 kDa) hindered the crystallization of the soft segments, as commonly seen in segmented polyurethanes and polyamides.27,33,34 Higher polyether incorporations led to lower crystallinities and melting temperatures, mainly due to a dilution effect of crystallizable hard segments.35,36 Hydrogen bonding in close proximity typically proves more effective in forming ordered structure due to cooperative effect. Flexible polyether segments between the hydrogen bonding groups resulted in shorter hard segment lengths and therefore weaker neighboring cooperative effects. This interference strengthened when the hard segments phase-mixed with polyether segments.
Amorphous PAHU-PTMO100 showed a single Tg at −63 °C, nearly 20 °C higher than the PTMO diamine (Tg = −85 °C), indicative of complete phase-mixing of polyether segments and urethane/amide-containing segments. Hydrogen bonding of the urethanes and amides resulted in higher Tg of PAHU-PTMO100 relative to PTMO diamine. Tg values of segmented PA12HU-PTMOs increased and broadened with increasing hard segment content, indicative of phase-mixing (Fig. S15†). This phase-mixing likely resulted from relatively short polyether segment (low degree of polymerization, N), similar polarity of hard segments and polyether segments (small Flory–Higgins interaction parameter, χ), or both.37 Yu et al. observed similar phase-mixing properties of segmented polyamides with 460 g mol−1 poly(propylene oxide) soft segment.31 The aliphatic spacers in CC-ME monomer and 1,12-diaminododecane contributed to more hydrophobic hard segments of PA12HU-PTMOs than conventional segmented polyurethanes, which commonly utilize butane diol or ethylene glycol as chain extenders.
DMA investigated the thermomechanical properties of segmented PA12HU-PTMOs from −100 °C to 100 °C (Fig. 4). All compression molded films showed similar moduli in the glassy region below −70 °C. Modulus curves started to drop near −60 °C, representing onset of the glass transition. The following gradual decreases of moduli and broad tan delta peaks indicated significant phase-mixing of the soft segments and the amorphous hard segments, which agreed with the DSC results. Tan delta peaks shifted to lower temperature with increasing polyether contents, attributed to increasing low-Tg component of the amorphous phase. The crystalline phase maintained the mechanical integrity for PA12HU-PTMO films, affording small plateau regimes from 0 °C to 40 °C, which became more distinguishable at higher polyether contents. Crystalline domains of segmented PA12HU-PTMO films served as reinforcing fillers to preserve the modulus level with increasing temperature until the onset of melting. The final drop in moduli corresponded to crystallites melting, and the observed temperature agreed well with melting temperatures from DSC.
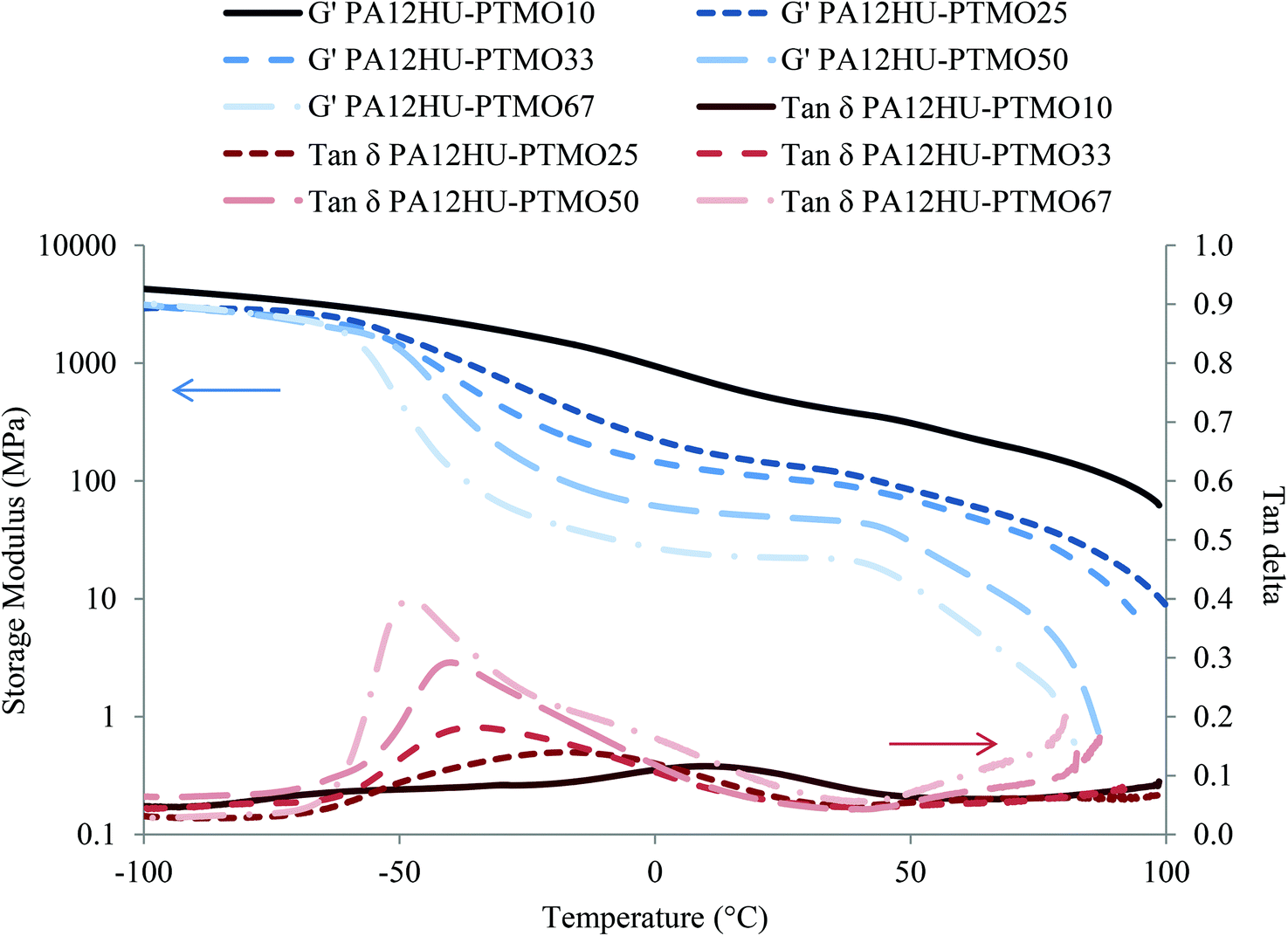 | ||
| Fig. 4 Dynamic mechanical temperature ramps for compression molded segmented PA12HU-PTMO films. | ||
Comparing the series of PA12HU-PTMOs with varying soft segment contents, samples with higher polyether mol% exhibited lower moduli and bigger tan delta peaks. The magnitude of tan delta peak generally correlates to the relative amount of the amorphous phase, which undergoes glass transition.31 PA12HU-PTMOs with higher polyether contents proved less crystalline, demonstrating a similar trend to the DSC results (Table 1). Increasing soft segment also led to earlier onset of flow due to reduction in both crystallinity and hydrogen bonding. Two broad tan delta peaks of PA12HU-PTMO10 near −70 °C and 10 °C suggested the presence of phase-separation. The tan delta peak of PA12HU-PTMO67 also contained a shoulder around 0 °C. However, phase-mixing still dominated the thermomechanical properties of PA12HU-PTMOs. Annealing at 130 °C afforded insignificant effects on the DMA results for PA12HU-PTMO films.
Morphology characterization
X-ray scattering elucidated the bulk morphology of annealed PA12HU and PA12HU-PTMO films. Annealing the samples at 130 °C, above the Tg of all PA12HU-PTMOs and near the Tm of nonsegmented PA12HU, and subsequent slow, step-wise cooling facilitated crystallization of segmented PA12HU-PTMOs. Small angle X-ray scattering (SAXS) revealed the microphase-separated bulk morphology of these semicrystalline PA12HU-PTMO films in Fig. 5. SAXS determined that the films contained both crystalline and amorphous phases. The amorphous phase consisted of intermixed polyether segments and non-crystallizable portions of the hard segments. SAXS profiles exhibited relatively broad scattering maxima, attributed to interlamellar scattering from PA12HU crystallites. Increasing the polyether content shifted the scattering peak from 0.6 nm−1 to 0.4 nm−1, corresponding to a shift in the long period (interlamellar spacing between crystallites) from 10.5 nm to 15.7 nm, respectively. The decrease in scattering peak intensity related to an overall decrease in % crystallinity (Xc), consistent with DSC (Fig. 3) and WAXD (Fig. 6 and Table 1). Additionally, the decrease in intensity likely resulted from a decrease in long range interlamellar ordering as amorphous content filled spaces between the lamellae.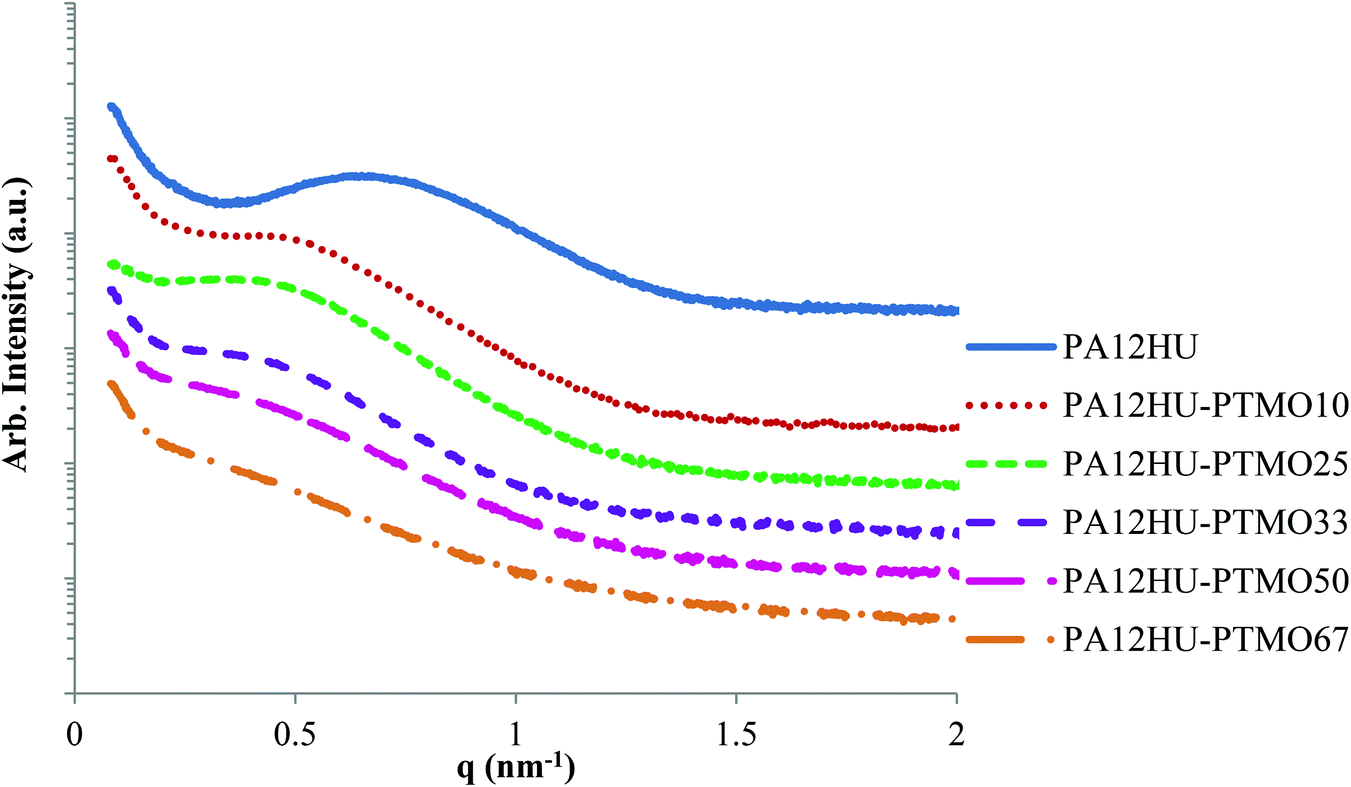 | ||
| Fig. 5 SAXS for compression molded films of nonsegmented PA12HU and segmented PA12HU-PTMOs after annealing at 130 °C for 12 h. | ||
 | ||
| Fig. 6 WAXD for compression molded films of nonsegmented PA12HU and segmented PA12HU-PTMOs after annealing at 130 °C for 12 h. Curves were shifted vertically for visual clarity. | ||
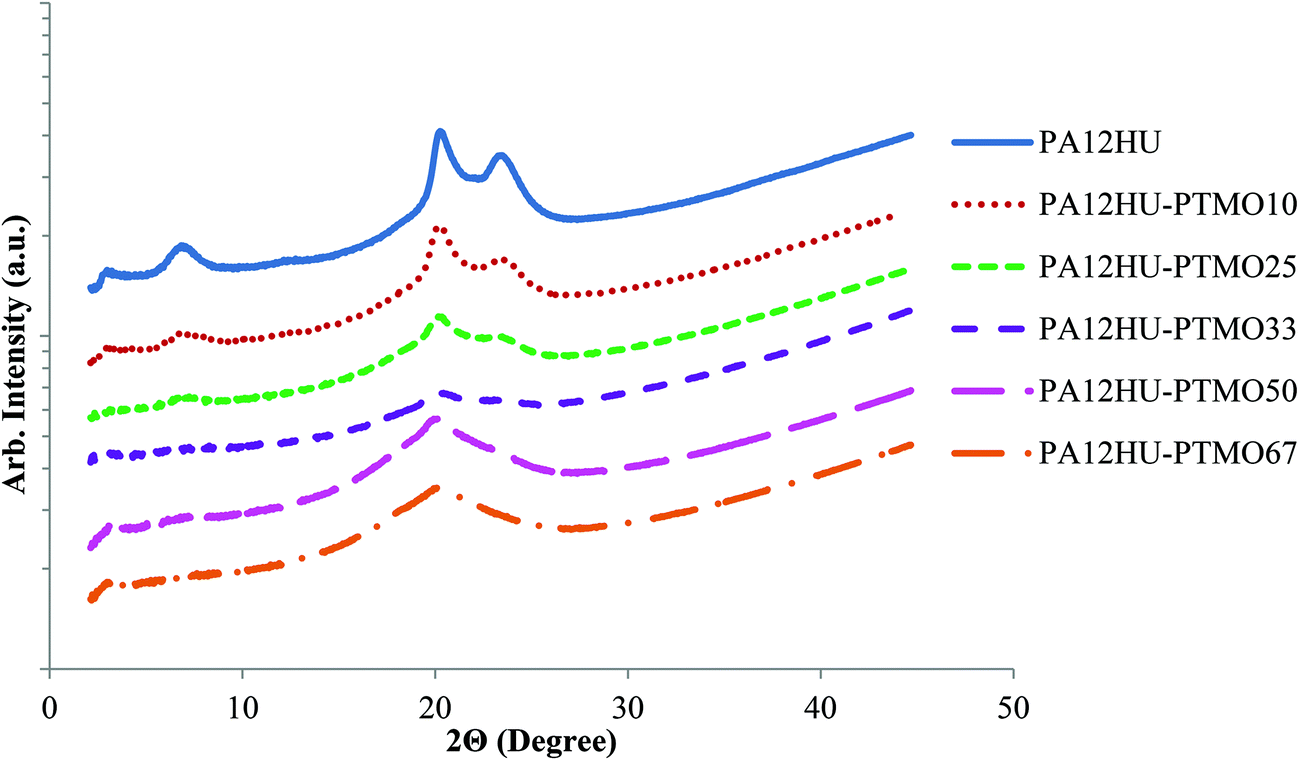
WAXD further enabled characterization of the degree of crystallinity of annealed PA12HU and PA12HU-PTMO films (Fig. 6). Prominent crystalline peaks were observed for nonsegmented PA12HU, and WAXD measurements were taken at room temperature, above the Tm of pure PTMO (Table 1). Therefore, these peaks corresponded to the crystalline order of the poly(amide-hydroxyurethane) segment in PA12HU-PTMO films. As shown in Fig. 6, these crystalline reflections appeared at 6.5°, 20.0°, and 23.0°. Table 1 summarizes the degree of crystallinity, Xc, calculated for each copolymer using the WAXD data. Crystalline peaks were deconvoluted using a fit of Gaussian and Lorentzian functions with minimized residuals (Fig. S18†). The ratio of combined area under the three crystalline peaks to total area determined the degree of crystallinity. As expected, the degree of crystallinity in the segmented block copolymer decreased with increasing polyether content (i.e. a decrease in the amount of the crystallizable hard segment).
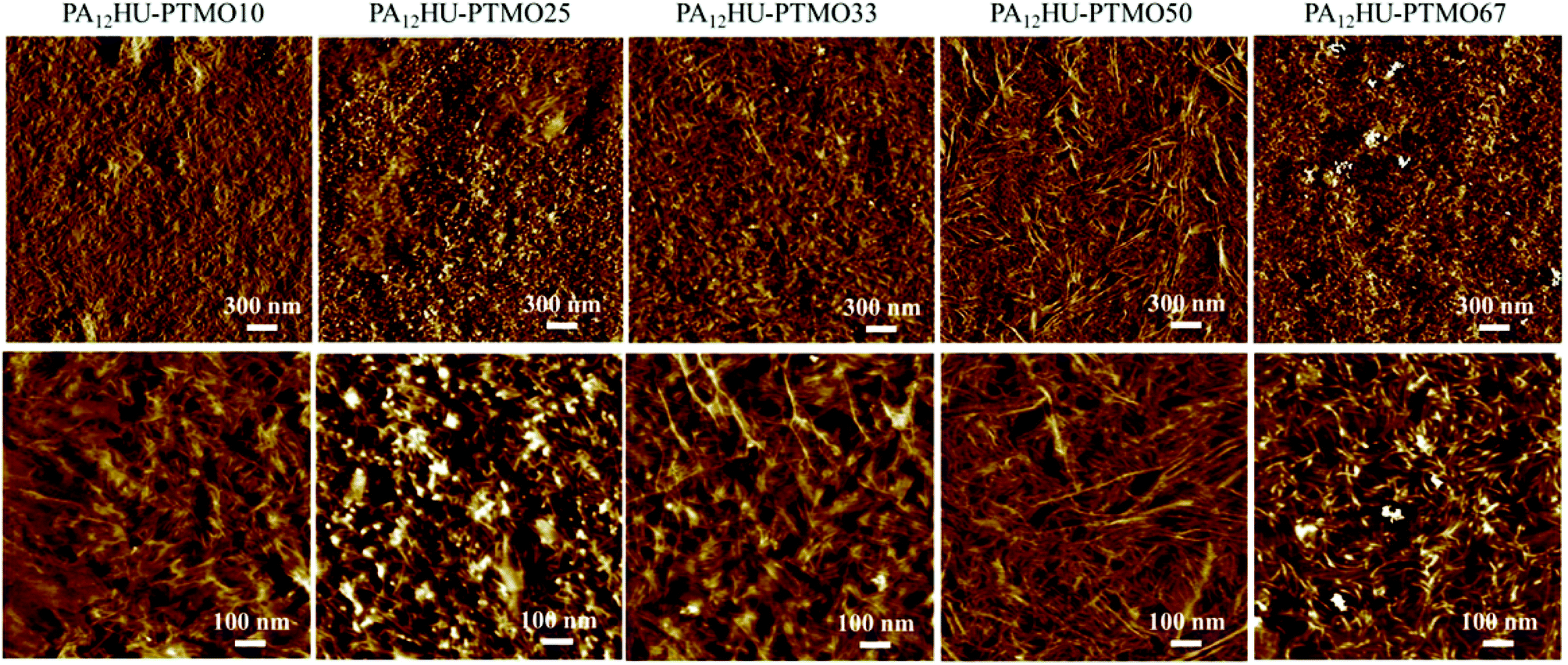
Atomic force microscopy (AFM) commonly applies to surface morphological characterization of segmented polyurethanes and polyamides.38Fig. 7 reveals the microphase-separated surface morphologies of annealed PA12HU-PTMO films. The light areas represented the hard, crystalline domains, and the dark areas represented the amorphous, soft domains, which contained intermixed polyether segments and non-crystallized hard segments. All PA12HU-PTMO samples displayed ribbon-like crystalline domains on the surface. AFM images of nonsegmented PA12HU showed a continuous crystalline matrix with isolated amorphous domains (Fig. S17†), which explained its rigidity. The incorporation of polyether segments contributed to densely packed ribbons for PA12HU-PTMO25 and PA12HU-PTMO33. With increasing soft segment content, the crystalline phase formed more loosely packed ribbons randomly dispersed in the amorphous matrix for PA12HU-PTMO50 and PA12HU-PTMO67. The wider-field images more clearly depicted the general trend of light, crystalline phase shrinking with increasing polyether incorporation, agreeing with both DSC and WAXD results. The width of these ribbons appeared rather constant with changing hard segment contents, likely due to the chain-folding mechanism.39 However, the lengths of crystalline ribbons appeared to increase with decreasing hard segment content from PA12HU-PTMO10 to 50. Increasing the content of polyether incorporation in the mixed amorphous phase led to lower Tg and higher chain mobility, which facilitated long-range packing of the crystallizable hard segments. The increasing lengths of the crystalline domains compensated for their loosening distribution as polyether content increased, which explained the relatively constant interlamellar spacing observed from SAXS. When polyether content increased beyond 50 mol%, the hard segment content proved insufficient to form one-micron size ribbons, accounting for the shorter crystalline domains in PA12HU-PTMO67. The crystalline domains of PA12HU-PTMO67 displayed more curvature, which explained the disappearance of the scattering peak in Fig. 5 for the SAXS profile of PA12HU-PTMO67.
 | ||
| Fig. 7 AFM phase images of compression molded segmented PA12HU-PTMOs after annealing at 130 °C for 12 h. | ||
Variable temperature FTIR spectroscopy (VT-FTIR)
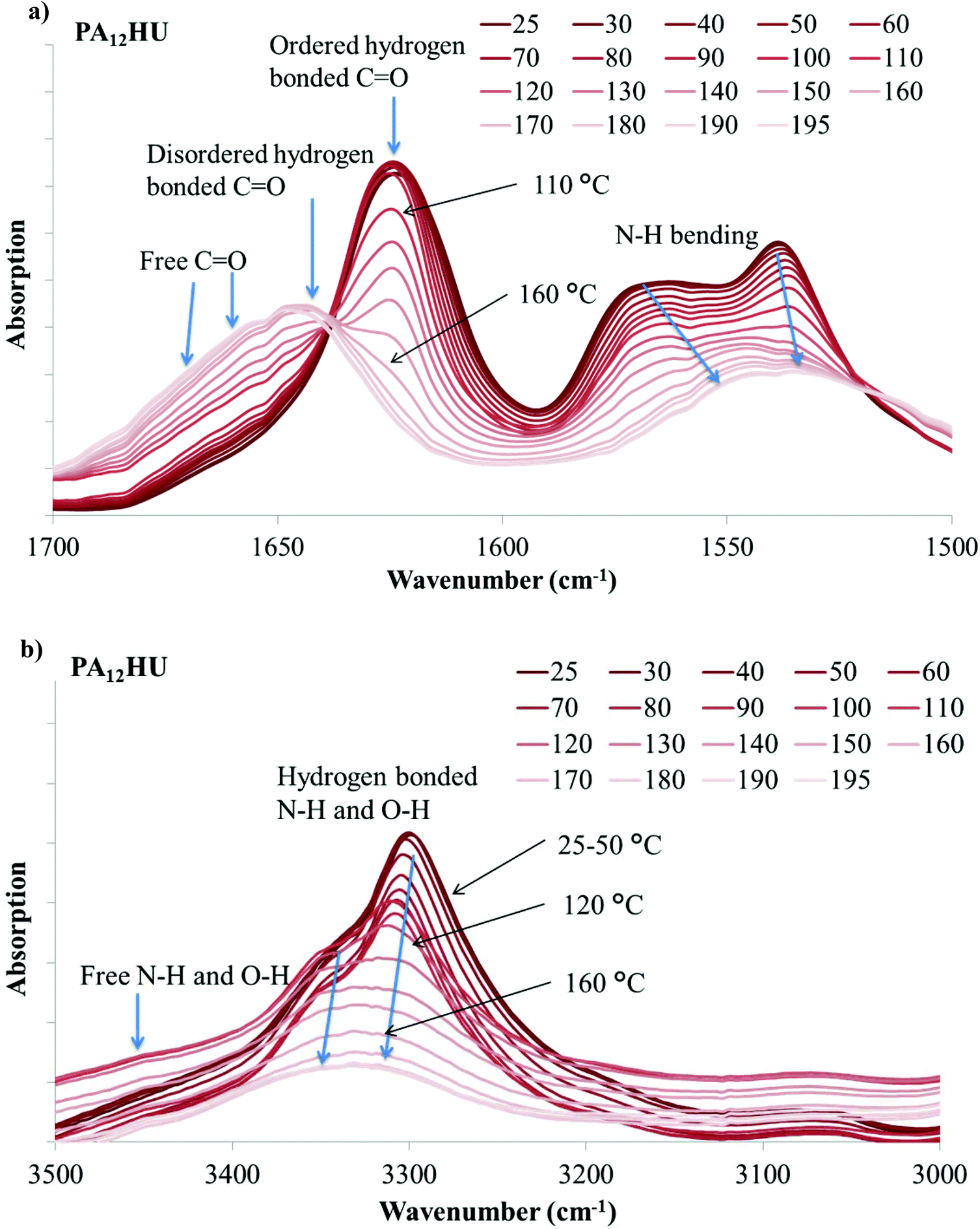
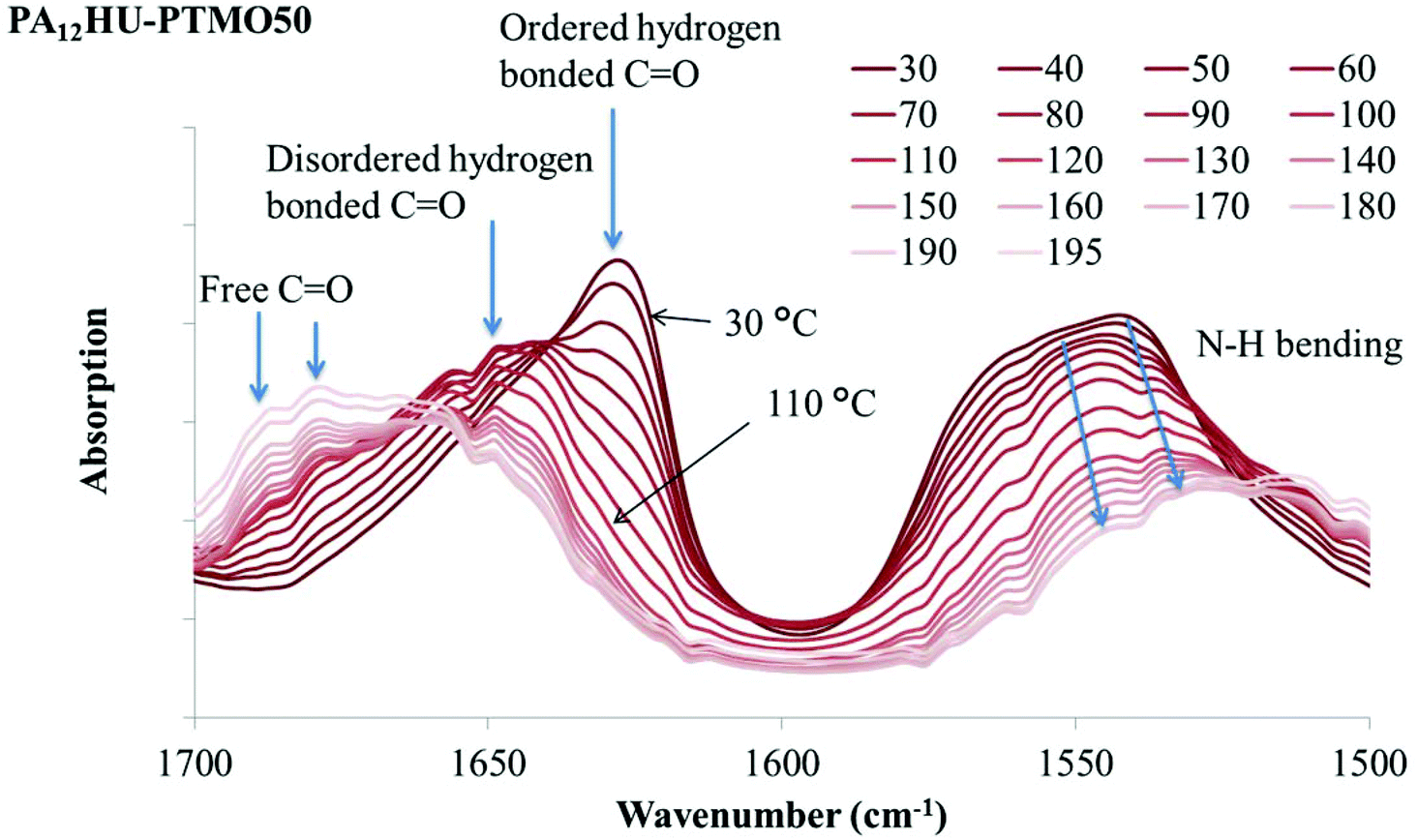
Hydrogen bonding plays a significant role in the crystallization and morphology of polyamides and polyurethanes. When C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and N–H units of amides and urethanes form hydrogen bonds, their signature infrared absorption frequencies shift due to bond length changes. In poly(amide-hydroxyurethane), the hydroxyls also provide hydrogen bonding donors and acceptors. VT-FTIR is a valuable tool for monitoring hydrogen bonding association and dissociation with changing temperature.40–42Fig. 8a demonstrates VT-FTIR spectra of nonsegmented PA12HU in the 1500–1700 cm−1 region with increasing temperature from dark to light curves. This carbonyl stretching region contains three distinctive contributions: ordered hydrogen bonded, disordered hydrogen bonded, and free (non-hydrogen bonded) carbonyls near 1624 cm−1, 1645 cm−1, and 1670 cm−1, respectively. Hydrogen bonding formation results in the red shift of CO stretching vibration band, due to bond lengthening.40 Increasing temperature leads to a blue shift of the CO stretching vibration band to higher frequency when hydrogen bond weakens or dissociates. In Fig. 8a, a sharp absorption band near 1624 cm−1 decreased with increasing temperature and a broad band gradually increased at higher frequency, indicating a blue shift due to hydrogen bond weakening and dissociation. This onset occurred near 100 °C, which correlated to the melting onset of nonsegmented PA12HU in Fig. 3. Melting of PA12HU finished near 170 °C in DSC, when the ordered hydrogen bonded carbonyl absorption disappeared completely. This indicated that ordered hydrogen bonded carbonyls existed mostly within the crystalline domains. Disordered hydrogen bonded and free carbonyls became predominant above 170 °C due to melting of the crystallites. N–H bending absorption red-shifted to lower frequency with decreasing intensity due to weakening hydrogen bonding as temperature increased.40,42,43Fig. 8b illustrates that N–H and O–H stretching vibration bands shifted to higher wavenumbers with increasing temperature, attributed to hydrogen bond weakening. A decrease in the absorption coefficient accounted for the dramatic band intensity decrease due to hydrogen bonding weakening.42 Segmented PA12HU-PTMO50 exhibited a similar phenomenon in Fig. 9. The absorption band of ordered hydrogen bonded carbonyls blue-shifted with increasing temperature. This shift started above 30 °C until ordered hydrogen bonded carbonyl band disappeared near 120 °C, which matched closely to the melting range of PA12HU-PTMO50 in Fig. 3. VT-FTIR revealed the melting point difference between segmented PA12HU-PTMO50 and nonsegmented PA12HU. The FTIR spectrum of PA12HU-PTMO50 at 30 °C contained a more obvious, relatively broader shoulder from the disordered hydrogen bonded and free carbonyls, compared to nonsegmented PA12HU at 30 °C. Polyether incorporation disrupted ordered hydrogen bonding formation, and resulted in a higher percentage of disordered and free hydrogen bonding donors and acceptors for PA12HU-PTMO50.
O and N–H units of amides and urethanes form hydrogen bonds, their signature infrared absorption frequencies shift due to bond length changes. In poly(amide-hydroxyurethane), the hydroxyls also provide hydrogen bonding donors and acceptors. VT-FTIR is a valuable tool for monitoring hydrogen bonding association and dissociation with changing temperature.40–42Fig. 8a demonstrates VT-FTIR spectra of nonsegmented PA12HU in the 1500–1700 cm−1 region with increasing temperature from dark to light curves. This carbonyl stretching region contains three distinctive contributions: ordered hydrogen bonded, disordered hydrogen bonded, and free (non-hydrogen bonded) carbonyls near 1624 cm−1, 1645 cm−1, and 1670 cm−1, respectively. Hydrogen bonding formation results in the red shift of CO stretching vibration band, due to bond lengthening.40 Increasing temperature leads to a blue shift of the CO stretching vibration band to higher frequency when hydrogen bond weakens or dissociates. In Fig. 8a, a sharp absorption band near 1624 cm−1 decreased with increasing temperature and a broad band gradually increased at higher frequency, indicating a blue shift due to hydrogen bond weakening and dissociation. This onset occurred near 100 °C, which correlated to the melting onset of nonsegmented PA12HU in Fig. 3. Melting of PA12HU finished near 170 °C in DSC, when the ordered hydrogen bonded carbonyl absorption disappeared completely. This indicated that ordered hydrogen bonded carbonyls existed mostly within the crystalline domains. Disordered hydrogen bonded and free carbonyls became predominant above 170 °C due to melting of the crystallites. N–H bending absorption red-shifted to lower frequency with decreasing intensity due to weakening hydrogen bonding as temperature increased.40,42,43Fig. 8b illustrates that N–H and O–H stretching vibration bands shifted to higher wavenumbers with increasing temperature, attributed to hydrogen bond weakening. A decrease in the absorption coefficient accounted for the dramatic band intensity decrease due to hydrogen bonding weakening.42 Segmented PA12HU-PTMO50 exhibited a similar phenomenon in Fig. 9. The absorption band of ordered hydrogen bonded carbonyls blue-shifted with increasing temperature. This shift started above 30 °C until ordered hydrogen bonded carbonyl band disappeared near 120 °C, which matched closely to the melting range of PA12HU-PTMO50 in Fig. 3. VT-FTIR revealed the melting point difference between segmented PA12HU-PTMO50 and nonsegmented PA12HU. The FTIR spectrum of PA12HU-PTMO50 at 30 °C contained a more obvious, relatively broader shoulder from the disordered hydrogen bonded and free carbonyls, compared to nonsegmented PA12HU at 30 °C. Polyether incorporation disrupted ordered hydrogen bonding formation, and resulted in a higher percentage of disordered and free hydrogen bonding donors and acceptors for PA12HU-PTMO50.
 | ||
| Fig. 8 Variable temperature FT-IR spectra of nonsegmented PA12HU (a) in the 1500–1700 cm−1 region and (b) in the 3000–3500 cm−1 region. | ||
 | ||
| Fig. 9 Variable temperature FT-IR spectra of segmented PA12HU-PTMO50 (a) in the 1500–1700 cm−1 region. | ||
Water sorption
Fig. 10 depicts water uptake of nonsegmented PA12HU and segmented PA12HU-PTMOs from 0% to 95% relative humidity (RH) at 25 °C. PA12HU lacked detectable water uptake until 90% RH and only showed 0.5 wt% water uptake at 95% RH, possibly due to condensed water. The low water uptake of PA12HU likely resulted from its relatively high crystallinity and Tg near room temperature (11 °C). Segmented PA12HU-PTMOs exhibited much higher water uptake compared to PA12HU due to lower crystallinity and lower Tg values (Table 1). Water uptake of PA12HU-PTMOs increased with increasing RH, reaching approximately 3.6–4.9 wt% at 95% RH and 1.6–2.0% at 65% RH, which was slightly higher than commercial Spandex® (thermoplastic polyurethane 0.75–1.3% at 65% RH), due to the additional hydroxyls.44 Water uptake of PA12HU-PTMOs proved independent of the polyether weight content, suggesting a similar polarity of the hard segments compared to the polyether segments.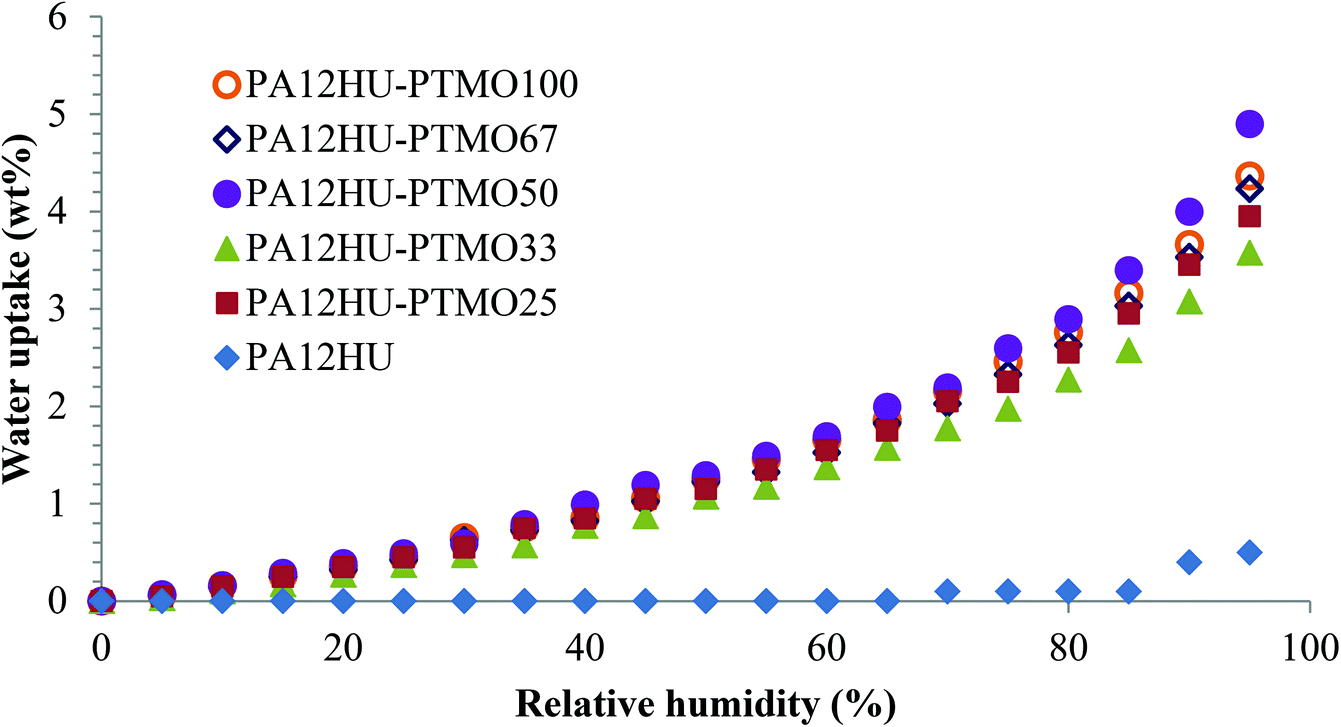 | ||
| Fig. 10 Equilibrium water sorption of nonsegmented PA12HU and segmented PA12HU-PTMOs in relative humidity steps from 0% to 95%. | ||
Tensile properties
Fig. 11 displays representative engineering stress-stain curves of PA12HU-PTMOs. Table 2 summarizes their tensile properties, including stress at break, strain at break, and Young's modulus. Compression molded films of PA12HU-PTMO25–50 exhibited sufficient mechanical integrity to yield statistically reliable tensile testing results. However, their tensile properties proved weaker than conventional segmented polyurethanes or segmented polyamides, likely due to the phase-mixed nature of the hard segments and polyether soft segments. Multiple thermal, dynamic mechanical and morphological analyses confirmed that the amorphous phase of each PA12HU-PTMO contained both hard and soft segments with a broad glass transition. The lack of a well-defined plateau regime in DMA curves agreed with the low strain at break values of PA12HU-PTMOs due to the broad Tg. Crystallinities of PA12HU-PTMOs also significantly limited the strains at break, resulting in thermoplastic solids instead of elastomers. The molecular weights of these PA12HU-PTMOs proved adequate for compression molding into free-stranding films, but insufficient to reach comparable toughness to commercial thermoplastic polyurethanes. Further optimization of PAHUs requires two main considerations: improving the phase-separation of hard and soft segments, and establishing SEC systems for molecular weight determination. | ||
| Fig. 11 Stress–strain curves of compression molded segmented PA12HU-PTMOs with 25–50 mol% soft segments. | ||
| Young's modulus (MPa) | Strain at break (%) | Stress at break (MPa) | |
|---|---|---|---|
| PA12HU-PTMO25 | 94.2 ± 24.5 | 25 ± 8 | 5.7 ± 0.4 |
| PA12HU-PTMO33 | 108.8 ± 8.1 | 21 ± 6 | 5.3 ± 0.5 |
| PA12HU-PTMO50 | 26.5 ± 2.3 | 51 ± 4 | 2.1 ± 0.0 |
Conclusion
This manuscript details an unprecedented synthetic platform for nonsegmented and segmented poly(amide-hydroxyurethane)s using sustainable resources. These PAHUs represent the first examples of film-forming, isocyanate-free polyurethanes with both melt and solution processability. A variety of physical characterizations established the structure–property–morphology relationship of PAHUs with varying compositions.A scalable, two-step synthesis converted methyl 9-decenoate to CC-ME monomer using CO2. A one-pot melt polymerization of CC-ME with diamines afforded synthesis of isocyanate-free PAHUs. Diaminododecane reacted with CC-ME to synthesize nonsegmented PA12HU, a semicrystalline copolymer with Tg of 11 °C and multiple melting exotherms at 100–150 °C. Incorporation of a low Tg 1 kDa polyether diamine added flexibility to segmented PA12HU-PTMOs. Segmented PA12HU-PTMOs exhibited broad glass transitions near −60 to –50 °C and lower crystallinity compared to nonsegmented PA12HU. Increasing ratio of polyether diamine to dodecane diamine resulted in lower crystallinity and lower melting point of PA12HU-PTMO due to shorter and fewer hard segments. Both DSC and DMA results indicated significant phase-mixing of polyether segments and the hard segments. Crystallizable hard segments formed crystalline domains, supporting mechanical strength of compression-molded PA12HU-PTMO films. The ribbon-like crystalline domains of PA12HU-PTMOs displayed constant width and varying lengths with different extent of polyether incorporation. VT-FTIR proved that most ordered hydrogen bonded carbonyls existed within the crystalline domains. The carbonyl vibration band shifts occurred in the same temperature range with melting of the crystalline domains of PA12HU or PA12HU-PTMOs. PA12HU exhibited minimal water uptake due to relatively high crystallinity. PA12HU-PTMOs showed slightly higher water uptake than conventional segmented polyurethanes. Reported poly(amide-hydroxyurethane)s displayed similar thermal degradation temperature as conventional polyurethanes, however lacked tensile strength due to their phase-mixed morphologies and possibly low molecular weights.
Acknowledgements
This research was supported by Elevance Renewable Science, Inc. This material is also partially based upon work supported by the National Science Foundation under Grant No. DMR-0923107. We also thank Professor Garth L. Wilkes for insightful discussion and Huntsman Corporation for material support.References
- B. Ochiai, S.-I. Sato and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 3408 CrossRef CAS.
- B. Tamami, S. Sohn and G. L. Wilkes, J. Appl. Polym. Sci., 2004, 92, 883 CrossRef CAS.
- B. Ochiai and T. Utsuno, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 525 CrossRef CAS.
- L. Ubaghs, N. Fricke, H. Keul and H. Höcker, Macromol. Rapid Commun., 2004, 25, 517 CrossRef CAS.
- D. V. Palaskar, A. Boyer, E. Cloutet, C. Alfos and H. Cramail, Biomacromolecules, 2010, 11, 1202 CrossRef CAS PubMed.
- N. Kihara and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 2765 CrossRef CAS.
- D. C. Webster, Prog. Org. Coat., 2003, 47, 77 CrossRef CAS.
- J. Guan, Y. Song, Y. Lin, X. Yin, M. Zuo, Y. Zhao, X. Tao and Q. Zheng, Ind. Eng. Chem. Res., 2011, 50, 6517 CrossRef CAS.
- G. Rokicki, P. G. Parzuchowski and M. Mazurek, Polym. Adv. Technol., 2015, 26, 707 CrossRef CAS.
- H. Blattmann, M. Fleischer, M. Bähr and R. Mülhaupt, Macromol. Rapid Commun., 2014, 35, 1238 CrossRef CAS PubMed.
- H. Sardon, A. Pascual, D. Mecerreyes, D. Taton, H. Cramail and J. L. Hedrick, Macromolecules, 2015, 48, 3153 CrossRef CAS.
- N. Kihara and T. Endo, Die Makromol. Chem., 1992, 193, 1481 CrossRef CAS.
- D. Couvret, J.-C. Brosse, S. Chevalier and J.-P. Senet, Die Makromol. Chem., 1990, 191, 1311 CrossRef CAS.
- D. C. Webster and A. L. Crain, Prog. Org. Coat., 2000, 40, 275 CrossRef CAS.
- S. Sohn, B. Tamami and G. Wilkes, US Pat., 7045577 B2, 2004 Search PubMed.
- I. Javni, D. P. Hong and Z. S. Petrović, J. Appl. Polym. Sci., 2008, 108, 3867 CrossRef CAS.
- I. Javni, D. P. Hong and Z. S. Petrović, J. Appl. Polym. Sci., 2013, 128, 566 CrossRef CAS.
- P. G. Parzuchowski, M. Jurczyk-Kowalska, J. Ryszkowska and G. Rokicki, J. Appl. Polym. Sci., 2006, 102, 2904 CrossRef CAS.
- B. Ochiai, S. Inoue and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 6613 CrossRef CAS.
- H. Tomita, F. Sanda and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 851 CrossRef CAS.
- M.-R. Kim, H.-S. Kim, C.-S. Ha, D.-W. Park and J.-K. Lee, J. Appl. Polym. Sci., 2001, 81, 2735 CrossRef CAS.
- L. Maisonneuve, A. S. More, S. Foltran, C. Alfos, F. Robert, Y. Landais, T. Tassaing, E. Grau and H. Cramail, RSC Adv., 2014, 4, 25795 RSC.
- A. Boyer, E. Cloutet, T. Tassaing, B. Gadenne, C. Alfos and H. Cramail, Green Chem., 2010, 12, 2205 RSC.
- J. Nanclares, Z. S. Petrović, I. Javni, M. Ionescu and F. Jaramillo, J. Appl. Polym. Sci., 2015, 132, 42492 CrossRef.
- S. G. Davies, A. M. Fletcher, W. Kurosawa, J. A. Lee, G. Poce, P. M. Roberts, J. E. Thomson and D. M. Williamson, J. Org. Chem., 2010, 75, 7745 CrossRef CAS PubMed.
- G. G. Odian, Principles of polymerization, Wiley-Interscience, Hoboken, N.J, 2004, vol. 4 Search PubMed.
- L. T. J. Korley, B. D. Pate, E. L. Thomas and P. T. Hammond, Polymer, 2006, 47, 3073 CrossRef CAS.
- D. J. Buckwalter, J. M. Dennis and T. E. Long, Prog. Polym. Sci., 2015, 45, 1–22 CrossRef CAS.
- M. Ravey and E. M. Pearce, J. Appl. Polym. Sci., 1997, 63, 47 CrossRef CAS.
- S. V. Levchik, E. D. Weil and M. Lewin, Polym. Int., 1999, 48, 532 CrossRef CAS.
- Y. C. Yu and W. H. Jo, J. Appl. Polym. Sci., 1995, 56, 895 CrossRef CAS.
- Thermal Transitions of Homopolymers, Sigma Aldrich https://www.sigmaaldrich.com/content/dam/sigma-aldrich/docs/Aldrich/General_Information/thermal_transitions_of_homopolymers.pdf.
- C. S. Paik Sung, C. B. Hu and C. S. Wu, Macromolecules, 1980, 13, 111 CrossRef.
- M. C. E. J. Niesten, J. Feijen and R. J. Gaymans, Polymer, 2000, 41, 8487 CrossRef CAS.
- M. Van Der Schuur and R. J. Gaymans, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 4769 CrossRef CAS.
- P. J. Flory, Principles of polymer chemistry, Conell Univerisity Press, 1953 Search PubMed.
- L. M. Robeson, A Comprehensive Review, Leseprobe, 2007 Search PubMed.
- R. S. McLean and B. B. Sauer, Macromolecules, 1997, 30, 8314 CrossRef CAS.
- Y. Chool Yu, W. H. Jo and M. S. Lee, J. Appl. Polym. Sci., 1997, 64, 2155 CrossRef.
- K. Zhang, M. Aiba, G. B. Fahs, A. G. Hudson, W. D. Chiang, R. B. Moore, M. Ueda and T. E. Long, Polym. Chem., 2015, 6, 2434 RSC.
- D. J. Skrovanek, P. C. Painter and M. M. Coleman, Macromolecules, 1986, 19, 699 CrossRef CAS.
- M. M. Coleman, K. H. Lee, D. J. Skrovanek and P. C. Painter, Macromolecules, 1986, 19, 2149 CrossRef CAS.
- M. Tamami, K. Zhang, N. Dixit, R. B. Moore and T. E. Long, Macromol. Chem. Phys., 2014, 215, 2337 CrossRef CAS.
- J. G. Cook, Handbook of textile fibres: man-made fibres, Elsevier, 1984, vol. 2 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6gc01096b |
| This journal is © The Royal Society of Chemistry 2016 |