
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalyst- and solvent-free hydrophosphination and multicomponent hydrothiophosphination of alkenes and alkynes†
Yanina
Moglie‡
a,
María José
González-Soria
a,
Iris
Martín-García
a,
Gabriel
Radivoy
b and
Francisco
Alonso
*a
aInstituto de Síntesis Orgánica (ISO) and Departamento de Química Orgánica, Facultad de Ciencias, Universidad de Alicante, Apdo. 99, 03080 Alicante, Spain. E-mail: falonso@ua.es
bDepartamento de Química, Instituto de Química del Sur (INQUISUR-CONICET), Universidad Nacional del Sur, Avenida Alem 1253, 8000 Bahía Blanca, Argentina
First published on 24th May 2016
Abstract
The hydrophosphination of carbon–carbon multiple bonds has been generally performed under acid, base or metal catalysis in different solvents. Herein, alkyl and alkenyl tertiary phosphines are obtained by the addition of diphenylphosphine to alkenes and alkynes, respectively, in the absence of a solvent and a catalyst. In the presence of elemental sulfur, the corresponding alkyl and alkenyl tertiary phosphine sulfides are synthesized in a three-component process. These simple methods, which meet most of the principles of Green Chemistry, are highly regioselective towards the anti-Markovnikov products and diastereoselective towards the Z alkenyl phosphines. The mechanistic aspects of the reactions are also tackled and the efficiency of the latter is compared with that of the catalytic methods.
Introduction
The deployment of phosphorus compounds in industry has become widespread because of their manifold applications.1 Nowadays, many chemical, agrochemical and pharmaceutical substances containing phosphorus are routinely produced to control some natural or human-triggered processes and to upgrade the quality of our lives. Research laboratories also make extensive use of organophosphorus compounds, particularly as ligands for homogeneous, heterogeneous and asymmetric catalysis,2a–i but also as organocatalysts.2j,k Metal–phosphine complexes with anticancer activity is another field of recent interest aimed at developing substitutes for the current platinum drugs.3 Tertiary phosphines can be synthesized using different procedures (Scheme 1), such as (a) the displacement of a good leaving group on a phosphine by an organometallic reagent (eqn (1)), (b) the reaction of a metal phosphide with an organic electrophile (eqn (2)), (c) the reduction of a phosphorus halide or oxide (eqn (3)), (d) from elemental phosphorus (eqn (4)) and (e) by phosphination (i.e., the metal-catalysed cross-coupling of aryl halides or triflates with P–H bonds; eqn (5)).4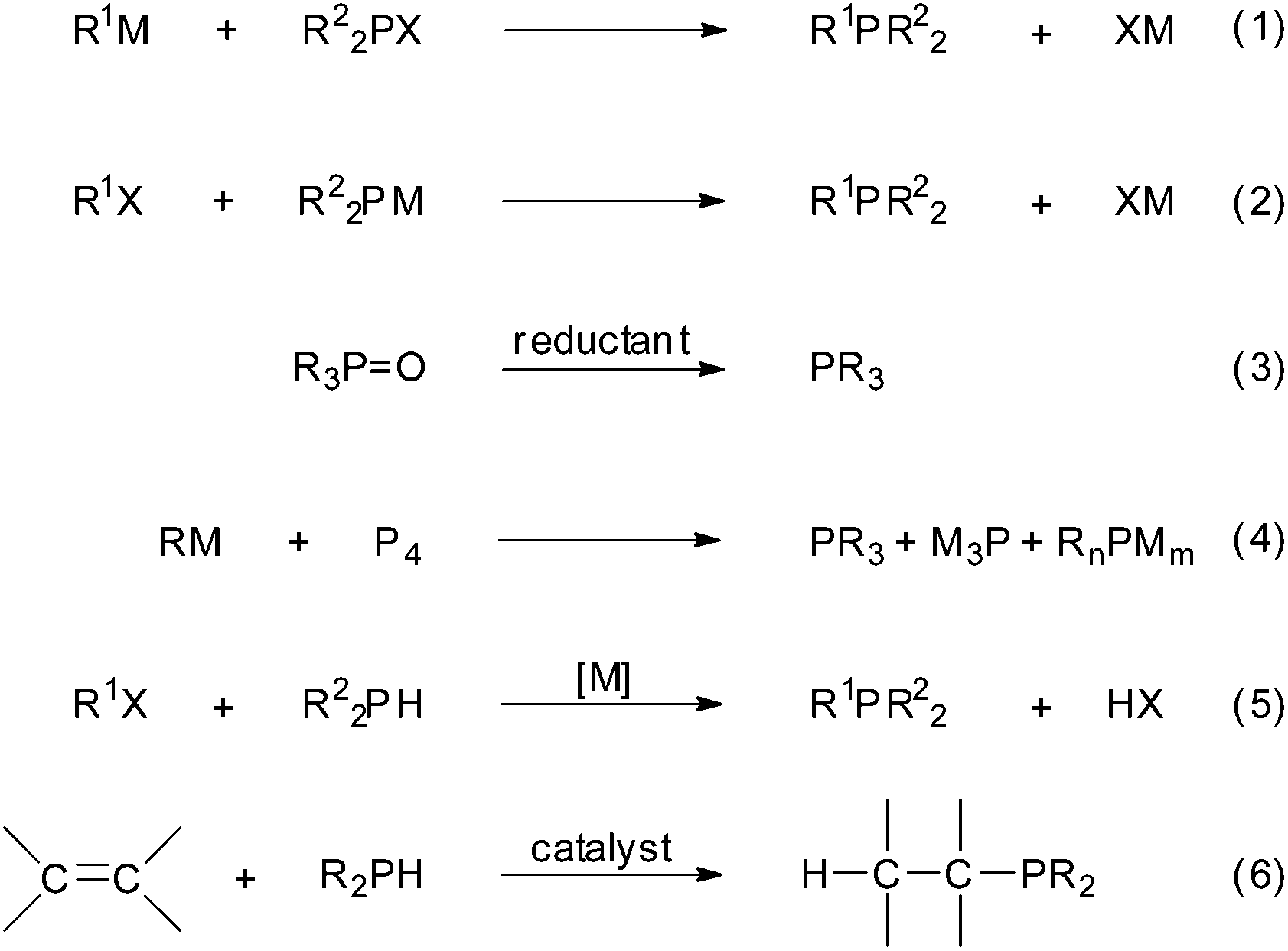 | ||
| Scheme 1 Some general methods for tertiary phosphine synthesis; M = metal, X = leaving group; n = 2, m = 1 and n = 1, m = 2. | ||
Modern chemical research and production must advance on the basis of sustainable and environmentally benign practices.5 In this vein, the hydrophosphination of unsaturated compounds (i.e., alkenes and alkynes) appears as the most straightforward approach to form C–P bonds from readily accessible starting materials (Scheme 1, eqn (6));6 maximum atom economy is attained with no by-product formation.
Closely related to phosphines are the phosphine sulfides,7 a type of compound with multiple applications in different disciplines. Among others, phosphine sulfides have been utilized in anion-selective electrodes,8 lanthanide extraction from a nitrate medium,9 as sensor fluorescent materials for metal ions of environmental concern,10 as anchor units for single molecule junctions,11 in polymer chemistry,12 and as ligands for gold,13 catalysis14 and asymmetric synthesis.15 Base-promoted16 and free-radical17 initiated addition of secondary phosphine sulfides to alkenes (or alkynes)7 are the most practiced methods to prepare tertiary phosphine sulfides. The reaction of secondary phosphine sulfides with carbonyl compounds can provide tertiary α-hydroxy phosphine sulfides.7 The transformation of pre-formed tertiary phosphines into the corresponding sulfides can be readily accomplished by reaction with sulphur; in this case, however, hazardous solvents such as benzene, chloroform and dichloromethane are required.7 More recently, Trofimov et al. reported the one-pot synthesis of tertiary phosphine sulfides from styrenes, red phosphorus and elemental sulfur in a superbasic system containing hydroquinone under microwave irradiation.18 In addition to these, transition-metal catalyzed procedures have recently been published.19
On the other hand, both solvent-free reactions20 and catalyst-free organic synthesis21 notably simplify the reaction mixtures and experimentally, at the same time reduce the amount of waste which, in turn, depletes the environmental impact.
By virtue of our current interest in phosphorus chemistry,22 we found out that tertiary phosphines can be obtained in a very straightforward manner by addition of secondary phosphines to carbon–carbon double bonds under solvent- and catalyst-free conditions.22a Moreover, under these conditions but in the presence of sulfur, α,β-unsaturated carbonyl compounds have been converted into the corresponding β-substituted tertiary phosphine sulfide derivatives through a three-component approach (Scheme 2).22b Our intention is to present herein a comprehensive study on the substrate scope and mechanism of these environmentally friendly protocols, including the hydrophosphination and multicomponent hydrothiophosphination of alkenes and alkynes.
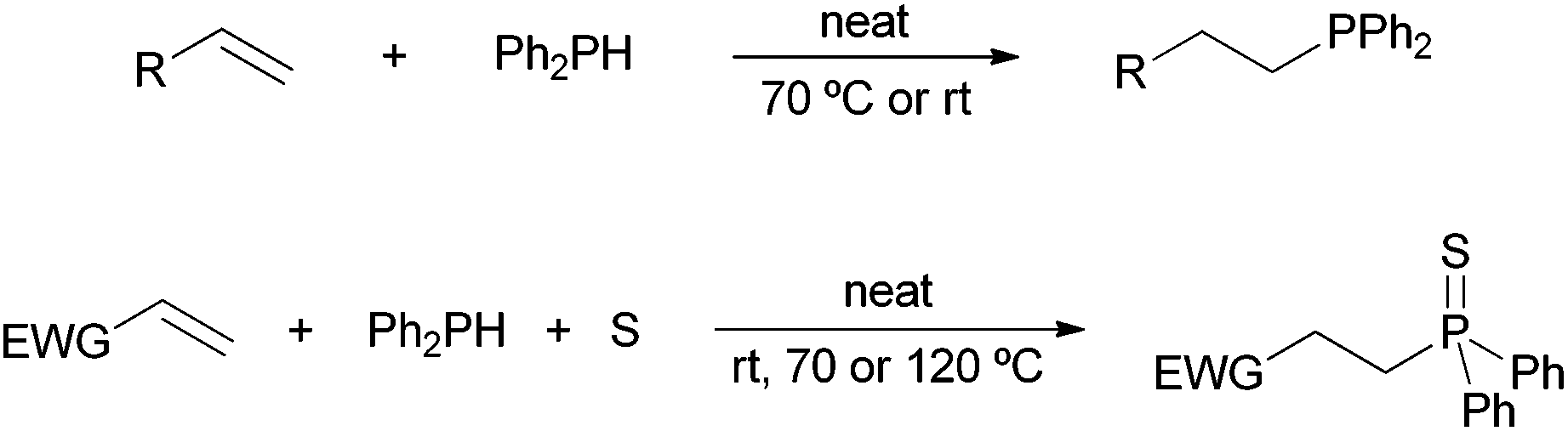 | ||
| Scheme 2 Solvent- and catalyst-free hydrophosphination22a and hydrothiophosphination22b of alkenes; EWG = electron-withdrawing group. | ||
Results and discussion
Alkene hydrophosphination
Originally, alkene hydrophosphination6 was induced by base catalysis or radical activation to form the anti-Markovnikov products,6c whereas the Markovnikov counterparts were better obtained by acid treatment.23 More advantageous were, however, the methodologies based on catalysis by metal6 complexes, such as those of platinum,24 lanthanides,25 alkaline-earth metals,25e,f,26 nickel,27 palladium27 or iron.28 Of particular interest is the catalytic asymmetric hydrophosphination of certain substrates, which was achieved by the use of chiral organopalladium(II) complexes.29 Not only metal complexes but also metal salts of copper30 or iron31 have been utilized as catalysts in alkene hydrophosphination. With the exception of the Pd- and Ni-catalysed hydrophosphination of vinyl ethers,27c,d the aforementioned methodologies afford the anti-Markovnikov products.In spite of the higher selectivity achieved by metal catalysis, most of its applications in alkene hydrophosphination does not meet some of the stringent criteria demanded for green and sustainable production because of the use of non-reusable precious metals or noxious solvents (e.g., benzene). In addition, transition metals can accelerate the undesired phosphine oxidation to the phosphine oxide. Ideally, this reaction should be conducted under metal-free and neutral conditions, with the latter also preventing side-reactions and use of aqueous work-up (which can favour oxidation). To the best of our knowledge, Gaumont's group was the first to report the uncatalysed hydrophosphination of alkenes: phosphine–borane complexes were added to inactivated alkenes under neutral conditions followed by either conventional or microwave heating.32
Following our recent discovery on the uncatalysed addition of secondary phosphines to carbon–carbon double bonds under solvent-free conditions,22a we expanded this practice to the addition of diphenylphosphine to a wide range of alkenes, including not only styrenes but also α,β-unsaturated carbonyl compounds and inactivated alkenes (Tables 1–3). All reactions were executed under an inert atmosphere of argon.
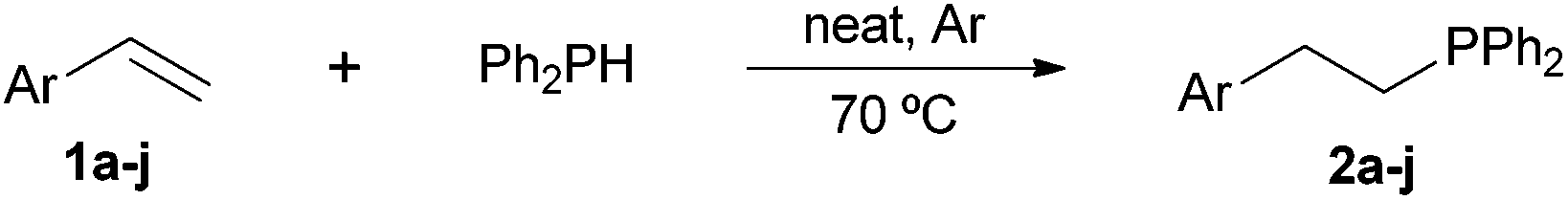
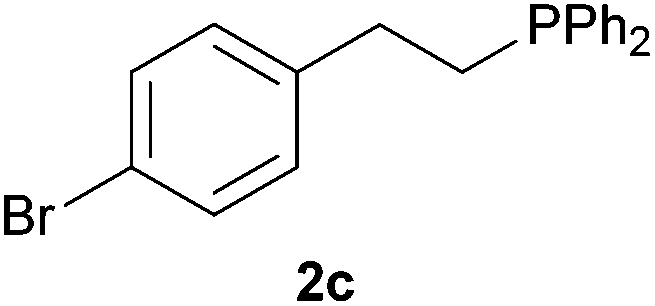
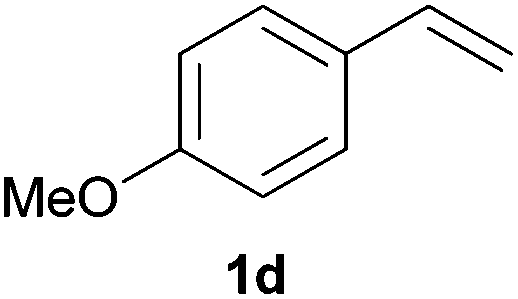
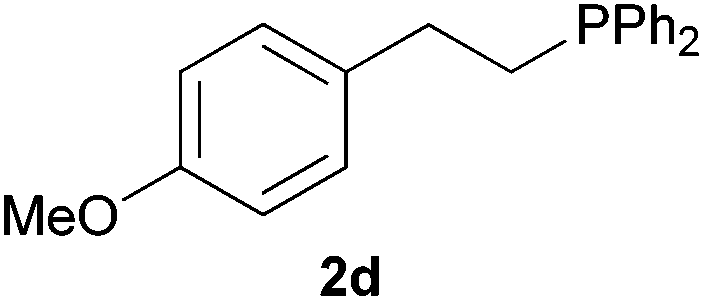
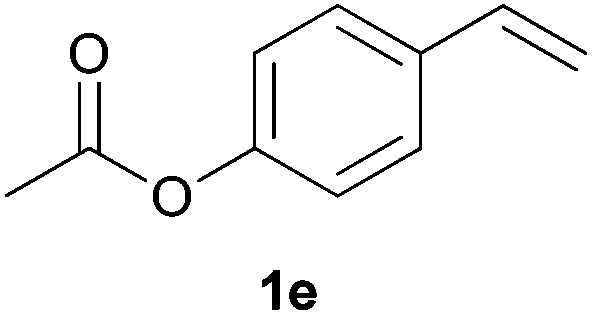
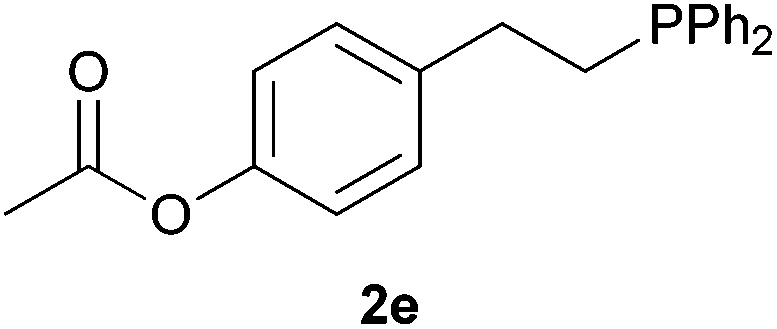
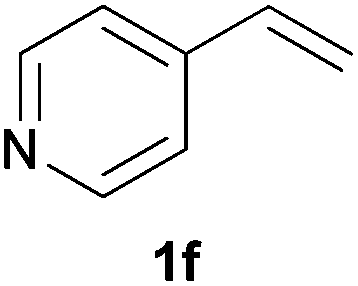
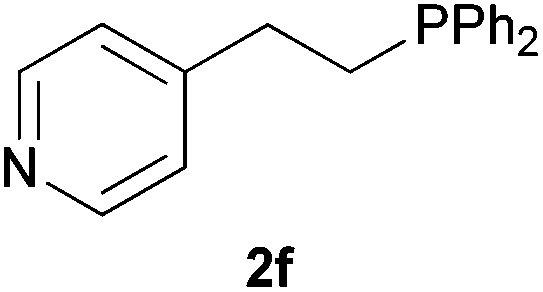
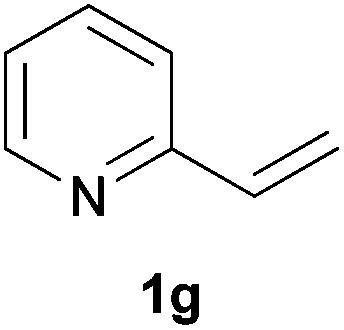
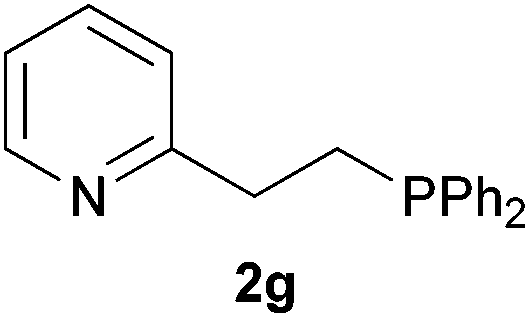
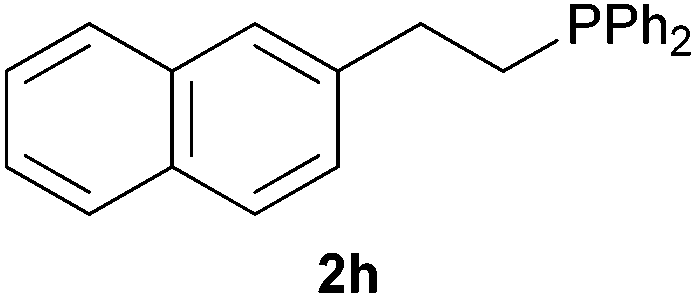
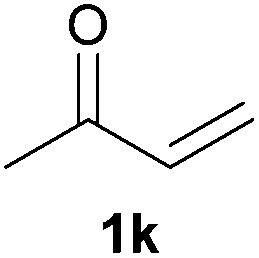
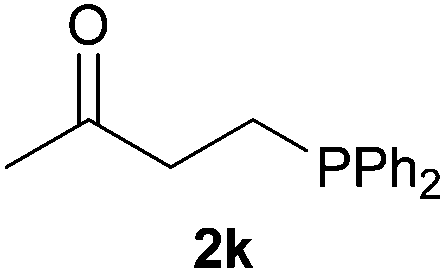
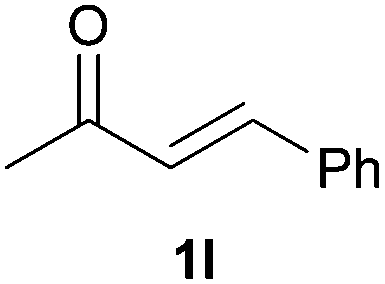
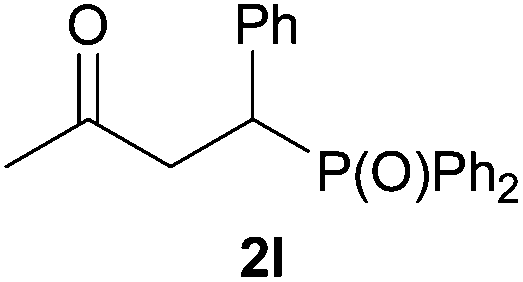
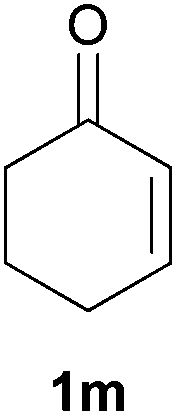
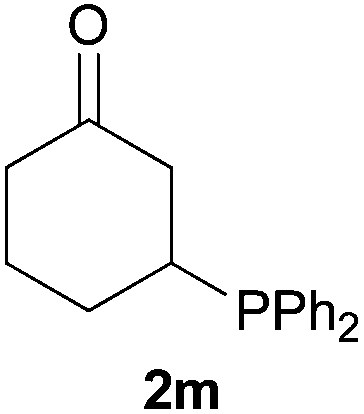
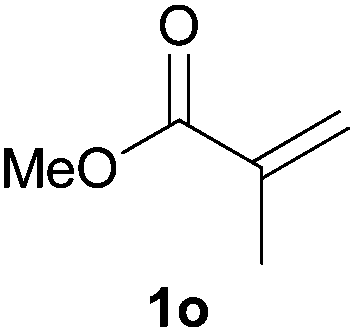
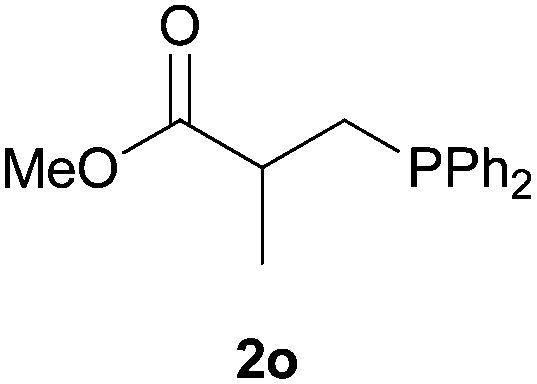
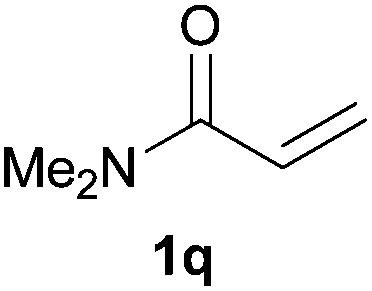
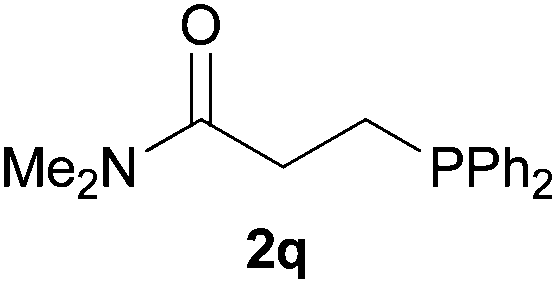
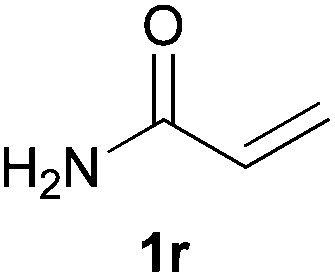
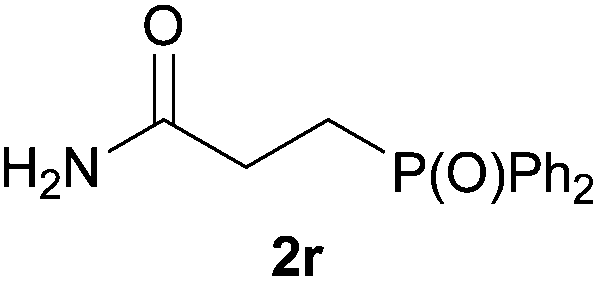
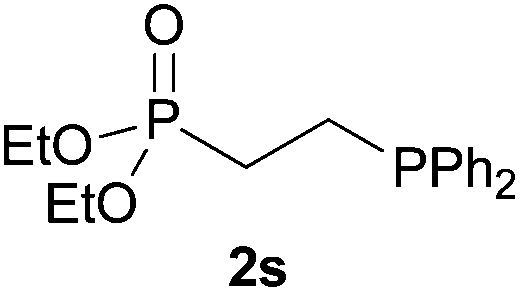
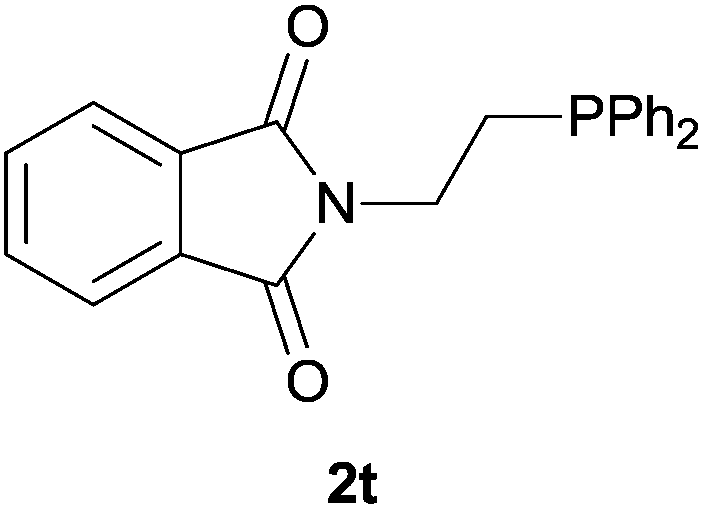
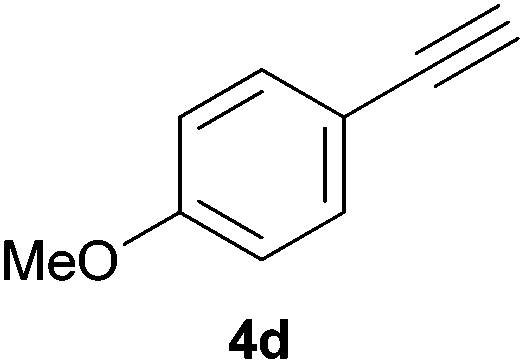
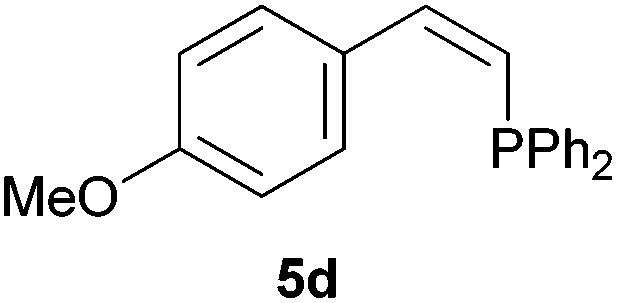
As regards styrenes, the simplest one (1a) reacted the fastest and resulted in good yield. p-Halostyrenes (1b and 1c), p-methoxy- and p-acetoxystyrene (1d and 1e, respectively) behaved similarly in terms of yield (around 85%), with a shorter reaction time for 2d. This hydrophosphination was also appropriate for vinyl pyridines (1f and 1g), permitting the synthesis of the P,N-bidentate ligand pyphos (2g), used in homogeneous transition-metal catalysis.33 Other vinyl aromatics, such as 2-vinylnaphthalene (1h) and 4-vinyl-1,1′-biphenyl (1i) were transformed into the tertiary phosphines in high yields. Furthermore, the standard conditions were also effectual for the less reactive 1,1-disubstituted alkene isopropenyl benzene (1j). We must underline that all products 2a–2j were produced with exclusively anti-Markovnikov regioselectivity.
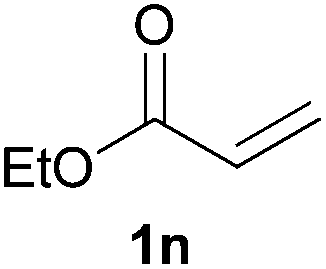
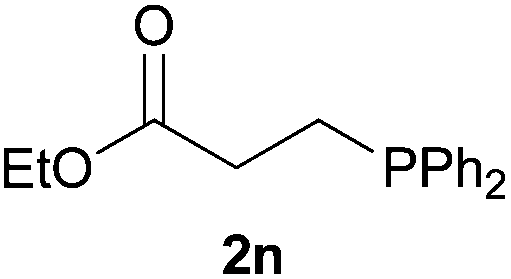
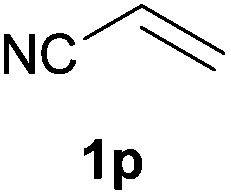
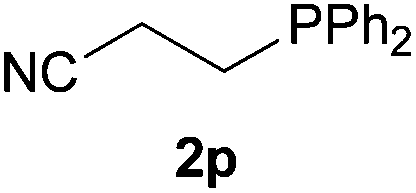
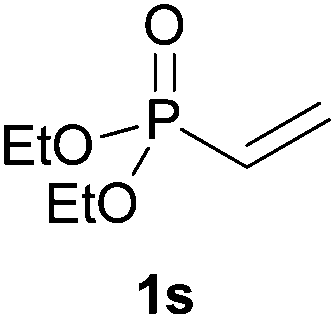
We next explored the diphenylphosphine addition to α,β-unsaturated carbonyl and related compounds (Table 2). These substrates can be considered more activated alkenes than the aforesaid styrenes and, hence, more reactive under milder conditions. Indeed, most of the starting alkenes (except 1l and 1q) experienced hydrophosphination at room temperature (7–12 h); alternatively, the reaction times could be decreased (1–3 h) by warming at 70 °C with comparable yields (see 1k, 1n and 1p). Diverse functional groups were compatible with these conditions, whereby the synthesis of β-diphenylphosphino ketones (1k–1m), esters (1n and 1o), nitrile (1p), amides (1q and 1r) and phosphonate (1s) was carried out in good yields; products 2l and 2r were isolated as the phosphine oxides due to easy oxidation of the phosphine precursors when exposed to air. It is worth noting that the platinum-catalysed24 addition of diphenylphosphine to acrylonitrile (1p) can instead be effectuated in the absence of a catalyst at room temperature.
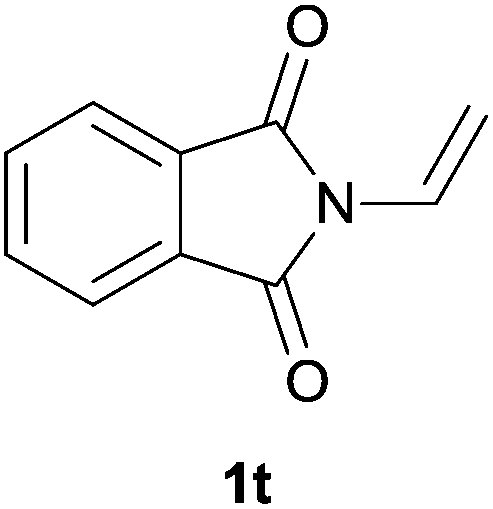
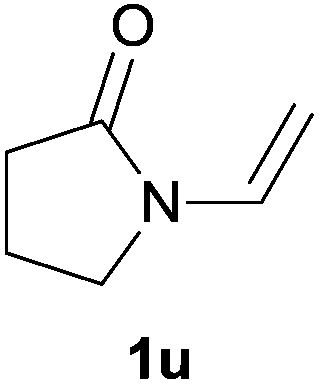
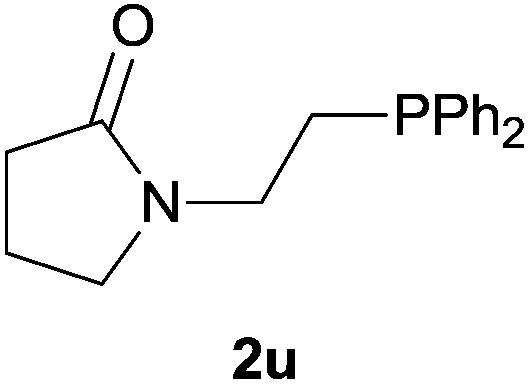
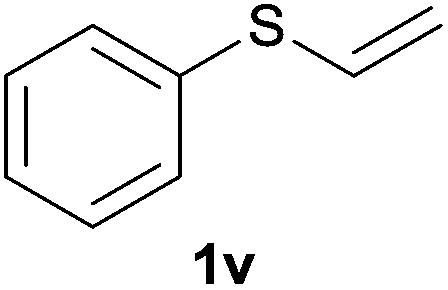
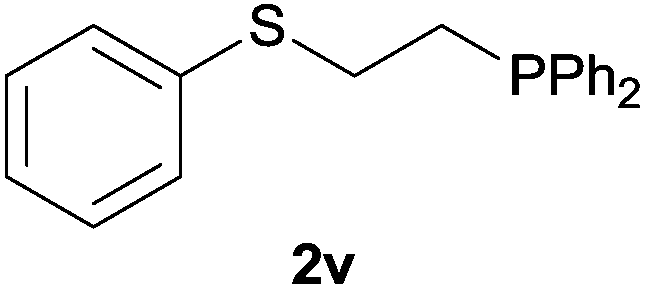
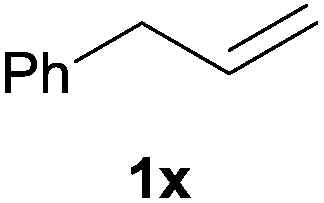
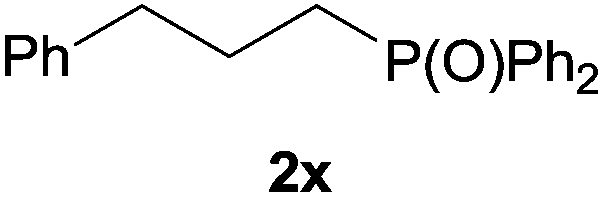
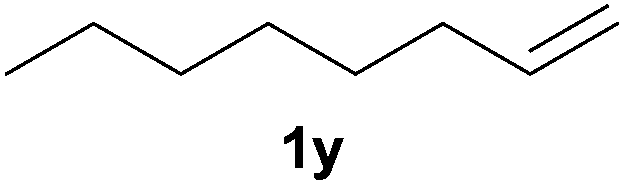
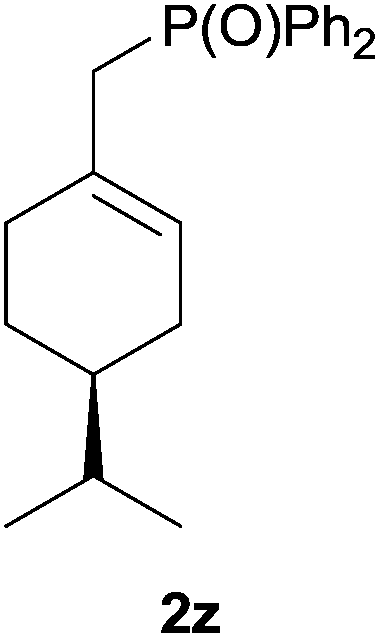
Heteroatom-bonded vinyl compounds, such as N-vinylphthalimide (1t), N-vinylpyrrolidin-2-one (1u) and phenyl vinyl sulfide (1v) gave the expected (diphenylphosphino)ethyl heteroatom products under the conventional conditions (70 °C) (Table 3). Compound 2v is a P,S-bidentate ligand also employed in catalysis.34 The usefulness of this protocol was validated by its exploitation in the hydrophosphination of inactivated alkenes such as allylbenzene (1x) and oct-1-ene (1y). The anti-Markovnivov regioselectivity was consistent with the trend displayed by all the substrates in Tables 1–3. Still, the tertiary phosphines originated from 1x and 1y were susceptible to oxidation and were isolated as the phosphine oxides 2x and 2y, respectively. It was gratifying to verify that the hydrophosphination of (–)-β-pinene (1z) followed an anti-Markovnivov addition and involved the opening of the cyclobutane ring to supply the enantiomerically pure p-menth-1-ene derived phosphine oxide 2z. An unmistakable assignment of the structure was done by X-ray crystallographic analysis (Fig. 1).35
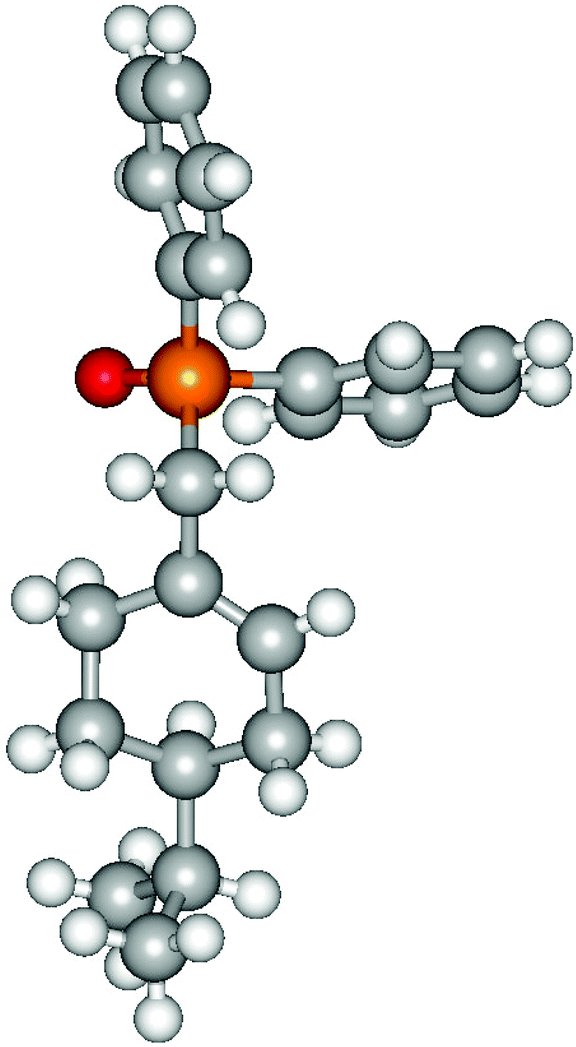 | ||
| Fig. 1 X-ray crystallographic structure of phosphine oxide 2z. | ||
Alkene hydrothiophosphination
Apart from the base-promoted16 and free-radical17 initiated addition of secondary phosphine sulfides to alkenes, and related to the title topic, is the recent synthesis of tertiary phosphine sulfides by addition of secondary phosphine sulfides to alkenes in the absence of a solvent and catalyst.36a,b Reactions were performed at 80 °C under an inert atmosphere to give the anti-Markovnikov products.In a previous study,22b we evidenced that alkene hydrothiophosphination could be achieved through a three-component approach involving the alkene, diphenylphosphine and sulfur, under solvent- and catalyst-free conditions in air. This is an advantageous strategy not only from the environmental point of view, but also because it circumvents the preparation of the secondary phosphine sulfide, which is generated in situ.36c Only α,β-unsaturated carbonyl compounds were covered in that preliminary study; the results obtained for other alkenes, either activated or not activated, are depicted in Table 4.
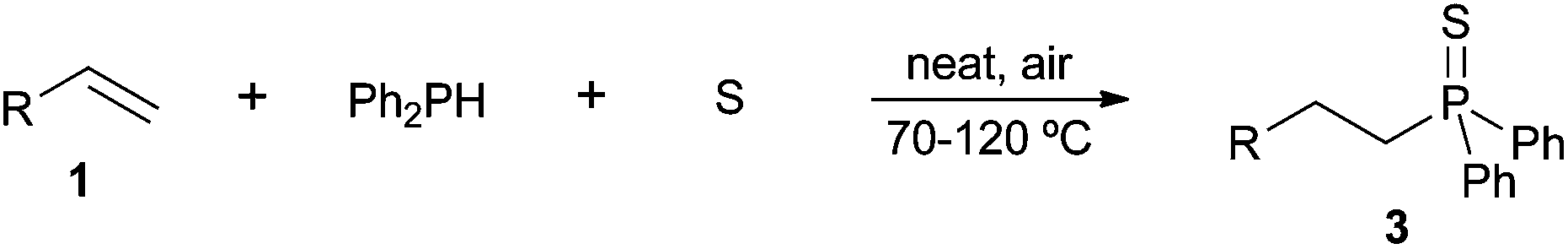
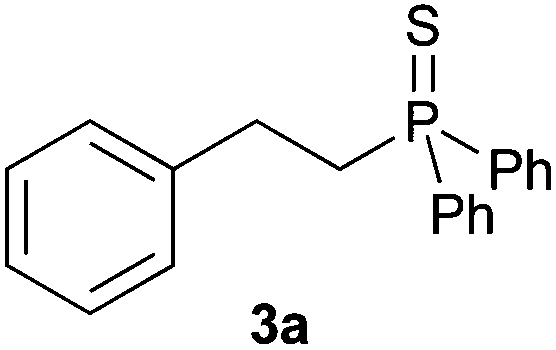
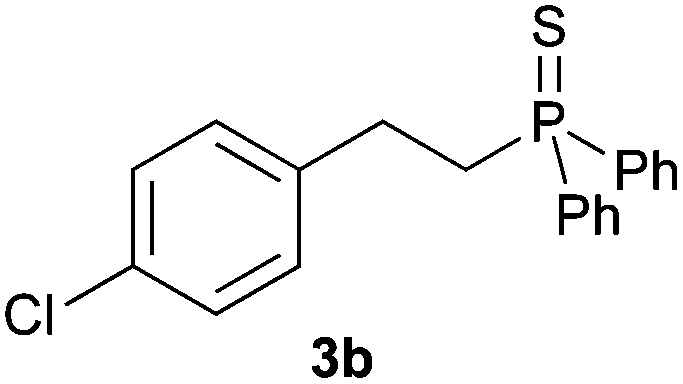
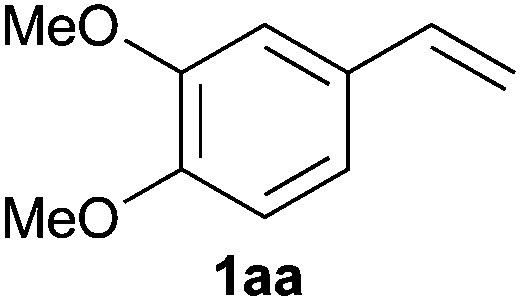
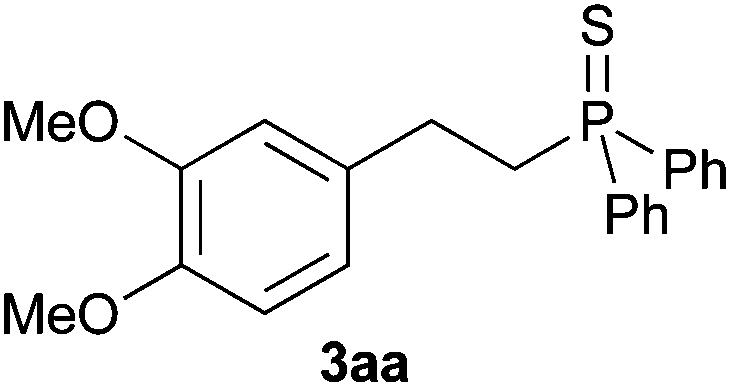
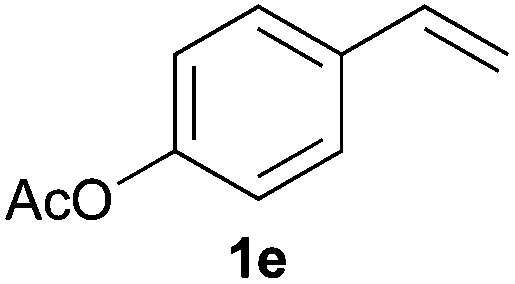
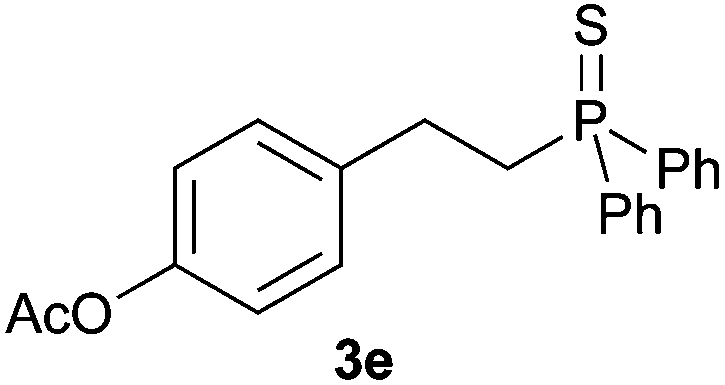
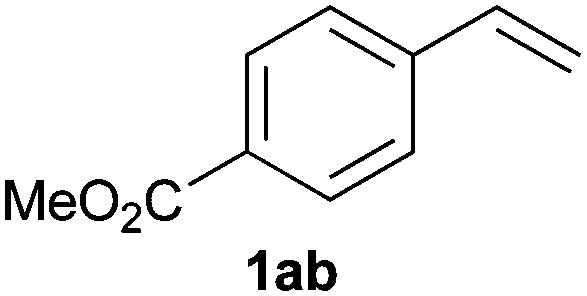
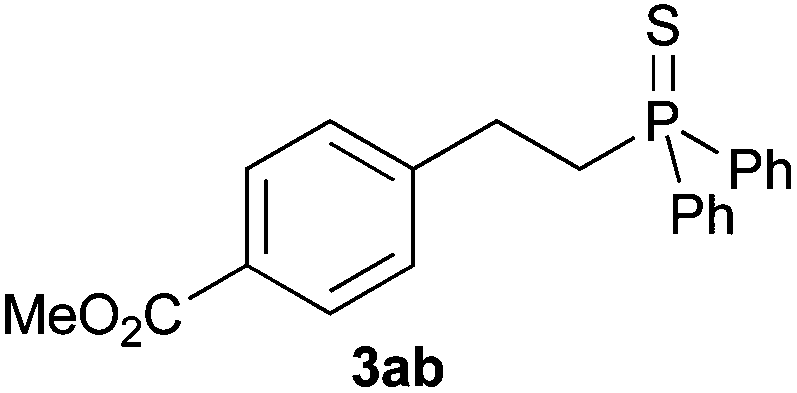
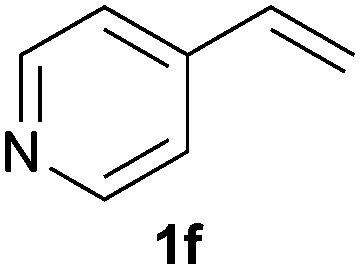
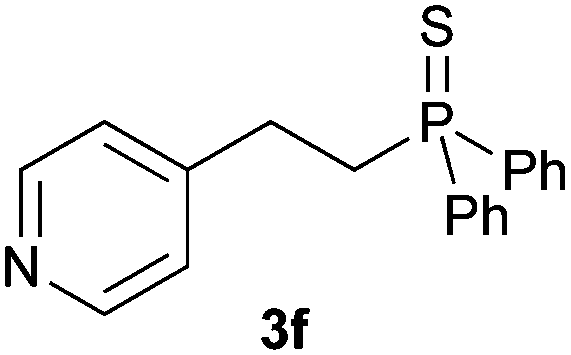
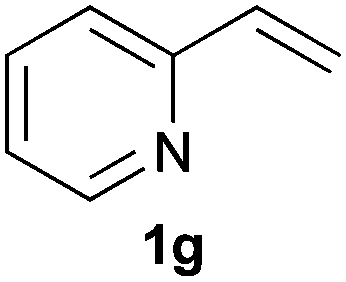
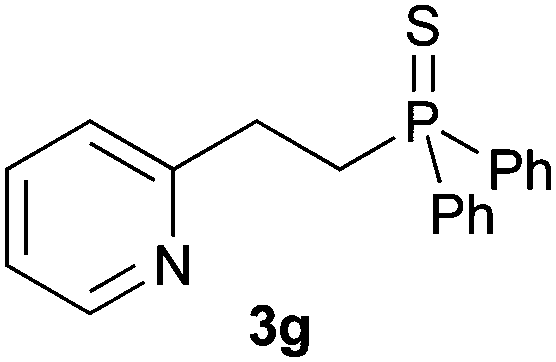
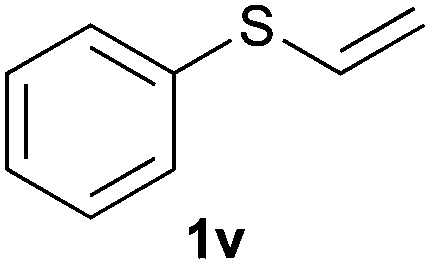
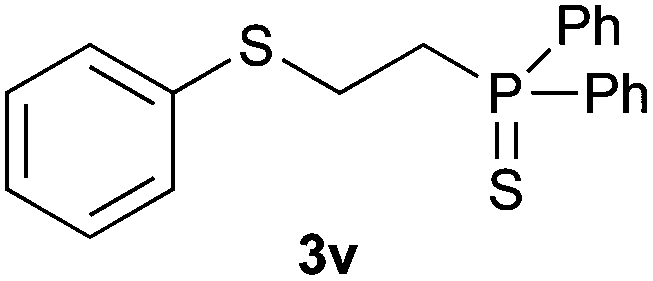
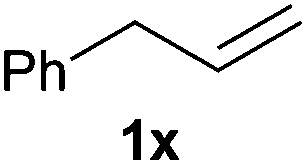
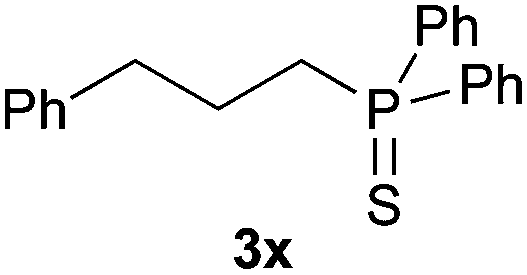
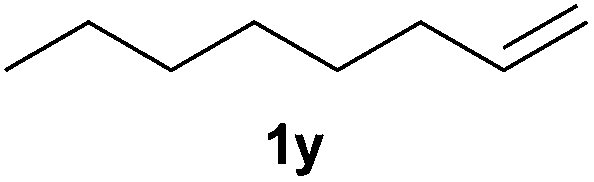
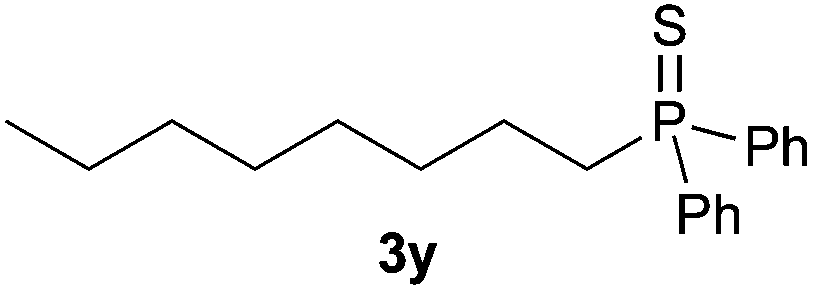
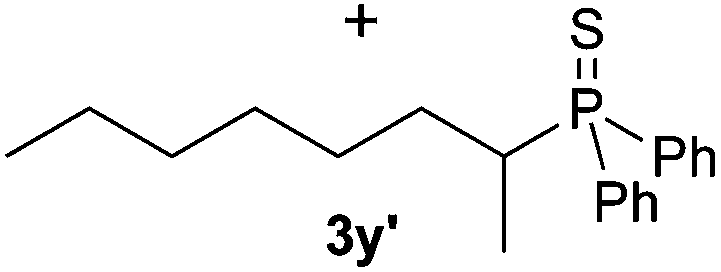
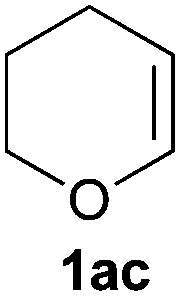
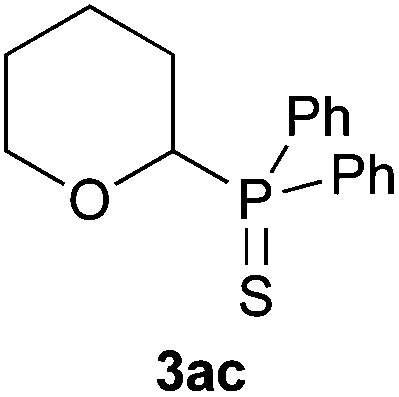
A variety of electron-neutral, -rich and -deficient styrenes underwent the hydrothiophosphination reaction at 70 °C in air; the corresponding products (3a, 3b, 3aa, 3e and 3ab) were obtained in moderate-to-excellent isolated yields (Table 4). Likewise, styrenes bearing N and S atoms, as well as allylbenzene, were successfully converted into phosphine sulfides (3f, 3g, 3v and 3x). It is worth noting that the process was highly regioselective, giving rise in all cases to the anti-Markovnikov products. The same conditions were applicable to aliphatic substrates, such as oct-1-ene (1y) and 3,4-dihydro-2H-pyran (1ac). Despite the lack of regioselectivity observed for oct-1-ene, both regioisomers (3y and 3y′) could be separated by chromatography in reasonable yields, as a result of the quantitative reaction conversion. In contrast, the hydrothiophosphination of 3,4-dihydro-2H-pyran (1ac) led to a single regioisomer (3ac), derived from the addition of P to the α-O carbon atom.
Alkyne hydrophosphination
Comparatively, alkyne hydrophosphination has been much less studied than alkene hydrophosphination. Beletskaya et al. reported, for the first time, the hydrophosphination of alkynes catalyzed by palladium and nickel complexes; interestingly, depending on the metal used the α- or β-adduct was mainly formed, with predominant syn addition.37 Other metal complexes or salts (i.e., Yb, Pd, Ni, Cu, Ca, Fe and Y) have been deployed for the same purpose with variable selectivity.25e,38In our preliminary communication,22a we observed that phenylacetylene could also undergo diphenylphosphine addition under solvent- and catalyst-free conditions. A variety of alkynes has been subjected to this simple procedure to produce vinylphosphines in a regio- and stereoselective manner (Table 5).
|
|
||
|---|---|---|
| Starting alkyne | Product | Yieldb (%) |
a Alkyne 4 (0.5 mmol) and Ph2PH (0.5 mmol) at 70 °C under argon, overnight.
b Isolated yield.
c As a 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 Z/E diastereomeric mixture.
d GLC yield.
e As a 90:10 E/Z diastereomeric mixture.
f As a 97:3 Z/E diastereomeric mixture.
g Yield of the major diastereoisomer, isolated from a 60:40 Z/E diastereomeric mixture. :5 Z/E diastereomeric mixture.
d GLC yield.
e As a 90:10 E/Z diastereomeric mixture.
f As a 97:3 Z/E diastereomeric mixture.
g Yield of the major diastereoisomer, isolated from a 60:40 Z/E diastereomeric mixture.
|
||
|
|
80c |
|
|
73 |
|
|
88 |
|
|
86 |
|
|
87 |
|
|
13d |
|
|
64e |
|
|
62f |
|
|
61g |
|
|
60 |
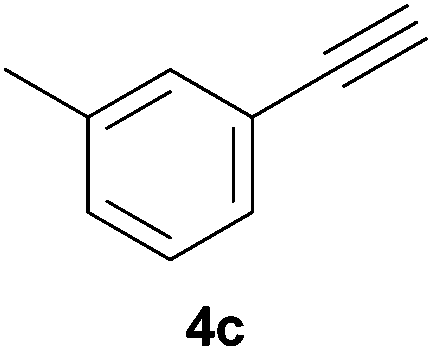
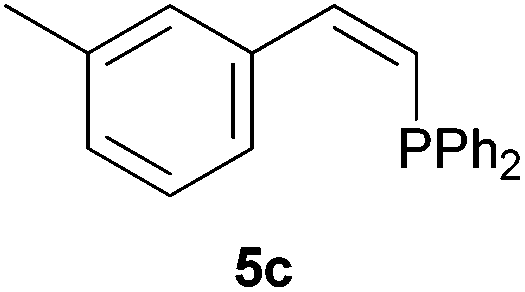
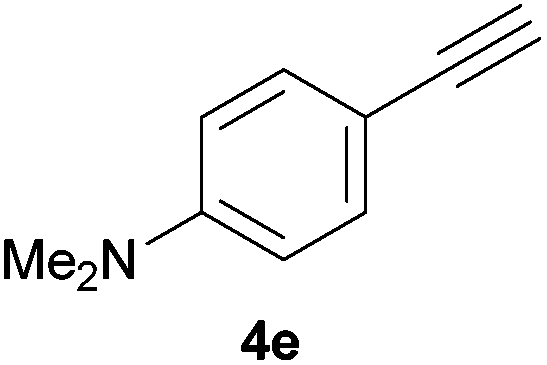
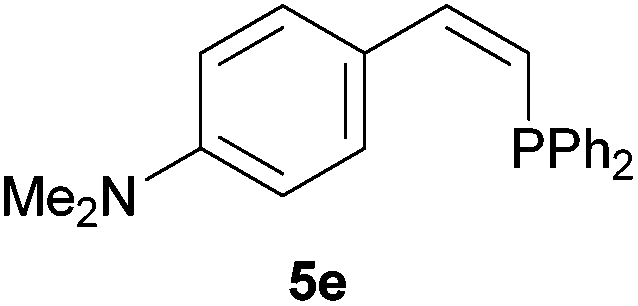
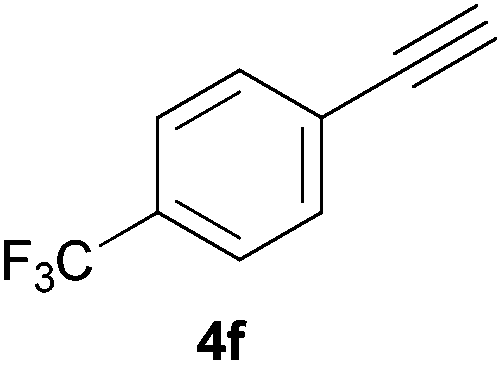
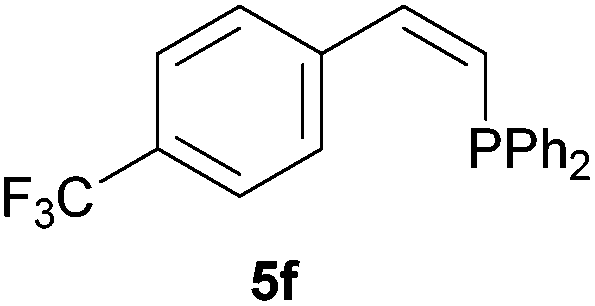
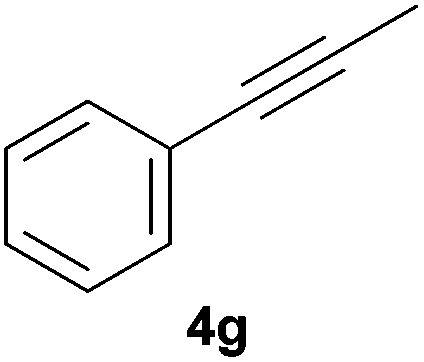
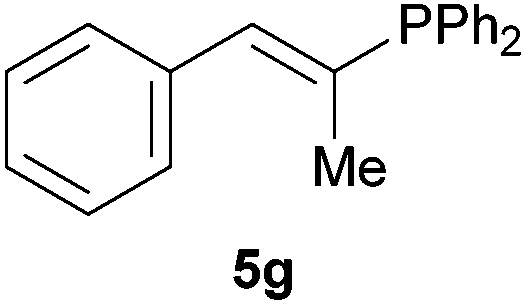
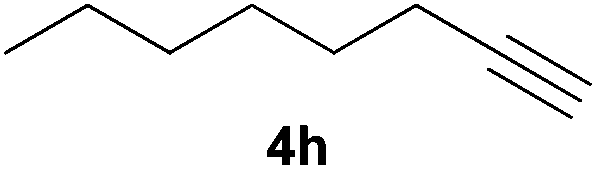
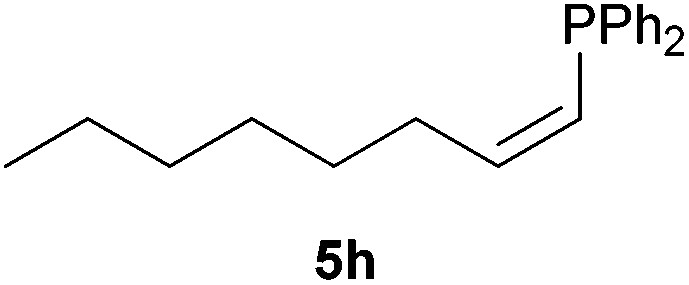
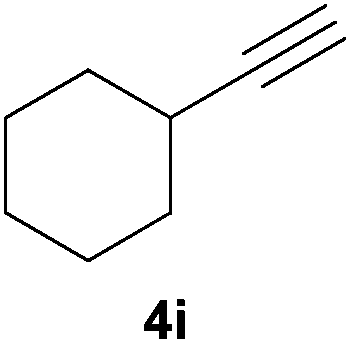
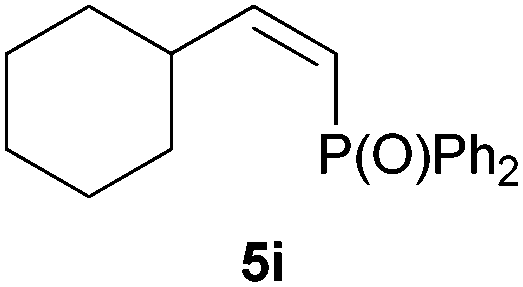
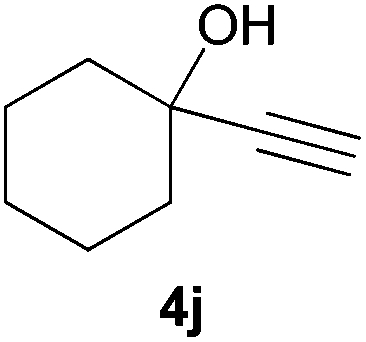
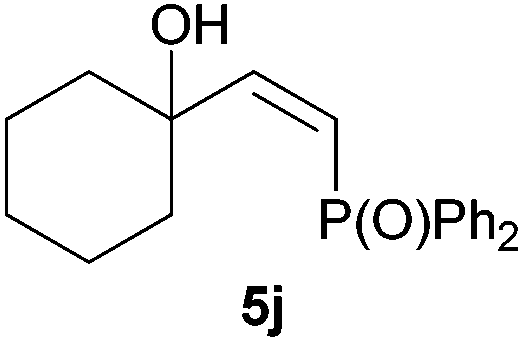
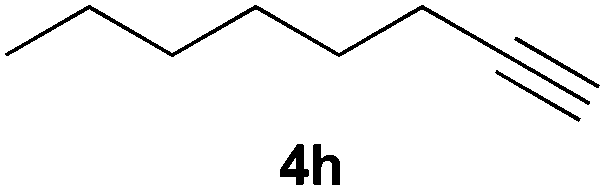
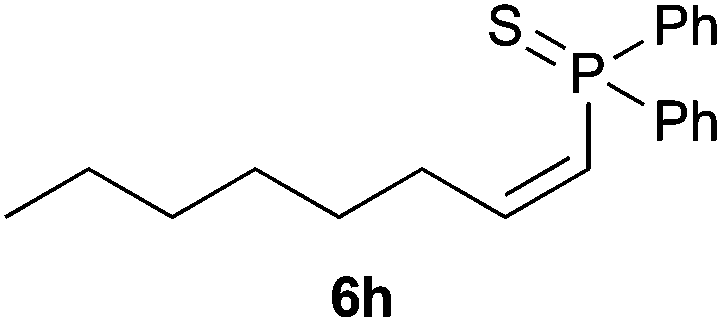
Electron-neutral and -rich arylacetylenes reacted nicely with diphenylphosphine at 70 °C yielding the expected anti-Markovnikov alkenylphosphines (5a–5e) as Z diastereoisomers.39 Converse behaviour was observed for arylacetylenes bearing electron-withdrawing substituents (e.g., 4f), which were reluctant to react under the same conditions. When methyl(phenyl)acetylene (4g) was subjected to the standard reaction conditions, the corresponding vinylphosphine was formed as a 90:10 E/Z diastereomeric mixture, with opposite regioselectivity to that reported with a calcium complex.38d Besides arylacetylenes, aliphatic alkynes also experienced hydrophosphination. The alkyl-chain alkyne 4h was converted into the phosphine 5h with a high degree of stereocontrol (97:3 Z/E ratio), whereas the tertiary phosphines resulting from the cyclohexyl derivatives 4i and 4j were prone to rapid oxidation, being obtained as the phosphine oxides 5i and 5j, respectively. A 60:40 Z/E ratio was recorded for 5i, though the major Z isomer could be isolated in moderate yield. Different outcome arose for the hydroxyl derivative 5j, with absolute control of both the regio- and the stereochemistry.
Alkyne hydrothiophosphination
Trofimov et al. have recently extended their addition of pre-formed secondary phosphine sulfides to alkenes, under catalyst- and solvent-free conditions, to alkynes, giving the tertiary anti-Markovnikov alkenylphosphine sulfides with Z stereochemistry.40 With a more straightforward approach in hand, and based on our previously reported three-component alkene hydrothiophosphination,22b hydrothiophosphination was undertaken for a series of alkynes (Table 6). The standard reaction conditions, in air, were applied to obtain alkenylphosphine sulfides in high yields from diphenylphosphine, sulfur and phenylacetylenes bearing electron-neutral, -donating and -withdrawing substituents (4a, 4b, 4d, 4k and 4l). As occurred in the alkyne hydrophosphination, the hydrothiophosphination of 4-trifluoromethyl(phenyl)acetylene was difficult, giving the expected product 6f in low yield. The same conditions were suitable for aliphatic alkynes, either linear-alkyl or cycloalkyl-substituted ones (4h and 4i).
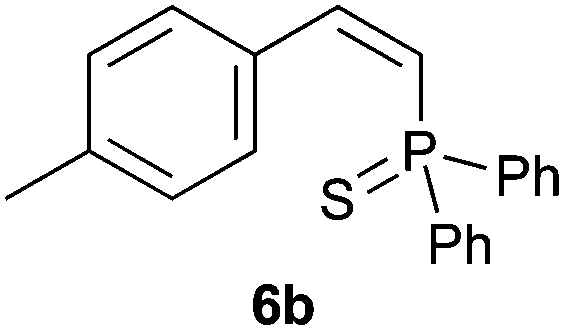
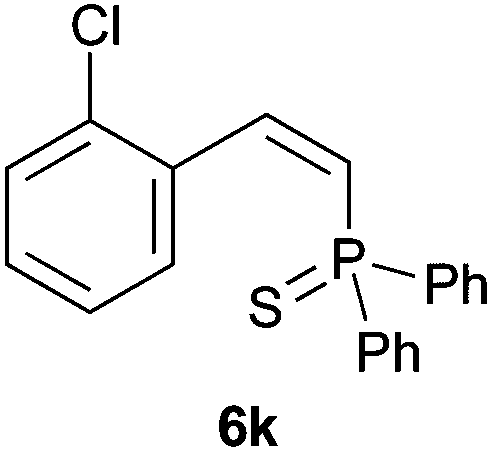
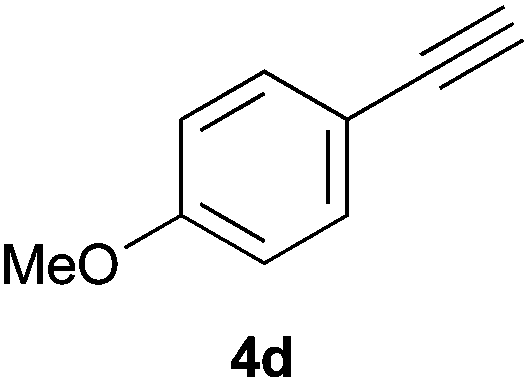
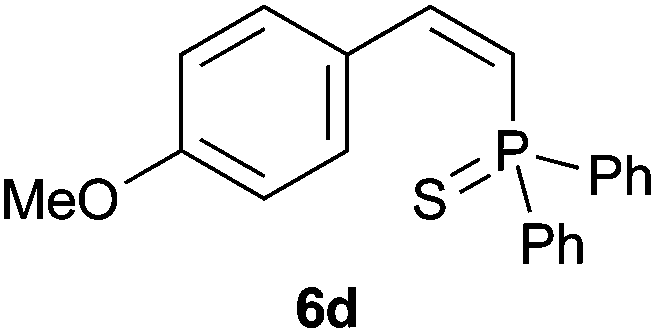
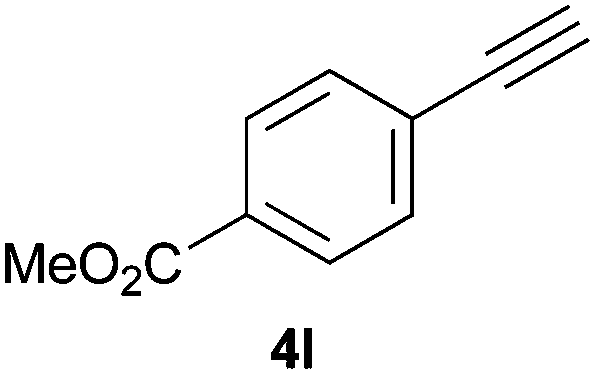
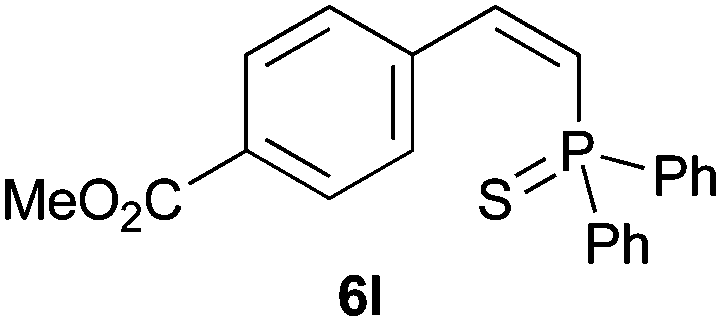
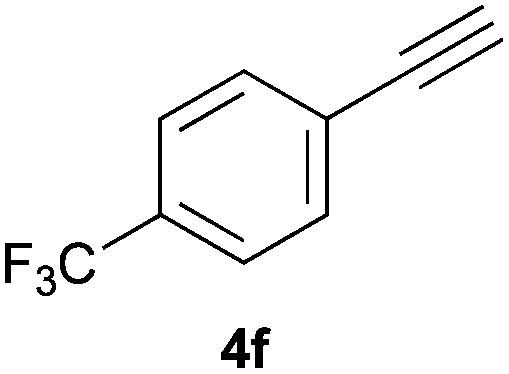
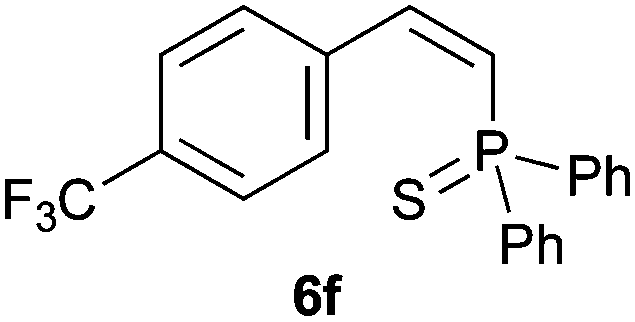
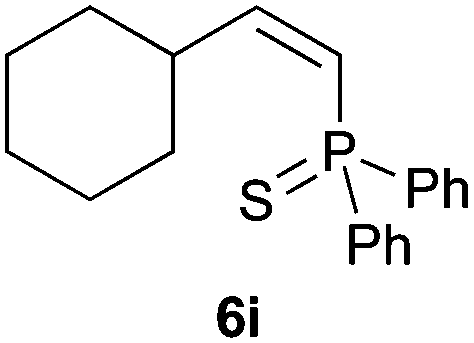
It must be pointed out that all the alkenylphosphine sulfides were synthesised as single anti-Markovnikov regioisomers and Z stereoisomers. The stereochemistry of the alkyne hydrothiophosphination and, therefore, that of the alkyne hydrophosphination was unequivocally established by X-ray crystallographic analysis of alkenylphosphine sulfide 6a (Fig. 2).41
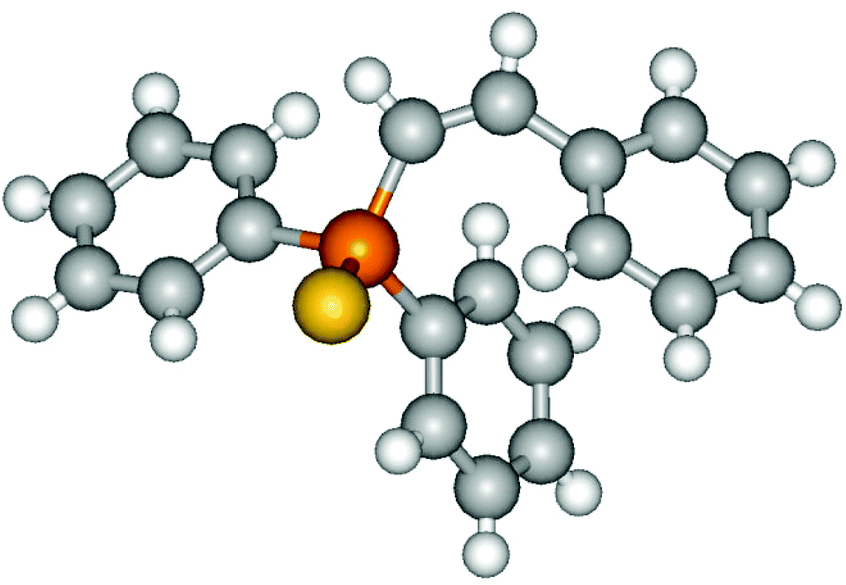 | ||
| Fig. 2 X-ray crystallographic structure of phosphine sulfide 6a. | ||
Mechanistic aspects
Koenig et al. concluded that the radical or ionic mechanism of addition of organophosphorus compounds containing a labile P–H bond to alkenes and alkynes was dependent on the nature of the unsaturated substrate (activated or inactivated), the nature of the organophosphorus compound (with P–H, O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) P–H or SP–H bonds) and the mode of activation (dry alkaline medium, alkaline medium/classical heating, US irradiation, alkaline medium/US irradiation, radical initiator/classical heating or photochemical irradiation).42
P–H or SP–H bonds) and the mode of activation (dry alkaline medium, alkaline medium/classical heating, US irradiation, alkaline medium/US irradiation, radical initiator/classical heating or photochemical irradiation).42
In order to gain an insight into the reaction mechanism, the hydrophosphination of styrene was performed in the presence of radical traps, such as cumene, TEMPO or 2,6-di-tert-butylphenol [Scheme 3, eqn (1)]. All reactions proceeded with >96% conversion and products derived from the radical traps and diphenylphosphinyl radicals were not detected. These results, together with the fact that the addition of diphenylphosphine to hepta-1,6-diene did not yield the corresponding cyclopentane derivative [Scheme 3, eqn (2)],43 point to a radical-free process. This hypothesis is consistent with the Z to E isomerisation observed for alkenylphosphine derivatives 5a and 5i under radical conditions (Scheme 4); that is, the thermodynamic E isomers, instead of the Z counterparts, should be largely formed if the hydrophosphination was driven by radicals. It must be highlighted that E/Z equilibration did not occur when the alkenyl phosphines 5a and 5i were subjected to prolonged heating either in the absence or presence of iodine.
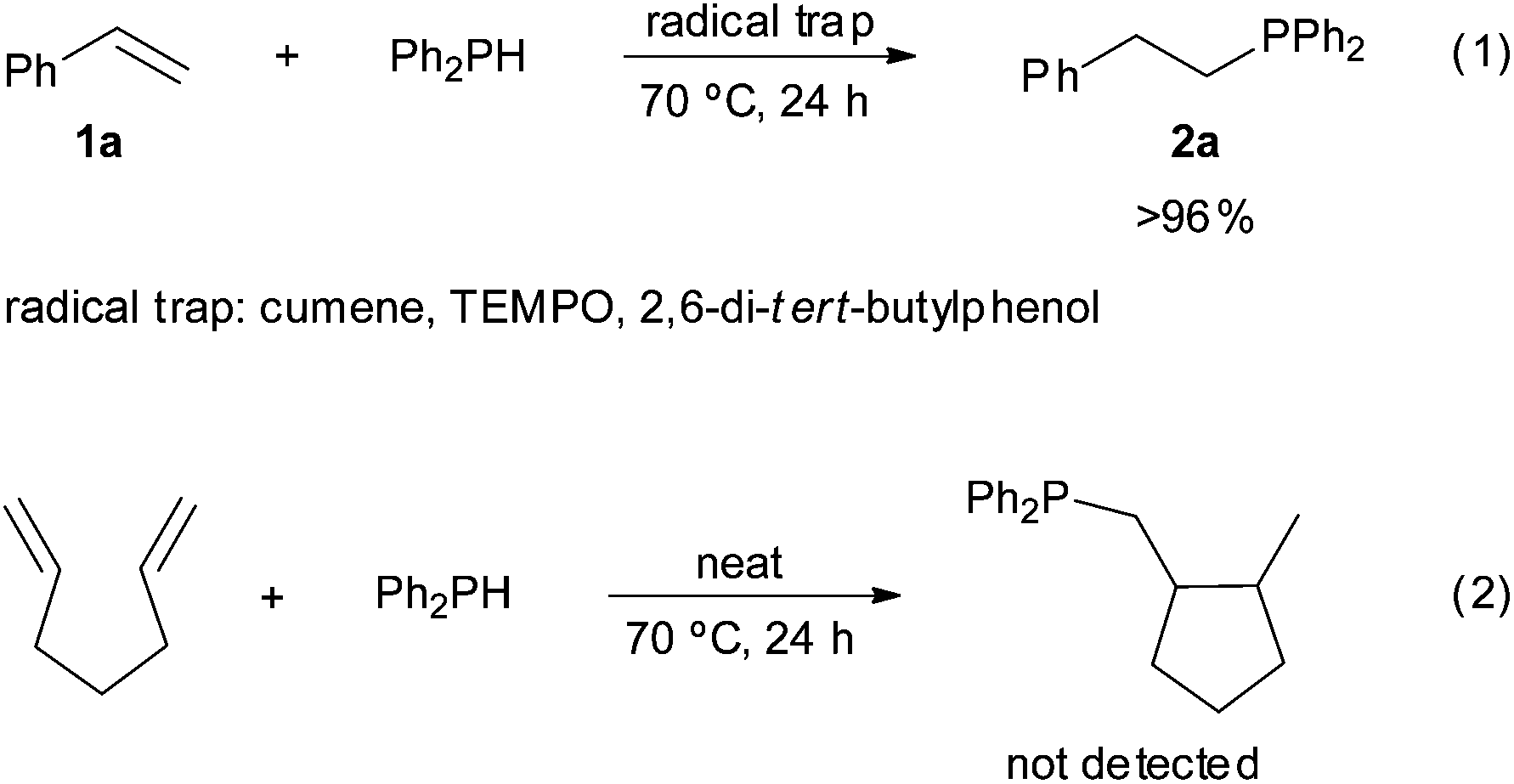 | ||
| Scheme 3 Experiments demonstrating the radical-free addition of diphenylphosphine to alkenes. | ||
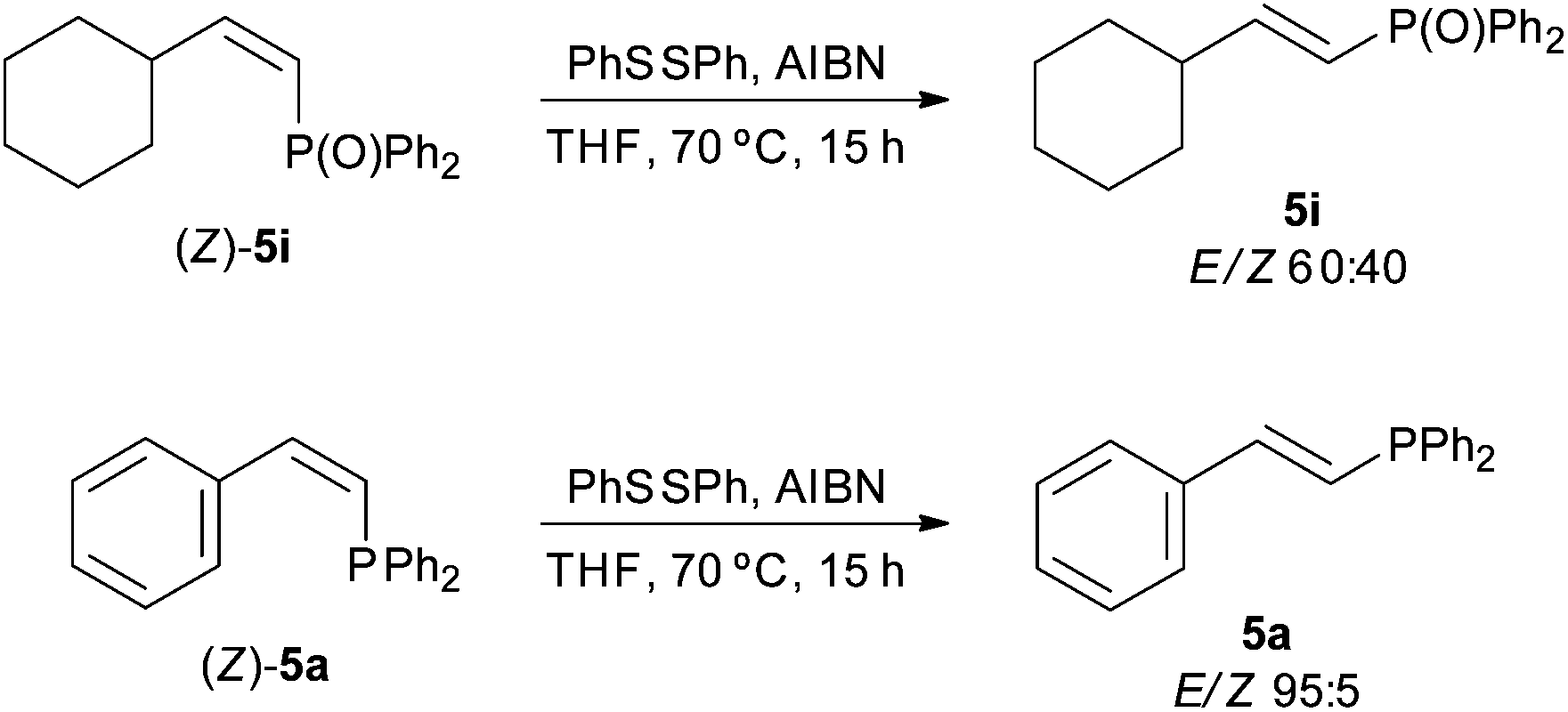 | ||
| Scheme 4 Radical-promoted Z/E isomerisation of alkenyl phosphine derivatives. | ||
A series of deuterium-labelling experiments have provided additional evidence on the reaction course. The addition of diphenylphosphine to deuterated cyclohexylacetylene D1-4i gave the Z α-deuteriovinylphosphine oxide D1-5i with 80% D incorporation. This essay ratifies the anti-addition of the P–H bond across the carbon–carbon triple bond which, apparently, can involve some H/D scrambling (Scheme 5).
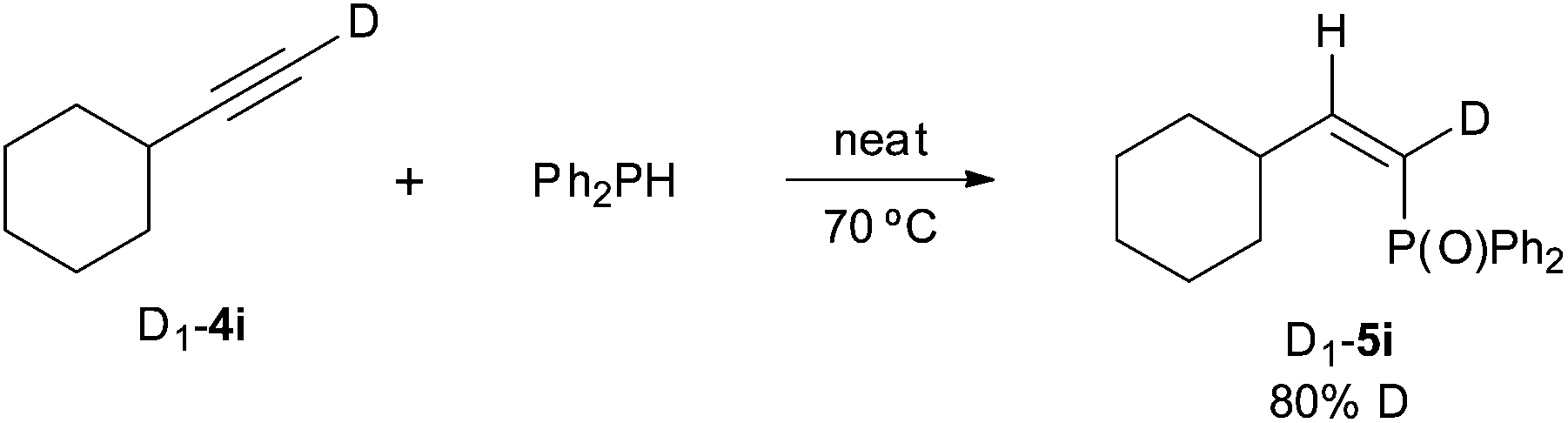 | ||
| Scheme 5 Deuterium-labelling experiments in the hydrophosphination of 4i. | ||
We also compared the rate of addition of Ph2PH to styrene at 70 °C with that of Ph2PD (Fig. 3). A kinetic isotopic effect was manifested, which was especially dramatic in the range 0–50 min (induction period for Ph2PD) and points to the cleavage of the P–H bond as being the rate-determining step of the reaction.
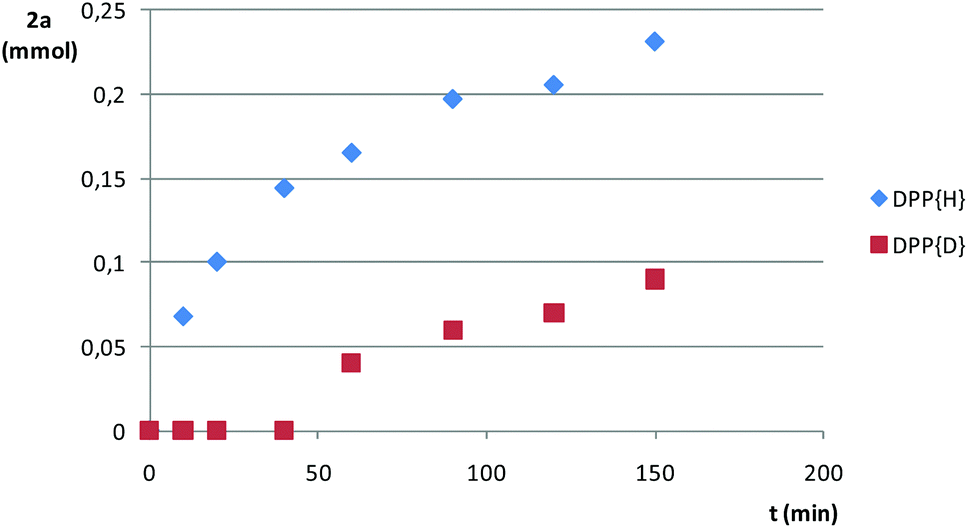 | ||
| Fig. 3 Kinetic profiles for the reaction of styrene (1a) with protio- and deuteriodiphenylphosphine, DPP{H} and DPP{D}, respectively, at 70 °C under Ar. | ||
The effect of the stoichiometry of the reactants was also investigated for the addition of diphenylphosphine to styrene, in this case, in the range 10–60 min (Fig. 4). It is clear that, with respect to a 1:1 stoichiometric ratio, the excess of styrene has a negligible effect on the formation of 2a, whereas an excess of diphenylphosphine speeds up its formation (e.g., >5-fold at 10 min by doubling the amount of diphenylphosphine). This effect was expected to be more prominent at shorter reaction times.44 Larger amounts of diphenylphosphine (e.g., 1:3 ratio) were shown to be unproductive with respect to the 1:2 ratio, displaying a very similar trend.
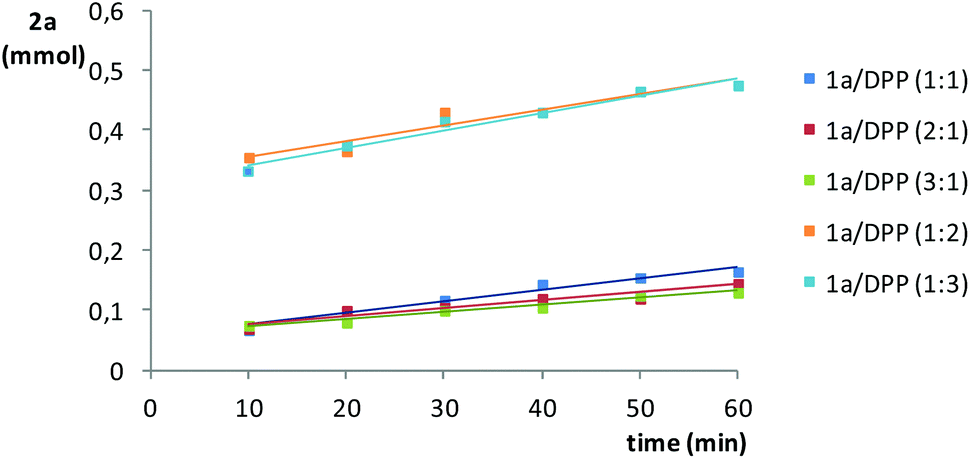 | ||
| Fig. 4 Effect of the styrene (1a)/diphenylphosphine (DPP) ratio on the formation of 2a in the 10–60 min range, at 70 °C under Ar. The amount of 2a is referenced to the limiting substrate. | ||
Considering the close electronegativities of P (χP = 2.19) and H (χH = 2.20), in principle it might be presumed that the anti-Markovnikov regioselectivity for the hydrophosphination (particularly, that of inactivated substrates) is primarily governed by steric factors rather than by electronic factors. In this context, substrates leading to more sterically hindered products, such as stilbene of diphenylacetylene, reacted sluggishly with diphenylphosphine either in the presence or absence of sulfur. A hydroboration-type model, in which the P–H bond is added through a four-membered ring transition state must be disregarded because (a) the B–H bond (χB = 2.04) is more polarized than the P–H bond and this circumstance works in the same direction as the steric factor in the former, and (b) the hydroboration is syn whereas the hydrophosphination is anti [Scheme 6, eqn (a)].
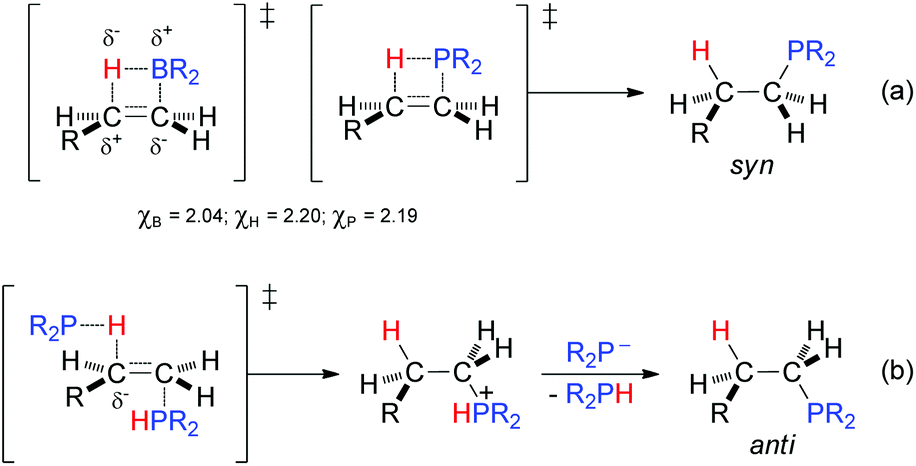 | ||
| Scheme 6 Mechanistic proposals for the hydrophosphination of carbon–carbon double bonds following (a) a syn addition (hydroboration-type model) and (b) an anti addition. | ||
The above experiments and considerations lend weight to the argument that the hydrophosphination reaction follows an ionic pathway with anti-addition, where each of the P and H atoms emerges from two different phosphine molecules. The ability of tertiary phosphines to act as nucleophilic catalysts in the addition to alkenes (especially, α,β-unsaturated carbonyl compounds) is well known.45 Therefore, it seems reasonable to propose that one phosphine molecule has a nucleophilic role, by addition to the terminal carbon atom of the alkene, while a second molecule behaves as the electrophilic partner through its hydrogen atom [Scheme 6, eqn (b)].
With the proposed transition state model, electronic factors also come into play as the negative charge density developed in the transition state is especially stabilised at the benzylic position (e.g., in the styrenes) as well as at the α-position with respect to heteroatoms (in Het-vinyl substrates) and carbonyl groups (in α,β-unsaturated carbonyl compounds). In the case of 3,4-dihydro-2H-pyran (1ac), with a near symmetric carbon–carbon double bond, electronic factors seem to prevail with the addition of the P atom to the most electrophilic α-C atom (Table 4). Further support for the interpretation made of the hydrophosphination comes from the addition of Ph2PH to β-pinene (1z): the formation of the ring-opened product 2z (Table 3) seems more feasible if two molecules of diphenylphosphine are implicated in the reaction.
Concerning the three-component syntheses of alkyl and alkenyl phosphine sulfides, we earlier confirmed that in situ formation of diphenylphosphine sulfide comes off prior to the addition (Scheme 7).22b
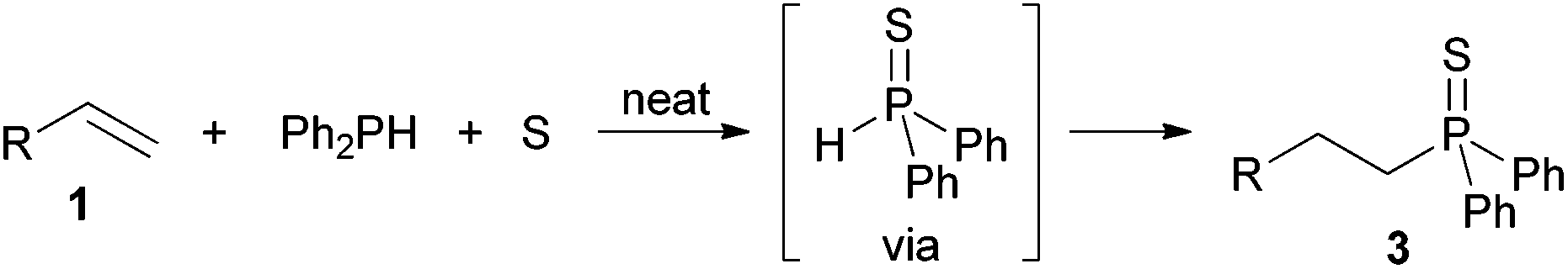 | ||
| Scheme 7 Diphenylphosphine sulfide as an intermediate in the three-component synthesis of alkylphosphine sulfides. | ||
Comparison with other catalysts
Although an array of effective catalysts has been designed for the hydrophosphination of alkenes, some operating even at room temperature, in practice, the results do not differ so much from those attained in the absence of a solvent and a catalyst (Table 7). Moreover, most of these results are based on NMR conversions (instead of isolated yields) and small-scale reactions performed in NMR tubes, which curtail the potential scope of the method. Similarly, the usage of an excess of the starting alkene in some of the methods has a negative effect on the E-factor.|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst (mol%) | Solvent | T (°C) | Time (h) | Yielda (%) |
| a Isolated yield unless otherwise stated; conversion in parentheses. b 2.0 equiv. of styrene; 1 equiv. of Et3N. c Isolated yield of the phosphine oxide. d Conversion determined by NMR. e Isolated as the borane complex. f 1.82 equiv. of alkene. g This work. | |||||
| 146 | t-BuOK (20) | DMSO | rt | 1 | 83 |
| 227b | Ni[P(OEt)3]4 (5) | C6H6b |
130 | 20 | 92 |
| 326a | Ca-amide complex (10) | C6H6 | 75 | 20 | 64c (95)d |
| 430 | Cu(OTf)2·PhMe (10) | Dioxane-d8 | 100 | 18–24 | 83 |
| 531 | FeCl2 (30) | MeCN | 60 | 12 | 87e |
| 625e | Ca-amide complex (2) | C6D6 | 25 | 3 | (100)d |
| 725f | Ba-amide complex (2) | C6D6 | 60 | 0.4 | (>96)d |
| 825g | Yb(II) complex (1) | C6D6 | 60 | 4 | (92)d |
| 928 | Fe(III) complex (0.5) | MeCNf | rt | 24 | 89 |
| 10 | None | None | 70 | 4 | 82g |

Similar comments can be extended to the hydrophosphination of alkynes, in this case exemplified by the addition of diphenylphosphine to phenylacetylene (Table 8). The vinyl phosphine 5a can be obtained without a catalyst and a solvent, not only in good isolated yield but also in the highest E/Z diastereoselectivity (entry 6). Taking into account different parameters (catalyst, solvent, temperature, time, yield and selectivity), together with the simplicity of the procedure, we can state that this approach towards the addition of the P–H bond to alkenes and alkynes distinctly outperforms others based on catalytic approaches.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst (mol%) | Solvent | T (°C) | Time | Yielda (%) |
|
a
Z/E ratio in parenthesis.
b 31P NMR yield.
c GC yield of the phosphine oxide.
d Conversion determined by NMR.
e This work; isolated yield.
|
|||||
| 137 | Pd(PPh3)4 (1.2) | MeCN | 130 | 18 h | 95b (86/14) |
| 238a | Yb-imine complex (5) | THF | rt | 5 min | >99c (24/76) |
| 325e | Ca-amide complex (5) | C6D6 | 75 | 38 h | 78d (76/24) |
| 425e | Yb-amide complex (5) | C6D6 | 75 | 38 h | 91d (10/90) |
| 538f | Y complex (2) | C6D6 | 70 | 72 h | 100d (42/58) |
| 6 | None | None | 70 | 10 | 80e (95/5) |
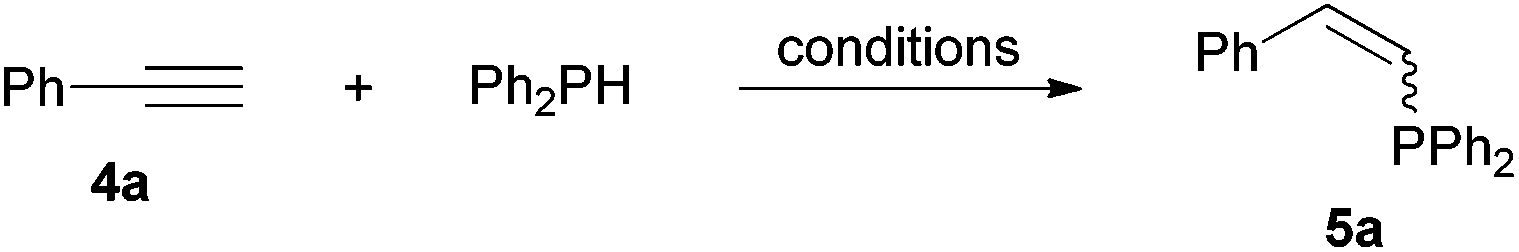
Conclusions
For decades, the addition of the P–H bond across multiple carbon–carbon bonds has been associated with activation by bases, acids, radicals or metals. We have demonstrated that tertiary phosphines can be readily prepared by thermal treatment of secondary phosphines and alkenes in the absence of solvents and catalysts, under an inert atmosphere. This method is applicable to α,β-unsaturated carbonyl compounds, styrenes, N- and S-vinyl compounds, as well as inactivated alkenes. The process is highly regioselective producing the tertiary phosphines in an anti-Markovnikov fashion. Alkynes undergo the same type of addition to furnish anti-Markovnikov alkenyl phosphines with Z stereoselectivity.Furthermore, by introducing elemental sulfur into the reaction mixture in air, alkyl and alkenyl phosphine sulfides can be readily synthesised through a one-pot three-component approach in a regio- and stereoselective manner. Compelling experimental evidence suggests an ionic-type anti addition reaction mechanism governed by steric and electronic effects different from that of the alkene/alkyne hydroboration.
These methods are in agreement with some of the twelve principles introduced by Anastas et al.,5a namely: (a) waste is prevented because of no by-product formation, (b) high atom economy, as all the starting materials are incorporated into the final products, (c) unnecessary use of solvents, (d) some reactions proceed at room temperature and most of them at 70 °C, (e) unnecessary derivatisation (the secondary phosphine sulfides are generated in situ) and (f) neither stoichiometric nor catalytic reagents are employed because the processes are catalyst free. In addition to this, similar or better results are obtained with respect to the catalytic methods.
In short, this study supports the statements by Sheldon that “the best catalyst is no catalyst” and “the best solvent is no solvent”.47
Experimental
General procedure for the hydrophosphination of alkenes 1
All reactions were performed using tubes in a multi-reactor system under argon. Diphenylphosphine (0.5 mmol, 87 μL) and alkene (1, 0.5 mmol) were stirred during the specified time at room temperature, 70 or 100 °C (Tables 1–3). The progress of the reaction was monitored by TLC and/or GLC until total or steady conversion was achieved. The resulting mixture was dissolved in EtOAc (20 mL) followed by the addition of silica gel and removal of the excess of solvent in vacuo. The reaction crude absorbed on silica gel was subjected to column chromatography (silica gel, hexane/EtOAc) to give the pure tertiary phosphines 2. The phosphine oxides 2x, 2y and 2z were purified by preparative chromatography (silica gel, hexane/EtOAc).General procedure for the hydrothiophosphination of alkenes 1
All reactions were performed using tubes in a multi-reactor system under air. Diphenylphosphine (0.5 mmol, 87 μL), alkene (1, 0.5 mmol) and elemental sulfur (0.5 mmol, 16.0 mg) were stirred at 70, 100 or 120 °C overnight (Table 4). The progress of the reaction was monitored by TLC and/or GLC until total or steady conversion was achieved. The resulting mixture was dissolved in EtOAc (20 mL) followed by the addition of silica gel and removal of the excess of solvent in vacuo. The reaction crude absorbed on silica gel was subjected to column chromatography (silica gel, hexane/EtOAc) to give the pure tertiary phosphine sulfides 3. Compounds 3b, 3e, 3n and 3y were purified by preparative chromatography (silica gel, hexane/EtOAc).General procedure for the hydrophosphination of alkynes 4
All reactions were performed using tubes in a multi-reactor system under argon. Diphenylphosphine (0.5 mmol, 87 μL) and alkyne (4, 0.5 mmol) were stirred at 70 °C overnight (Table 5). The progress of the reaction was monitored by TLC and/or GLC until total or steady conversion was achieved. The resulting mixture was dissolved in EtOAc (20 mL) followed by the addition of silica gel and removal of the excess of solvent in vacuo. The reaction crude absorbed on silica gel was subjected to column chromatography (silica gel, hexane/EtOAc) to give the pure alkenyl phosphines 5.General procedure for the hydrothiophosphination of alkynes 4
All reactions were performed using tubes in a multi-reactor system under air. Diphenylphosphine (0.5 mmol, 87 μL), alkyne (4, 0.5 mmol) and elemental sulfur (0.5 mmol, 16.0 mg) were stirred at 70 °C overnight (Table 6). The progress of the reaction was monitored by TLC and/or GLC until total or steady conversion was achieved. The resulting mixture was dissolved in EtOAc (20 mL) followed by the addition of silica gel and removal of the excess of solvent in vacuo. The reaction crude absorbed on silica gel was subjected to column chromatography (silica gel, hexane/EtOAc) to give the pure alkenyl phosphine sulfides 6.Acknowledgements
This work was generously supported by the Spanish Ministerio de Economía y Competitividad (MINECO; CTQ2011-24151). Y. M. and I. M.-G. acknowledge the Instituto de Síntesis Orgánica (ISO) of the Universidad de Alicante for the grants. M. J. G.-S. Thanks the ISO and the Generalitat Valenciana (grant no. APOTIP/2015/014) for financial support.Notes and references
- L. D. Quin, A Guide to Organophosphorus Chemistry, Wiley-Interscience, New York, 2000 Search PubMed.
- For recent reviews and monographs, see: (a) Phosphorus Ligands in Asymmetric Catalysis: Synthesis and Applications, ed. A. Börner, Wiley-VCH, Weinheim, 2008 Search PubMed; (b) P. E. Goudriann, P. W. N. M. van Leeuwen, M.-N. Birkholz and J. N. H. Reek, Eur. J. Org. Chem., 2008, 2939–2958 CrossRef; (c) S. Lühr, J. Holz and A. Börner, ChemCatChem, 2011, 3, 1708–1730 CrossRef; (d) J. Wassenaar and J. N. H. Reek, Org. Biomol. Chem., 2011, 9, 1704 RSC; (e) F. L. Lam, F. K. Kwong and A. S. C. Chan, Top. Organomet. Chem., 2011, 36, 29–66 CrossRef CAS; (f) S. Lühr, J. Holz and A. Börner, ChemCatChem, 2011, 3, 1708–1730 CrossRef; (g) H. Fernández-Pérez, P. Etayo, A. Panossian and A. Vidal-Ferran, Chem. Rev., 2011, 111, 2119–2176 CrossRef PubMed; (h) Privileged Chiral Ligands and Catalysis, ed. Q.-L. Zhou, Wiley-VCH, Weinheim, 2011 Search PubMed; (i) Phosphorus(III) Ligands in Homogeneous Catalysis: Design and Synthesis, ed. P. C. J. Kamer and P. W. N. M. van Leeuwen, John Wiley & Sons, Chichester, 2012 Search PubMed; (j) Y. C. Fan and O. Kwon, Chem. Commun., 2013, 49, 11588–11619 RSC; (k) Y. Xiao, H. Guo and O. Kwon, Aldrichimica Acta, 2016, 49, 3–13 Search PubMed.
- A. A. Nazarov and P. J. Dyson, in Phosphorus Compounds, Advanced Tools in Catalysis and Materials Science, Catalysis by Metal Complexes, ed. M. Peruzzini and L. Gonsalvi, Springer, Dordrecht, 2011, ch. 13, vol. 37 Search PubMed.
- For reviews, see: (a) I. P. Beletskaya and M. A. Kazankova, Russ. J. Org. Chem., 2002, 38, 1391–1430 CrossRef CAS; (b) M. L. Clarke and J. M. J. Williams, in Organophosphorus Reagents. A Practical Approach in Chemistry, ed. P. J. Murphy, Oxford University Press, Oxford, 2004, ch. 2 Search PubMed; (c) E. Hey-Hawkins and A. A. Karasik, in Science of Synthesis, ed. F. Mathey, Georg Thieme Verlag, Stuttgart, 2009, vol. 42, pp. 71–108 Search PubMed; (d) B. A. Trofimov and N. K. Gusarova, Mendeleev Commun., 2009, 19, 295–302 CrossRef CAS; (e) D. S. Glueck, Top. Organomet. Chem., 2010, 31, 65–100 CrossRef CAS.
- (a) P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC. For reviews on organophosphorus compounds and Green Chemistry, see: (b) I. L. Odinets and E. V. Matveeva, Russ. Chem. Rev., 2012, 81, 221–238 CrossRef CAS; (c) G. Keglevich and I. Greiner, Curr. Green Chem., 2014, 1, 2–16 CrossRef CAS.
- For reviews, see: (a) C. Baillie and J. Xiao, Curr. Org. Chem., 2003, 7, 477–514 CrossRef CAS; (b) F. Alonso, I. P. Beletskaya and M. Yus, Chem. Rev., 2004, 104, 3079–3159 CrossRef CAS PubMed; (c) O. Delacroix and A. C. Gaumont, Curr. Org. Chem., 2005, 9, 1851–1882 CrossRef CAS; (d) S. A. Pullarkat and P.-H. Leung, Top. Organomet. Chem., 2013, 43, 145–166 CrossRef CAS; (e) V. Koshti, S. Gaikwad and S. H. Chikkali, Coord. Chem. Rev., 2014, 265, 52–73 CrossRef CAS.
- N. L. Kilah and S. B. Wild, in Science of Synthesis, ed. F. Mathey, Georg Thieme Verlag, Stuttgart, 2009, vol. 42, pp. 595–632 Search PubMed.
- A. Florido, L. G. Bachas, M. Valiente and I. Villaescusa, Analyst, 1994, 119, 2421–2425 RSC.
- Q. Jia, Q. Shang and W. Zhou, Ind. Eng. Chem. Res., 2004, 43, 6703–6707 CrossRef CAS.
- M. Castillo and I. A. Rivero, ARKIVOC, 2003, xi, 193–202 Search PubMed.
- A. Fukazawa, M. Kiguchi, S. Tange, Y. Ichihashi, Q. Zhao, T. Takahashi, T. Konishi, K. Murakoshi, Y. Tsuji, A. Staykov, K. Yoshizawa and S. Yamaguchi, Chem. Lett., 2011, 40, 174–176 CrossRef CAS.
- (a) T. Eto, H. Nakajima and H. Kobayashi, JP Patent, 2002265759, 2002 Search PubMed; (b) A. Jallouli, Y. Turshani, S. Wanigatunga and M. Rickwood, WO Patent, 2004085447, 2004 Search PubMed.
- (a) M. Preisenberger, A. Bauer and H. Schmidbaur, Chem. Ber./Recl., 1997, 130, 955–958 CrossRef CAS; (b) S. Ahmad, A. A. Isab, H. P. Perzanowski, M. Sakhawat Hussain and M. Naseem Akhtar, Transition Met. Chem., 2002, 27, 177–183 CrossRef CAS; (c) A. T. Breshears, A. C. Behrle, C. L. Barnes, C. H. Laber, G. A. Baker and J. R. Walensky, Polyhedron, 2015, 100, 333–343 CrossRef CAS.
- (a) N. K. Skvortsov, V. N. Spevak, L. V. Pashnova and V. K. Bel'skii, Russ. J. Gen. Chem., 1997, 67, 487 CAS; (b) M. Hayashi, H. Takezaki, Y. Hashimoto, K. Takaoki and K. Saigo, Tetrahedron Lett., 1998, 39, 7529–7532 CrossRef CAS; (c) M. Hayashi, Y. Hashimoto, Y. Yamamoto, J. Usuki and K. Saigo, Angew. Chem., Int. Ed., 2000, 39, 631–633 CrossRef CAS.
- (a) C. J. Chapman, C. G. Frost, M. P. Gill-Carey, G. Kociok-Köhn, M. F. Mahon, A. S. Weller and M. C. Willis, Tetrahedron: Asymmetry, 2003, 14, 705–710 CrossRef CAS; (b) E. Ichikawa, M. Suzuki, K. Yabu, M. Albert, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2004, 126, 11808–11809 CrossRef CAS PubMed.
- (a) S. R. Gilberston and X. Wang, J. Org. Chem., 1996, 61, 434–435 CrossRef; (b) N. K. Gusarova, M. V. Bogdanova, N. I. Ivanova, N. A. Chernysheva, B. G. Sukhov, L. M. Sinegovskaya, O. N. Kazheva, G. G. Alexandrov, O. A. D'yachenko and B. A. Trofimov, Synthesis, 2005, 3103–3106 CAS; (c) N. K. Gusarova, M. V. Bogdanova, N. I. Ivanova, N. A. Chernysheva, A. A. Tatarinova and B. A. Trofimov, Russ. J. Gen. Chem., 2006, 76, 1201–1204 CrossRef CAS; (d) N. K. Gusarova, S. F. Malysheva, N. A. Belogorlova, V. A. Kuimov, I. A. Ushakov and B. A. Trofimov, Russ. Chem. Bull., 2009, 58, 234–237 CrossRef CAS.
- (a) A. F. Parsons, D. J. Sharpe and P. Taylor, Synlett, 2005, 2981–2983 CrossRef CAS; (b) N. K. Gusarova, M. V. Bogdanova, N. I. Ivanova, N. A. Chernysheva, S. V. Yas'ko, V. G. Samoilov, S. M. Markosyan and B. A. Trofimov, Russ. J. Gen. Chem., 2006, 76, 1514–1515 CrossRef CAS; (c) B. A. Trofimov, S. F. Malysheva, N. K. Gusarova, N. A. Belogorlova, S. F. Vasilevsky, V. B. Kobychev, B. G. Sukhov and I. A. Ushakov, Mendeleev Commun., 2007, 17, 181–182 CrossRef CAS; (d) L. A. Oparina, S. F. Malysheva, N. K. Gusarova, N. A. Belogorlova, O. V. Vysotskaya, A. V. Stepanov, A. I. Albanov and B. A. Trofimov, Synthesis, 2009, 3427–3432 CAS; (e) N. A. Chernysheva, S. V. Yas'ko, N. K. Gusarova, A. A. Tatarinova, L. V. Klyba and B. A. Trofimov, Mendeleev Commun., 2010, 20, 346–347 CrossRef CAS; (f) N. K. Gusarova, S. F. Malysheva, N. A. Belogorlova, L. N. Parshina and B. A. Trofimov, Synthesis, 2011, 1777–1782 CAS.
- V. A. Kuimov, S. F. Malysheva, N. K. Gusarova, A. O. Korocheva and B. A. Trofimov, J. Sulfur Chem., 2014, 35, 137–144 CrossRef CAS.
- (a) M. Arisawa and M. Yamaguchi, Tetrahedron Lett., 2009, 50, 45–47 CrossRef CAS; (b) M. Hayashi, T. Matsuura, I. Tanaka, H. Ohta and Y. Watanabe, Org. Lett., 2013, 15, 628–631 CrossRef CAS PubMed.
- (a) K. Tanaka and F. Toda, Chem. Rev., 2000, 100, 1025–1074 CrossRef CAS PubMed; (b) K. Tanaka and F. Toda, Solvent-free Organic Synthesis, Wiley-VCH, Weinheim, 2003 Search PubMed.
- Review: G. Brahmachari and B. Banerjee, Curr. Green Chem., 2015, 2, 274–305 CrossRef CAS.
- (a) F. Alonso, Y. Moglie, G. Radivoy and M. Yus, Green Chem., 2012, 14, 2699–2702 RSC; (b) F. Alonso and Y. Moglie, Curr. Green Chem., 2014, 1, 87–93 CrossRef CAS; (c) V. Gutiérrez, E. Mascaró, F. Alonso, Y. Moglie and G. Radivoy, RSC Adv., 2015, 5, 65739–65744 RSC; (d) Y. Moglie, E. Mascaró, V. Gutiérrez, F. Alonso and G. Radivoy, J. Org. Chem., 2016, 81, 1813–1818 CrossRef CAS PubMed.
- (a) M. C. Hoff and P. Hill, J. Org. Chem., 1959, 24, 356–359 CrossRef CAS; (b) B. D. Dombek, J. Org. Chem., 1978, 43, 3408–3409 CrossRef CAS.
- (a) D. K. Wicht, I. V. Kourkine, B. M. Lew, J. Mulei Nthenge and D. S. Glueck, J. Am. Chem. Soc., 1997, 119, 5039–5040 CrossRef CAS; (b) D. K. Wicht, I. V. Kourkine, I. Kovacik and D. S. Glueck, Organometallics, 1999, 18, 5381–5394 CrossRef CAS; (c) C. Scriban, I. Kovacik and D. S. Glueck, Organometallics, 2005, 24, 4871–4874 CrossRef CAS; (d) I. Kovacik, C. Scriban and D. S. Glueck, Organometallics, 2006, 25, 536–539 CrossRef CAS; (e) C. Scriban, D. S. Glueck, L. N. Zakharov, W. Scott Kassel, A. G. DiPasquale, J. A. Golen and A. L. Rheingold, Organometallics, 2006, 25, 5757–5767 CrossRef CAS.
- (a) M. R. Douglass and T. J. Marks, J. Am. Chem. Soc., 2000, 122, 1824–1825 CrossRef CAS; (b) M. R. Douglass, C. L. Stern and T. J. Marks, J. Am. Chem. Soc., 2001, 123, 10221–10238 CAS; (c) K. Takaki, M. Takeda, G. Koshoji, T. Shishido and K. Takehira, Tetrahedron Lett., 2001, 42, 6357–6360 CrossRef CAS; (d) A. Motta, I. L. Fragalà and T. J. Marks, Organometallics, 2005, 24, 4995–5003 CrossRef CAS; (e) H. Hu and C. Cui, Organometallics, 2012, 31, 1208–1211 CrossRef CAS; (f) B. Liu, T. Roisnel, J.-F. Carpentier and Y. Sarazin, Chem. – Eur. J., 2013, 19, 13445–13462 CrossRef CAS PubMed; (g) I. V. Basalov, S. C. Rosca, D. M. Lyubov, A. N. Selikhov, G. K. Fukin, Y. Sarazin, J.-F. Carpentier and A. A. Trifonov, Inorg. Chem., 2014, 53, 1654–1661 CrossRef CAS PubMed.
- (a) M. R. Crimmin, A. G. M. Barret, M. S. Hill, P. B. Hitchcock and P. A. Procopiu, Organometallics, 2007, 26, 2953–2956 CrossRef CAS; (b) S.-C. Rosca, T. Roisnel, V. Dorcet, J.-F. Carpentier and Y. Sarazin, Organometallics, 2014, 33, 5630–5642 CrossRef CAS.
- (a) M. O. Shulyupin, M. A. Kazankova and I. P. Beletskaya, Org. Lett., 2002, 4, 761–763 CrossRef CAS PubMed; (b) M. A. Kazankova, M. O. Shulyupin, A. A. Borisenko and I. P. Beletskaya, Russ. J. Org. Chem., 2002, 38, 1479–1484 CrossRef CAS. For the Pd- and Ni-catalysed hydrophosphination of vinyl ethers, see: (c) M. A. Kazankova, M. O. Shulyupin and I. P. Beletskaya, Synlett, 2003, 2155–2158 CAS; (d) M. O. Shulyupin, I. G. Trostyanskaya, M. A. Kazankova and I. P. Beletskaya, Russ. J. Org. Chem., 2006, 42, 17–22 CrossRef CAS.
- K. J. Gallager and R. L. Webster, Chem. Commun., 2014, 50, 12109–12111 RSC.
- (a) W.-C. Yeo, L. Tang, B. Yan, S.-Y. Tee, L. L. Koh, G.-K. Tan and P.-H. Leung, Organometallics, 2005, 24, 5581–5585 CrossRef CAS; (b) Y. Zhang, L. Tang, S. A. Pullarkat, F. Liu, Y. Li and P.-H. Leung, J. Organomet. Chem., 2009, 694, 3500–3505 CrossRef CAS; (c) Y. Huang, S. A. Pullarkat, Y. Li and P.-H. Leung, Chem. Commun., 2010, 46, 6950–6952 RSC; (d) Y. Huang, S. A. Pullarkat, M. Yuan, Y. Ding, Y. Li and P.-H. Leung, Organometallics, 2010, 29, 536–542 CrossRef CAS; (e) C. Xu, G. J. H. Kennard, F. Hennersdorf, Y. Li, S. A. Pullarkat and P.-H. Leung, Organometallics, 2012, 31, 3022–3026 CrossRef CAS.
- A. Leyva-Pérez, J. A. Vidal-Moya, J. R. Cabrero-Antonino, S. S. Al-Deyab, S. I. Al-Resayes and A. Corma, J. Organomet. Chem., 2011, 696, 362–367 CrossRef.
- (a) M. Taillefer, L. Routaboul and A. C. Gaumont, FR Patent, 2965809, 2012 Search PubMed; (b) L. Routaboul, F. Toulgoat, J. Gatignol, J.-F. Lohier, B. Norah, O. Delacroix, C. Alayrac, M. Taillefer and A.-C. Gaumond, Chem. – Eur. J., 2013, 19, 8760–8764 CrossRef CAS PubMed.
- (a) D. Mimeau, O. Delacroix and A.-C. Gaumont, Chem. Commun., 2003, 2928–2929 RSC; (b) D. Mimeau, O. Delacroix, B. Join and A.-C. Gaumont, C. R. Chim., 2004, 7, 845–854 CrossRef CAS.
- See, for instance: R. L. Rogers, J. L. Moore and T. Rovis, Angew. Chem., Int. Ed., 2007, 46, 9301–9304 CrossRef CAS PubMed.
- See, for instance: C. Pisano, A. Mezzetti and G. Consiglio, Organometallics, 1992, 11, 20–22 CrossRef CAS.
- CCDC 1468810 (2z) contains the supplementary crystallographic data for this paper (see the ESI†).
- (a) S. F. Malysheva, N. K. Gusarova, A. V. Artem'ev, N. A. Belogorlova, A. I. Albanov, T. N. Borodina, V. I. Smirnov and B. A. Trofimov, Eur. J. Org. Chem., 2014, 2516–2521 CrossRef CAS; (b) N. K. Gurasova, N. A. Chernysheva, S. V. Yas'ko and B. A. Trofimov, J. Sulfur Chem., 2015, 36, 526–534 CrossRef; (c) For a related example published during the preparation of this manuscript, see: A. V. Artem'ev, N. A. Chernysheva, S. V. Yas'ko, N. K. Gusarova, I. Y. Bagryanskaya and B. A. Trofimov, Heteroat. Chem., 2016, 27, 48–53 CrossRef.
- M. A. Kazankova, I. V. Efimova, A. N. Kochetkov, V. V. Afanas'ev and I. P. Beletskaya, Russ. J. Org. Chem., 2002, 38, 1465–1474 CrossRef CAS.
- (a) Yb: K. Takaki, G. Koshoji, K. Komeyama, M. Takeda, T. Shishido, A. Kitani and K. Takehira, J. Org. Chem., 2003, 68, 6554–6565 CrossRef CAS PubMed; (b) Pd and Ni: B. Join, D. Mimeau, O. Delacroix and A.-C. Gaumont, Chem. Commun., 2006, 3249–3251 RSC; (c) Cu: A. Kondoh, H. Yorimitsu and K. Oshima, J. Am. Chem. Soc., 2007, 129, 4099–4104 CrossRef CAS PubMed; (d) Ca: T. M. A. Al-Shboul, V. K. Pálfi, L. Yu, R. Kretschmer, K. Wimmer, R. Fischer, H. Görls, M. Reiher and M. Westerhausen, J. Organomet. Chem., 2011, 696, 216–227 CrossRef CAS; (e) Fe: M. Kamitami, M. Itazaki, C. Tamiya and H. Nakazawa, J. Am. Chem. Soc., 2012, 134, 11932–11935 CrossRef PubMed; (f) Y: A. A. Kissel, T. V. Mahrova, D. M. Lyubov, A. V. Cherkasov, G. K. Fukin, A. A. Trifonov, I. Del Rosal and L. Maron, Dalton Trans., 2015, 44, 12137–12148 RSC.
- The first attempt of diphenylphosphine addition to phenylacetylene reported by us at 70 °C in the absence of a solvent and a catalyst gave a 90:10 Z/E diastereomeric ratio.22a Any further experiment conducted with the same starting materials has led to Z/E ratios of about 95:5.
- A. V. Artem'ev, S. F. Malysheva, N. K. Gusarova, N. A. Belogorlova, V. A. Shagun, A. I. Albanov and B. A. Trofimov, Synthesis, 2015, 263–271 Search PubMed.
- CCDC 1437629 (6a) contains the supplementary crystallographic data for this paper (see the ESI†).
- D. Semenzin, G. Etemad-Moghadam, D. Albouy, O. Diallo and M. Koenig, J. Org. Chem., 1997, 62, 2414–2422 CrossRef CAS PubMed.
- (a) C. M. Jessop, A. F. Parsons, A. Routledgea and D. Irvine, Tetrahedron Lett., 2003, 44, 479–483 CrossRef CAS; (b) Viewpoint: A. N. Hancock and C. H. Schiesser, Chem. Commun., 2013, 49, 9892–9895 RSC.
- A near second-order kinetics with respect to Ph2PH has been determined (i.e., 1.7 order). However, to obtain accurate kinetic data at very short reaction times has been troublesome and, therefore, not very reliable. This is in part due to the fact that the amounts of diphenylphosphine oxide and the oxidised product formed while handling the samples for gas chromatography analysis can mask the results.
- Y. C. Fan and O. Kwon, Chem. Commun., 2013, 49, 11588–11619 RSC.
- T. Bunlaksananusorn and P. Knochel, Tetrahedron Lett., 2002, 43, 5817–5819 CrossRef CAS . According to Corma et al.,30 the corresponding phosphine oxide was the major product isolated under those conditions.
- R. A. Sheldon, Chem. Commun., 2008, 3352–3365 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, compound characterisation, NMR spectra, kinetic graphics and X-ray data. CCDC 1468810 and 1437629. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6gc00903d |
| ‡ Present address: Departamento de Química, Instituto de Química del Sur (INQUISUR-CONICET), Universidad Nacional del Sur, Avenida Alem 1253, 8000 Bahía Blanca, Argentina. |
| This journal is © The Royal Society of Chemistry 2016 |