DOI:
10.1039/C6GC00586A
(Paper)
Green Chem., 2016,
18, 4116-4127
Purification of indium by solvent extraction with undiluted ionic liquids†
Received
29th February 2016
, Accepted 9th May 2016
First published on 9th May 2016
Abstract
A sustainable solvent extraction process for purification of indium has been developed from a chloride aqueous feed solution using the ionic liquids Cyphos® IL 101 and Aliquat® 336. The high affinity of indium(III) for the ionic liquid phase gave extraction percentages above 95% over the HCl concentration range from 0.5 to 12 M. Attention was paid to the loading capacity of the ionic liquid phase and the kinetics of the extraction process. An extraction mechanism was proposed based on the relationship between the viscosity of the ionic liquid phase and the loading with indium(III) ions. Even for loadings as high as 100 g L−1, equilibrium was reached within 10 min. Due to the very high distribution ratio for indium(III), stripping of indium(III) from the ionic liquid phase was very difficult with water or acid solutions. However, indium could conveniently be recovered as In(OH)3 by precipitation stripping with a NaOH solution. Precipitation stripping has the advantage that no ionic liquid components are lost to the aqueous phase and that the ionic liquid is regenerated for direct re-use. The extraction of some metal ions that are commonly found as impurities in industrial indium process solutions, i.e. cadmium(II), copper(II), iron(III), manganese(II), nickel(II), tin(IV) and zinc(II), has been investigated. The distribution ratios for the different metal ions show that indium(III) can be purified efficiently by a combination of extraction, scrubbing and stripping stages. This new ionic liquid process avoids the use of volatile organic solvents.
Introduction
Indium is a scarce metal with a 0.1 ppm abundance in the earth's crust comparable to that of silver.1,2 It is labeled as a critical raw material by the European Commission, due to its high supply risk.3,4 This risk is due to two supply and demand factors, namely, the increasing demand for applications in photovoltaics, flat screen TVs, laptops and mobile phones as well as the Chinese production monopoly. In 2008, China had a 58% share in the primary global indium production.5 The two factors are further compounded by low substitutability and low recycling rates, resulting in higher prices and the need to raise production.3,4
Indium is derived as a by-product from ores and is most commonly found in association with zinc ores such as sphalerite, although it also occurs in lead, copper, iron and tin ores.1–3 Indium is accumulated in low concentrations in residues formed during the processing of these ores. Therefore, indium is often regarded as an impurity that increases the production costs.5 Furthermore, indium recycling is mainly limited to production scrap. To keep up with the rising demand for high-tech applications, the development of industrial processes for the successful recovery of indium from various primary and secondary sources is crucial. Possible sources of indium are by-products of zinc refining, flue dusts, slags and metallic intermediates, e-waste and impure indium (<99%).1,2 Hydrometallurgical separation methods are very suitable for extracting indium from these sources. They compromise an essential part of extractive metallurgy utilized for treating complex and low-grade materials. Typically, hydrometallurgical solutions are generated by leaching the metals present in concentrates with strong acids or bases. Metals are already separated partially or completely in the leaching step, if one or more of the metals does not solubilize. Further concentration or separation generally takes place via solvent extraction or ion exchange. Once the metals have been separated, pure metals or metallic compounds can be produced by, for example, precipitation, cementation or electrolysis.
Solvent extraction (SX) has been widely used as a process for separation, purification and recovery of metals, due to its simplicity of equipment and operation. Solvent extraction is based on selective and efficient transfer of the desired metal species from one phase to another, usually from an aqueous to an organic phase.6,7 The aqueous phase, in which the metals are present, is brought into contact with a water-immiscible organic phase, consisting of one or more extractants, a diluent and sometimes a modifier. The separation is based on differences in the solubility of the metal ions in both the organic and aqueous phases due to the variations in the strength of the chemical interaction between the metal ion and the extractant. The efficiency of the extraction process depends on several parameters such as the pH, the temperature, the concentration of the metal ions in the aqueous feed and the concentration of the extractant in the organic phase.
The traditional solvent extraction process for indium makes use of water-immiscible organic solvents, many of which are flammable, volatile or toxic.8 Ionic liquids are good alternatives as an extraction medium for the development of sustainable separation processes.9–11 Ionic liquids are solvents consisting completely of ions, mostly an organic cation and an inorganic anion.12,13 The physicochemical properties of ionic liquids can be tuned adequately for a given application.14 Therefore, it is not surprising that water-immiscible ionic liquids have already been investigated as extractants for metals from aqueous solutions.11,15–21 These ionic liquids often contain fluorinated anions, such as the hexafluorophosphate (PF6−) or the bis(trifluoromethylsulfonyl)imide (Tf2N−) anion.22,23 Besides their high prices, in some cases, these type of ionic liquids pose a severe risk due to hydrolysis and formation of hydrofluoric acid.24 Therefore it is better from an environmental and economical point of view to use ionic liquids with long alkyl chains instead of fluorinated anions.22 Several non-fluorinated hydrophobic ionic liquids have already been used for the extraction of metal ions but generally not in a pure state. These ionic liquids such as Aliquat® 33625–27 and tri(hexyl)tetradecylphosphonium chloride (Cyphos® IL 101)27–32 are usually diluted in molecular solvents prior to use. Diluents such as toluene, kerosene or chloroform are added to decrease the viscosity of the organic phase leading to an increase in mass transfer and faster kinetics. However, recently in some cases, the problem of viscosity was overcome by saturating the ionic liquid with water, by working at slightly elevated temperatures and/or by using intermediate metal feed concentrations.33–36
Major advantages of ionic liquids for application in solvent extraction processes, in comparison with traditionally used water-immiscible organic solvents, are their negligible vapor pressure and low flammability.12–14 Due to their low volatility, they can be considered as more environmental-friendly and safer alternatives to organic solvents. While the use of ionic liquids for metal extraction does offer many advantages, there are some disadvantages as well. First of all, not all types of ionic liquids can be used for extraction. Water-immiscible ionic liquids are required for solvent extraction. Secondly, extraction often takes place through an ion-exchange mechanism which leads to losses of ionic liquid components to the aqueous phase.37–40 Ionic liquids are also quite expensive, so that their use for conventional solvent extraction processes cannot be justified solely from an economical point of view. Furthermore, their generally high viscosity often leads to slow mass transfer so that long contact times are required to reach equilibrium.33,34
In this paper, an efficient solvent extraction process for the purification of indium from a chloride medium was developed based on the use of quaternary phosphonium and ammonium ionic liquids. The feasibility of the quaternary phosphonium ionic liquid trihexyl(tetradecyl)phosphonium chloride (Cyphos® IL 101) and the quaternary ammonium ionic liquid Aliquat® 336 to serve as a possible undiluted organic phase/extractant for the extraction of indium is investigated. Special attention was paid to the difficult stripping of indium from the ionic liquid phase after extraction. Is it shown that precipitation stripping of In(OH)3 with NaOH is a convenient sustainable method for recovery of indium and for regeneration of the ionic liquid.
Experimental
Chemicals
Trihexyl(tetradecyl)phosphonium chloride (Cyphos® IL 101, purity >97%) was purchased from Cytec Industries Inc. (Niagara Falls, Ontario, Canada). Aliquat® 336 (mixture of quaternary ammonium chlorides, with 88.2–90.6% quaternary ammonium content), InCl3·4H2O (97%), MnCl2 (98%) and phosphor standard (1000 ppm) were obtained from Fluka (Sigma-Aldrich, Diegem, Belgium). CdCl2 (99%), CuCl2 (99%), NiCl2 (98%), PbCl2 (99%), SnCl4 (99.99%), Na2EDTA·2H2O (99+%), Na2SO4 (99%, extra pure) and ethanol (99.8+%, absolute) were purchased from Acros Organics (Thermo Fisher Scientific, Geel, Belgium). As2O3 (99.5%) was obtained from Alfa Aesar (Karlsruhe, Germany), FeCl3 (98.5%) from Carl Roth (Karlsruhe, Germany) and ZnCl2 (98–100.5%), CaCl2·2H2O (99.5+%) and lanthanum standard (1000 ppm) from Chem-Lab (Zedelgem, Belgium). NaOH pellets (>99% AnalaR Normapur®) were purchased from VWR (Leuven, Belgium). The silicone solution in isopropanol was obtained from SERVA Electrophoresis GmbH (Heidelberg, Germany) and the rhodium standard (1000 ppm) from Merck (Overijse, Belgium). Hydrochloric acid solutions were prepared from HCl (37%, Acros Organics) and Milli-Q® water. All chemicals were used as received, without further purification.
Instrumentation and analysis methods
After each extraction, the mixtures were centrifuged with a Heraeus Megafuge 1.0 centrifuge. Metal concentrations were determined using a bench top total reflection X-ray fluorescence spectrometer (TXRF; Bruker S2 Picofox) for simple matrices (mono-element system) or inductively coupled plasma optical emission spectrometer (ICP-OES; Agilent, type E730) with an axial plasma configuration for the more complex matrices (multi-element system) due to the spectral overlap and matrix effects that were encountered with TXRF. For analysis of the aqueous phase by TXRF, part of the aqueous phase was mixed with a lanthanum internal standard and Milli-Q® water until a total volume of 1 mL was obtained. The quartz glass sample carriers were first treated with 20 μL of a silicone solution in isopropanol, dried for 5 min in a hot air oven at 60 °C, followed by the addition of 5 μL of the sample and drying for 30 min at the same temperature. The metal concentrations in the aqueous phase were measured for 1000 s. For the organic phase, the lanthanum internal standard was added to a small amount of the organic phase (27 mg) and was further diluted with absolute ethanol until 1 mL. The sample carrier pre-treatment, the drying procedure and the measuring time were performed in the same way for the organic phase as described for the aqueous phase but the sampling volume was reduced to 2 μL. For analysis of the aqueous phase by ICP-OES, a calibration curve was prepared with multi-element solutions over a concentration range of 0–10 mg L−1 with a quality control of 2 mg L−1 and scandium as an internal standard. The calibration solutions and the samples were diluted in 10 vol% HCl. The spectra were measured with a power of 1.4 kW, an argon flow of 15 L min−1 and an auxiliary argon flow of 1.5 L min−1. For the organic phase, the sample was first digested with a mixture of H2SO4 and HNO3 in a quartz beaker on a heating plate. H2SO4 and HNO3 were evaporated (∼400 °C) and the residue was dissolved in 10 vol% HCl before measurement. The calibration and measuring procedure have been performed in the same way for the organic phase as described for the aqueous phase.
The viscosity of the organic phase was measured using a falling-ball type viscometer (Anton Paar, Lovis 2000 ME), densities were determined using a density meter with an oscillating U-tube sensor (Anton Paar, DMA 4500 M) and pH measurements were performed with an S220 SevenCompact™ pH/Ion meter (Mettler-Toledo) and a Slimtrode (Hamilton) electrode. A Mettler-Toledo DL39 coulometric Karl Fischer titrator was used with Hydranal® AG reagent to determine the water content of the ionic liquid. 31P NMR spectra have been recorded on a Bruker® Avance™ II 600 MHz Spectrometer, operating at 242.94 MHz. The samples for 31P NMR measurements were dissolved in toluene-d8 and measured with respect to a H3PO4 external reference. A delay time (d1) of 60 s was applied in the NMR pulse sequence to avoid saturation effects in the 31P NMR spectra. All NMR spectra were analyzed with the TOPSPIN software package.
The loss of Cyphos® IL 101 to the aqueous phase was determined by ICP-OES. All measurements were carried out using an ICP-OES spectrometer, model Optima 8300 DV (Perkin Elmer) equipped with axially viewed plasma. 31P was analyzed at 177.434 nm. The internal standard was measured at 343.489 nm (Rh). Standard solutions of 31P (3, 1, 0.5, 0.1, 0.05 ppm) were prepared in 5 vol% HCl. 1 ppm of Rh was added as an internal standard. 5 vol% HCl was used as a blank solution for calibration. All samples were diluted 10 times with a 5 vol% HCl solution and 1 ppm of Rh was added as an internal standard. All aqueous solutions were prepared with Milli-Q® water. A reagent blank solution of 5 vol% HCl containing 1 ppm of Rh as an internal standard was used for correcting the Ca-free samples. The Ca-containing samples were corrected using a reagent blank having the same Ca concentration as the samples and containing 1 ppm of Rh as an internal standard. The loss of Aliquat® 336 to the aqueous phase was determined using TOC measurements. The total organic carbon content (TOC) was analyzed with a HiPerTOC TOC analyser (Thermo Scientific) using the non-purgeable organic carbon (NPOC) method. The total inorganic carbon (TIC; dissolved CO2, HCO3− and CO32−) of the sample (30 mL) was converted to CO2 by addition of 400 μL of a 10 vol% H3PO4 solution. The formed CO2 gas was removed from the solution by flushing with O2 gas. Subsequently, 1 mL of the sample was transported to the UV reactor together with 1.5 mL of an oxidizing solution (120 g L−1 Na2S2O8) under O2 atmosphere (99.999%) with an O2 flow rate of 250 mL min−1. The UV reactor converted the remaining carbon to CO2. Next, the formed gas was dried by a Peltier cooler and separated from the liquid components by a gas–liquid separator. Afterwards, the dried CO2 was carried to a non-dispersive infrared detector, where CO2 was measured. A calibration curve was prepared to relate the detector signal to the CO2 concentration and hence to the corresponding total carbon concentration of the sample.
Extraction experiments
Extraction experiments were performed using two phases: metal chloride salts in an HCl acidified water phase and undiluted ionic liquid comprising the organic phase. The IL was presaturated with Milli-Q® water and hydrochloric acid in a volume ratio of 4.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, having the same chloride concentration as the aqueous phase before extraction, at 60 °C to prevent co-extraction of Milli-Q® water and hydrochloric acid. Therefore, the ratio of the volume of the aqueous and the ionic liquid phase remains constant. Extractions were performed by intensive stirring at 500 rpm for 60 min at 60 °C with a magnetic stirring bar. Hydrochloric acid was used as the chloride source. After the extraction, separation of the phases was assisted by centrifugation for 15 min at 3500 rpm.
:1, having the same chloride concentration as the aqueous phase before extraction, at 60 °C to prevent co-extraction of Milli-Q® water and hydrochloric acid. Therefore, the ratio of the volume of the aqueous and the ionic liquid phase remains constant. Extractions were performed by intensive stirring at 500 rpm for 60 min at 60 °C with a magnetic stirring bar. Hydrochloric acid was used as the chloride source. After the extraction, separation of the phases was assisted by centrifugation for 15 min at 3500 rpm.
Mono-element system
Distribution ratio studies.
The distribution ratios of indium(III) at different HCl concentrations were determined using equal volumes of ionic liquid and an acidified water phase (5 mL) containing 5 g L−1 of indium(III). Further experiments studied the distribution ratios of indium(III) as a function of feed concentration at the optimal hydrochloric acid concentration of 0.5 M and using feed solution concentrations ranging from 5 to 120 g L−1. The viscosity of the organic phase was determined for the various feed solution concentrations.
Kinetics.
Reaction kinetics were studied by shaking equal volumes of ionic liquid and an acidified water phase (5 mL) containing 5 to 100 g L−1 of indium(III) for different times ranging from 1 to 180 min.
Stripping.
First, extractions were performed from a feed solution containing 5 g L−1 indium(III) at the optimal hydrochloric acid concentration of 0.5 M. Afterwards, the loaded ionic liquid phase was stripped with a water phase (5 mL) containing different stripping agents: Milli-Q® water, ethylenediaminetetraacetic acid disodium salt dihydrate (Na2EDTA·2H2O) and sodium hydroxide. All stripping experiments were executed using equal volumes of ionic liquid and a water phase at 60 °C, except for the stripping with sodium hydroxide which was performed at room temperature. After stripping, separation of the phases was assisted by centrifugation for 15 min at 3500 rpm.
Recycling of the ionic liquid for reuse.
Extractions were performed from a feed solution containing 5 g L−1 indium(III) at the optimal HCl concentration of 0.5 M. Afterwards, the loaded ionic liquid phase was stripped with an aqueous NaOH solution. The loaded Cyphos® IL 101 and Aliquat® 336 were stripped by 4.5 and 4 equivalents of NaOH, respectively. All extraction and stripping experiments were executed with equal volumes of ionic liquid and water phase. After extraction and stripping the ionic liquid was equilibrated by a 0.5 M HCl solution in a volume ratio 1:5. The equilibrated ionic liquid was reused in a second extraction step using a feed solution containing 5 g L−1 indium(III) at the optimal HCl concentration of 0.5 M.
Loss of ionic liquid to the aqueous phase.
First, Cyphos® IL 101 was washed 4 times with a 0.05 M NaCl solution in a 1:25 volume ratio at room temperature until the water soluble phosphor-containing impurities present in the ionic liquid were removed. The P content of the aqueous phase after washing remained constant. A 0.05 M NaCl solution was used as a washing solution instead of Milli-Q® water due to emulsion formation with Milli-Q® water. Subsequently, extractions were performed using equal volumes of ionic liquid and aqueous phase (5 mL) at 60 °C. The solubility of the ionic liquid in the aqueous phase was determined at different HCl (0.5 M, 6 M and 12 M), indium(III) (5 g L−1, 40 g L−1 and 120 g L−1) and CaCl2 (0.25 M, 3 M and 6 M) concentrations in the aqueous phase. The loss of Cyphos® IL 101 to the aqueous phase was determined by ICP-OES. Aliquat® 336 was first saturated with a 0.05 M NaCl solution in a 1:1 volume ratio at room temperature. Afterwards, extractions were performed using equal volumes of ionic liquid and aqueous phase (250 mL) at 60 °C. The solubility of the ionic liquid in the aqueous phase was determined at different HCl (0.5 M, 6 M and 12 M) and CaCl2 (0.25 M, 3 M and 6 M) concentrations in the aqueous phase. The loss of Aliquat® 336 to the aqueous phase was determined by TOC measurements. Aliquat® 336 is considered to be a mixture quaternary ammonium chloride with a 2:1 molar ratio of octyl groups to decyl groups. A molar weight of 432 g mol−1 was used instead of 404 g mol−1 which is valid for pure trioctylmethylammonium chloride. Based on the molar ratio, the following composition of quaternary ammonium chloride was assumed (not including water and other impurities) according to random statistical distribution: trioctylmethylammonium chloride (33 wt%), dioctyldecylmethylammonium chloride (28 wt%), octyldidecylmethylammonium chloride (22 wt%) and tridecylmethylammonium chloride (17 wt%).41 These assumptions were made to calculate the solubility of Aliquat® 336 in the aqueous phase based on TOC analyses results.
Multi-element system
Distribution ratio studies.
The distribution ratio of cadmium(II), copper(II), indium(III), iron(III), manganese(II), nickel(II), tin(IV) and zinc(II) at different HCl concentrations was studied using equal volumes of ionic liquid and an acidified water phase (10 mL) containing 5 g L−1 of each metal added as chloride salts. Additionally, 1 g L−1 of lead(II) as a chloride salt and arsenic(III) as an oxide were added to the water phase. These elements are commonly found in primary and secondary sources of indium. Extraction experiments of the multi-element system were carried out only once.
Distribution ratio and separation factor
The distribution ratio D of a metal M is defined as| |  | (1) |
where [M]IL is the metal concentration in the ionic liquid and [M]aq is the metal concentration in the aqueous phase after extraction. For metals which are strongly extracted to the ionic liquid phase (%E ≥ 50%) only the remaining metal concentration in the aqueous phase was measured after extraction and eqn (1) can be rewritten as| | 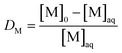 | (2) |
where [M]0 is the initial metal concentration in the aqueous phase before extraction. The metal concentration of the ionic liquid phase was measured for metals which are poorly extracted (%E < 50%), and eqn (1) becomes| |  | (3) |
The percentage extraction (%E) is defined as the amount of metal extracted to the ionic liquid phase over the initial amount of metal present in the aqueous phase:
| |  | (4) |
The separation efficiency between two metals can be described by the separation factor α, which is defined as the ratio of the respective distribution ratios of two extractable solutes measured under the same conditions:
| |  | (5) |
where
DM1 and
DM2 are the distribution ratios
D of metal M
1 and M
2, respectively. By definition, the value of the separation factor is always greater than unity.
Metals are removed after extraction from the organic phase by a stripping agent. The percentage stripping (%S) in the stripping phase can be defined as the amount of metal stripped from the organic phase to the total amount of metal in the organic phase before stripping:
| | 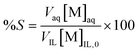 | (6) |
where [M]
IL,0 is the metal concentration in the organic phase after extraction or before stripping.
Results and discussion
Mono-element system
Distribution ratio studies.
In the first series of experiments, the distribution ratios and the extraction percentages of indium(III) between the ionic liquid and aqueous phase were determined as a function of hydrochloric acid concentration (Fig. 1, S1,† and Table 1). Fig. 1 shows bell-shaped curves; i.e., the distribution ratios increase as a function of HCl concentration until a value from where they begin to decrease. Bell-shaped curves of the distribution ratio versus the HCl content have also been observed by other authors for extraction of Cu(II), Co(II), Fe(III), Ga(III), In(III), Mn(II) and Zn(II) by quaternary ammonium salts.42–45 The average distribution ratio of indium(III) between Cyphos® IL 101 and hydrochloric acid aqueous solution, increases with increasing chloride concentration, with a maximum DIn = 4966 ± 262, at a HCl concentration of approximately 5 M. The same trend but with lower values was observed for the distribution ratio for the HCl–Aliquat® 336 system, where a maximum distribution ratio of DIn = 340 ± 28 was found at the same 5 M HCl concentration. In both cases, the initial indium(III) concentration in the aqueous phase was 5 g L−1.
 |
| | Fig. 1 Distribution ratios of indium(III) (DIn) as a function of HCl concentration for the HCl–Cyphos® IL 101 (■) and HCl–Aliquat® 336 (▲) system at 60 °C. The volume ratio of the aqueous to the organic phase is 1:1. Aqueous phase: initial indium(III) concentration 5 g L−1. | |
Table 1 Distribution ratios of indium(III) (D), the sample standard deviation on the distribution ratio (s) and the percentages extraction (%E) of indium(III)
| HCl conc. (M) |
Cyphos® IL 101 |
Aliquat® 336 |
|
D
|
s
|
%E |
D
|
s
|
%E |
| 0 |
15.00 |
0.53 |
93.74 |
1.58 |
0.21 |
59.42 |
| 0.5 |
391 |
24 |
99.74 |
24.2 |
1.4 |
96.03 |
| 1 |
854 |
65 |
99.88 |
51.5 |
1.9 |
98.09 |
| 2 |
1.95 × 103 |
0.12 × 103 |
99.95 |
119.9 |
4.2 |
99.17 |
| 3 |
3.10 × 103 |
0.62 × 103 |
99.97 |
212 |
18 |
99.53 |
| 4 |
4.44 × 103 |
0.69 × 103 |
99.98 |
279 |
40 |
99.64 |
| 5 |
4.73 × 103 |
0.43 × 103 |
99.98 |
340 |
40 |
99.70 |
| 6 |
4.12 × 103 |
0.38 × 103 |
99.98 |
326 |
37 |
99.69 |
| 7 |
3.47 × 103 |
0.51 × 103 |
99.97 |
300 |
49 |
99.66 |
| 8 |
2.085 × 103 |
0.064 × 103 |
99.95 |
216.5 |
4.7 |
99.54 |
| 9 |
1.18 × 103 |
0.13 × 103 |
99.91 |
179.2 |
9.1 |
99.45 |
| 10 |
735 |
99 |
99.87 |
114.8 |
3.7 |
99.14 |
| 11 |
472 |
98 |
99.78 |
75.2 |
3.1 |
98.69 |
| 12 |
248 |
66 |
99.63 |
42.0 |
6.0 |
97.65 |
The bell-shaped curve of the distribution ratio is tentatively attributed to a change of the indium speciation in the aqueous and/or in the ionic liquid phase in combination with HCl extraction. Narita et al.46 already described that the indium speciation in the aqueous phase can change as a function of HCl concentration. Co-extraction of HCl by phosphonium and ammonium ionic liquids has also been previously observed by Komasawa et al.,47 Sato et al.48 and Wellens et al.49
The distribution ratios of indium(III) for the HCl–Aliquat® 336 system were significantly lower, minimum by a factor of 5, than those for the HCl–Cyphos® IL 101 system over the whole HCl concentration range (Fig. 1). This can be explained by the higher water uptake of Aliquat® 336 (21.27 wt%) compared to Cyphos® IL 101 (13.65 wt%). The hydrophobicity of ionic liquids with a common anion is dependent on the total number of carbon atoms in the alkyl chains attached to the corresponding central atom. Shorter alkyl chains attached to the ammonium cation core lead to a lower hydrophobicity of Aliquat® 336 compared to Cyphos® IL 101, and therefore to a higher mutual miscibility with water.
Furthermore, the charge delocalization at the ammonium cations makes the central part of these cations more charged, and thus overall more polar, than the corresponding phosphonium cations.50,51 Very high values for the distribution ratio were observed over the entire HCl concentration range for the HCl–Cyphos® IL 101 system indicating a strong affinity of indium(III) for the ionic liquid phase. The distribution ratio observed for the quaternary ammonium ionic liquid over the entire HCl concentration range are not exceptionally high. High distribution ratios are not desirable for solvent extraction since they make stripping in many cases very difficult. Following studies regarding the kinetics, maximal loading, the extraction mechanism and the stripping of the mono-element systems are executed at a low HCl concentration of 0.5 M where the distribution ratio is high enough to ensure almost complete extraction of indium(III), but not too high so that stripping difficulties are avoided.
Kinetics.
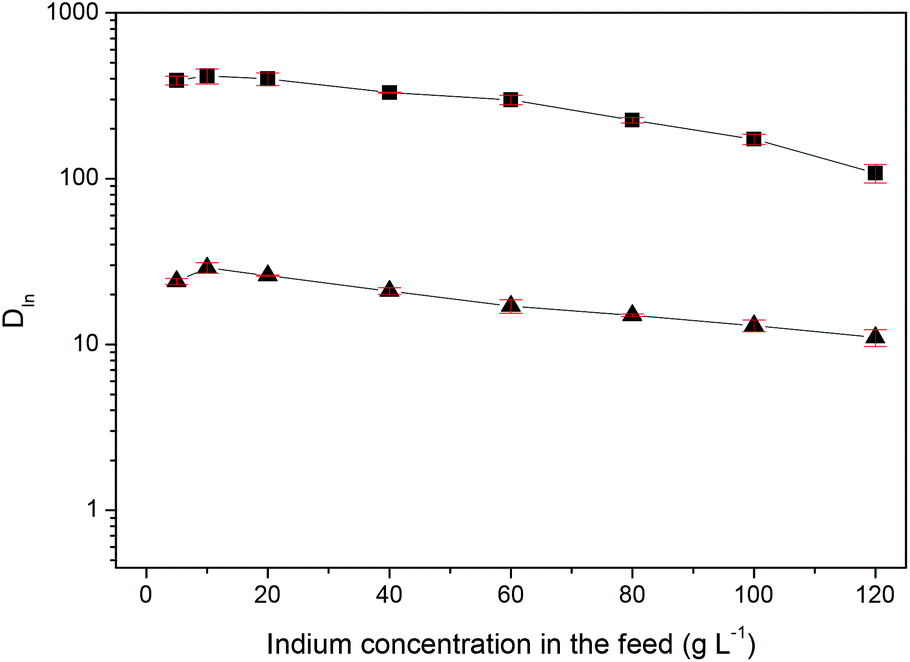
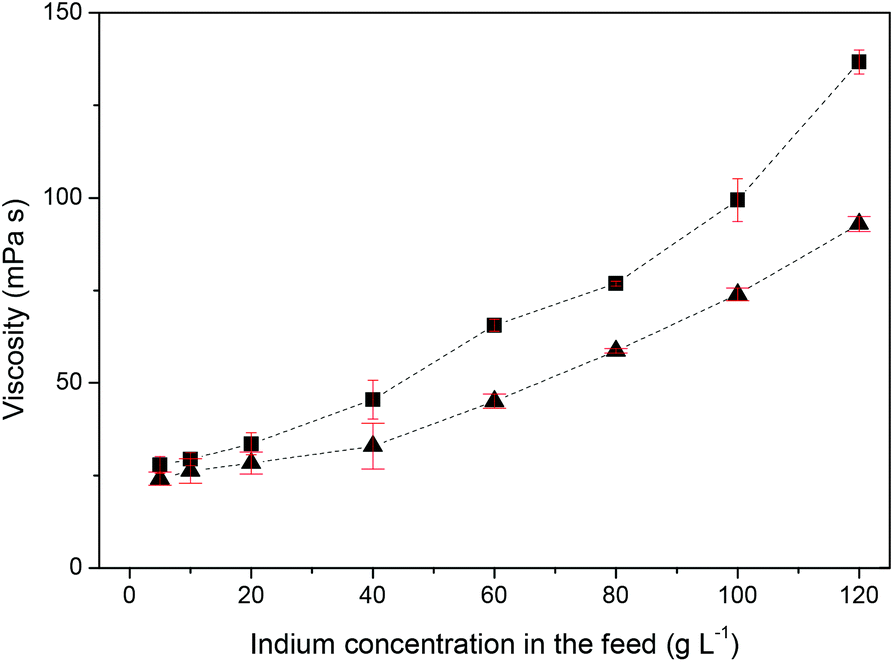
For industrial applications, it is more interesting to expand the indium(III) concentration range of the feed solution. The distribution ratios of indium(III) as a function of feed concentration were studied at the optimal HCl concentration of 0.5 M and with feed solution concentrations ranging from 5 to 120 g L−1 for both Cyphos® IL 101 and Aliquat® 336 (Fig. 2). A stirring time of 1 h was used to ensure that equilibrium was reached. Due to the strong affinity between the ionic liquid and indium(III), the organic phase can be loaded with high amounts of indium(III), while it is still possible to obtain a high percentage extraction (Fig. S2†). Phase inversion was observed for Cyphos® IL 101 and an initial indium(III) concentration of 100 g L−1 and higher and for Aliquat® 336 and an initial indium(III) concentration of 120 g L−1. At high loading of the ionic liquid phase, its density becomes larger than the density of the aqueous phase so that a phase inversion occurs. The density at 25 °C of the 100 g L−1 indium(III) loaded Cyphos® IL 101 and the 120 g L−1 indium(III) loaded Aliquat® 336 was 1.0242 ± 0.0023 g mL−1 and 1.0303 ± 0.0021 g mL−1, respectively The density of the corresponding aqueous phases was 1.0078 ± 0.0003 g mL−1 for the Cyphos® IL 101 system and 1.0303 ± 0.0021 g mL−1 for the Aliquat® 336 system at 25 °C. The avoidance of phase inversion is essential to industrial extraction processes, since the design of the contacting equipment is based on preferred direction of solute transfer, giving optimum mass transfer rates. Phase inversion, can cause considerable operating problems, especially in mixer-settlers, in which the change in properties of both the continuous phase and the drop size can lead to a delay in the settling process. Next, the influence of the extraction time was evaluated from 1 min to 60 min at the optimal HCl concentration of 0.5 M, with feed solution concentrations ranging from 5 to 100 g L−1 (Fig. 3 and S3†). The results show that equilibrium is reached after 10 min, regardless of the indium(III) concentration. However, longer stirring times to reach the equilibrium were expected at higher indium(III) concentrations due to an increased viscosity of the ionic liquid phase at high indium(III) loadings. This increase in viscosity as a function of indium(III) concentration in the aqueous feed solution is shown in Fig. 4. The influence of the viscosity on the reaction time is thus most probably negligible due to the high affinity of indium for the ionic liquid phase.
 |
| | Fig. 2 Distribution ratios of indium(III) (DIn) as a function of indium(III) concentration in the aqueous feed solution for the HCl–Cyphos® IL 101 (■) and HCl–Aliquat® 336 (▲) system at 60 °C. The volume ratio of the aqueous to the organic phase is 1:1. Aqueous phase: initial HCl concentration 0.5 M. | |
 |
| | Fig. 3 Distribution ratios of indium(III) (DIn) as a function of reaction time for different indium(III) concentrations in the aqueous feed solution at 60 °C for the HCl–Cyphos® IL 101 system: 5 g L−1 In (■), 10 g L−1 In (●), 60 g L−1 (▲) and 100 g L−1 (▼). The volume ratio of the aqueous to the organic phase is 1:1. Aqueous phase: initial HCl concentration 0.5 M. | |
 |
| | Fig. 4 Viscosity of the ionic liquid phase at equilibrium as function of the indium(III) concentration in the aqueous feed solution for the HCl–Cyphos® IL 101 (■) and HCl–Aliquat® 336 (▲) system at 60 °C. The volume ratio of the aqueous to the organic phase is 1:1. Aqueous phase: initial HCl concentration 0.5 M. | |
The viscosity of the ionic liquid phase increased only slightly when it was loaded with higher indium(III) concentrations (Fig. 4). This indicates that the chloroindate(III) anion formed during extraction has the same charge as the chloride anion in the pure ionic liquid, i.e. that the [InCl4]− anion is formed instead of higher charged species such as [InCl5]2− or [InCl6]3−. The presence of species with a higher charge are known to lead to a sharp increase of the viscosity at higher loadings.33,34,46,52 The same speciation was also observed in the organic phase for the extraction of indium(III) using long chain alkylamines and quaternary ammonium chlorides from chloride media.43,44,46,53,54 The proposed extraction mechanism is therefore:
| | 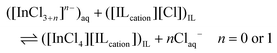 | (7) |
Indium(III) is probably extracted as a neutral InCl3 or the single negatively charged [InCl4]− complex. The more negatively charged chloroindate(III) complexes, [InCl5]2− and [InCl6]3−, are more strongly hydrated, thus more difficult to dehydrate and to convert to extractable anionic species.
Stripping.
The ease of recovery of the metal from the ionic liquid phase and regeneration of the ionic liquid are as important as obtaining a high extraction efficiency.7 Stripping of the ionic liquid phase after extraction was tested using several stripping agents. Cyphos® IL 101 and Aliquat® 336 are basic extractants (anion exchangers). The driving force for extraction is the presence of chloride anions. Stripping of metals is efficient if the distribution ratio is low. This can be achieved by decreasing the chloride concentration by addition of water. However, the stripping with water did not result in an efficient stripping; 6.1 ± 0.7% indium was stripped from the loaded Cyphos® IL 101 and 29 ± 3% from the loaded Aliquat® 336 phase. Furthermore, the stripping of the loaded Cyphos® IL 101 ionic liquid gave also rise to a difficult disengagement (formation of an emulsion). A small amount of Na2SO4 (±15 mg) was added as a salting-out agent to 5 mL of the aqueous phase. Although the stripping of the loaded ionic liquids with water resulted in a low percentage stripping, changing the temperature or the volume ratio could increase the stripping efficiency. By increasing the volume ratio of the aqueous to the organic phase from 1:1 to 3.5:1, stripping percentages of 19% and 78% were obtained for Cyphos® IL 101 and Aliquat® 336, respectively. In contrast to stripping with a 1:1 volume ratio, a white In(OH)3 precipitate was found in the aqueous phase after stripping. Ethylenediaminetetraacetic acid (EDTA) forms a very stable, highly water-soluble chelate complex with indium(III) (logβ = 24.90).55,56 Therefore, it was tested to strip indium(III) from the ionic liquid with an aqueous solution of Na2EDTA. Although reasonable percentages of stripping were acquired, 52 ± 1% for loaded Cyphos® IL 101 and 67 ± 1% for the loaded Aliquat® 336, the use of EDTA in the industry is not favored due to its low biodegradability and difficulties associated with recovery of EDTA in a continuous solvent extraction process due to its high level of complexing capacity with respect to heavy metals.57–60 If wanted, higher percentages stripping can probably be obtained by adjusting the pH with an alkaline solution, neutralizing the HCl present in the ionic liquid phase and removing the remaining protons from Na2EDTA. Care must be taken to avoid ionic liquid decomposition in alkaline environment61–64 and anion exchange with EDTA.34
Aside from the complexing agent EDTA, stripping was also tested with sodium hydroxide as a precipitation agent. Precipitation stripping from a metal-loaded ionic liquid phase has already been reported for the rare earths using oxalic acid as the stripping agent.65–67 Indium is directly stripped from the organic phase with sodium hydroxide forming an insoluble hydroxide. The precipitation stripping reaction can be represented as:
| |  | (8) |
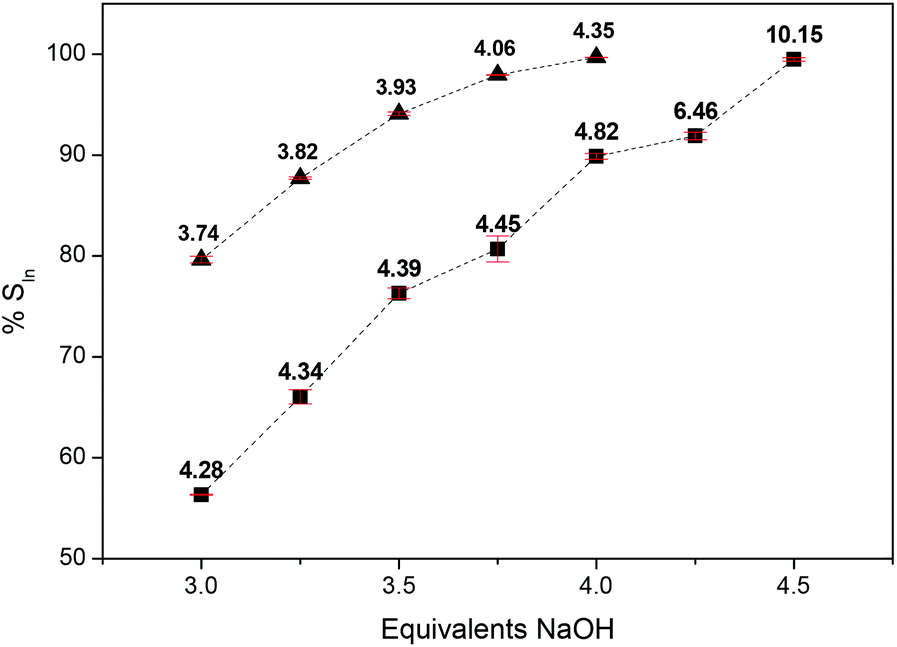
As the pH increases, the equilibrium of the hydrolysis reaction is shifted to the right and more indium is precipitated from the aqueous phase. According to Pourbaix, the solubility of In(OH)3 is minimal at pH 6.79.68 The loaded ionic liquid phase was stripped with an aqueous phase containing different equivalents of sodium hydroxide (Fig. 5). Using sodium hydroxide as a stripping agent, it was possible to obtain a percentage stripping close to 100%. The 0.5 M HCl–Aliquat® 336 system was easier to strip compared to the 0.5 M HCl–Cyphos® IL 101 system due to the lower indium distribution ratio obtained during extraction. 4 equivalents of NaOH were needed to achieve a percentage stripping over 99% for the 0.5 M HCl–Aliquat® 336 system. The equilibrium pH under these conditions was 4.35. However, at this point not all of the indium was precipitated yet as In(OH)3. Part of it is dissolved in the aqueous phase. 4.75 equivalents of NaOH were needed to fully precipitate all indium. A percentage stripping over 99% was reached for the 0.5 M HCl–Cyphos® IL 101 system if 4.5 equivalents of NaOH were used. Unlike the previous stripping experiments with water and Na2EDTA, the stripping was performed at room temperature instead of 60 °C, because quaternary phosphonium and ammonium ionic liquids tend to decompose in alkaline conditions, especially at elevated temperatures. Thus, in this context, quaternary phosphonium ionic liquids can decompose to yield a tertiary phosphine oxide and alkane under alkaline conditions.61–63
| | [R3P–CH2–R′] + OH− → R3P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O + CH3–R′ O + CH3–R′ | (9) |
 |
| | Fig. 5 Percentages stripping of indium(III) (%SIn) as a function of equivalents NaOH in the stripping solution for the 0.5 M HCl–Cyphos® IL 101 (■) and 0.5 M HCl–Aliquat® 336 (▲) system at RT. The volume ratio of the aqueous to the organic phase is 1:1. Ionic liquid phase: initial indium(III) concentration 5 g L−1. The corresponding equilibrium pH values are given above the data points. | |
Quaternary ammonium ionic liquids can easily undergo Hofmann elimination or β-elimination to yield a tertiary amine and an alkene in the presence of a base (Fig. 6).61,62,64 Trihexyl(tetradecyl)phosphonium chloride stability was investigated by comparing the integration of the degradation products 31P resonance signal, i.e. phosphine oxide, formed after mixing for 1 h with an aqueous NaOH solution to the integration of the trihexyl(tetradecyl)phosphonium cation 31P resonance signal at 32.9 ppm. The resonance signal of phosphine oxide was situated at 45.8 ppm. 31P NMR spectra were measured for the ionic liquid before and after stripping since commercial Cyphos® IL 101 already contains small amounts of phosphine oxide impurities. Peak integration ratios were then compared and it was concluded that no decomposition occurs because no change of the ratio of the integration of the phosphine peak over the integration of the phosphonium cation peak took place when stripping with NaOH at room temperature.
 |
| | Fig. 6 Schematic representation of Hofmann elimination of a quaternary ammonium IL, yielding an alkene and a tertiary amine. | |
Recycling of the ionic liquid for reuse.
An important aspect of green chemistry is the recyclability and reusability of the ionic liquid. The recyclability was shown by reusing the ionic liquid in a second extraction step after stripping. The results of each individual step are given in Table 2. The results indicate that the ionic liquid can be recycled for reuse in liquid/liquid extraction of indium(III) without any loss of activity.
Table 2 Distribution ratios of indium(III) (D) and the percentages extraction (%E) and stripping (%S) of indium(III) with the corresponding standard deviation
| Process |
Cyphos® IL 101 |
Aliquat® 336 |
|
D
|
%E/%S |
D
|
%E/%S |
| Extraction 1 |
520 ± 64 |
99.81 ± 0.02% |
23.00 ± 0.81 |
95.8 ± 0.1% |
| Stripping 1 |
578 ± 19 |
99.82 ± 0.04% |
489 ± 72 |
99.79 ± 0.03% |
| Extraction 2 |
476 ± 35 |
99.79 ± 0.02% |
21.82 ± 0.88 |
95.6 ± 0.2% |
Loss of the ionic liquid to the aqueous phase.
Ionic liquids are often considered as green solvents primarily due to their negligible vapor pressure and low flammability. However, the avoidance of loss to the aqueous phase and the recyclability of the ionic liquid are equally as important in establishing a sustainable process. The loss of Cyphos® IL 101 and Aliquat® 336 to the aqueous phase was determined. First, the phosphor containing water soluble impurities present in Cyphos® IL 101 were removed by four consecutive washing steps with a 0.05 M NaCl solution using a 1:25 volume ratio at room temperature (Fig. S4†). If not removed these impurities will insinuate a higher solubility of Cyphos® IL 101 in the aqueous phase. Subsequently, the ionic liquid was brought into contact with several aqueous solutions at 60 °C: HCl (0.5 M, 6 M and 12 M), indium(III) (5 g L−1, 40 g L−1 and 120 g L−1) and CaCl2 (0.25 M, 3 M and 6 M). The solubility of Cyphos® IL 101 in the different aqueous phases is presented in Table 3. The purity of Aliquat® 336 (88.2–90.6%) is low in comparison with Cyphos® IL 101 (>97%). Therefore, no prior washing steps were executed to remove possible water soluble organic impurities. For result comparison reasons, Aliquat® 336 was saturated with a 0.05 M NaCl solution in a 1:1 volume ratio at room temperature. Afterwards, the solubility of Aliquat® 336 was determined in several HCl (0.5 M, 6 M and 12 M) and CaCl2 (0.25 M, 3 M and 6 M) solutions after contacting the ionic liquid with the aqueous solutions at 60 °C. The solubility results are displayed in Table 3. The solubility of Aliquat® 336 in aqueous phase is higher than the solubility of Cyphos® IL 101 due to it higher mutual miscibility with water. As the salt concentration in the aqueous phase increases, the solubility of the ionic liquid in the aqueous phase decreases due to the salting-out effect. In general, the solubility of the ionic liquids in the aqueous phase is very limited thereby having a low associated economical and environmental impact.
Table 3 Solubility of the ionic liquid in the aqueous phase with the corresponding standard deviation
| Aq phase |
Solubility Cyphos® IL 101 (ppm) |
Solubility Aliquat® 336 (ppm) |
|
Below detection limit.
Not determined.
|
| 0.5 M HCl |
248.4 ± 1.1 |
836.6 ± 6.2 |
| 6 M HCl |
183.1 ± 2.2 |
727 ± 14 |
| 12 M HCl |
186.8 ± 2.6 |
592.5 ± 2.7 |
| 0.25 M CaCl2 |
69.6 ± 6.9 |
708.7 ± 6.0 |
| 3 M CaCl2 |
<15a |
340 ± 19 |
| 6 M CaCl2 |
<15a |
339.7 ± 4.0 |
| 0.5 M HCl, 5 g L−1 In(III) |
275.0 ± 1.5 |
—b |
| 6 M HCl, 5 g L−1 In(III) |
181.2 ± 1.0 |
—b |
| 12 M HCl, 5 g L−1 In(III) |
198.7 ± 1.4 |
—b |
| 0.5 M HCl, 40 g L−1 In(III) |
305.9 ± 1.3 |
—b |
| 0.5 M HCl, 120 g L−1 In(III) |
187.6 ± 4.4 |
—b |
Multi-element system
Distribution ratio studies.
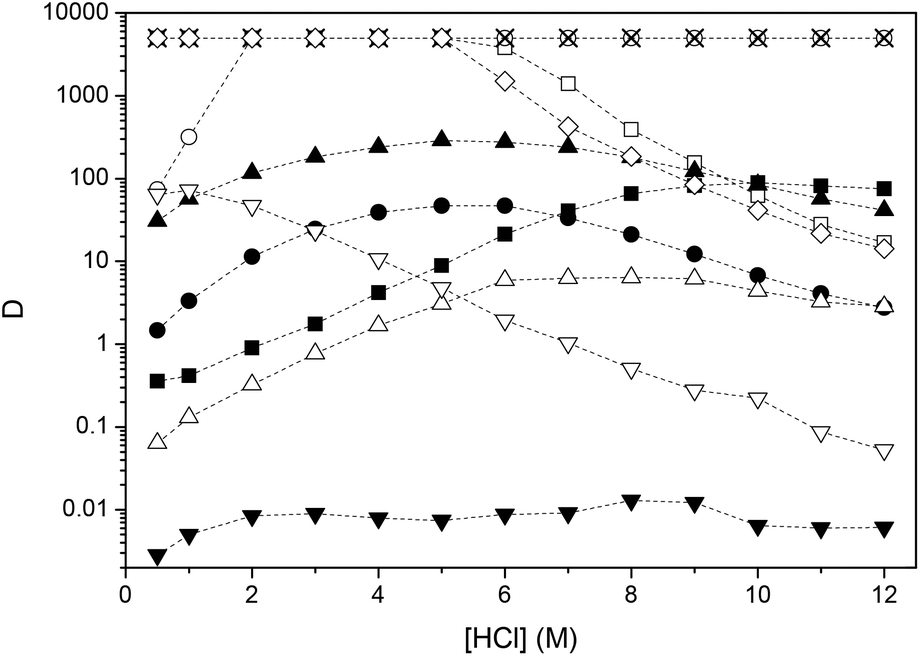
A useful purification process needs to have a reasonable selectivity for the targeted metal. All the experiments described above have been carried out on mono-element solutions but industrial solutions are generally complex mixtures of several elements. In a second series of experiments, the separation of indium(III) from arsenic(III), cadmium(II), copper(II), iron(III), lead(II), manganese(II), nickel(II), tin(IV) and zinc(II) was investigated. More elements are added to the feed solution compared to previous experiments, to a more industrial relevant elemental composition: 5 g L−1 of each of the other elements with the exception of arsenic(III) and lead(II) present in a concentration of 1 g L−1. A concentration of 1 g L−1 for arsenic(III) and lead(II) was chosen due to the limited solubility of arsenic(III) oxide and lead(II) chloride in the aqueous phase.69,70Fig. 7 and Fig. 8 show the distribution ratios between the ionic liquid phase and aqueous phase of the elements as a function of HCl concentration for the HCl–Cyphos® IL 101 and HCl–Aliquat® 336 systems. Percentage extraction as a function of HCl concentration for both systems is given in Fig. S5 and S6.† Many elements display a bell-shaped distribution ratio curve. Indium(III) has a maximum distribution ratio at 5 M in both systems in agreement with the previous experiments. For copper(II) and manganese(II), the maximum distribution ratio was found at a HCl concentration of 4 M and 6 M, respectively for the HCl–Cyphos® IL 101 system and at 5 M and 7 M, respectively for the HCl–Aliquat® 336 system. For nickel(II) and arsenic(III), the maximum distribution ratio was found at higher HCl concentration of 8 M and 10 M, respectively, independent of the extraction system. Moreover, the distribution ratios of nickel(II) were significantly lower than those of the other elements over the whole HCl concentration range, indicating a low affinity of nickel(II) for the ionic liquid phase. For lead(II) the behavior was distinctly different from that of the other elements. Here the distribution ratios were high at low HCl concentrations and decreased with increasing HCl concentration. It is very likely that only the right side of the bell-shaped curve is represented. The maximum value for the distribution ratios of cadmium(II), iron(III), tin(IV) and zinc(II) could not be determined with ICP-OES as the analysis method due to the lower detection limit of 1 mg L−1. However, from the partial shape of the distribution ratio curves of cadmium(II), iron(III) and zinc(II), it can be concluded that also a bell-shaped path is followed. The maximum distribution ratio and corresponding HCl concentration for each of the elements are given in Table 4. Advantage can be taken of the low affinity of arsenic(III), manganese(II) and nickel(II) for the ionic liquid phase at lower HCl concentrations to separate these ions from cadmium(II), iron(III), lead(II), tin(IV), zinc(II) and most importantly from indium(III). Due to the lower distribution ratios obtained for most of the elements, that allows an easier stripping of indium(III) from the ionic liquid phase after extraction, Aliquat® 336 is the preferred undiluted extractant.
 |
| | Fig. 7 Distribution ratios (D) as a function of HCl concentration for the HCl–Cyphos® IL 101 system at 60 °C. The volume ratio of the aqueous to the organic phase is 1:1. Aqueous phase initial metal concentrations: 5 g L−1 of cadmium(II) (□), copper(II) (●), iron(III) (○), indium(III) (▲), manganese(II) (△), nickel(II) (▼), tin(IV) (✗) and zinc(II) (◊) and 1 g L−1 of arsenic(III) (■) and lead(II) (∇). | |
 |
| | Fig. 8 Distribution ratios (D) as a function of HCl concentration for the HCl–Aliquat® 336 system at 60 °C. The volume ratio of the aqueous to the organic phase is 1:1. Aqueous phase initial metal concentrations: 5 g L−1 of cadmium(II) (□), copper(II) (●), iron(III) (○), indium(III) (▲), manganese(II) (△), nickel(II) (▼), tin(IV) (✗) and zinc(II) (◊) and 1 g L−1 of arsenic(III) (■) and lead(II) (∇). | |
Table 4 Maximum distribution ratio (D) with corresponding HCl concentration of various elements for the HCl–Cyphos® IL 101 and HCl–Aliquat® 336 system. Aqueous phase initial metal concentrations: 5 g L−1 of cadmium(II), copper(II), iron(III), indium(III), manganese(II), nickel(II), tin(IV) and zinc(II) and 1 g L−1 of arsenic(III) and lead(II)
| Element |
HCl–Cyphos® IL 101 |
HCl–Aliquat® 336 |
| HCl (M) |
D
max
|
HCl (M) |
D
max
|
|
The corresponding HCl concentration of the maximum distribution ratio for Cd(III), Fe(III), Sn(IV) and Zn(II) cannot be deduced from Fig. 7 and 8.
|
| As(III) |
10 |
197 |
10 |
89.0 |
| Cd(II) |
—a |
>5.00 × 103 |
—a |
>5.00 × 103 |
| Cu(II) |
4 |
472 |
5 |
47.2 |
| Fe(III) |
—a |
>5.00 × 103 |
—a |
>5.00 × 103 |
| In(III) |
5 |
2.90 × 103 |
5 |
289 |
| Mn(II) |
6 |
24.7 |
7 |
6.26 |
| Ni(II) |
8 |
7.06 × 10−3 |
8 |
9.75 × 10−3 |
| Pb(II) |
1 |
366 |
1 |
72.3 |
| Sn(IV) |
—a |
>5.00 × 103 |
—a |
>5.00 × 103 |
| Zn(II) |
—a |
>5.00 × 103 |
—a |
>5.00 × 103 |
In the following paragraphs, information is given on the capabilities of the 0.5 M HCl–Aliquat® 336 system for purification of indium(III), and possible process steps are suggested. The low distribution ratios of arsenic(III), manganese(II) and nickel(II) in combination with high separation factors between these elements and indium(III), imply an easy separation (Table 5). The contaminating ions will remain largely in the aqueous phase during extraction. A high separation factor was also obtained for the In(III)/Cu(II) couple, but due to the slightly elevated distribution ratio of copper(II) in comparison with arsenic(III), manganese(II) and nickel(II), extraction of copper(II) cannot be avoided completely (Table 5). However, reasonably low distribution ratio enables the scrubbing of copper(II) from the ionic liquid phase together with traces of arsenic(III), manganese(II) and nickel(II) extracted. A solution with a low HCl concentration has to be used for scrubbing to avoid loss of indium(III) to the aqueous phase. After extraction and scrubbing, cadmium(II), iron(III), lead(II), tin(IV), zinc(II) will still be present in the ionic liquid phase together with indium(III). The high separation factors for the couples In(III)/Cd(II), In(III)/Sn(IV) and In(III)/Zn(II) suggest an easy separation (Table 5). Although they all possess a high affinity for the ionic liquid phase (DIn = 30.7, DCd, DSn, DZn > 5.00 × 103), indium(III) will be easier to strip due to its distribution ratio being 160 times smaller. It was demonstrated previously that indium can be removed from the loaded Aliquat® 336 phase by stripping with water or NaOH. Both methods can be satisfactory, but stripping with water will require larger volume ratios of the aqueous to the organic phase. Also selectivity will play a vital role in choosing a stripping method. Selective stripping of indium(III) from iron(III) and lead(II) will be difficult due to the small separation factors, αFe(III)/In(III) and αPb(II)/In(III) (Table 5). Hydrolysis curves have to be constructed for the stripping of the ionic liquid phase with NaOH to get a better understanding about the hydrolysis behavior of the various metals, elaborating a more selective stripping. After stripping, the ionic liquid phase has to be scrubbed so that it can be reused. Scrubbing of the ionic liquid phase with water will have little effect due to the high distribution ratios of cadmium(II), tin(IV) and zinc(II). Scrubbing with a NaOH solution will in most cases be the best option.
Table 5 Metal distribution ratios (D) and separation factors (α) between indium(III) and various elements for the 0.5 M HCl–Cyphos® IL 101 and 0.5 M HCl–Aliquat® 336 system. Aqueous phase initial metal concentrations: 5 g L−1 of cadmium(II), copper(II), iron(III), indium(III), manganese(II), nickel(II), tin(IV) and zinc(II) and 1 g L−1 of arsenic(III) and lead(II)
| Element couple |
0.5 M HCl–Cyphos® IL 101 |
0.5 M HCl–Aliquat® 336 |
|
D
In
|
D
M
|
α
|
D
In
|
D
M
|
α
|
|
According to IUPAC, by convention the ratio of the respective distribution ratios has to be chosen so that α > 1.71
|
| In(III)/As(III) |
569 |
0.435 |
1.31 × 103 |
30.7 |
0.356 |
68.2 |
| In(III)/Cd(II) |
569 |
>5.00 × 103 |
>9.78a |
30.7 |
>5.00 × 103 |
>162a |
| In(III)/Cu(II) |
569 |
26.4 |
21.6 |
30.7 |
1.48 |
20.7 |
| In(III)/Fe(III) |
569 |
962 |
1.69a |
30.7 |
73.3 |
2.39a |
| In(III)/Mn(II) |
569 |
0.220 |
2.57 × 103 |
30.7 |
6.38 × 10−2 |
481 |
| In(III)/Ni(II) |
569 |
6.31 × 10−4 |
9.02 × 104 |
30.7 |
2.96 × 10−3 |
1.04 × 104 |
| In(III)/Pb(II) |
569 |
349 |
1.63 |
30.7 |
64.3 |
2.09a |
| In(III)/Sn(IV) |
569 |
>5.00 × 103 |
>9.78a |
30.7 |
>5.00 × 103 |
>162a |
| In(III)/Zn(II) |
569 |
>5.00 × 103 |
>9.78a |
30.7 |
>5.00 × 103 |
>162a |
Conclusions
It is shown that the commercial ionic liquids Cyphos® IL 101 and Aliquat® 336 are very efficient for extraction of indium(III) from chloride feed solutions. The ionic liquids are used in an undiluted form so that volatile molecular organic solvents can be avoided. The extraction process is selective for indium(III), over many other metal ions that are commonly found as impurities in process solutions of indium refineries. Due to the very high distribution ratios, co-extracted impurities can easily be scrubbed from the ionic liquid phase without affecting the extracted indium(III) ions. Indium could be recovered in the form of In(OH)3 by precipitation stripping with a NaOH solution. This stripping step also regenerates the ionic liquid. This work indicates that precipitation stripping with NaOH is a very convenient methods for recovering metal ions that show high affinities for the organic phase, such as indium(III) for undiluted chloride ionic liquids. The method is environmentally friendly because NaCl is the sole waste product and the ionic liquids can be reused.
Acknowledgements
This research was supported by the Flemish Institute for the Promotion of Innovation by Science and Technology (IWT Vlaanderen) via a Baekeland PhD fellowship to Clio Deferm (IWT 130305) and by the Umicore Group Research & Development. ICP-OES analyses performed on the multiple element system and TOC analyses were executed in the analytical laboratory of Umicore Group Research & Development.
Notes and references
- A. M. Alfantazi and R. R. Moskalyk, Miner. Eng., 2003, 16, 687–694 CrossRef CAS.
-
N. Felix, in Ullmann's Encyclopedia of Industrial Chemistry, ed. F. Ullmann, WILEY-VCH, Weinheim, 2012, vol. 19, pp. 65–74 Search PubMed.
-
U. Scwarz-Schampera and P. M. Herzig, Indium: Geology, Mineralogy, and Economics, Springer Verlag, New York, 2002 Search PubMed.
- Historical statistics for mineral and material commodities in the United States: U.S. Geological Survey Data Series 140, http://minerals.usgs.gov/ds/2005/140/ (accessed January 2015).
- Report on critical raw materials for the EU, http://ec.europa.eu/growth/sectors/raw-materials/specific-interest/critical/index_en.htm (accessed April 2016).
-
J. Stary, The Solvent Extraction of Metal Chelates, Pergamon Press, New York, 1964 Search PubMed.
-
J. Rydberg, M. Cox, C. Musikas and G. R. Choppin, Solvent Extraction: Principles and Practice, Marcel Dekker, New York, 2004 Search PubMed.
- A. P. Paiva, Sep. Sci. Technol., 2001, 36, 1395–1419 CrossRef CAS.
- J. G. Huddleston, H. D. Willauer, R. P. Swatloski, A. E. Visser and R. D. Rogers, Chem. Commun., 1998, 1765–1766 RSC.
- F. Kubota and M. Goto, Solvent Extr. Res. Dev., Jpn., 2006, 13, 23–36 CAS.
- M. L. Dietz, Sep. Sci. Technol., 2006, 41, 2047–2063 CrossRef CAS.
- N. V. Plechkova and K. R. Seddon, Chem. Soc. Rev., 2008, 37, 123–150 RSC.
- T. Welton, Chem. Rev., 1999, 99, 2071–2083 CrossRef CAS PubMed.
- R. D. Rogers and K. R. Seddon, Science, 2003, 302, 792–793 CrossRef PubMed.
- J. Park, Y. Jung, P. Kusumah, J. Lee, K. Kwon and C. K. Lee, Int. J. Mol. Sci., 2014, 15, 15320–15343 CrossRef CAS PubMed.
- M. L. Dietz, Sep. Sci. Technol., 2006, 41, 2047–2063 CrossRef CAS.
- D. Parmentier, S. J. Metz and M. C. Kroon, Green Chem., 2013, 15, 205–209 RSC.
- N. Papaiconomou, G. Vite, N. Goujon, J. M. Leveque and I. Billard, Green Chem., 2012, 14, 2050–2056 RSC.
- N. Papaiconomou, J.-M. Lee, J. Salminen, M. von Stosch and J. M. Prausnitz, Ind. Eng. Chem. Res., 2008, 47, 5080–5086 CrossRef CAS.
- I. Billard, A. Ouadi and C. Gaillard, Anal. Bioanal. Chem., 2011, 400, 1555–1566 CrossRef CAS PubMed.
- S. Génand-Pinaz, N. Papaiconomou and J.-M. Leveque, Green Chem., 2013, 15, 2493–2501 RSC.
- J. G. Huddleston, A. E. Visser, W. M. Reichert, H. D. Willauer, G. A. Broker and R. D. Rogers, Green Chem., 2001, 3, 156–164 RSC.
- P. G. Rickert, D. C. Stepinski, D. J. Rausch, R. M. Bergeron, S. Jakab and M. L. Dietz, Talanta, 2007, 72, 315–320 CrossRef CAS PubMed.
- R. P. Swatloski, J. D. Holbrey and R. D. Rogers, Green Chem., 2003, 5, 361–363 RSC.
- R. K. Mishra, P. C. Rout, K. Sarangi and K. C. Nathsarma, Hydrometallurgy, 2011, 108, 93–99 CrossRef CAS.
- F. d. M. Fábrega and M. B. Mansur, Hydrometallurgy, 2007, 87, 83–90 CrossRef.
- J. Castillo, M. T. Coll, A. Fortuny, P. N. Donoso, R. Sepúlveda and A. María Sastre, Hydrometallurgy, 2014, 141, 89–96 CrossRef CAS.
- M. Regel-Rosocka, Ł. Nowak and M. Wiśniewski, Sep. Purif. Technol., 2012, 97, 158–163 CrossRef CAS.
- A. Cieszynska and M. Wiśniewski, Sep. Purif. Technol., 2011, 80, 385–389 CrossRef CAS.
- M. Regel-Rosocka and M. Wiśniewski, Hydrometallurgy, 2011, 110, 85–90 CrossRef CAS.
- D. Kogelnig, A. Stojanovic, F. Jirsa, W. Körner, R. Krachler and B. K. Keppler, Sep. Purif. Technol., 2010, 72, 56–60 CrossRef CAS.
- P. Rybka and M. Regel-Rosock, Sep. Sci. Technol., 2012, 47, 1296–1302 CrossRef CAS.
- S. Wellens, R. Goovaerts, C. Möller, J. Luyten, B. Thijs and K. Binnemans, Green Chem., 2013, 15, 3160–3164 RSC.
- T. Vander Hoogerstraete, S. Wellens, K. Verachtert and K. Binnemans, Green Chem., 2013, 15, 919–927 RSC.
- K. Larsson and K. Binnemans, Green Chem., 2014, 16, 4595–4603 RSC.
- D. Dupont and K. Binnemans, Green Chem., 2016, 18, 176–185 RSC.
- M. P. Jensen, J. Neuefeind, J. V. Beitz, S. Skanthakumar and L. Soderholm, J. Am. Chem. Soc., 2003, 125, 15466–15473 CrossRef CAS PubMed.
- M. L. Dietz and J. A. Dzielawa, Chem. Commun., 2001, 20, 2124–2125 RSC.
- M. P. Jensen, J. A. Dzielawa, P. Rickert and M. L. Dietz, J. Am. Chem. Soc., 2002, 124, 10664–10665 CrossRef CAS PubMed.
- M. L. Dietz, J. A. Dzielawa, I. Laszak, B. A. Young and M. P. Jensen, Green Chem., 2003, 5, 682–685 RSC.
-
M. Halpern, http://phasetransfer.com/WhatisAliquat336andAdogen464.pdf (accessed April 2016).
- M. L. Good, S. E. Bryan, F. F. Holland Jr. and G. J. Maus, J. Inorg. Nucl. Chem., 1963, 25, 1167–1173 CrossRef CAS.
- M. L. Good, S. C. Srivastava and F. F. Holland Jr., Anal. Chim. Acta, 1964, 31, 534–544 CrossRef CAS.
- M. L. Good and F. F. Holland Jr., J. Inorg. Nucl. Chem., 1964, 26, 321–327 CrossRef CAS.
- F. G. Seeley and D. J. Crouse, J. Chem. Eng. Data, 1966, 11, 424–429 CrossRef CAS.
- H. Narita, M. Tanaka, H. Shiwaku, Y. Okamoto, S. Suzuki, A. Ikeda-Ohnob and T. Yaitab, Dalton Trans., 2014, 43, 1630 RSC.
- I. Komasawa, N. Kurokawa, Y. Maekawa and T. Otake, J. Chem. Eng. Jpn., 1986, 19, 592–598 CrossRef CAS.
- T. Sato, H. Watanabe and S. Kikuchi, J. Appl. Chem. Biotechnol., 1975, 25, 63–72 CAS.
- S. Wellens, B. Thijs and K. Binnemans, Green Chem., 2012, 14, 1657–1665 RSC.
- P. J. Carvalho, S. P. M. Ventura, M. L. S. Batista, B. Schröder, F. Gonçalves, J. Esperança, F. Mutelet and J. A. P. Coutinho, J. Chem. Phys., 2014, 140, 064505 CrossRef PubMed.
- K. A. Kurnia, M. V. Quental, L. M. N. B. F. Santos, M. G. Freire and J. A. P. Coutinhoa, Phys. Chem. Chem. Phys., 2015, 17, 4569–4577 RSC.
- T. Vander Hoogerstraete and K. Binnemans, Green Chem., 2014, 16, 1594–1606 RSC.
- C. Fischer and H. Wagner, Inorg. Nucl. Chem., 1977, 39, 513–517 CrossRef CAS.
- H. M. N. H. Irving and A. D. Damodaran, Anal. Chim. Acta, 1970, 50, 277–285 CrossRef CAS.
-
A. E. Martell and R. M. Smith, Critical Stability Constants, Volume-I: Amino Acids, Plenum Press, New York, 1974 Search PubMed.
-
R. Pribil, Analytical Applications of EDTA and Related Compounds, Pergamon Press, New York, 1972 Search PubMed.
- M. Pociecha and D. Lestan, J. Hazard. Mater., 2012, 201–202, 273–279 CrossRef CAS PubMed.
- M. Pociecha and D. Lestan, Chemosphere, 2012, 86, 843–846 CrossRef CAS PubMed.
- L. Di Palma, P. Ferrantelli, C. Merli and F. Biancifiori, J. Hazard. Mater., 2003, 103, 153–168 CrossRef CAS PubMed.
- O. Gylienė, J. Aikaitė and O. Nivinskienė, J. Hazard. Mater., 2004, 116, 119–124 CrossRef PubMed.
- K. J. Fraser and D. R. MacFarlane, Aust. J. Chem., 2009, 62, 309–321 CrossRef CAS.
- S. Sowmiah, V. Srinivasadesikan, M. C. Tseng and Y. H. Chu, Molecules, 2009, 14, 3780–3813 CrossRef CAS PubMed.
- M. C. Tseng, H. C. Kan and Y. H. Chu, Tetrahedron Lett., 2007, 48, 9085–9089 CrossRef CAS.
- D. Landini, A. Maia and A. Rampoldi, J. Org. Chem., 1986, 51, 3187–3191 CrossRef CAS.
- T. Vander Hoogerstraete, B. Blanpain, T. Van Gerven and K. Binnemans, RSC Adv., 2014, 4, 64099–64111 RSC.
- D. Dupont and K. Binnemans, Green Chem., 2015, 17, 856–868 RSC.
- B. Onghena and K. Binnemans, Ind. Eng. Chem. Res., 2015, 54, 1887–1898 CrossRef CAS.
-
M. Pourbaix, Atlas d’équilibres électrochimiques, Gauthier-Villars, Paris, 1963 Search PubMed.
-
A. Seidell and W. F. Linke, Solubilities, Inorganic and Metal Organic Compounds, American Chemical Society, Washington D.C., 4th edn, 1958, vol. 1 Search PubMed.
-
A. Seidell and W. F. Linke, Solubilities, Inorganic and Metal Organic Compounds, American Chemical Society, Washington D.C., 4th edn, 1958, vol. 2 Search PubMed.
- N. M. Rice, H. M. N. H. Irving and M. A. Leonard, Pure Appl. Chem., 2009, 65, 2373–2396 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Figures with additional extraction results. See DOI: 10.1039/c6gc00586a |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a