
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence“On water” reactivity between carbohydrate-derived nitroalkenes and furans
Verónica
Luque-Agudo
,
María Victoria
Gil
*,
Emilio
Román
and
José Antonio
Serrano
IACYS-Unidad de Química Verde y Desarrollo Sostenible, Departamento de Química Orgánica e Inorgánica, Facultad de Ciencias, Universidad de Extremadura, 06006, Badajoz, España. E-mail: vgil@unex.es; Fax: +34 924 271 149
First published on 4th April 2016
Abstract
Eco-friendly “on water” reactions of carbohydrate-derived nitroalkenes with furan, 2-methylfuran and furfural N,N-dimethylhydrazone have been investigated under different mixing methods, such as a magnetic stirrer and a wrist-action shaker. Cyclic and acyclic furyl derivatives from carbohydrates were obtained with high diastereoselectivity.
Introduction
Within the last decade, synthetic organic chemistry has sought safer reaction conditions through more efficient protocols that avoid the use of toxic or hazardous reagents. The so-called “green chemistry” also takes into account economic and environmental factors such as energy efficiency, atom economy and sustainability of the chemical processes.1 Consequently, it should be noted that solvents are usually the major components in the reactions and often constitute more than half of the total material used in most chemical processes.2 As a result, research in new solvents and reaction media is considered as a priority because of the benefits that could have a direct and substantial change in this respect.3In this context, the use of water as a medium for organic reactions is one of the latest challenges for modern chemists, opening a new and advantageous way4 for carrying out transformations with remarkable increases in speed and performance over the use of organic solvents and with improved chemo-, regio- and enantioselectivity.5 When organic and industrial chemists seek to choose a solvent for synthesis, water should be high or at the top of the list.4a Thus, literature describing organic reactions “on water” is rapidly growing and has recently been reviewed.4a–c The term “on water” was first used by Sharpless et al.6 in 2005 to refer to those reactions involving water-insoluble reagents in which a vigorous stirring generated aqueous suspensions in the absence of organic cosolvents. In this method, the water plays both roles as the reaction medium and catalyst, having become one of the most recent and promising innovations in organic synthesis.7 In order to assess the influence of the mixing of the reactants, Pirrung et al.8 tested for different stirring methods in Passerini, Ugi and Alder–Ene reactions. In particular, they used magnetic stirring, an ultrasonic bath thermostated at room temperature and a wrist-action shaker. The authors concluded that the most effective stirring method to address organic reactions “on water” should be empirically determined for each type of reaction.
On the other hand, 2-nitro-D-glycals had been extensively used in Michael additions of alcohols, thiophenols or carbohydrates,9 as well as in the addition of phenols for the synthesis of arylglycosides.10 This synthetic methodology has been extended to the preparation of oligosaccharides,11 β-N-glycosides12 and bicyclic lactams obtained through the addition of stabilized carbanions.13 Recent reviews (Awan and Werz13b and Delaunay et al.12c) summarize most of the reactivity of the 2-nitroglycals investigated until now. However, neither the reactivity of these nitroalkenes nor any other carbohydrate derivatives has been reported under the “on water” conditions.
In this paper, we describe the reactions of three carbohydrate derived nitroalkenes with furan, 2-methylfuran and a furfural hydrazone under “on water” conditions. In order to optimize the results, we have tested three modes of agitation: magnetic stirring of a non-emulsionated two-phase system, vigorous magnetic stirring of a self-generated suspension and stirring of self-generated suspension with a wrist-action shaker (see details in the Experimental section). It is worth noting that, as far as we are aware, there were no previous reports on reactions describing the formation of new carbon bonds on C-3 of 2-nitroglycals. Until now, it should be considered that there are only two general routes for the synthesis of furyl derivatives from carbohydrates: the first is based on treating simple derivatives of furan activated with the corresponding acceptors (such as 2- and 3-furyllitium with aldehydes14 or 2-nitroglycals12c), and the second is based on the use of acid catalysis15 or high pressure to prepare heterocyclic rings from cyclic16 or acyclic precursors.17
Experimental
Preparative TLC was performed using silica gel (Merck 60 GF254). TLC was carried out with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 hexane–ethyl acetate as the eluent, on precoated Merck Kieselgel 60 GF254 aluminum backed plates; spots were visualized by UV light or iodine vapour. NMR spectra were taken either on Bruker AC/PC instruments with tetramethylsilane as internal reference and deuteriochloroform as the solvent. Coupling constant values are recorded in Hz. When reported, characterization of NMR signals is based on homonuclear double-resonance and DEPT experiments. High resolution mass spectra were recorded on an Autoespec (Micromass) spectrometer, at the Centro de Investigación Tecnológica e Innovación (CITIUS) from Universidad de Sevilla. Infrared spectra were registered on an IR3000 Thermo Electron Corporation spectrophotometer in the range between 4000 and 600 cm−1. Magnetic stirring was carried out at 750 rpm with a Heidolph plate; for generating the non-emulsionated biphasic system, the flask was placed on the center of the plate, while the effect of vigorous stirring was achieved placing the flask about 5 cm away from the center of the stirring plate. The wrist-action shaker was programmed at 750 oscillations per minute. Unless otherwise specified, all the reactions were carried out in 10 mL round bottom glass flasks with minimum volumes of furan derivatives that were necessary to dissolve the corresponding nitroalkenes, and then adding distilled water. The size of the magnetic stirring bar used for the first two modes of stirring was 2.0 cm long and 0.7 cm diameter.
:1 hexane–ethyl acetate as the eluent, on precoated Merck Kieselgel 60 GF254 aluminum backed plates; spots were visualized by UV light or iodine vapour. NMR spectra were taken either on Bruker AC/PC instruments with tetramethylsilane as internal reference and deuteriochloroform as the solvent. Coupling constant values are recorded in Hz. When reported, characterization of NMR signals is based on homonuclear double-resonance and DEPT experiments. High resolution mass spectra were recorded on an Autoespec (Micromass) spectrometer, at the Centro de Investigación Tecnológica e Innovación (CITIUS) from Universidad de Sevilla. Infrared spectra were registered on an IR3000 Thermo Electron Corporation spectrophotometer in the range between 4000 and 600 cm−1. Magnetic stirring was carried out at 750 rpm with a Heidolph plate; for generating the non-emulsionated biphasic system, the flask was placed on the center of the plate, while the effect of vigorous stirring was achieved placing the flask about 5 cm away from the center of the stirring plate. The wrist-action shaker was programmed at 750 oscillations per minute. Unless otherwise specified, all the reactions were carried out in 10 mL round bottom glass flasks with minimum volumes of furan derivatives that were necessary to dissolve the corresponding nitroalkenes, and then adding distilled water. The size of the magnetic stirring bar used for the first two modes of stirring was 2.0 cm long and 0.7 cm diameter.
Synthetic procedures
:1). 1H-NMR (CDCl3, 500 MHz) δ: 8.08 (s, 1H, OCHO), 7.38 (d, 1H, J4′,5′ = 1.5 Hz, H-5′), 6.35 (dd, 1H, J3′,4′ = 3.5 Hz, J4′,5′ = 2.0 Hz, H-4′), 6.24 (d, 1H, H-3′), 5.42 (dd, 1H, J2,3 = 3.0 Hz, J3,4 = 8.5 Hz, H-3), 5.10 (ddd, 1H, H-4), 4.73 (dd, 1H, J1a,2 = 6.5 Hz, J1a,1b = 14.0 Hz, H-1a), 4.69 (dd, 1H, J1b,2 = 8.5 Hz, H-1b), 4.21 (dd, 1H, J4,5a = 2.5 Hz, J5a,5b = 13.0 Hz, H-5a), 4.14 (dd, 1H, J4,5b = 4.5 Hz, H-5b), 4.12 (ddd, 1H, H-2), 2.11 (s, 3H, OCOCH3), 2.05 (s, 3H, OCOCH3). 13C-NMR (CDCl3, 125 MHz) δ: 170.5, 169.9 (OCOCH3), 159.4 (OCHO), 147.4 (C-2′), 143.1 (C-5′), 110.7 (C-4′), 109.6 (C-3′), 74.8 (C-1), 69.6, 69.1 (C-3,4), 61.2 (C-5), 38.6 (C-2), 20.6 (OCOCH3). HRMS (CI): m/z calcd for C14H18NO9 ([M + H]+): 344.0982. Found 344.0986.
:0.9 mixture of 10a and 11a. An analytical sample of compound 10a was isolated pure by PTLC (light petroleum–ethyl ether, 1:1, two elutions).
(B) To a solution of (E)-3,5-di-O-acetyl-4-O-formyl-D-erythro-1-nitropent-1-enitol 7 (0.53 g, 1.93 mmol) in 2-methylfuran 9 (1.0 mL, 11.16 mmol) distilled water was added (3.0 mL) and the mixture was subjected to vigorous magnetic stirring. After 5 days, TLC and 1H-NMR showed completion of the reaction and the crude was treated with brine (5 mL) and extracted with dichloromethane (3 × 5 mL). The organic phase was dried over anhydrous sodium sulfate, filtered and evaporated to yield 0.37 g (54%) of a 1.0:1.5 oily mixture of 10a and 11a.
10a: 1H-NMR (CDCl3, 500 MHz) δ: 8.11 (s, 1H, OCHO), 6.12 (d, 1H, J3′,4′ = 3.0 Hz, H-3′), 5.94 (m, 1H, J4′,CH3 < 1.0 Hz, H-4′), 5.42 (dd, 1H, J2,3 = 3.0 Hz, J3,4 = 8.5 Hz, H-3), 5.12 (ddd, 1H, H-4), 4.71 (dd, 1H, J1a,2 = 7.0 Hz, J1a,1b = 13.5 Hz, H-1a), 4.67 (dd, 1H, J1b,2 = 8.5 Hz, H-1b), 4.24 (dd, 1H, J4,5a = 2.5 Hz, J5a,5b = 12.5 Hz, H-5a), 4.16 (dd, 1H, J4,5b = 4.5 Hz, H-5b), 4.08 (ddd, 1H, H-2), 2.26 (s, 3H, CH3-5′), 2.13 (s, 3H, OCOCH3), 2.07 (s, 3H, OCOCH3). 13C-NMR (CDCl3, 125 MHz) δ: 170.5, 169.9 (OCOCH3), 159.3 (OCHO), 152.9 (C-2′), 145.3 (C-5′), 110.3 (C-4′), 106.6 (C-3′), 74.9 (C-1), 69.6, 69.2 (C-3,4), 61.3 (C-5), 38.6 (C-2), 20.6 (OCOCH3), 13.5 (CH3-5′). HRMS (CI): m/z calcd for C15H20NO9 ([M + H]+): 358.1138. Found 358.1132.
11a (data from mixture 10a + 11a): 1H-NMR (CDCl3, 500 MHz) δ: 8.02 (s, 1H, OCHO), 6.19 (d, 1H, J3′,4′ = 3.0 Hz, H-3′), 5.89 (m, 1H, H-4′), 5.50 (dd, 1H, J2,3 = 4.0 Hz, J3,4 = 8.5 Hz, H-3), 5.26 (ddd, 1H, H-4), 4.60 (d, 2H, H-1a, H-1b), 2.27 (s, 3H, CH3-5′), 2.09 (s, 3H, OCOCH3), 2.04 (s, 3H, OCOCH3).
:0.3:1.0 oily mixture of 10a, 11a and 12. A fraction of this mixture (0.1 g) was acetylated with acetic anhydride and pyridine, thus resulting in 0.08 g of compound 12 as the only detected product.
1H-NMR (CDCl3, 500 MHz) δ: 8.37 (s, 1H, H-1), 6.04 (dd, 1H, J3′,4′ = 2.5 Hz, J2,3′ < 1.0 Hz, H-3′), 5.92 (m, 1H, J4′,CH3 < 1.0 Hz, H-4′), 5.58 (t, 1H, J3,4 = 2.5 Hz, J4,5 = 2.5 Hz, H-4), 4.61 (ddd, 1H, H-5), 4.45 (bs, 1H, H-3), 4.09 (dd, 1H, J5,6a = 8.5 Hz, J6a,6b = 12.5 Hz, H-6a), 3.76 (dd, 1H, J5,6b = 4.5 Hz, H-6b), 2.27 (s, 3H, CH3-5′), 2.14 (s, 3H, OCOCH3), 2.05 (s, 3H, OCOCH3). 13C-NMR (CDCl3, 125 MHz) δ: 170.1, 169.5 (OCOCH3), 153.3 (C-1), 152.4, 146.3 (C-2′,5′), 128.9 (C-2), 108.8, 106.8 (C-3′,4′), 76.8 (C-5), 66.7 (C-4), 61.3 (C-6), 35.7 (C-3), 20.9, 29.6 (OCOCH3), 13.5 (CH3-5′). HRMS (CI): m/z calcd for C15H18NO8 ([M + H]+): 340.1032. Found 340.1024.
:1.0 oily mixture of 10b and 11b.
:1).
(B) To a solution of (E)-3,5-di-O-acetyl-4-O-formyl-D-erythro-1-nitropent-1-enitol 7 (0.30 g, 1.11 mmol) in furfural N,N-dimethylhydrazone 13 (0.25 mL, 1.89 mmol) distilled water was added (2.0 mL). After 3 h of vigorous magnetic stirring, the reaction mixture was treated with dichloromethane (10 mL) and then filtered and evaporated to yield 0.44 g of an oily 1.0:1.0 mixture of 14a and 15a, unpurified with the hydrazone in excess.
14a: 1H-NMR (CDCl3, 400 MHz) δ: 8.09 (bs, 1H, OCHO), 6.99 (bs, 1H, CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), 6.32 (d, 1H, J3′,4′ = 3.2 Hz, H-4′), 6.25 (d, 1H, H-3′), 5.41 (dd, 1H, J2,3 = 3.2 Hz, J3,4 = 8.4 Hz, H-3), 5.16 (m, 1H, H-4), 4.72 (dd, 2H, J1a,2 = 7.6 Hz, H-1a, H-1b), 4.25 (dd, 1H, J4,5a = 2.4 Hz, J5a,5b = 12.4 Hz, H-5a), 4.17 (dd, 1H, J4,5b = 4.8 Hz, H-5b), 4.11 (m, 1H, H-2), 2.96 (s, 6H, N(CH3)2), 2.13 (s, 3H, OCOCH3), 2.10 (s, 3H, OCOCH3).
N), 6.32 (d, 1H, J3′,4′ = 3.2 Hz, H-4′), 6.25 (d, 1H, H-3′), 5.41 (dd, 1H, J2,3 = 3.2 Hz, J3,4 = 8.4 Hz, H-3), 5.16 (m, 1H, H-4), 4.72 (dd, 2H, J1a,2 = 7.6 Hz, H-1a, H-1b), 4.25 (dd, 1H, J4,5a = 2.4 Hz, J5a,5b = 12.4 Hz, H-5a), 4.17 (dd, 1H, J4,5b = 4.8 Hz, H-5b), 4.11 (m, 1H, H-2), 2.96 (s, 6H, N(CH3)2), 2.13 (s, 3H, OCOCH3), 2.10 (s, 3H, OCOCH3).
15a: (data from mixture 14a + 15a) 1H-NMR (CDCl3, 400 MHz) δ: 8.03 (bs, 1H, OCHO), 7.00 (bs, 1H, CHN), 6.34 (d, 1H, J3′,4′ = 2.8 Hz, H-3′), 6.32 (d, 1H, H-4′), 5.57 (dd, 1H, J2,3 = 3.2 Hz, J3,4 = 6.4 Hz, H-3), 5.28 (m, 1H, H-4), 4.66 (dd, 2H, J1a,2 = 5.6 Hz, H-1a, H-1b), 4.25–4.17 (m, 2H, H-2, H-5a), 4.04 (dd, 1H, H-5b), 2.95 (s, 6H, N(CH3)2), 2.05 (s, 3H, OCOCH3), 2.04 (s, 3H, OCOCH3).
:1.0 oily mixture (1.31 g) of 14b and 15b, unpurified by the hydrazone. Diastereoisomers could be separated by PTLC (light petroleum–ethyl ether, 5:1, two elutions).
14b: 1H-NMR (CDCl3, 400 MHz) δ: 6.99 (s, 1H, CHN), 6.27 (d, 1H, J3′,4′ = 3.6 Hz, H-4′), 6.24 (d, 1H, H-3′), 5.46 (dd, 1H, J4,5 = 8.4 Hz, J5,6 = 5.6 Hz, H-5), 5.40 (dd, J2,3 = 9.2 Hz, J3,4 = 2.0 Hz, H-3), 5.24 (dd, 1H, H-4), 5.04 (m, 1H, J6,7a = 2.4 Hz, J6,7b = 4.8 Hz, H-6), 4.77 (dd, 1H, J1a,2 = 5.2 Hz, J1a,1b = 13.6 Hz, H-1a), 4.68 (dd, 1H, J1b,2 = 8.8 Hz, H-1b), 4.17 (dd, 1H, J6,7a = 2.8 Hz, J7a,7b = 12.4 Hz, H-7a), 4.02 (dd, 1H, J6,7b = 4.8 Hz, H-7b), 3.97 (m, 1H, H-2), 2.93 (s, 6H, N(CH3)2), 2.09 (s, 3H, OCOCH3), 2.07 (s, 3H, OCOCH3), 2.04 (s, 3H, OCOCH3), 2.04 (s, 3H, OCOCH3), 2.01 (s, 3H, OCOCH3). 13C-NMR (CDCl3, 100 MHz) δ: 170.5, 170.2, 169.8, 169.7, 169.3 (OCOCH3), 153.0 (C-2′), 147.8 (C-5′), 122.2 (CN), 110.1 (C-4′), 107.0 (C-3′), 74.0 (C-1), 69.4, 68.3, 67.8, 66.9 (C-3,4,5,6), 61.8 (C-7), 42.5 N(CH3)2, 38.3 (C-2), 20.8, 20.7, 20.6, 20.6 (OCOCH3). HRMS (ESI): m/z calcd for C24H34N3O13: 572.2086. Found 572.2077.
15b: 1H-NMR (CDCl3, 400 MHz) δ: 7.00 (s, 1H, CHN), 6.35 (d, 1H, J3′,4′ = 3.2 Hz, H-4′), 6.32 (d, 1H, H-3′), 5.36 (d, 2H, J2,3 = 9.2 Hz, H-5, H-3), 5.10 (dd, 1H, J4,5 = 9.6 Hz, J3,4 = 1.2 Hz, H-4), 4.98 (m, 1H, H-6), 4.78 (dd, 1H, J1a,2 = 7.6 Hz, J1a,1b = 14.0 Hz, H-1a), 4.69 (dd, 1H, J1b,2 = 7.2 Hz, H-1b), 4.16 (dd, 1H, J6,7a = 2.8 Hz, J7a,7b = 12.4 Hz, H-7a), 4.00 (dd, 1H, J6,7b = 4.8 Hz, H-7b), 3.97 (td, 1H, H-2), 2.94 (s, 6H, N(CH3)2), 2.12 (s, 3H, OCOCH3), 2.10 (s, 3H, OCOCH3), 2.07 (s, 3H, OCOCH3), 2.03 (s, 3H, OCOCH3), 2.02 (s, 3H, OCOCH3). 13C-NMR (CDCl3, 100 MHz) δ: 170.6, 170.5, 170.1, 169.9, 169.7 (OCOCH3), 153.2 (C-2′), 146.2 (C-5′), 122.1 (CN), 111.5 (C-4′), 106.7 (C-3′), 74.9 (C-1), 68.6, 68.3, 67.8, 67.4 (C-3,4,5,6), 61.7 (C-7), 42.6 N(CH3)2, 38.7 (C-2), 20.8, 20.8, 20.7, 20.7, 20.6 (OCOCH3). HRMS (ESI): m/z calcd for C24H34N3O13: 572.2086. Found 572.2077.
Results & discussion
Reaction between 2-nitro-D-glucal 1 and furan 2
The reaction between 3,4,6-tri-O-acetyl-2-nitro-D-glucal 1 and furan 2 (Scheme 1) was performed by using the “on water” methodology with vigorous magnetic stirring for 3 days. After the work-up, a syrup containing nitroadduct 3 and a very small quantity of 4 was obtained; however, when this same process was carried out by stirring the non-emulsionated biphasic system for 80 h, the starting material disappeared, the product being different from that prepared by the above method. In the second case, the resulting product was a complex mixture in which we did not detect anything of adducts 3 or 4, although PTLC analysis showed the presence of traces of a compound similar to 3, but without carrying the formate group.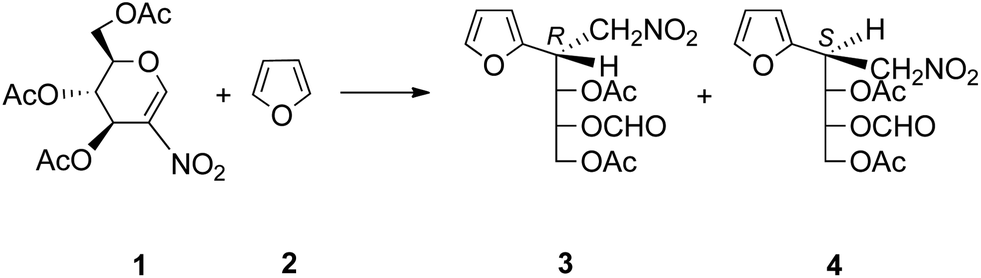 | ||
| Scheme 1 Reaction between 1 and 2. | ||
To discard this method of agitation, other assays with analogous results were performed because, in our view, a biphasic system is not suitable to generate “on water” reaction conditions, promoting hydrolysis of the formate groups in this case.
With respect to the stereochemistry of the reaction, the R configuration at C-2 in 3 was assigned by an analogy between the 1H-NMR signals of its diastereotopic nitromethylene protons with those of the same group in compounds of similar structures.16 When the above reaction was carried out under “on water” conditions, but using a wrist-action shaker, the starting material disappeared after 2 days and, by acetylation of the resulting crude product, cyclic nitroalkene 5 (Scheme 2) was obtained. It was noteworthy that the 1H-NMR spectrum showed that 5 was the major compound in the mixture before acetylation (with 3 and 4 also present); thus, this latter process showed that 5 was the only product. Structural assignment for nitroalkene 5 was supported by the similarity between its 1H-NMR spectrum and that of cyclic nitroalkene 1; moreover, comparing the spectra, the upfield shift for H-4, H-6a and H-6b signals in 5 (of about 0.40 ppm) could be attributed to the shielding caused by the furan ring, considering that this fragment is in a cis-relationship with these three protons. In the same way, it is worth noting that chemical shifts21 of H-4, H-6a and H-6b for compound 6 (Fig. 1) are as could be expected (considering the lack of effect of β-acetylation on H-4) when compared with these same data in 1. Once again, the chemical shift of H-5 (cis-relationship with respect to the furan ring) in 6 appeared shielded by about 0.6 ppm.
 | ||
| Scheme 2 Reaction between 1 and 2 followed by conventional acetylation. | ||
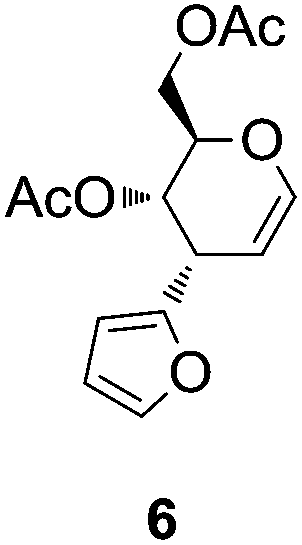 | ||
| Fig. 1 Structure of compound 6. | ||
Reactions of nitroalkenes 1, 7 or 8 with 2-methylfuran 9
The reaction between cyclic nitroalkene 1 and 2-methylfuran 9 led to a mixture of the acyclic nitro Michael adducts 10a and 11a, together with cyclic nitroalkene 12, (0.3:0.2:1.0 respective ratio; Scheme 3). After 4.5 days with vigorous magnetic stirring, TLC of the reaction mixture revealed that the starting material had disappeared; moreover, 1H-NMR showed that derivative 12 was the first to be formed, and then the acyclic nitro Michael adducts 10a and 11a appeared; after work-up, conventional acetylation of the crude product led exclusively to compound 12.
 | ||
| Scheme 3 Reaction between 1 or 7 or 8 and 9. | ||
Absolute configurations at C-2 for 10a and 11a were supported based on the 1H-NMR spectra similarities that were found between these compounds and their analogs 3 and 4. Assignment of the stereochemistry at C-3 in the adduct 12 was based on shielding effects, that were in parallel to those described above for 5.
When reaction between 1 and 9 was performed by the wrist-action shaker, the process was completed after 2 days, and the ratios of the products and reaction times are shown in Table 1; as can be seen, proportion of the acyclic products 10a and 11a in the crude mixture increases, extending the reaction times.
:(10a + 11a) and reaction times
| t (h) | Ratio 12:(10a + 11a) |
|---|---|
| 24.5 | 25:1 |
| 48 | 12.5:1 |
| 72 | 11.4:1 |
| 95 | 2.8:1 |
To check if nitroalkene 7 is an intermediate of the reaction between 1 and 2 or 9, we carried out the reaction between this acyclic nitroalkene (Scheme 3) and 2-methylfuran 9 with vigorous magnetic stirring for 2 days, the only product being a 1.0:1.5 mixture of the nitro Michael adducts 10a and 11a. In addition, to test the “on water” methodology using a solid nitroalkene, the mixture of crystalline 820 and 9 was subjected to vigorous magnetic stirring; thus, the reaction was completed after 6.5 days, leading to a 1.7:1.0 mixture of 10b and 11b. We had synthesized16 this same Michael adducts through a ring-opening reaction from 7-oxabicyclic compounds prepared by ultra-high pressure Diels–Alder cycloaddition between 2-methylfuran 9 and nitroalkene 8. It is noteworthy that the formation of such compounds, which had not been detected in our “on water” reactions, required conditions of 13000 kbar for 3 days.
Neither time nor the reaction products match those observed in the reactions of nitroalkene 1, thus discarding common intermediates and evidencing different reaction mechanisms for reactions of 1 and 7 (or 8) with 2 and 9. In the cases of compounds 7 and 8, the reaction should be an addition of the type Michael-aromatic electrophilic substitution of 2 or 9 on the less hindered C1-si face of 7, or the C1-re face of 8. In agreement with the experimental results, both these processes would lead, respectively, to the major adducts 11a or 10b, with a relative 2,3-erythro configuration.
Reactions of nitroalkenes 1, 7 or 8 with furfural N,N-dimethylhydrazone 13
The reaction between cyclic nitroalkene 1 and furfural hydrazone 13 was carried out under vigorous magnetic stirring for 6.5 h, leading exclusively to the Michael adduct 14a (Scheme 4); however, when acyclic nitroalkene 7 was used, a 1.2:1.0 mixture of 14a + 15a was obtained after 3 h. Assignment of the absolute configurations at C-2 was based on the close resemblance between the 1H-NMR spectra of 14a with those of 3 and 10a, the same occurring by comparing 1H-NMR spectra of 15a with that of 11a (Schemes 1 and 3). Once again, stereochemical outcomes and the different reaction times are suggesting different mechanisms; however, they would be in the same sense as in the reactions with 2-methylfuran 9, with the difference that in the reaction between 1 and 13, we did not detect products similar to 12.
 | ||
| Scheme 4 Reaction between 1 or 7 or 8 and 13. | ||
When treatment of the solid nitroalkene 8 and furfural hydrazone 13 was conducted with the wrist-action shaker, the reaction was completed after 1 day, obtaining a 1.2:1.0 mixture of the Michael adducts 14b + 15b (Scheme 4). These same compounds were formed in the same ratio by using vigorous magnetic stirring, although in this case, after 6 h reaction time.
Configurations at C-2 of compounds 14 and 15 have been assigned using the same theoretical arguments mentioned above for the Michael adducts derived from 2-methylfuran 9. The reaction times are significantly shortened, which should be due to the higher reactivity of the hydrazone 13 when it is compared22 with those from 2 or 9.
Influence of stirring methods
As it has been observed for all processes involving cyclic nitroalkenes 1, the stirring method markedly influences the reaction times and the products obtained as well as their proportions. The case of furan 2 is particularly noticeable (Table 2), since it was possible to direct the transformation towards one or the other of the products, simply by varying the stirring method. For reactions with 2-methylfuran 9, it is worth noting the change in the ratio of the products, although 12 was the major compound in both cases. With vigorous magnetic stirring, the ratio between the acyclic nitro adducts 10a + 11a and 12 did not suffer change with time, whereas this increased when the wrist-action shaker was used (Table 1).| Reaction | Stirring method | t reaction (days) | Product(s) | Ratio |
|---|---|---|---|---|
| a Time at which the starting material disappeared. b Unreproducible product ratio. | ||||
| 1 + 2 | Magnetic stirring | 2 | 3 + 5 | (3:1) |
| 3 | 3 | |||
| Wrist-action shaker | 2 | 5 | ||
| 3 | 3 + 5 | (2.5:1) |
||
| 1 + 9 | Magnetic stirring | 2 | 12 + 1 | (2.6:1) |
| 4.5 | 10a + 11a + 12 | (1.3:1:4.8) |
||
| Wrist-action shaker | 2a,b | 10a + 11a + 12 | (1:1:16.7) |
|
| 4.5 | 10a + 11a + 12 | (1:1.3:5) |
||
As shown in Table 2, differences in reaction times are substantial depending on the stirring method. Furthermore, after conducting a lot of experiments, we found that, with the exception of the synthesis of 5 and the reaction with 9, the results with the wrist-action shaker were poorly reproducible in product ratios; sometimes, the results were different by varying the location of the flask along the wrist-action shaker.
Difference in the reaction time between the two stirring methods could be justified considering that the magnetic stirring generates a greater surface interface, resulting in a suspension with smaller discrete droplets7c which would improve the efficiency of “on water” conditions.
The reactions described above with nitroalkene 1 have also been carried out under the same conditions of vigorous magnetic stirring, but by adding 2.0 mL of dichloromethane or replacing the water with an equal volume of aqueous 5 M lithium chloride. Additionally, we also performed these reactions in refluxing toluene for 4 days, and under ultra-high pressure (13000 kbar) for 3 days. In all the cases, no reaction was observed, and nitroalkene 1 was recovered unaltered. Hence, these facts underline the usefulness of the “on water” methodology as a method of activation for reactions involving compound 1.
Proposed mechanism for “on water” reactions involving 2-nitro-D-glucal 1
To explain the experimental results shown above, it would be necessary to consider that 2-nitro-D-glucal 1 could lead to acyclic nitroalkene 7 through a Grob-type fragmentation23 of intermediate 16, produced by addition of water on 1, as reported by Zajac et al.18 (Scheme 5). These authors proposed a mechanism for the fragmentation of glycals in alkaline media in which the first step would involve the attack of the base on the carbon–carbon double bond; however, under the “on water” conditions, we cannot exclude addition of water yielding intermediate 16 that, after a Grob-type fragmentation, would lead to nitroalkene 7. After carrying out many experiments, we could not reproduce the yields that had been described18 for the preparation of 7 from compound 1 and, therefore we have modified the method of synthesis, thus obtaining 7 in 50% yield (see Experimental). | ||
| Scheme 5 Grob-type fragmentation proposed for 1. | ||
Acyclic nitroalkene 7 has not been detected in any of the reaction media in which cyclic nitroalkene 1 was used. This fact would suggest that compounds 3, 4, 10a and 11a have not been formed through a Michael addition–aromatic electrophilic substitution of furan 2 or 2-methylfuran 9 on nitroalkene 7 arising from 1. When identical conditions of vigorous stirring “on water” were used, compound 7 did not alter; however, this was not the case for 1.
A similar transformation to that shown in Scheme 6, known as the Ferrier rearrangement, was reported by Ferrier and Prasad24 in 3,4,6-tri-O-acetyl-D-glycals. In our case, the rearrangement would transform nitroalkene 1 into 17 by migration of an acetate group (Scheme 6). This rearrangement has also been described for 2,3,4,6-tetra-O-acetyl-D-glycals in the presence of acetic acid.25 The authors propose that the acetate at C-3 provides anchimeric assistance for the rearrangement, although they did not exclude the SN1′ and SN2′ mechanisms, always in the presence of BF3. Otherwise, Szczerek et al.26 even suggest the formation of an intermediate carbocation, as 18, in ether as the solvent and in the presence of iodine; through this intermediate, Guthrie and Irvine27 explained that although addition on glycals preferentially takes place on C-1, it also could occur on C-3.
 | ||
| Scheme 6 Ferrier rearrangement proposed for 1. | ||
Additional support for the proposed mechanism (Scheme 7) was obtained from the acetylation reactions of mixtures of 3 and 4 (Scheme 1) or 10a and 11a (Scheme 3). In both cases, we obtained cyclic nitroalkenes 19 as the only products; hence, the justification of the stereochemical results necessarily implies that, under these conditions, the mechanism should be reversible until 17, where a carbanion at C-1 of 21 should be generated, which intramolecularly would attack the formyl group, thus leading to 20 (Henry reaction). This product would suffer acetylation of the anomeric hydroxyl group, allowing elimination of acetic acid to yield 19, then regenerating 17 upon addition of acetic acid and elimination of the furan framework.
 | ||
| Scheme 7 Proposed mechanism of reaction. | ||
The role of the aqueous phase under the “on water” conditions remains a mystery.4d Nevertheless, to explain the results summarized in Table 2, the mechanism proposed by Jung and Marcus7a as well as studies about the droplet sizes and saturation of the interface developed by Mellouli et al.7c and Guo et al.28 should be considered. Thus, magnetic stirring provides smaller droplet sizes, thereby increasing the interfacial area, where hydrogen-bond catalysis occurs more effectively. Consequently, magnetic stirring would promote addition of water to 19, leading to 20 (Scheme 7). When the wrist-action shaker is used, the catalytic surface is smaller because droplets are greater and the interface is saturated (because it is not been effectively renewed), preventing the water surface from catalyzing addition of water to 19. This argument is supported by data shown in Table 1: with larger reaction times, ratio 10a + 11a/12 increases (21 and 19 in Scheme 7, respectively). Although the effects of the magnetic field on water are still under investigation,29 it is known that the presence of a magnetic field influences the hydrogen-bond strengthening, determining their formation or reorientation and the restructure of water cluster based on the change of water intramolecular energy30 and on the polarization effects of water.31 They also perturb the gas/liquid and liquid/liquid interfaces, because these changes modify the structure and the reactivity of the bulk and interfacial water.32
Conclusions
“On water” methodology with vigorous magnetic stirring and, in minor extension, with stirring by a wrist-action shaker, first applied to some carbohydrate derived nitroalkenes constitutes a new, easy and, in some cases, highly diastereoselective method to prepare cyclic and acyclic furyl derivatives of nitro carbohydrates. This methodology also allows developing a new 2-nitroglycals chemistry: the formation of new carbon–carbon bonds on C-3.Acknowledgements
This work was supported by Gobierno de Extremadura-Ayuda a Grupos de Investigación Catalogados, and FEDER (Grant GR15022). We also thank the Universidad de Extremadura, Plan de Iniciación a la Investigación, Desarrollo Tecnológico e Innovación 2015 for a fellowship to Verónica Luque-Agudo.Notes and references
- M. B. Gawande, V. D. B. Bonifácio, R. Luque, P. S. Branco and R. S. Varma, Chem. Soc. Rev., 2013, 42, 5522 RSC.
- D. J. C. Constable, C. Jiménez-González and R. K. Henderson, Org. Process Res. Dev., 2007, 11, 133 CrossRef CAS.
- H. E. Eastman, C. Jamieson and A. J. B. Watson, Aldrichimica Acta, 2015, 48, 51 Search PubMed.
- (a) A. Chanda and V. V. Fokin, Chem. Rev., 2009, 109, 725 CrossRef CAS PubMed; (b) R. N. Butler and A. G. Coyne, Chem. Rev., 2010, 110, 6302 CrossRef CAS PubMed; (c) M. O. Simon and C.-J. Li, Chem. Soc. Rev., 2012, 41, 1415 RSC; (d) T. Sela and A. Vigalok, Org. Lett., 2014, 16, 1964 CrossRef CAS PubMed; (e) P. Norcott and C. S. P. McErlean, Org. Biomol. Chem., 2015, 13, 6866 RSC; (f) Y. Zhang, B.-W. Wei, H. Lin, L. Zhang, J.-X. Liu, H.-Q. Luo and X.-L. Fan, Green Chem., 2015, 17, 3266 RSC; (g) M. Oliverio, M. Nardi, L. Cariati, E. Vitale, S. Bonacci and A. Procopio, ACS Sustainable Chem. Eng., 2016, 4(3), 661 CrossRef CAS.
- J. Pinto, V. L. M. Silva, A. M. G. Silva, A. M. S. Silva, J. C. S. Costa, L. M. N. B. F. Santos, R. Enes, J. A. S. Cavaleiro, A. A. M. O. S. Vicente and J. A. C. Teixeira, Green Chem., 2013, 15, 970 RSC.
- S. Narayan, J. Muldoon, M. G. Finn, V. V. Fokin, H. C. Kolb and K. B. Sharpless, Angew. Chem., Int. Ed., 2005, 44, 3275 CrossRef CAS PubMed.
- (a) Y. Jung and R. A. Marcus, J. Am. Chem. Soc., 2007, 129, 5492 CrossRef CAS PubMed; (b) J. K. Beattie, C. S. P. McErlean and C. B. W. Phippen, Chem. – Eur. J., 2010, 16, 8972 CrossRef CAS PubMed; (c) S. Mellouli, L. Bousekkine, A. B. Theberge and W. T. S. Huck, Angew. Chem., Int. Ed., 2012, 51, 7981 CrossRef CAS PubMed.
- M. C. Pirrung, K. D. Sarma and J. Wang, J. Org. Chem., 2008, 73, 8723 CrossRef CAS PubMed.
- W. Xue, J. Sun and B. Yu, J. Org. Chem., 2009, 74, 5079 CrossRef CAS PubMed.
- A. I. Khodair, G. A. Winterfeld and R. R. Schmidt, Eur. J. Org. Chem., 2003, 1847 CrossRef CAS.
- R. Balamurugan, K. Pachamuthu and R. R. Schmidt, Synlett, 2005, 134 CAS.
- (a) G. A. Winterfeld, J. Das and R. R. Schmidt, Eur. J. Org. Chem., 2000, 3047 CrossRef CAS; (b) S. Xiang, J. Ma, B. K. Gorityyala and X. W. Liu, J. Org. Chem., 2010, 75, 8457 CrossRef PubMed; (c) T. Delaunay, T. Poisson, P. Jubault and X. Pannecoucke, Eur. J. Org. Chem., 2014, 7525 CrossRef CAS.
- (a) K. Pachamuthu, A. Gupta, J. Das, R. R. Schmidt and Y. D. Vankar, Eur. J. Org. Chem., 2002, 1479 CrossRef CAS; (b) S. I. Awan and D. B. Werz, Bioorg. Med. Chem., 2012, 20, 1846 CrossRef CAS PubMed; (c) Q. Zhang, J. Sun, F. Zhang and B. Yu, Eur. J. Org. Chem., 2010, 3579 CrossRef CAS.
- A. Dondoni, A. Marra and M.-C. Scherrmann, Tetrahedron Lett., 1993, 34, 7323 CrossRef CAS.
- K. Dziewiszek, M. Chmielewski and A. Zamojski, Carbohydr. Res., 1982, 104, C1 CrossRef CAS.
- N. Araújo, M. V. Gil, E. Román and J. A. Serrano, Tetrahedron: Asymmetry, 2009, 20, 1999 CrossRef.
- J. Jurczak and S. Pikul, Tetrahedron, 1988, 44, 4569 CrossRef CAS.
- W. W. Zajac Jr., P. Dampawan and D. Ditrow, J. Org. Chem., 1986, 51, 2618 CrossRef.
- P. K. Kancharla, Y. S. Reddy, S. Dharuman and Y. D. Vankar, J. Org. Chem., 2011, 76, 5832 CrossRef CAS PubMed.
- J. C. Sowden and R. Schaffer, J. Am. Chem. Soc., 1950, 73, 4662 CrossRef.
- J. S. Yadav, B. V. S. Reddy, J. V. Raman, N. Niranjan, S. K. Kumar and A. C. Kunwar, Tetrahedron Lett., 2002, 43, 2095 CrossRef CAS.
- M. V. Gil, V. Luque-Agudo, E. Román and J. A. Serrano, Synlett, 2014, 2179 CAS.
- C. A. Grob and P. W. Schiess, Angew. Chem., Int. Ed. Engl., 1967, 6, 1 CrossRef CAS.
- R. J. Ferrier and N. Prasad, J. Chem. Soc. C, 1969, 570 RSC.
- R. U. Lemieux and R. J. Bose, Can. J. Chem., 1966, 44, 1855 CrossRef CAS.
- I. Szczerek, J. S. Jewell, R. G. S. Ritchie, W. A. Szarek and J. K. N. Jones, Carbohydr. Res., 1972, 22, 163 CrossRef CAS.
- R. D. Guthrie and R. W. Irvine, Carbohydr. Res., 1980, 82, 207 CrossRef CAS.
- D. Guo, D. Zhu, X. Zhou and B. Zheng, Langmuir, 2015, 31, 13759 CrossRef CAS PubMed.
- Y.-Z. Guo, D.-C. Yin, H.-L. Cao, J.-Y. Shi, C.-Y. Zhang, Y.-M. Liu, H.-H. Huang, Y. Liu, Y. Wang, W.-H. Guo, A.-R. Qian and P. Shang, Int. J. Mol. Sci., 2012, 13, 16916 CrossRef CAS PubMed.
- R. Cai, H. Yang, J. He and W. Zhu, J. Mol. Struct., 2009, 938, 15 CrossRef CAS.
- A. Szczes, E. Chibowski, L. Holysz and P. Rafalski, Chem. Eng. Process., 2011, 50, 124 CrossRef CAS.
- M. Colic and D. Morse, Colloid Surf., A, 1999, 154, 167 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |