
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolvent-free synthesis of quaternary α-hydroxy α-trifluoromethyl diazenes: the key step of a nucleophilic formylation strategy†
Esteban
Matador
a,
David
Monge
*a,
Rosario
Fernández
*a and
José M.
Lassaletta
*b
aDepartamento de Química Orgánica, Universidad de Sevilla and Centro de Innovación en Química Avanzada (ORFEO-CINQA), C/ Prof. García González, 1, 41012 Sevilla, Spain. E-mail: dmonge@us.es; ffernan@us.es
bInstituto de Investigaciones Químicas (CSIC-US) and Centro de Innovación en Química Avanzada (ORFEO-CINQA), Avda. Américo Vespucio, 49, 41092 Sevilla, Spain. E-mail: jmlassa@iiq.csic.es
First published on 18th April 2016
Abstract
An efficient, scalable and operationally simple one-pot, 2-step strategy for the nucleophilic formylation of trifluoromethyl ketones is presented. The key step is an unprecedented diaza-carbonyl-ene reaction of formaldehyde tert-butyl hydrazone and trifluoromethyl ketones under solvent-free conditions. This reaction proved to be very fast, clean and high-yielding, affording densely functionalised α-hydroxy α-trifluoromethyl diazenes. The ensuing diazene-to-aldehyde transformation, avoiding protection/deprotection reactions and chromatographic purifications, and subsequent derivatizations in a one-pot fashion provide a direct entry to a variety of useful trifluoromethylated building blocks.
Introduction
Organofluorine compounds have attracted the interest of academia and industry from the viewpoint of their fruitful applications in pharmaceutical (approximately 20% of the market, including some of the most selling drugs)1 and materials sciences.2 Therefore, the development of synthetic methods for accessing new fluorinated compounds is an increasingly important issue in modern organic chemistry.3 In recent years, trifluoromethylated compounds have received considerable attention due to their unique chemical, physical and biological properties.4 In particular, trifluoromethyl carbinols and derivatives are present in a plethora of biologically active compounds. The selected examples shown in Scheme 1 include aminoalcohols I![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 5 and II,6 α-hydroxy amides III and IV,7 the marketed anti-HIV agent efavirenz V,8 matrix metalloproteinase (MMP) peptidomimetic inhibitors such as VI,9 and the neurokinin 1 receptor antagonist CJ-17493 VII.10
5 and II,6 α-hydroxy amides III and IV,7 the marketed anti-HIV agent efavirenz V,8 matrix metalloproteinase (MMP) peptidomimetic inhibitors such as VI,9 and the neurokinin 1 receptor antagonist CJ-17493 VII.10
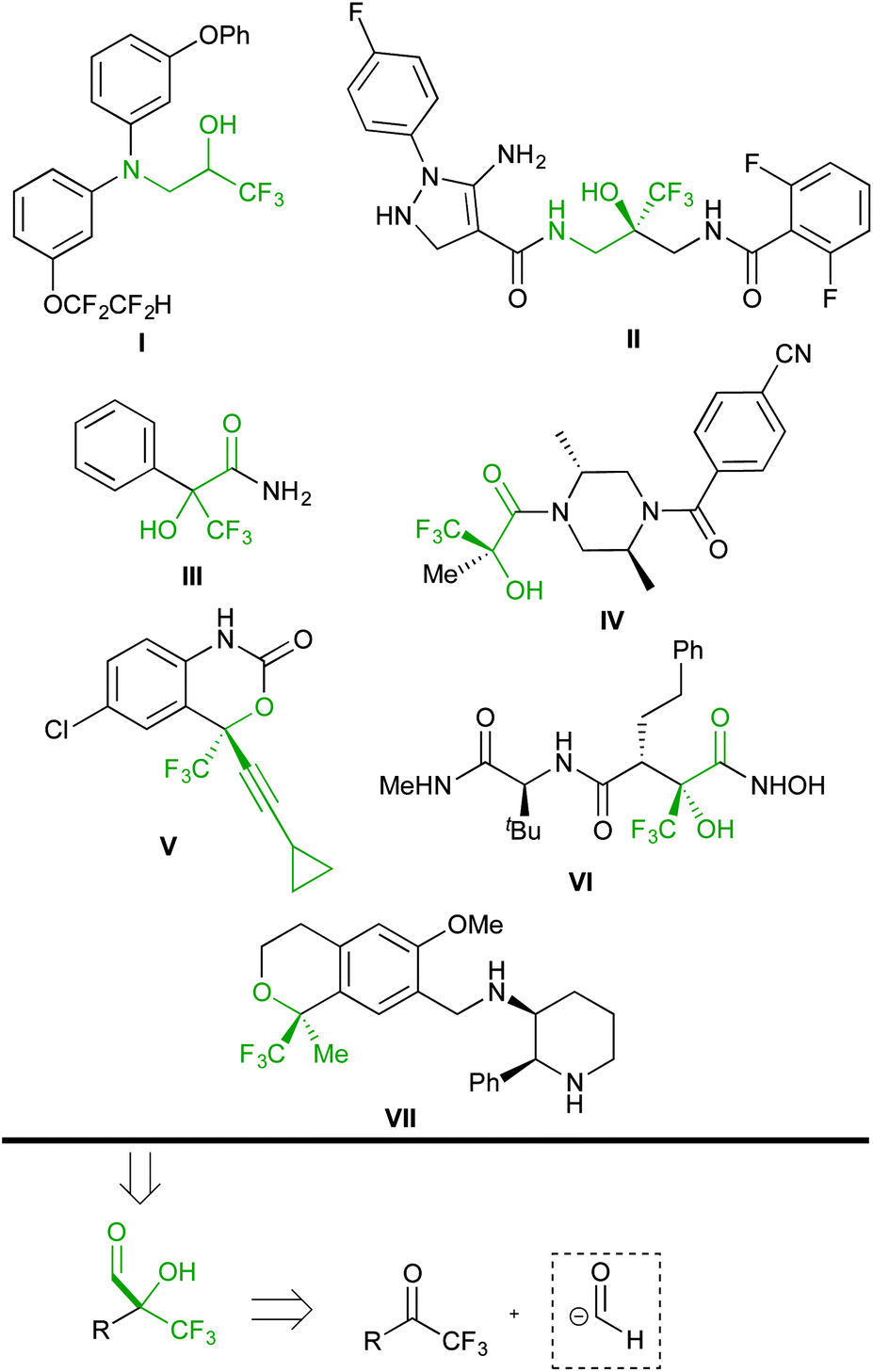 | ||
| Scheme 1 Retrosynthetic analysis of biologically relevant functionalized trifluoromethyl carbinols and derivatives. | ||
Accordingly, two general approaches to the synthesis of such compounds have been developed. The first one is the nucleophilic trifluoromethylation of carbonyl compounds,11 and the second strategy is based on the addition of carbon nucleophiles to trifluoromethyl ketones.12 The retrosynthetic analysis of the selected targets suggests the use of α-hydroxy α-trifluoromethyl aldehydes as common building blocks by virtue of the versatility of the formyl group.13 These intermediates might be conveniently built by employing trifluoromethyl ketones as electrophiles for the attack of d1 reagents (Scheme 1). This approach, however, has been scarcely investigated.14
Our research team has intensively explored the nucleophilic reactivity of formaldehyde N,N-dialkylhydrazones (FDAHs),15 which behave as formyl anion equivalents (d1 synthons) in their reactions with a variety of electrophiles, including carbonyl compounds.16 In particular, pyrrolidine derivatives spontaneously add to trifluoromethyl ketones affording α-hydroxy α-trifluoromethyl hydrazones (Scheme 2, top).16a,b This reaction, combined with hydroxyl protection and hydrazone cleavage by ozone, constitutes a convenient entry (3-steps) to both racemic or enantiomerically enriched α-alkoxy-α-trifluoromethyl aldehydes and derivatives thereof. More recently, we have exploited the superior reactivity of formaldehyde tert-butyl hydrazone (FTBH) with carbonyl compounds (α-keto esters,17 isatins18 and α-keto phosphonates)19 as the key step of a formylation strategy. In the seeking of greener methodologies, herein we present a more concise one-pot, 2-step approach to α-hydroxy α-trifluoromethyl aldehydes employing FTBH (Scheme 2, bottom) based on the positive effect that solvent-free conditions have on the reaction rate of the first step, allowing subsequent transformations in a one-pot fashion.
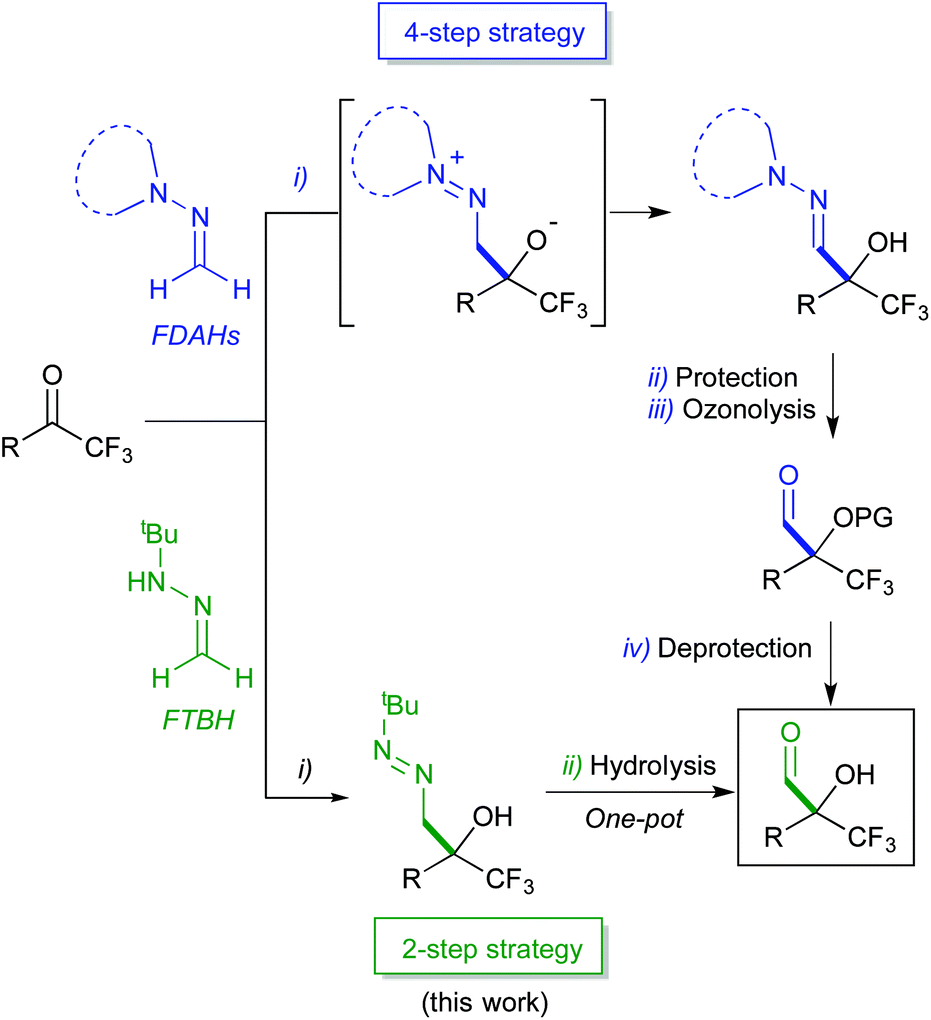 | ||
| Scheme 2 Nucleophilic formylations of trifluoromethyl ketones employing formaldehyde hydrazones as d1 reagents. | ||
Results and discussion
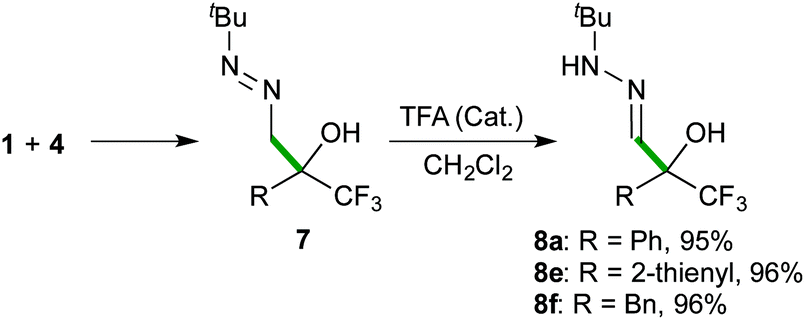
Preliminary experiments were performed with commercially available 2,2,2-trifluoroacetophenone (1a) as the model substrate. For comparative purposes, the reactivity of different simple formaldehyde hydrazones was analysed (Scheme 3). The simplest formaldehyde dimethylhydrazone 2 was very slowly added to the carbonyl compound (employed in a 2-fold excess) to afford the corresponding α-hydroxy α-trifluoromethyl-hydrazone 5a in a modest 55% yield (after 20 days in CH2Cl2 at room temperature) along with small amounts of the hydrazo transfer by-product 6a. It was observed that the reaction rates are highly dependent on the reaction media (32% after 3 days (neat); 10 and 40% yields after 5 days in CH3CN and H2O, respectively). The anisyl-substituted derivative 3 showed no reactivity, while formaldehyde N-tert-butyl hydrazone 4, employed in a 1.5-fold excess, readily added to 1a in CH2Cl2 with complete C-selectivity, affording the desired α-hydroxy α-trifluoromethyl diazene 7a in 83% yield after 9 hours (entry 1, Table 1). Further optimization experiments were conducted in different solvents and the E-factor‡20 was used as a descriptor of the environmental impact. Using CHCl3 instead of CH2Cl2 provided full conversions in shorter reaction times, albeit yielding an 8:2 mixture of azocompound 7a and its tautomeric hydrazone form 8a, presumably induced by acid traces in the reaction media (entry 2). In general, lower reactivities were observed in hydrocarbons and ethereal solvents (entries 3–5), while conducting the reaction in a polar aprotic solvent such as CH3CN had a positive effect, affording 7a in a higher yield (E-factor = 6.68, entry 6) and shorter reaction time (6 hours).
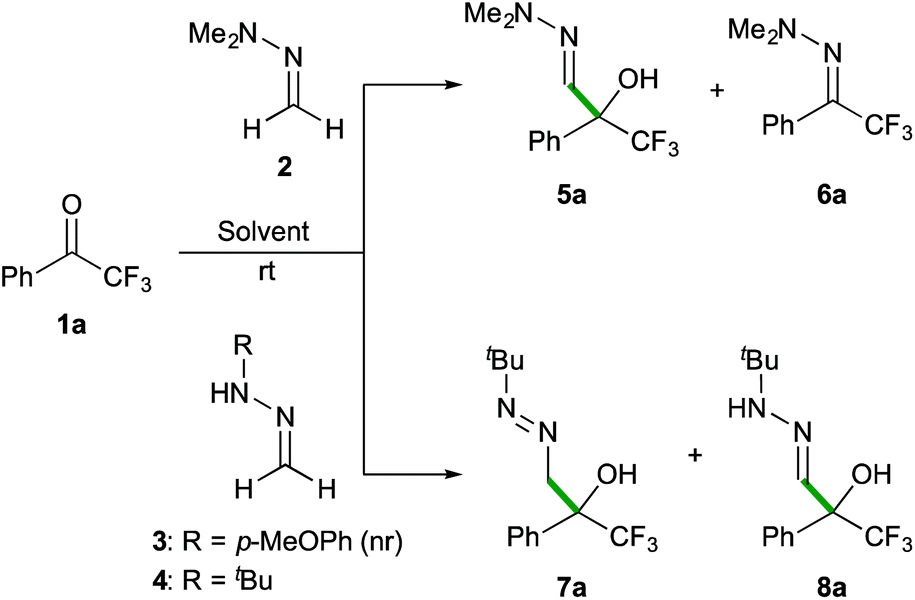 | ||
| Scheme 3 Preliminary reactivity experiments. | ||
a
| Entry | Solvent | T |
7a:8ab |
Yieldc (%) | E-factord (g g−1) |
|---|---|---|---|---|---|
|
a Reactions performed on a 0.5 mmol scale (0.5 M) using 1a (0.5 mmol) and 4 (0.75 mmol).
b Determined by 1H NMR.
c Isolated yield after column chromatography (entries 1–6), after L–L extraction (entry 7), after removing the excess of hydrazone 4 under reduced pressure (entries 8 and 9).
d
E-factor = Waste (g)/7a (g); without considering potential recycling of 4 and solvents.
e Reactions performed on a 6 mmol scale.
f 1:1 1a/4 molar ratio.
|
|||||
| 1 | CH2Cl2 | 9 h | 10:0 |
83 | 12.08 |
| 2 | CHCl3 | 7 h | 8:2 |
70 | 16.31 |
| 3 | n-Hexane | 31 h | 10:0 |
80 | 6.49 |
| 4 | Toluene | 31 h | 10:0 |
81 | 8.27 |
| 5 | Et2O | 31 h | 10:0 |
74 | 7.63 |
| 6 | CH3CN | 6 h | 10:0 |
90 | 6.69 |
| 7 | H2O | 45 min | 10:0 |
90 | 1.31 |
| 8 | — | 20 min | 10:0 |
>99 | 0.18 |
| 9e | — | 10 min | 10:0 |
>99 | 0.18 |
| 10 , | — | 20 min |
10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :0 :0
|
>99 | 0.0004 |
Next, we decided to explore the possibility of performing the reaction “on water”, exploiting the rate acceleration previously observed for FDAHs in reactions with α-keto esters.16e When pure water was used as the reaction medium, full conversion was observed in only 45 minutes, giving 7a in 90% isolated yield after simple L–L extraction with Et2O, with an E-factor of 1.31§ (entry 7). Finally, we were delighted that the reaction carried out in the absence of a solvent¶21,22 proceeded cleanly and at a high rate, reaching completion in only 20 minutes. These conditions efficiently afforded analytically pure 7a in quantitative yield after removing the excess of hydrazone 4 under reduced pressure and without the need for chromatographic purification (E-factor = 0.18, entry 8). Finally, scaling-up from 0.5 to 6 mmol made the reaction proceed even faster, reaching completion in 10 minutes (entry 9). Under these optimal conditions, the reaction was performed with a 1:1 ratio of ketone 1a and reagent 4 to afford 7a without any further elaboration and, therefore, in a very high overall efficiency, quantified by an E-factor close to zero (entry 10).
The scope of the reaction was then explored with a range of trifluoromethyl ketones 1, including aromatic (1a–1d), heteroaromatic (1e), aliphatic derivatives (1f–1i) and the densely functionalized ethyl 3,3,3-trifluoropyruvate (1j), as outlined in Scheme 4. The collected data indicate that the reaction is highly efficient (5–300 minutes of reaction time) for all types of substrates, proceeding at room temperature to afford α-hydroxy α-trifluoromethyl diazenes 7 in quantitative yields (>99%) and high purity (>95% by NMR), without the need for chromatographic purification. The reaction rates correlate with stereoelectronic properties of the substituents, with the more reactive 1d (R = 4-F-C6H4), 1h (R = Me), and 1j (R = CO2Et) reaching completion in less than 10 minutes. The solid ketone 1c, bearing an electron-rich aryl group, appeared as the most challenging substrate but, although requiring extra time for complete solubilisation, also afforded a satisfactory result. The mild and simple reaction conditions (room temperature, no need of oxygen and/or moisture exclusion) offer a practical way to scale-up the production (see pictures in the ESI†), as illustrated by a 10 gram (36 mmol) synthesis of 7a. Moreover, the simplicity of the solvent-free methodology allowed the development of some transformations of diazenes 7 into useful building blocks in a one-pot fashion. For example, applying an acid-catalysed isomerization reaction, α-hydroxy α-trifluoromethyl hydrazones 823 were obtained in high yields (Scheme 5).
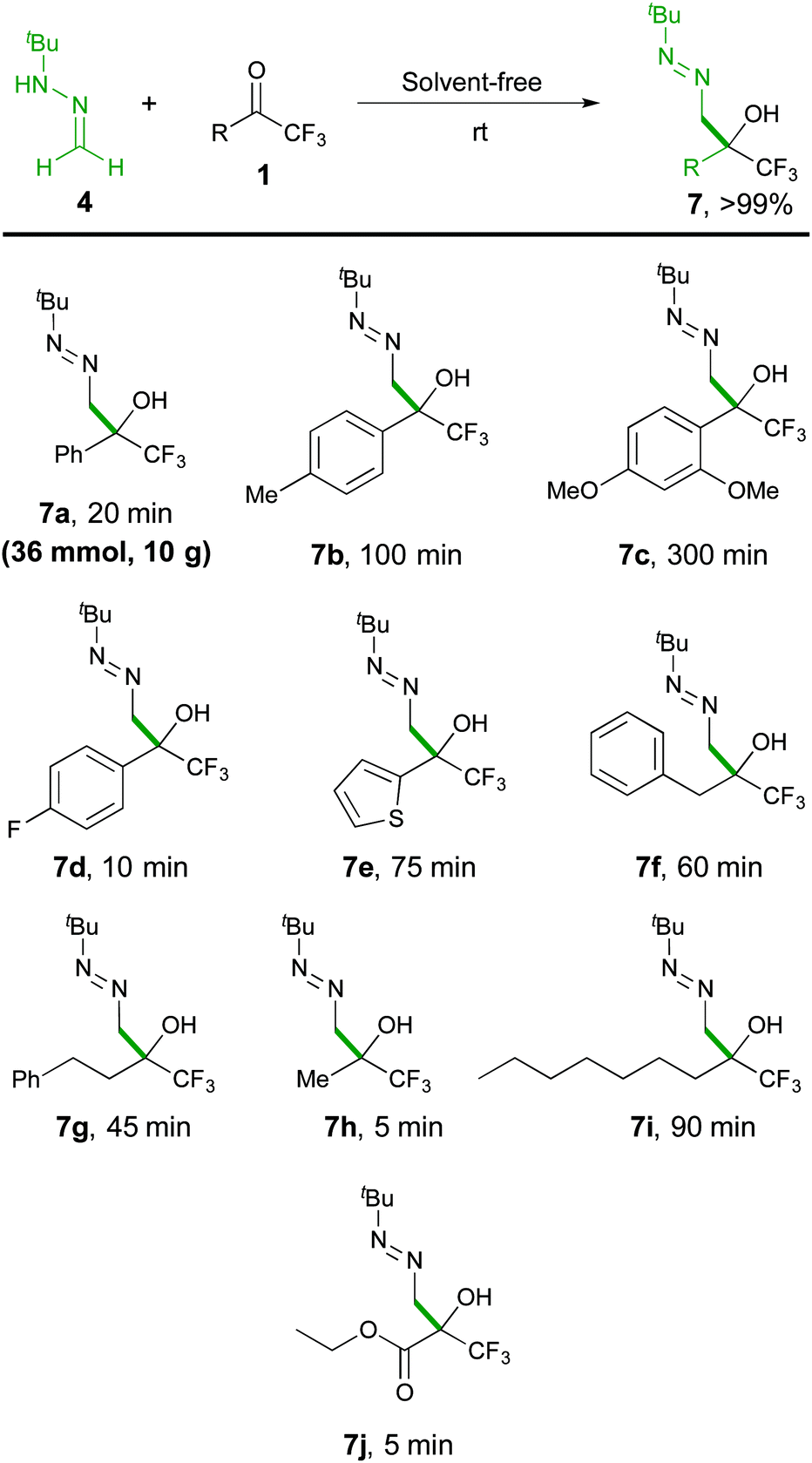 | ||
| Scheme 4 Synthesis of diazenes 7. | ||
 | ||
| Scheme 5 Synthesis of α-hydroxy α-trifluoromethyl hydrazones. | ||
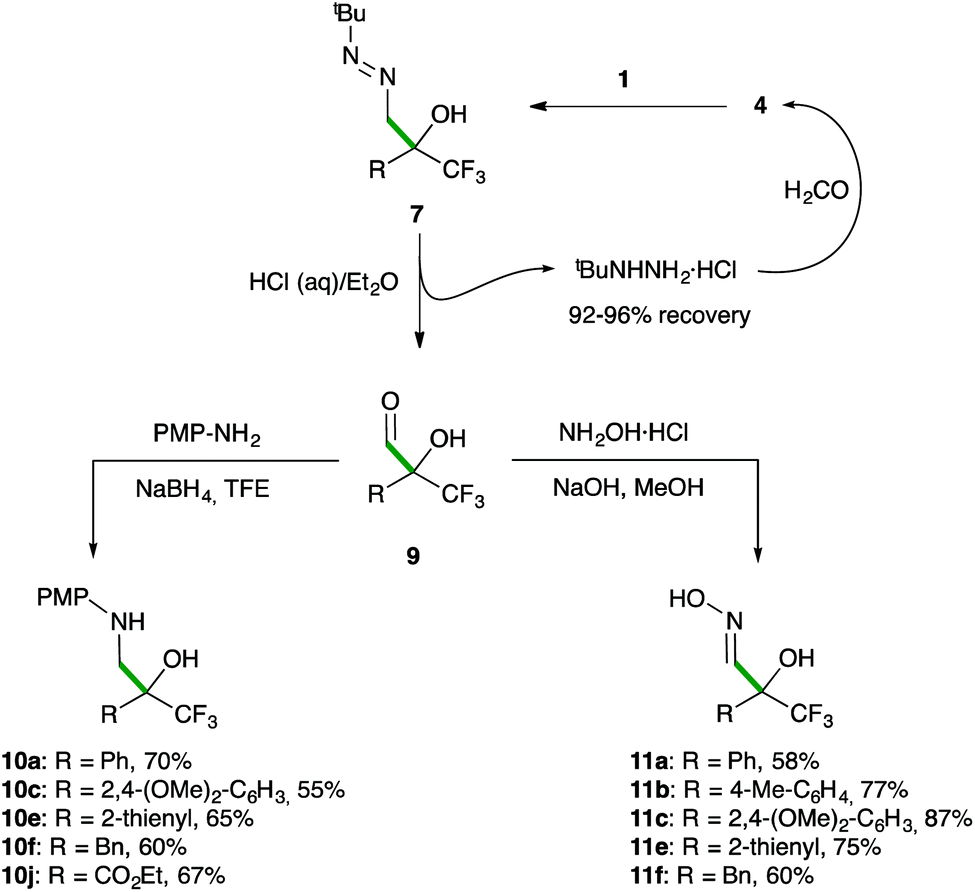
To validate the announced formylation strategy, the subsequent one-pot diazene-to-aldehyde transformation from 7 was easily performed (Scheme 6). Thus, upon completion of the addition step, a simple treatment with HCl in a biphasic H2O/Et2O or H2O/MTBE medium|| afforded the desired α-hydroxy α-trifluoromethyl aldehydes 9 in good yields with a high degree of purity (>95% estimated by 1H NMR, see the ESI†).
 | ||
| Scheme 6 Synthesis of β-aminoalcohols 10 and α-hydroxy aldoximes 11. Overall yield for the three-step sequence. | ||
Remarkably, the tert-butyl hydrazine was recovered (92–96%) as its hydrochloride salt and reused for the synthesis of 4, thus minimizing waste production in the formylation procedure.20c Sensitive aldehydes 9 were directly used in subsequent reductive aminations or condensations with hydroxylamine to yield valuable trifluoromethylated β-aminoalcohols 10 and α-hydroxy aldoximes 11 in satisfactory overall yields for the three-step transformations. To again demonstrate the preparative utility of this methodology, the synthesis of 10a and 11a was performed on an 18 mmol scale without compromising the chemical yield.
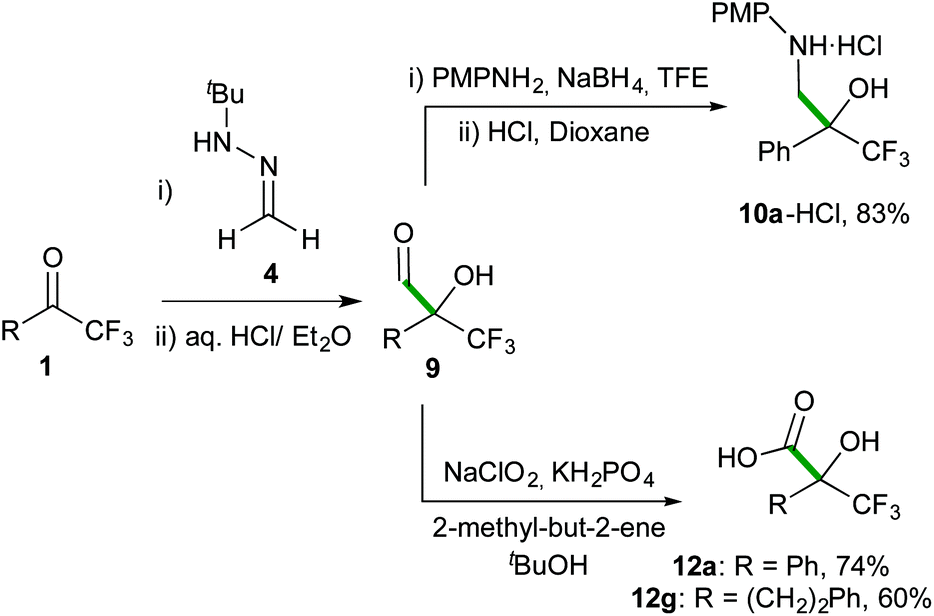
Finally, the efficiency and simplicity of the present methodology are highlighted with 3-step protocols outlined in Scheme 7 for the synthesis of representative trifluoromethylated β-aminoalcohol hydrochloride 10a-HCl and α-hydroxy acids 12a and 12g in good overall yields, without the need for further chromatographic purifications of these products.
 | ||
| Scheme 7 Chromatography-free synthesis of β-aminoalcohol hydrochloride 10a-HCl and α-hydroxy acids 12a and 12g. | ||
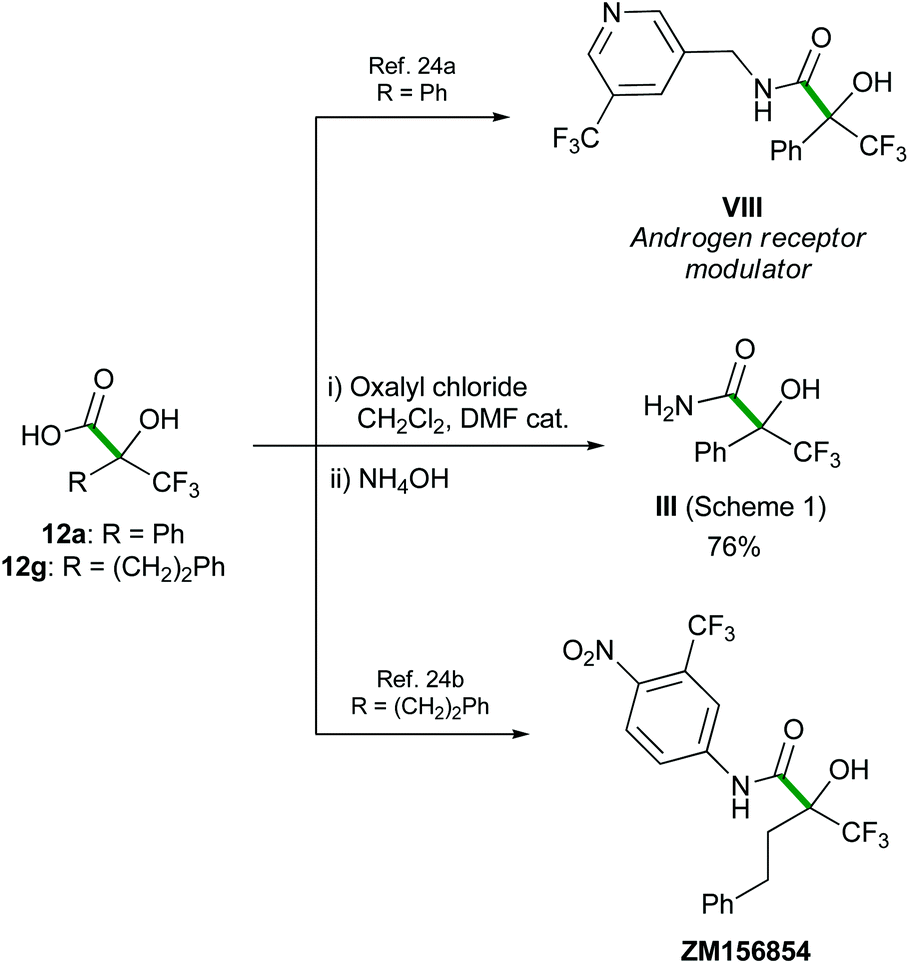
These α-hydroxy α-trifluoromethyl carboxylic acids 12 are valuable building blocks for target oriented synthesis, as illustrated with their transformation into amide III,7a,b and the formal synthesis of several biologically active α-hydroxy α-trifluoromethyl amides (Scheme 8).24
 | ||
| Scheme 8 Synthesis of amide III and the formal synthesis of rac-VIII and ZM156854. | ||
Experimental
Spectra were recorded at 300 or 500 MHz (1H NMR); 75.5 or 125 MHz (13C NMR); and 470.6 MHz (19F NMR) with the solvent peak used as the internal reference (7.26 and 77.0 ppm for 1H and 13C respectively). Column chromatography was performed on silica gel (Merck Kieselgel 60). Analytical TLC was performed on aluminum backed plates (1.5 × 5 cm) pre-coated (0.25 mm) with silica gel (Merck, Silica Gel 60 F254). Compounds were visualized by exposure to UV light or by dipping the plates in solutions of KMnO4, vanillin or phosphomolybdic acid stains followed by heating. Melting points were recorded in a metal block and are uncorrected. Unless otherwise noted, analytical grade solvents and commercially available reagents were used without further purification. Formaldehyde hydrazones 2–425 and not commercially available trifluoromethyl ketones 126 were synthesized according to literature procedures.General procedure for the synthesis of α-hydroxy α-trifluoromethyl diazenes 7
Freshly distilled formaldehyde tert-butylhydrazone 4 (0.75 mL, 6 mmol) was added to trifluoromethyl ketone 1 (6 mmol) at room temperature. The mixture was stirred for the time specified (Scheme 4, TLC monitoring) to afford pure diazene 7.General procedure for the ‘one-pot’ synthesis of α-hydroxy α-trifluoromethyl hydrazones 8
Freshly distilled formaldehyde tert-butylhydrazone 4 (0.13 mL, 1 mmol) was added to trifluoromethyl ketone 1 (1 mmol) at room temperature. The mixture was stirred for the time specified (Scheme 4, TLC monitoring) to afford pure diazene 7. Subsequently, a solution of TFA (0.1 mmol) in CH2Cl2 (10 mL) was added to a solution of diazene 7 (1 mmol) in CH2Cl2 (0.5 mL) at 0 °C. The mixture was allowed to warm to room temperature and stirred for 12 h. A saturated NaHCO3 solution (15 mL) was added and the organic phase was separated. The remaining aqueous phase was extracted with Et2O (3 × 10 mL), and the combined organic layer was dried over anhydrous MgSO4, filtered and concentrated to afford pure α-hydroxy hydrazone 8.General procedure for the one-pot synthesis of α-hydroxy α-trifluoromethyl aldehydes 9
Freshly distilled formaldehyde tert-butylhydrazone 4 (0.75 mL, 6 mmol) was added to trifluoromethyl ketone 1 (6 mmol) at room temperature. The mixture was stirred for the time specified (Scheme 4, TLC monitoring) to afford pure diazene 7. Subsequently, aq. HCl (30 mL, 6 M) was added to a solution of diazene 7 (6 mmol) in Et2O (55 mL) at 0 °C. The mixture was allowed to warm to room temperature and stirred until consumption of the starting material (TLC monitoring, approx. 6 h). The organic phase was separated and the aqueous phase was extracted with Et2O (2 × 30 mL). The combined organic layers were dried over anhydrous MgSO4, filtered and the solvent was removed under reduced pressure (35 mmHg, 5 °C) to afford α-hydroxy aldehyde 9 (purity > 95% estimated by 1H-NMR). This material was used in subsequent transformations without further purification. 9c was synthesized employing MTBE as the solvent, which was removed under reduced pressure (15 mmHg, 15 °C).General procedure for the synthesis of β-aminoalcohols 10
p-Methoxyphenylaniline (0.73 g, 6 mmol) was added to a solution of crude aldehyde 9 (6 mmol) in TFE (15 mL). The mixture was stirred at 30 °C for 20 minutes. NaBH4 (0.28 g, 7.2 mmol) was then added and the reaction was stirred vigorously until hydrogen evolution ceased (approx. 30 min). The mixture was filtered through a Celite pad, concentrated and the residue was purified by flash chromatography (pentane/CH2Cl2) to afford products 10.General procedure for the synthesis of α-hydroxy aldoximes 11
Hydroxylamine hydrochloride (0.50 g, 7.2 mmol) and sodium hydroxide (0.29 g, 7.2 mmol) were sequentially added to a solution of crude aldehyde 9 (6 mmol) in MeOH (45 mL). The mixture was stirred at room temperature overnight. The mixture was then diluted with water (15 mL) and the organic phase was extracted with CH2Cl2 (2 × 30 mL) and Et2O (2 × 30 mL). The combined organic layer was dried (MgSO4), filtered and concentrated. The product was purified by flash chromatography (3:1 cyclohexane/AcOEt) to afford α-hydroxy aldoximes 11.
Synthesis of β-aminoalcohol hydrochloride 10a-HCl
p-Methoxyphenylaniline (0.37 g, 3 mmol) was added to a solution of crude aldehyde 9a (3 mmol) in TFE (7.5 mL). The mixture was stirred at 30 °C for 20 minutes. After this time, NaBH4 (0.14 g, 3.6 mmol) was added and the reaction was stirred vigorously until the end of hydrogen evolution (approximately 30 minutes). The solvent was removed under reduced pressure and the crude was dissolved in CH2Cl2 (5 mL). The mixture was filtered through silica and Celite pad (height: 1 cm), and washed with a mixture of pentane/CH2Cl2 (2/1, 10 mL). Solvents were removed under reduced pressure and the product was dissolved in dry Et2O (15 mL). HCl (1 M in dioxane, 3.8 mL) was added and the mixture was stirred at room temperature until the appearance of a white solid (approximately 1 h). The solid was filtered and washed with Et2O (2 mL) to afford the pure amine hydrochloride 10a-HCl (0.72 g, 83%). 1H NMR (300 MHz, DMSO-d6): δ 7.69–7.60 (m, 2H), 7.44–7.33 (m, 3H), 7.02 (d, 2H, J = 8.8 Hz), 6.81 (d, 2H, J = 8.8 Hz), 6.70 (br s, 2H), 4.01 (d, 1H, J = 13.4 Hz), 3.77 (d, 1H, J = 13.4 Hz), 3.68 (s, 3H). 13C NMR (75.5 MHz, CDCl3): δ 156.9, 134.9, 133.4, 128.9, 128.2, 127.1, 125.0 (q, JC,F = 285.3 Hz), 121.8, 114.5, 75.2 (q, JC,F = 27.8 Hz), 55.5, 53.0. 19F NMR (470.6 MHz, CDCl3): δ −77.07 (s, CF3). HRMS (ESI): m/z calcd for C16H17F3NO2+ [M+] 312.1206, found 312.1198.General procedure for the synthesis of α-hydroxy acids 12
A solution of NaClO2 (20 mmol) and KH2PO4 (18 mmol) in H2O (70 mL) was added dropwise to a solution of crude aldehyde 9 (6 mmol) in tBuOH (70 mL) and 2-methyl-but-2-ene (60 mL) at 0 °C. The mixture was allowed to warm to room temperature and stirred for 16 h. The solvents were removed under reduced pressure and the residue was treated with 2 M NaOH and extracted with Et2O. The aqueous layer was acidified to pH 1 (2 M HCl) and extracted with EtOAc. The combined organic layer was dried (MgSO4), filtered and the solvent was removed under reduced pressure to afford pure α-hydroxy acid.Synthesis of α-hydroxy α- trifluoromethyl amide III
Oxalyl chloride (0.2 mL, 2.16 mmol) and a drop of DMF were subsequently added to a stirred solution of the acid 12a (264 mg, 1.2 mmol) in dichloromethane (10 mL) at 0 °C. The reaction mixture was stirred for 3 h, slowly warming to rt, then NH3 (28% in water, 40 mL) was added. The resulting solution was stirred at rt for 6 h, diluted with water (40 mL), and extracted with ethyl acetate (3 × 100 mL). The combined organic phase was washed with brine (100 mL), dried over MgSO4, filtered and concentrated under reduced pressure. The product was purified by flash chromatography (1:1 cyclohexane/AcOEt) to afford α-hydroxy amide III as a white solid (0.2 g, 76%). Characterization data are in agreement with those reported in the literature.27
Conclusions
In summary, the high diaza–ene reactivity of formaldehyde tert-butyl hydrazone (FTBH) with trifluoromethyl ketones under solvent-free conditions afforded analytically pure α-hydroxy α-trifluoromethyl diazenes in a quantitative way. From these products, operationally simple diazene-to-aldehyde transformations and subsequent derivatizations in a ‘one-pot’ fashion provide a simple and environmentally friendly entry to trifluoromethylated β-aminoalcohols, α-hydroxy aldoximes and α-hydroxy acids.Acknowledgements
This work was supported by the Ministerio de Economía y Competitividad of Spain (CTQ2013-48164-C2-1-P, CTQ2013-48164-C2-2-P, and predoctoral fellowship to E. M.), European FEDER funds and the Junta de Andalucía (Grant 2012/FQM 1078). D. M. acknowledges Universidad de Sevilla for a postdoctoral contract.Notes and references
- A. M. Thayer, Chem. Eng. News, 2006, 84, 15 Search PubMed.
- (a) T. Hiyama, in Organofluorine Compounds: Chemistry Applications, Springer, New York, 2000 Search PubMed; (b) P. Kirsch, in Modern Fluoroorganic Chemistry, Wiley-VCH, Weinheim, 2004 Search PubMed; (c) R. D. Chambers, in Fluorine in Organic Chemistry, Blackwell, Oxford, 2004 Search PubMed; (d) T. Yamazaki, T. Taguchi and I. Ojima, in Fluorine in Medicinal Chemistry and Chemical Biology, ed. I. Ojima, Wiley-Blackwell, Chichester, 2009 Search PubMed; (e) V. A. Petrov, Fluorinated Heterocyclic Compounds: Synthesis, Chemistry, and Applications, Wiley, Hoboken, New Jersey, 2009 Search PubMed; (f) C. Isanbor and D. ÓHagan, J. Fluorine Chem., 2006, 127, 303 CrossRef CAS; (g) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881 CrossRef PubMed; (h) M. Hird, Chem. Soc. Rev., 2007, 36, 2070 RSC; (i) D. ÓHagan, Chem. Soc. Rev., 2008, 37, 308 RSC; (j) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC; (k) W. K. Hagmann, J. Med. Chem., 2008, 51, 4359 CrossRef CAS PubMed; (l) D. O'Hagan, J. Fluorine Chem., 2010, 131, 1071 CrossRef.
- (a) J.-A. Ma and D. Cahard, Chem. Rev., 2008, 108, PR1 CrossRef CAS PubMed and references cited therein; (b) G. Valero, X. Company and R. Rios, Chem. – Eur. J., 2011, 17, 2018 CrossRef CAS PubMed; (c) T. Furuya, A. S. Kamlet and T. Ritter, Nature, 2011, 473, 470 CrossRef CAS PubMed.
- (a) J. Nie, H.-C. Guo, D. Cahard and J.-A. Ma, Chem. Rev., 2011, 111, 455 CrossRef CAS PubMed; (b) O. A. Tomashenko and V. V. Grushin, Chem. Rev., 2011, 111, 4475 CrossRef CAS PubMed and references therein. (c) H. Kawai and N. Shibata, Chem. Rec., 2014, 14, 1024 CrossRef CAS PubMed.
- Cholesteryl ester transfer protein inhibitor I: M. A. Massa, D. P. Spangler, R. C. Durley, B. S. Hickory, D. T. Connolly, B. J. Witherbee, M. E. Smith and J. A. Sikorski, Bioorg. Med. Chem. Lett., 2001, 11, 1625 CrossRef CAS PubMed.
- Non-steroidal selective glucocorticoid receptor agonist II: H. A. Barnett, D. M. Coe, T. W. J. Cooper, T. I. Jack, H. T. Jones, S. J. f. Macdonald, I. M. McLay, N. Rayner, R. Z. Sasse, T. J. Shipley, P. A. Skone, G. I. Somers, S. Taylor, I. J. Uings, J. M. Woolven and G. G. Weingarten, Bioorg. Med. Chem. Lett., 2009, 19, 158 CrossRef CAS PubMed.
- Anesthetic and anti-convulsant III: (a) I. Choudhury-Mukherjee, H. A. Schenck, S. Cechova, T. N. Pajewski, J. Kapur, J. Ellena, D. S. Cafiso and M. L. Brown, J. Med. Chem., 2003, 46, 2494 CrossRef CAS PubMed; (b) H. A. Schenck, P. W. Lenkowski, I. Choudhury-Mukherjee, S. –H. Ko, J. P. Stables, M. K. Patel and M. L. Brown, Bioorg. Med. Chem., 2004, 12, 979 CrossRef CAS PubMed. Pyruvate dehydrogenase kinase inhibitor IV: (c) T. D. Aicher, R. C. Anderson, G. R. Bebernitz, G. M. Coppola, C. F. Jewell, D. C. Knorr, C. Liu, D. M. Sperbeck, L. J. Brand, R. J. Strohschein, J. Gao, C. C. Vinluan, S. S. Shetty, C. Dragland, E. L. Kaplan, D. DelGrande, A. Islam, X. Liu, R. J. Lozito, W. M. Maniara, R. E. Walterand and W. R. Mann, J. Med. Chem., 1999, 42, 2741 CrossRef CAS PubMed.
- (a) J. W. Corbett, S. S. Ko, J. D. Rodgers, L. A. Gearhart, N. A. Magnus, L. T. Bacheler, S. Diamond, S. Jeffrey, R. M. Klabe, B. C. Cordova, S. Garber, L. Logue, G. L. Trainor, P. S. Anderson and S. K. Erickson-Viitanen, J. Med. Chem., 2000, 43, 2019 CrossRef CAS PubMed; (b) M. E. Pierce, L. P. Rodney, L. A. Radesca, Y. S. Lo, S. Silverman, J. R. Moore, Q. Islam, A. Choudhury, J. M. D. Fortunak, D. Nguyen, C. Luo, S. J. Morgan, W. P. Davis, P. N. Confalone, C.-Y. Chen, R. D. Tillyer, L. Frey, L. Tan, F. Xu, D. Zhao, A. S. Thompson, E. G. Corley, E. J. J. Grabowski, R. Reamer and P. J. Reider, J. Org. Chem., 1998, 63, 8536 CrossRef CAS; (c) E. J. J. Grabowski, Chirality, 2005, 17, S249 CrossRef CAS PubMed; (d) M. M. Bastos, C. C. P. Costa, T. C. Bezerra, F. C. Da Silva and N. Boechat, Eur. J. Med. Chem., 2016, 108, 455 CrossRef CAS PubMed.
- M. Sani, D. Belotti, R. Giavazzi, W. Panzeri, A. Volonterio and M. Zanda, Tetrahedron Lett., 2004, 45, 1611 CrossRef CAS.
- S. Caron, N. M. Do, J. E. Sieser, P. Arpin and E. Vazquez, Org. Process Res. Dev., 2007, 11, 1015 CrossRef CAS.
- Recent review: (a) G. Rubiales, C. Alonso, E. Martínez de Marigorta and F. Palacios, ARKIVOC, 2014, ii, 362 Search PubMed. Selected examples: (b) J. J. Song, Z. Tan, J. T. Reeves, F. Gallou, N. K. Yee and C. H. Senanayake, Org. Lett., 2005, 7, 2193 CrossRef CAS PubMed; (c) G. K. S. Prakash, Z. Zhang, F. Wang, S. Munoz and G. A. Olah, J. Org. Chem., 2013, 78, 3300 CrossRef CAS PubMed.
- Selected examples: (a) R. Motoki, M. Kanai and M. Shibasaki, Org. Lett., 2007, 9, 5079 CrossRef PubMed; (b) F. Tur and J. M. Saá, Org. Lett., 2007, 9, 5079 CrossRef CAS PubMed; (c) G. Blay, I. Fernández, A. Monleón, José R. Pedro and C. Vila, Org. Lett., 2009, 11, 441 CrossRef CAS PubMed; (d) C. Palacio and S. J. Connon, Org. Lett., 2011, 13, 1298 CrossRef CAS PubMed; (e) N. Duangdee, W. Harnying, G. Rulli, J.–M. Neudörfl, H. Gröger and A. Berkessel, J. Am. Chem. Soc., 2012, 134, 11196 CrossRef CAS PubMed.
- M. B. Smith, in March, Jerry, Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, Wiley-Interscience, New York, 6th edn, 2007 Search PubMed.
- Some examples: (a) P. Bravo, M. Frigerio and G. Resnati, J. Org. Chem., 1990, 55, 4216 CrossRef CAS; (b) A. Dondoni, A. Boscarato, P. Formaglio, J.-P. Bégué and F. Benayoud, Synthesis, 1995, 654 CrossRef CAS.
- Reviews: (a) R. Fernańdez and J. M. Lassaletta, Synlett, 2000, 1228 Search PubMed; (b) R. Brehme, D. Enders, R. Fernańdez and J. M. Lassaletta, Eur. J. Org. Chem., 2007, 5629 CrossRef CAS.
- Diastereoselective 1,2 additions to trifluoromethyl ketones: (a) R. Fernández, E. Martín-Zamora, C. Pareja, J. Vázquez, E. Díez, A. Monge and J. M. Lassaletta, Angew. Chem., Int. Ed., 1998, 37, 3428 CrossRef; (b) C. Pareja, E. Martín-Zamora, R. Fernández and J. M. Lassaletta, J. Org. Chem., 1999, 64, 8846 CrossRef CAS PubMed. α-Alkoxy-(amino)aldehydes: (c) R. Fernández, E. Martín-Zamora, C. Pareja and J. M. Lassaletta, J. Org. Chem., 2001, 66, 5201 CrossRef. Simple aldehydes: (d) R. Fernández, E. Martín-Zamora, C. Pareja, M. Alcarazo, J. Martín and J. M. Lassaletta, Synlett, 2001, 1158 CrossRef. Racemic 1,2-additions to α-keto esters: (e) A. Crespo-Peña, E. Martín-Zamora, R. Fernández and J. M. Lassaletta, Chem. – Asian J., 2011, 6, 2287 CrossRef PubMed. Enantioselective 1,4-additions to α-hydroxyenones: (f) D. Monge, E. Martín-Zamora, J. Vaźquez, M. Alcarazo, E. Álvarez, R. Fernańdez and J. M. Lassaletta, Org. Lett., 2007, 9, 2867 CrossRef CAS PubMed; (g) R. P. Herrera, D. Monge, E. Martín-Zamora, R. Fernańdez and J. M. Lassaletta, Org. Lett., 2007, 9, 3303 CrossRef CAS PubMed.
- A. Crespo-Peña, D. Monge, E. Martín-Zamora, E. Álvarez, R. Fernández and J. M. Lassaletta, J. Am. Chem. Soc., 2012, 134, 12912 CrossRef PubMed.
- D. Monge, A. Crespo-Peña, E. Martín-Zamora, E. Álvarez, R. Fernández and J. M. Lassaletta, Chem. – Eur. J., 2013, 19, 8421 CrossRef CAS PubMed.
- I. Serrano, D. Monge, E. Álvarez, R. Fernández and J. M. Lassalettta, Chem. Commun., 2015, 51, 4077 RSC.
- (a) R. A. Sheldon, Pure Appl. Chem., 2000, 72, 1233 CrossRef CAS; (b) A. D. Curzons, D. J. C. Constable, D. N. Mortimer and V. L. Cunningham, Green Chem., 2001, 3, 1 RSC; (c) R. A. Sheldon, Green Chem., 2007, 9, 1273 RSC; (d) R. A. Sheldon, Green Chem., 2012, 41, 1437 CAS.
- (a) K. Tanaka and F. Toda, Chem. Rev., 2000, 100, 1025 CrossRef CAS PubMed; (b) K. Tanaka, in Solvent-Free Organic Synthesis, Wiley-VCH, Weinheim, 2003 Search PubMed.
- A. Loupy, Top. Curr. Chem., 1999, 206, 153 CrossRef.
- For an example of the C-coupling of aryl bromides with N-tert-butyl hydrazones, see: A. Takemiya and J. F. Hartwig, J. Am. Chem. Soc., 2006, 128, 14800 CrossRef CAS PubMed.
- (a) Y. Kim, R. S. Meissner, H. J. Mitchell, J. J. Perkins, M. A. Rossi and J. Wang, US2009/0275515A1 Search PubMed; (b) P. Wang, L.-W. Feng, L. Wang, J.-F. Li, S. Liao and Y. Tang, J. Am. Chem. Soc., 2015, 137, 4626 CrossRef CAS PubMed.
- (a) Y. Kamitori, M. Hojo, R. Masuda, T. Yoshida, S. Ohara, K. Yamada and N. Yoshikawa, J. Org. Chem., 1988, 53, 129 CrossRef CAS; (b) J.-S. M. Lehn, S. Javed and D. M. Hoffman, Inorg. Chem., 2007, 46, 993 CrossRef CAS PubMed.
- Trifluoromethyl ketones 1 were prepared by Grignard reagent addition to 2,2,2-ethyltrifluoroacetate: (a) For characterization of trifluoromethyl ketone 1g see: K. Fuchibe, H. Jyono, M. Fujiwara, T. Kudo, M. Yokota and J. Ichiwaka, Chem. – Eur. J., 2011, 17, 12175 CrossRef CAS PubMed . For characterization of trifluoromethyl ketone 1i see: E. Massolo, M. Benaglia, M. Orlandini, S. Rossi and G. Celentano, Chem. – Eur. J., 2015, 21, 3589 CrossRef PubMed.
- H. A. Schenck, P. W. Leukowski, I. Choudhury-Mukherjee, S.-H. Ko, J. P. Stables, M. K. Patel and M. L. Brown, Bioorg. Med. Chem., 2014, 12, 979 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, characterization data and copies of NMR spectra. See DOI: 10.1039/c6gc00408c |
| ‡ The E-factor is defined as the mass ratio of the waste to the desired product. For E-factors including solvents after chromatographic purifications or L–L extractions see the ESI.† |
| § Water amounts are normally not included in the E-factor; even though additional amounts of organic solvents are required in the subsequent L–L extractions. |
| ¶ Solvent-free methodologies (ref. 21) are among the most promising strategies towards waste prevention and environmental protection, which also lead to milder conditions, very high volumetric productivity, increased safety and cost reduction (ref. 22). |
| || For products 9 of relatively low volatility, Et2O can be replaced by less hazardous MTBE, as illustrated for 9c (see crude 1H-NMR in the ESI†). |
| This journal is © The Royal Society of Chemistry 2016 |