
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Highly efficient CO2 capture with simultaneous iron and CaO recycling for the iron and steel industry†
Sicong
Tian
a,
Jianguo
Jiang
*abc,
Feng
Yan
a,
Kaimin
Li
a,
Xuejing
Chen
a and
Vasilije
Manovic
*d
aSchool of Environment, Tsinghua University, Beijing 100084, P. R. China. E-mail: jianguoj@tsinghua.edu.cn
bKey Laboratory for Solid Waste Management and Environment Safety, Ministry of Education of China, P. R. China
cCollaborative Innovation Center for Regional Environmental Quality, Tsinghua University, Beijing, P. R. China
dCombustion and CCS Centre, Cranfield University, Bedford, Bedfordshire, MK43 0AL, UK. E-mail: v.manovic@cranfield.ac.uk
First published on 25th April 2016
Abstract
An efficient CO2 capture process has been developed by integrating calcium looping (CaL) and waste recycling technologies into iron and steel production. A key advantage of such a process is that CO2 capture is accompanied by simultaneous iron and CaO recycling from waste steel slag. High-purity CaO-based CO2 sorbents, with CaO content as high as 90 wt%, were prepared easily via acid extraction of steel slag using acetic acid. The steel slag-derived CO2 sorbents exhibited better CO2 reactivity and slower (linear) deactivation than commercial CaO during calcium looping cycles. Importantly, the recycling efficiency of iron from steel slag with an acid extraction is improved significantly due to a simultaneous increase in the recovery of iron-rich materials and the iron content of the materials recovered. High-quality iron ore with iron content of 55.1–70.6% has been recovered from waste slag in this study. Although costing nearly six times as much as naturally derived CaO in the purchase of feedstock, the final cost of the steel slag-derived, CaO-based sorbent developed is compensated by the byproducts recovered, i.e., high-purity CaO, high-quality iron ore, and acetone. This could reduce the cost of the steel slag-derived CO2 sorbent to 57.7 € t−1, appreciably lower than that of the naturally derived CaO. The proposed integrated CO2 capture process using steel slag-derived, CaO-based CO2 sorbents developed appears to be cost-effective and promising for CO2 abatement from the iron and steel industry.
1. Introduction
CO2 emissions from industry are increasing, having reached 13.14 Gt in 2010 and making up 40% of the total anthropogenic CO2 emissions worldwide.1,2 In addition, industrial energy consumption worldwide is expected to grow from 211 EJ in 2010 to 324 EJ in 2040 due to global industrialization, mainly in Asia, Africa, South America, and the Arab countries.3 This will result in an increase in total industrial CO2 emissions of 74–91% by 2050 compared to 2007.4 However, to limit the increase in the global average temperature to 2 °C, direct CO2 emissions from industry must be 24% lower by 2050 than those in 2007.4,5 Therefore, large energy-intensive industries manufacturing iron and steel, cement, and petrochemicals, which account for more than half of total industrial CO2 emissions, should be the first targets to be completely decarbonized by 2050.Implementation of CO2 capture technologies in existing industrial sectors is urgently required, as carbon capture and storage (CCS) is the only current technology that would allow industrial sectors to realize a deep reduction in CO2 emissions.6 The development of CCS in various industrial sectors is springing up worldwide, leaving much room for deploying applicable technologies for industrial CO2 capture.7 As a promising alternative to conventional amine scrubbing and oxy-fuel combustion CO2 capture technologies, calcium looping (CaL), which relies on CaO as a regenerable CO2 sorbent upon cyclic carbonation and calcination reactions,8 has been widely investigated recently through bench- and pilot-scale demonstrations at sizes of 1 kWth–1.7 MWth.9–12 A recent techno-economic analysis of post-combustion CO2 capture technologies found that CaL is more energy-efficient and cost-effective than other emerging CO2 capture technologies, including chilled ammonia absorption, alkali–metal carbonate adsorption, and membrane separation;13 a further reduction in the CO2 capture cost can be achieved by integrating CaL with thermal energy storage.14
However, the dramatic decay in the cyclic CO2 capture performance of naturally occurring precursors-derived CaO, triggered mainly by sintering of CaCO3 during the CaL-based CO2 capture process, remains as an obstacle for the development and implementation of this technology. To overcome this limitation, various strategies have been investigated to date. Several measures, including thermal activation,15–17 steam hydration,18–21 and recarbonation,22 have been demonstrated to be effective in stabilizing the CO2 capture performance of naturally occurring sorbents. Another approach involves developing synthetic CaO-based CO2 sorbents with thermally stable surface areas and pore volumes, thereby minimizing performance deterioration for CO2 capture. This can be achieved by employing organic-carbon templates23–25 during sorbent preparation and introducing refractory supports26–28 into the CaO matrix. Broda and colleagues29 developed a highly effective MgO-stabilized, CaO-based CO2 sorbent via recrystallization of calcium and magnesium acetates in organic solvents. The as-prepared material exhibited an excellent CO2 uptake of 10.71 mmolCO2 gsorbent−1 after 10 harsh CaL cycles, with only 8 wt% of MgO required. However, the significantly increased cost for sorbent preparation due to the employment of organic acids should be considered before practical application.30,31
The iron and steel industry is the largest energy-consuming manufacturing industry in the world.32 Currently, more than 2.5 Gt of CO2 is emitted annually from the global iron and steel industry without implementation of any effective CO2 reduction measures.33 In addition to an urgent demand for CO2 abatement in the iron and steel industry, valorization of steel slag, a Ca-rich and Fe-containing industrial waste, is another challenge faced in iron and steel production. Global annual generation of steel slag is almost 200 Mt a−1, half of which is contributed by China.34 However, only 20% of the steel slag produced annually in China is utilized, and the other 80% is stockpiled in the open air.
The CaL-based CO2 capture process can be operated readily in a dual circulating fluidized bed system with regard to practical applications.10 Importantly, limestone is an essential feedstock for many energy-intensive manufacturing industries (e.g., the iron-and-steel and cement industries),32,35 making it feasible for integration into industrial processes for CO2 abatement. The potential synergy among power generation, cement production, and CO2 abatement in the cement industry has been investigated by integrating CaL into cement manufacturing processes, where spent CaO from the CaL cycle is reused directly as a “carbon-free” feedstock for cement production, resulting in almost zero sorbent cost and waste production for CO2 capture from the cement industry.35–38 However, no research on the integration of CaL into the iron and steel industry for CO2 abatement has been reported to date. Therefore, we have developed an efficient CO2 capture process (Fig. 1) by integrating CaL and waste recycling technologies into iron and steel production to help realize simultaneous CO2 abatement and steel slag minimization in the iron and steel industry. In this system, Ca and Fe in the steel slag are separated via the leaching of steel slag using acetic acid solution (acid extraction), with Ca extracted into the leachate and Fe concentrated in the residues. The Fe-rich materials can be recycled directly into the blast furnace, substituting for iron ore as the feedstock for iron production after magnetic separation. The CaO-based sorbent, derived from the precipitation of ions in the Ca-rich leachate and a subsequent dry-distillation of the resulting precipitate, is delivered to the CaL unit for CO2 capture. The spent sorbent can also be recycled into the blast furnace as a substitute for natural limestone to remove impurities in the iron ore during iron production. Such an integrated CO2 capture process is demonstrated to be highly efficient and cost-effective in this study, which is promising for application to CO2 abatement in the iron and steel industry.
 | ||
| Fig. 1 General schematic of integrated CO2 capture and steel slag valorization process proposed for use in iron and steel industry. | ||
2. Experimental section
2.1 Material preparation
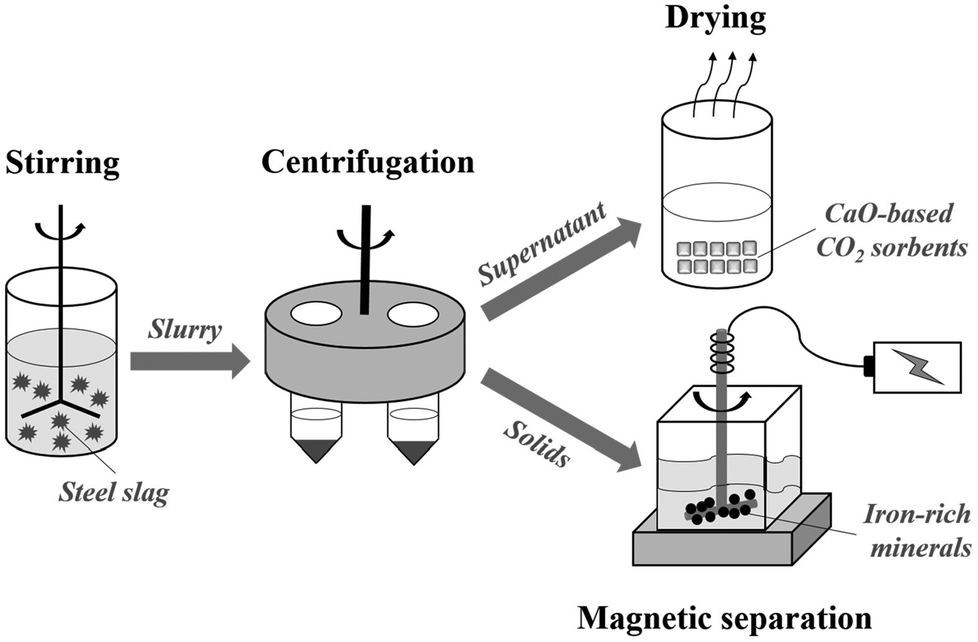 | ||
| Scheme 1 Sketch of operation route for preparing CaO-based CO2 sorbents and recovering iron-rich minerals from steel slag. | ||
| Material | Acid concentration [M] | Extraction time [h] | Solid/liquid ratio [g![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :mL] :mL] |
|---|---|---|---|
| 1 M–0.5 h–1:10 |
1 | 0.5 | 1:10 |
| 1 M–2 h–1:10 |
1 | 2 | 1:10 |
| 2 M–0.5 h–1:5 |
2 | 0.5 | 1:5 |
| 2 M–2 h–1:5 |
2 | 2 | 1:5 |
| 3 M–2 h–1:10 |
3 | 2 | 1:10 |
| 5 M–2 h–1:10 |
5 | 2 | 1:10 |
2.2 Material characterization
Concentrations of Ca, Mg, Fe, Mn, Al, and Si in the steel slag leachate were detected using an inductively coupled plasma-atomic emission spectrometer (ICP-AES, iCAP7400, Thermo Scientific, USA). The iron content in the magnetically separated materials was determined using an X-ray Fluorescence (XRF) analyzer (Niton XL2, Thermo Scientific, USA). Powder X-ray diffraction (XRD) was performed to determine the phase composition of the materials synthesized using an X-ray diffractometer (Smartlab, Rigaku, Japan) with the operating parameters of Cu Kα radiation (λ = 1.5418 Å) and a 40 kV/150 mA power generator. An angular range of 10–90° 2θ was measured, with a step size of 0.02° and counting time of 2 s per step. The morphology of the materials synthesized was observed using a Merlin scanning electron microscope (SEM, Zeiss, Germany), all samples were sputter-coated with an approximately 5 nm-thick layer of platinum before observation.2.3 CO2 sorbent test
The nitrogen temperature-programmed decomposition (N2-TPD) experiments were performed in a thermo-gravimetric analyzer (TGA, TGA/DSC 2, Mettler Toledo). A small amount of material (∼25 mg) was placed in a 150 μL alumina pan and heated to 900 °C at a rate of 10 °C min−1 under a N2 flow of 50 mL min−1. A mass spectrograph (MS, ThermoStar, Pfeiffer Vacuum, Germany) was connected to the TGA to simultaneously monitor the change in gas compositions in the reaction atmosphere.The cyclic CO2 capture experiments were performed in the TGA with a precision of 0.001 mg. During each run, a small amount (∼25 mg) of sorbent was placed in a 150 μL alumina pan and heated to 900 °C at a rate of 20 °C min−1 under a N2 flow of 50 mL min−1. The temperature was held at 900 °C to calcine the sorbent for 10 min. Subsequently, the temperature was decreased to 650 °C at a rate of 50 °C min−1. Once the reaction temperature was reached, a gas flow of 75 mL min−1 containing 20 vol% CO2 and 80 vol% N2 was introduced into the reaction chamber to carbonate the sample for 10 min. Then, the reaction atmosphere was switched to a gas flow of 75 mL min−1 containing 80 vol% CO2 and 20 vol% N2, and the sample was heated to 900 °C at a rate of 50 °C min−1. After calcination at 900 °C for 5 min, a new CaL cycle was started by decreasing the reaction temperature to 650 °C under a N2 flow of 75 mL min−1. The carbonation–calcination cycle was repeated 20 times for each sorbent. The cyclic uptake of CO2, expressed in gCO2 gsorbent−1, was calculated from the continuously monitored weight change of the sample. Blank runs were performed to correct for the effects of buoyancy and change in gas density between different reaction steps.
CO2 capture characteristics of the steel slag-derived, CaO-based CO2 sorbents were also investigated by carbonating the material at 650 °C under a 20 vol% CO2/80 vol% N2 atmosphere for 120 min using TGA/DSC 2, after a pre-calcination of the sorbent at 900 °C under a N2 atmosphere for 10 min.
3. Results and discussion
3.1 Recycling of CaO and iron from steel slag
Ca and Mg are the main elements extracted from steel slag; other major elements include Fe, Si, Mn and Al (Fig. 2). This is in line with the elemental composition of the freshly calcined CO2 sorbents prepared from the steel slag leachate, determined by using XRF (Table S1, ESI†). It was also revealed from Fig. 2 that the extraction time and solid/liquid ratio did not result in any significant change in the amount of elements extracted from the slag sample, yielding approximately 0.27 g of CO2 sorbents per gram of steel slag used. This is encouraging, as a shorter extraction time and higher solid/liquid ratio are required for practical application. The amount of Ca and Mg extracted from slag increased gradually with an increasing mass ratio of acid to slag; however, this limited increase was at the cost of a higher dosage and lower conversion of acetic acid. In addition, the higher dosage (i.e., mass ratio of acid to slag) resulted in a significant increase in the amount of Fe, Si, and Al extracted, which in turn lowered the total mass fraction of CaO and MgO from 96.3% (1 M–2 h–1:10) to 74.0% (5 M–2 h–1:10) in the freshly calcined sorbents prepared. For the freshly calcined 1 M–0.5 h–1:10, 1 M–2 h–1:10, 2 M–0.5 h–1:5, and 2 M–2 h–1:5 materials with a low acid-to-slag mass ratio of 0.6:1, high CaO purities of ∼90% could be achieved (Table S1†), indicative of a promising alternative for natural limestone as feedstock for iron production.
 | ||
| Fig. 2 Elements recovered from steel slag during acid extraction. | ||
Fig. 3 plots the recycling efficiency of iron from the steel slag with an acid extraction, with raw steel slag included for comparison. It is shown in Fig. 3(a) that recovery of all magnetically-separated materials decreased with increasing centrifugal speed, regardless of whether raw slag or the slag with an acid extraction was employed. This is because a stronger magnetic force is required to separate phases at a higher centrifugal speed. However, there was a significant increase in the recovery of the magnetically-separated materials from the steel slag with an acid extraction compared with that from the raw slag. Increasing the dosage of acetic acid from 1 M to 3 M or 5 M during the acid extraction did not significantly increase the recovery of the magnetically-separated materials, probably because more iron was extracted into the leachate from the steel slag with higher concentrations of acetic acid as shown in Fig. 2. The iron content of the magnetically-separated materials from either the raw steel slag or the steel slag with an acid extraction increased with centrifugal speed (Fig. 3(b)); this is because the more magnetic phases could be separated at higher centrifugal speeds. Importantly, the iron content of all magnetically-separated materials from the steel slag with an acid extraction was appreciably higher than that from the raw slag, exceeding 55% at a centrifugal speed of 500 rpm. The magnetically separated materials corresponding to 2 M–0.5 h–1:5 and 5 M–2 h–1:10 exhibited iron contents as high as 70.0% and 70.6%, respectively. These values were close to the theoretical iron content of 72.4% in pure magnetite, indicating that the as-separated materials could be a high-quality alternative to iron ore available for iron production. Therefore, when acid extraction of the waste steel slag is used, not only is high-purity CaO obtained for use in CO2 capture (to be discussed below) and subsequent iron production, but the recycling efficiency of iron is also improved significantly by increasing both the recovery of iron-rich materials and the iron content of the materials recovered.
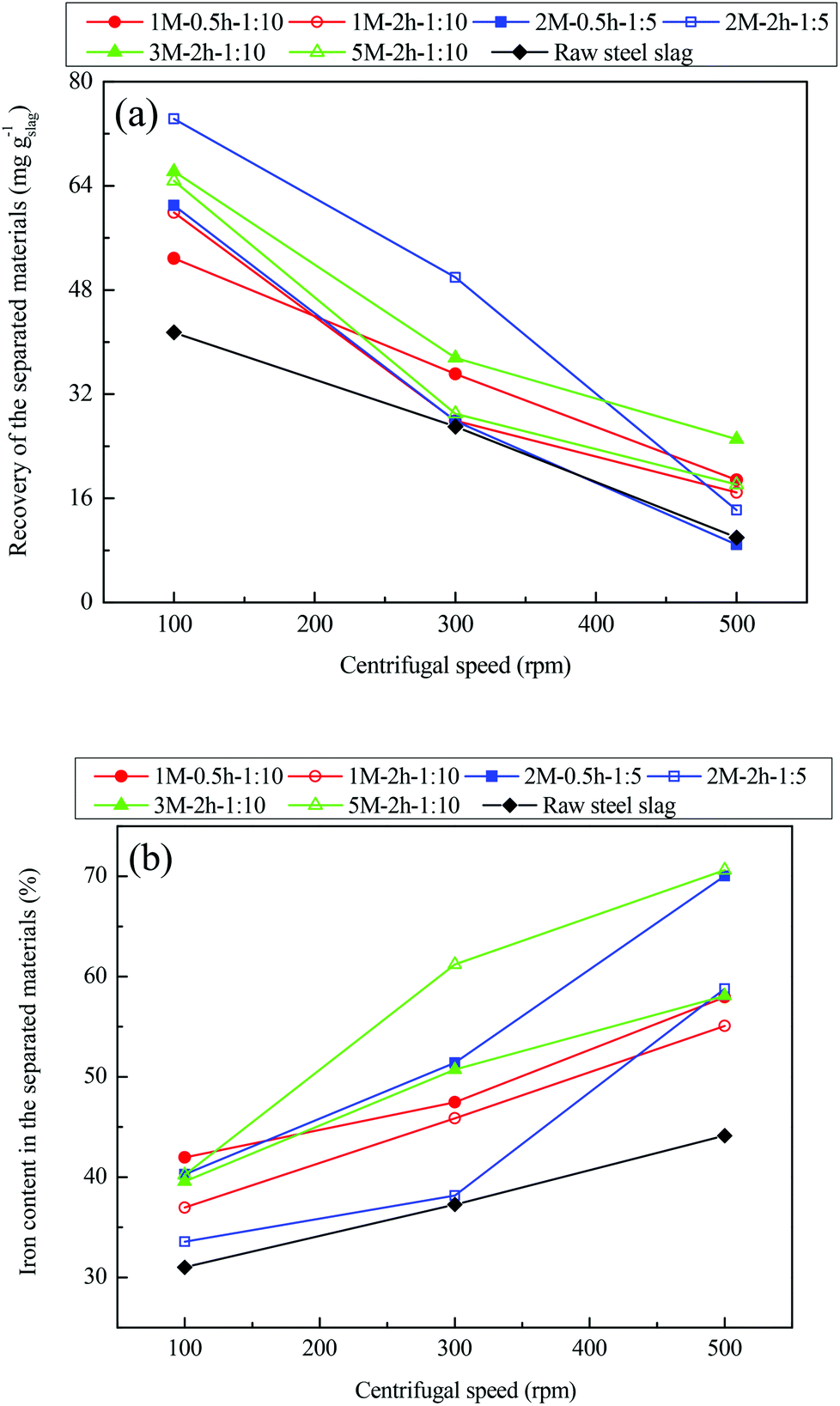 | ||
| Fig. 3 Recycling efficiency of iron from raw steel slag and the steel slag with an acid extraction using magnetic separation with a magnetic field intensity of 0.4 T: (a) recovery of the magnetically-separated materials and (b) the iron content of the magnetically-separated materials. | ||
The mechanism underlying the significant enhancement in the recycling efficiency of iron from steel slag was studied by characterizing the XRD patterns, as shown in Fig. 4. Ca12Al14O33 (mayenite), Ca(OH)2 (portlandite), (Ca1.92Fe1.08)Fe2(SiO4)3 (andradite ferroan), Ca2SiO4 (calcium silicate), and Mg6Fe2(OH)16CO3·4H2O (sjogrenite) were the major mineral phases in the raw steel slag sample. After acid extraction, the phases (Ca1.92Fe1.08)Fe2(SiO4)3 and Mg6Fe2(OH)16CO3·4H2O were no longer identified in the remaining steel slag residues, and a new phase, SiO2 (quartz), was present. This was likely associated with the reaction between acetic acid and these phases during the acid extraction process, where Ca and Mg were dissolved, leaving SiO2 in the residues. Upon a subsequent magnetic separation, Ca12Al14O33 became the major phase in the final steel slag solids. Fe3O4 (magnetite) and MgFe2O4 (magnesioferrite) were identified as the dominant phases in the magnetically-separated materials (at a centrifugal speed of 500 rpm), indicating that the formed Fe3O4 and MgFe2O4, with a theoretical iron content of 72.4% and 56.0%, respectively, are the Fe-rich phases available for magnetic separation from the steel slag with an acid extraction. Therefore, depending on the ratio of Fe3O4 to MgFe2O4, the iron content of the magnetically-separated materials should be 56.0–72.4%, which is in line with the results from samples at 500 rpm (Fig. 3(b)).
 | ||
| Fig. 4 X-ray diffraction (XRD) patterns of raw steel slag and the solids after different recycling steps corresponding to material 2 M–2 h–1:5. The following phases were identified: (■) mayenite, Ca12Al14O33; (▲) portlandite, Ca(OH)2; (◆) andradite ferroan, (Ca1.92Fe1.08)Fe2(SiO4)3; (●) calcium silicate, Ca2SiO4; (★) sjogrenite, Mg6Fe2(OH)16CO3·4H2O; (□) quartz, SiO2, (△) srebrodolskite, Ca2Fe2O5; and (◊) magnetite, Fe3O4 or magnesioferrite, MgFe2O4. | ||
3.2 CO2 capture performance of the CaO-based sorbents derived from steel slag
The results of N2 temperature-programmed decomposition (N2-TPD) analysis of the 2 M–2 h–1:5 material are presented in Fig. 5(a). Three distinctive decomposition steps were revealed in the weight (change) curve, corresponding to temperature regions of 130–225 °C, 360–500 °C, and 630–750 °C, respectively. These decomposition steps were associated with the three main endothermic peaks located at 185 °C and 215 °C, 435 °C, and 745 °C, respectively, in the heatflow signal. The first decomposition step, in which two H2O peaks occurred together in the MS signal, represented the dehydration of acetates in the material. During the second decomposition step, a strong acetone peak was associated with small H2O and CO2 peaks in the MS signal. This is mainly due to the decomposition of anhydrous calcium acetate into CaCO3.39,40 The third step, with a CO2 peak in the MS signal, represented the decomposition of CaCO3 into CaO. A comparison of the normalized weight change in all materials synthesized during N2-TPD is shown in Fig. 5(b). Materials 1 M–0.5 h–1:10, 1 M–2 h–1:10, 2 M–0.5 h–1:5, and 2 M–2 h–1:5 were fairly close in their normalized weight changes, likely due to their similar elemental compositions as shown in Table S1.† The weight loss of these materials during N2-TPD was ∼5.1%, ∼34.5%, and ∼22.9%, respectively, for the three decomposition steps. Compared with the above-mentioned materials, 3 M–2 h–1:10 and 5 M–2 h–1:10 did not present a clear platform between the dehydration and acetate-decomposition steps in the normalized weight curve, and more H2O was generated during their dehydration (Fig. S1 in the ESI†). In addition, weight loss during the CaCO3-decomposition step was less normalized for 3 M–2 h–1:10 and 5 M–2 h–1:10 than for the other four materials, indicative of a lower CaO content in the material, as is in line with the results shown in Table S1.†
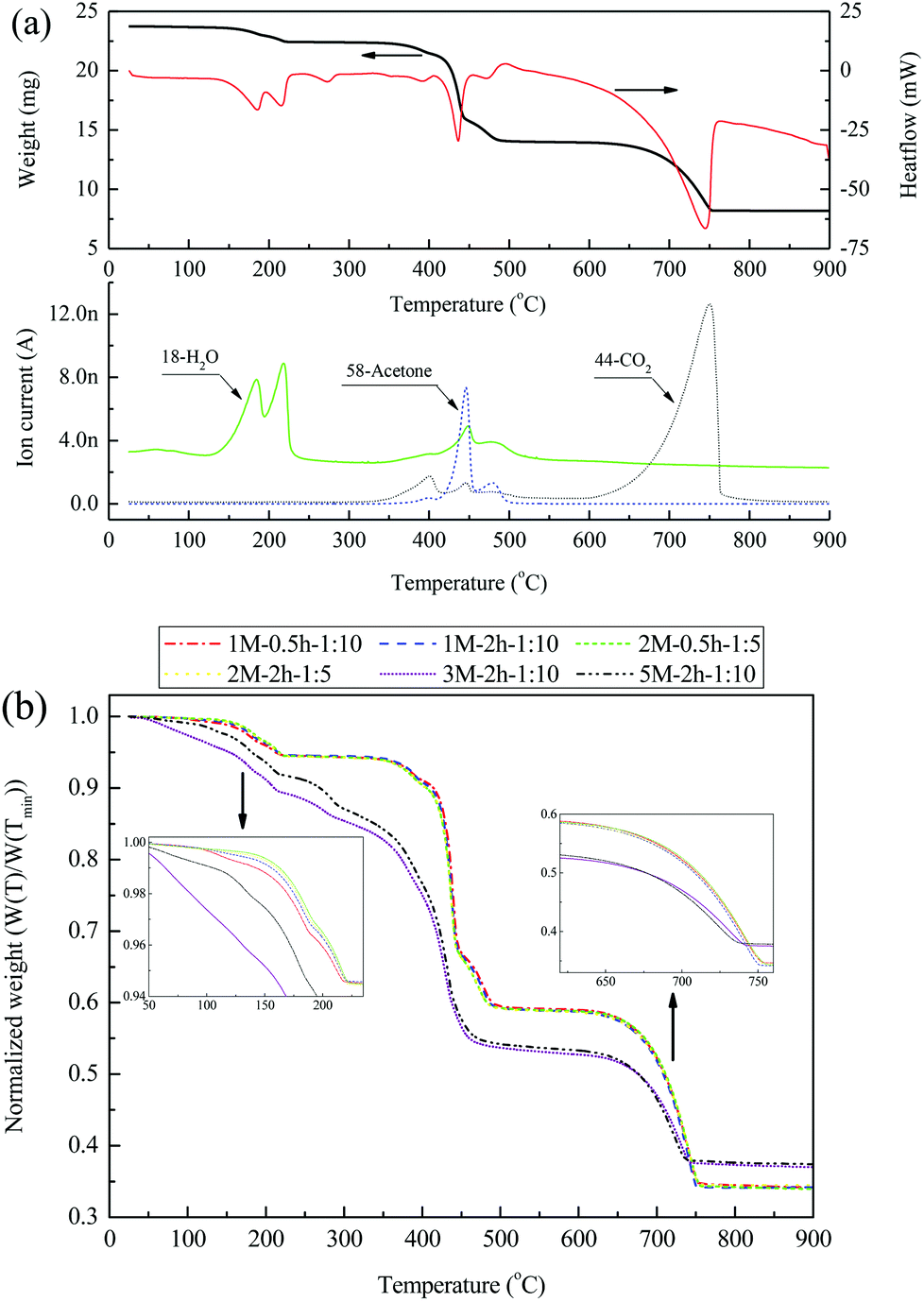 | ||
| Fig. 5 N2-TPD profiles of the freshly dried materials from the steel slag leachate: (a) TGA/DSC-MS curve of the material 2 M–2 h–1:5, (b) normalized weight change of all materials prepared as a function of temperature. | ||
The change in morphology of the material 1 M–2 h–1:10 during N2-TPD was further characterized using scanning electron microscopy (Fig. 6). The freshly dried 1 M–2 h–1:10 had a coarse structure with a comparatively compact surface (Fig. 6(a)). When heated to 600 °C, acetone was released due to the decomposition of calcium acetate, resulting in a slice-shaped morphology in the material (Fig. 6(b)). With subsequent decarbonation of CaCO3 before heating to 900 °C, a well-defined, porous structure, mainly comprised of fine CaO grains, formed in the freshly calcined 1 M–2 h–1:10 (Fig. 6(c)). XRD patterns of typical as-prepared CO2 sorbents in the freshly calcined state are shown in Fig. 7. The major phase, CaO, along with minor phases, MgO and CaS, were identified in 1 M–0.5 h–1:10, 1 M–2 h–1:10, 2 M–0.5 h–1:5, and 2 M–2 h–1:5, the materials with the same (low) mass ratio of acetic acid to steel slag during acid extraction (Fig. S2 in the ESI†). With increasing doses of acetic acid, small quantities of Ca2Fe2O5 and (MgFe)2SiO4 were detected in the 3 M–2 h–1:10 and 5 M–2 h–1:10 materials synthesized.
 | ||
| Fig. 6 SEM images of the material 1 M–2 h–1:10 (a) freshly dried at 105 °C, (b) heated to 600 °C, (c) heated to 900 °C, and (f) after 20 real CaL cycles; with (d) fresh and (e) cycled commercial CaO included for comparison. | ||
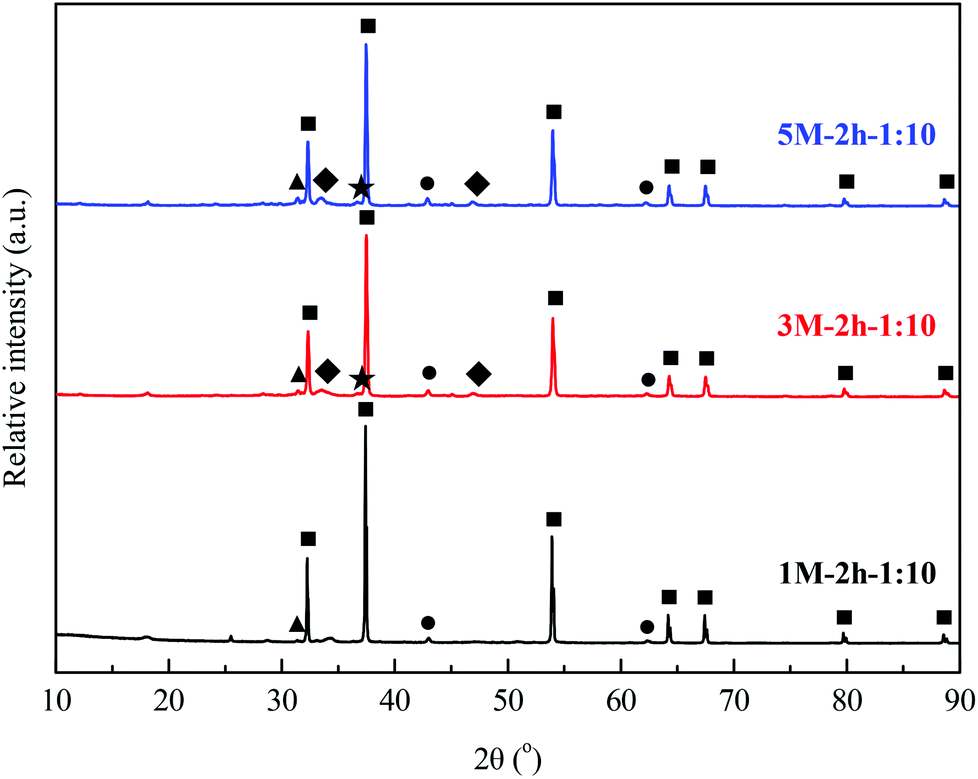 | ||
| Fig. 7 XRD patterns of typical steel slag-derived, CaO-based CO2 sorbents freshly calcined at 900 °C for 10 min. The following phases were identified: (■) lime, CaO; (●) periclase, MgO; (▲) calcium sulphide, CaS; (★) wadsleyite, (MgFe)2SiO4; and (◆) srebrodolskite, Ca2Fe2O5. | ||
CO2 capture performance as a function of reaction time was compared between commercial CaO and the various CaO-based CO2 sorbents synthesized in this study (Fig. 8). Commercial CaO presented a classical two-stage CO2 capture regime under the conditions studied here.41 In the first 5 min, the commercial CaO experienced a fast and kinetically-controlled reaction stage as pores with a diameter <100 nm were filled by newly formed CaCO3. This was followed by a substantially slower reaction stage controlled by the diffusion of CO2 through the CaCO3 product layer. Alvarez and Abanades42 concluded that the transition between the two reaction stages occurs at a critical product layer thickness of ∼50 nm, which was better explained later by Li and colleagues43 through a rate equation theory they developed. The carbonation rate of the synthesized CaO-based CO2 sorbents was not as fast as that of the commercial CaO during the kinetically-controlled reaction stage. However, materials 1 M–0.5 h–1:10, 1 M–2 h–1:10, 2 M–0.5 h–1:5, and 2 M–2 h–1:5 exhibited significantly faster carbonation rates than commercial CaO during the diffusion-controlled reaction stage, allowing their uptake of CO2 to surpass that of commercial CaO after a 20 min carbonation period. After 120 min, 1 M–2 h–1:10 achieved a CO2 uptake of 0.62 gCO2 gsorbent−1, which was 1.5 times as high as that of the commercial CaO, indicative of an activation effect of acetic acid on the CaO-based CO2 sorbents.44 Freshly calcined 1 M–0.5 h–1:10, 1 M–2 h–1:10, 2 M–0.5 h–1:5, and 2 M–2 h–1:5 exhibited similar uptakes of CO2, indicating that the extraction time and solid/liquid ratio did not influence significantly the isothermal CO2 capture performance of the steel slag-derived CO2 sorbents. This is probably because the extraction time and solid/liquid ratio had almost no effect on the elemental (Table S1†) and mineral (Fig. S2†) compositions of materials. However, the CO2 uptake experienced a significant decrease with increasing acetic acid dosage, which was likely associated with the lower CaO content in freshly calcined 3 M–2 h–1:10 and 5 M–2 h–1:10, as discussed in Fig. 2 and Table S1.†
 | ||
| Fig. 8 Carbonation of the CO2 sorbents synthesized, with commercial CaO included for comparison, under a 20 vol% CO2/80 vol% N2 atmosphere at 650 °C for 120 min. | ||
The multicyclic CO2 capture performance of commercial CaO and the steel slag-derived, CaO-based CO2 sorbents synthesized in this study under realistic CaL conditions are compared in Fig. 9. The commercial CaO presented a slightly higher CO2 uptake of 0.27 gCO2 gsorbent−1 compared with the steel slag-derived sorbents 1 M–0.5 h–1:10, 1 M–2 h–1:10, 2 M–0.5 h–1:5, and 2 M–2 h–1:5 in the first cycle, whereas 3 M–2 h–1:10 and 5 M–2 h–1:10 had lower CO2 uptakes of 0.16 gCO2 gsorbent−1 and 0.14 gCO2 gsorbent−1, respectively. However, the CO2 uptake of the commercial CaO experienced a drastic decrease within the first five cycles, and began to stabilize after falling to 0.07 gCO2 gsorbent−1 by the tenth cycle. For the steel slag-derived, CaO-based CO2 sorbents synthesized, two types of multicyclic CO2 capture characteristics were revealed. 3 M–2 h–1:10 and 5 M–2 h–1:10, although bearing a limited CO2 capture performance compared to commercial CaO, displayed a similar CO2 capture characteristic as the commercial CaO, i.e., the CO2 uptake dropped drastically during the first several cycles, followed by a comparatively stable uptake afterwards. However, 1 M–0.5 h–1:10, 1 M–2 h–1:10, 2 M–0.5 h–1:5, and 2 M–2 h–1:5, the materials with the same (low) mass ratio of acetic acid to steel slag during acid extraction, exhibited a different CO2 capture characteristic. A drastic decay in CO2 uptake occurred mainly between the first two cycles. Then, the CO2 uptake experienced a slow, linear decay with increasing CaL cycles. Because of this, the CO2 uptake of 1 M–0.5 h–1:10, 1 M–2 h–1:10, 2 M–0.5 h–1:5, and 2 M–2 h–1:5 exceeded that of the commercial CaO from the third cycle onward, and 1 M–2 h–1:10 captured almost twice as much CO2 as the commercial CaO by the end of the twentieth cycle. One would expect that the well-preserved porous structure of the cycled 1 M–2 h–1:10, compared to that of the commercial CaO, ensured its favorable CO2 capture characteristics (Fig. 6(c–f)). Therefore, the better carbonation reactivity (Fig. 8) and cyclic stability (Fig. 9) of the steel slag-derived, CaO-based CO2 sorbents 1 M–0.5 h–1:10, 1 M–2h–1:10, 2 M–0.5 h–1:5, and 2 M–2 h–1:5, when compared to commercial CaO, suggest that they are promising alternatives for use in CaL.
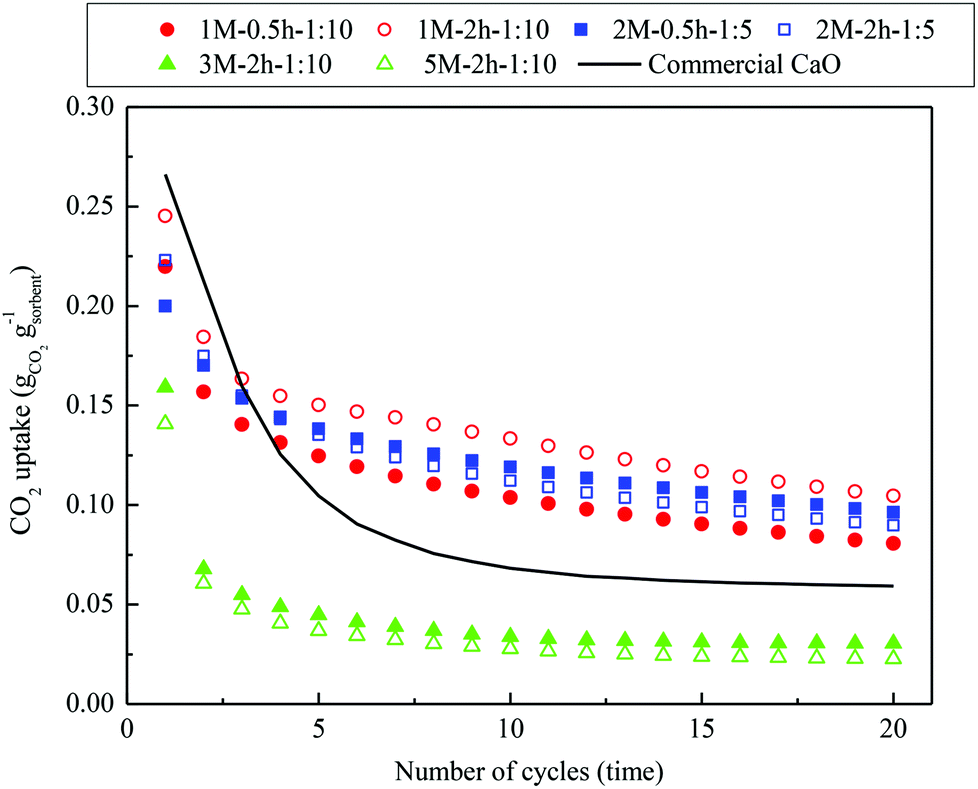 | ||
| Fig. 9 Multicyclic CO2 capture performance of the steel slag-derived, CaO-based sorbents synthesized, with commercial CaO included for comparison, under realistic CaL conditions. | ||
3.3 Preliminary cost structure of the proposed integrated CO2 capture process
One inherent advantage of CaL over any other current post-combustion CO2 capture technologies, when employed for CO2 abatement in the iron and steel industry, is that the spent CaO can be recycled directly as a flux into the blast furnace for iron production. This would result in an additional reduction in CO2 emissions associated with the calcination of limestone during conventional iron and steel production processes. With regard to the practical application of CaL-based CO2 capture technology, the cost structure can be affected by a variety of operating parameters. MacKenzie and colleagues45 performed a sensitivity analysis on eight potentially key variables influencing the cost of CaL-based CO2 capture technology used in practical applications. They confirmed that the CO2 capture cost of the CaL process is most affected by the cost of CaO-based sorbents and their deactivation rate when the internal solid circulation (molar ratio of CaO to CO2) and the make-up flow of solids (purge percentage of fresh CaO in the calciner) have been determined. The superior CO2 reactivity and favorable deactivation rate of the steel slag-derived, CaO-based materials 1 M–0.5 h–1:10, 1 M–2 h–1:10, 2 M–0.5 h–1:5, and 2 M–2 h–1:5, when compared to the commercial CaO, have been verified above. Now we attempt to compare the preliminary cost structure between limestone-derived CaO and typical steel slag-derived CO2 sorbents developed in this study, mainly based on the material flow analysis, as shown in Table 2. Naturally occurring limestone is the main feedstock of conventional CaO-based CO2 sorbent for CaL. For the steel slag-derived, CaO-based CO2 sorbents developed in this study, given that steel slag is an almost zero-cost industrial waste, acetic acid is the main consideration in the cost analysis. For materials 1 M–0.5 h–1:10 and 1 M–2 h–1:10, ∼2.5 g of acetic acid was consumed to produce 1 g of CO2 sorbent, which is near the theoretically lowest requirement of ∼2.1 gacetic acid gsorbent−1. Although bearing a higher expense for feedstock than naturally derived CaO, the final cost of the steel slag-derived, CaO-based sorbent was well compensated by the byproducts recovered, i.e., the high-purity CaO, high-quality iron ore, and acetone. The cost of materials 1 M–0.5 h–1:10 and 1 M–2 h–1:10 was 57.7 € t−1 and 62.2 € t−1, respectively, appreciably lower than that of the naturally derived CaO (102 € t−1).
| Material | Feedstock consumeda [t tsorbent−1] | Byproduct recovereda [t tsorbent−1] | Cost of sorbents [€ t−1] | |||
|---|---|---|---|---|---|---|
| Acetic acid | Limestone | CaO | Iron ore | Acetone | ||
| a Prices of the components mentioned were determined according to the average price from the past 3 years in the Chinese market, i.e., 231.9 € t−1, 51.5 € t−1, 103.1 € t−1, and 450.9 € t−1 for acetic acid (≥99.8 wt%), limestone (∼90 wt%), iron ore (∼62 wt%), and acetone (≥99.5 wt%), respectively. b The component was not consumed or recovered. | ||||||
| 1 M–0.5 h–1:10 |
2.50 | — | 0.91 | 0.05 | 0.94 | 57.7 |
| 1 M–2 h–1:10 |
2.50 | — | 0.90 | 0.06 | 0.93 | 62.2 |
| 2 M–0.5 h–1:5 |
2.86 | — | 0.90 | 0.06 | 0.93 | 145.7 |
| 2 M–2 h–1:5 |
2.73 | — | 0.89 | 0.07 | 0.92 | 120.0 |
| Naturally derived CaO | —b | 1.98 | — | — | — | 102.0 |
Given the enhanced CO2 capture performance of the steel slag-derived sorbents compared to naturally derived CaO, an increase in the cost of the as-prepared CO2 sorbents is acceptable as long as it remains below an upper limit, which largely depends on the CO2 capture capacity of the materials. Romeo and colleagues46 studied in detail the relationship between the increase in the maximum average CO2 capture capacity of CaO-based sorbents and the maximum acceptable investment in sorbents under different CaL operations, and proposed a useful approach to assess the availability of new CaO-based CO2 sorbents for use in CaL; we adapted this approach to assess the acceptable cost of the steel slag-derived CO2 sorbents developed in this study (Table 3). As expected, a more cost-effective CaO-based CO2 sorbent is required when CaL is performed under higher sorbent-to-CO2 ratios (R) or material make-up flows (fp), as more fresh sorbents are consumed under these operating conditions. Materials 1 M–0.5 h–1:10 and 1 M–2 h–1:10 exhibited appreciable superiority over naturally derived CaO with regard to the cost of sorbents under all sorbent/CO2 ratios and material make-up flows. Material 2 M–2 h–1:5 was comparable with naturally derived CaO under low sorbent/CO2 ratios and material make-up flows, while the cost of material 2 M–0.5 h–1:5 was marginally higher than naturally derived CaO. However, it should be pointed out that the cost structure of the proposed integrated CO2 capture process was assessed here mainly based on the material flow analysis. The capital costs and variable operating costs commonly considered in practical CaL projects have not been mentioned yet at this stage. Nevertheless, the proposed integrated CO2 capture process using materials 1 M–0.5 h–1:10 and 1 M–2 h–1:10 appears to be cost-effective, which is promising for CO2 abatement applications in the iron and steel industry.
| Material | X ave increasea [%] | Maximum acceptable cost of sorbents [€ t−1] | |||||
|---|---|---|---|---|---|---|---|
| R = 1.5 | R = 3 | R = 5 | |||||
|
f
pc = 1% |
f p = 2.5% | f p = 1% | f p = 2.5% | f p = 1% | f p = 2.5% | ||
| a The subtraction of average carbonation conversion during 20 cycles between the steel slag-derived CO2 sorbent and naturally derived CaO. b The molar ratio of the CaO-based sorbent to CO2 during the CaL process. c The make-up flow (purge percentage) of fresh CaO-based sorbents in the calciner. | |||||||
| 1 M–0.5 h–1:10 |
3.8 | 117.2 | 109.4 | 112.6 | 109.7 | 111.4 | 108.9 |
| 1 M–2 h–1:10 |
7.6 | 132.3 | 116.7 | 123.3 | 117.3 | 120.8 | 115.9 |
| 2 M–0.5 h–1:5 |
5.9 | 125.6 | 113.4 | 118.5 | 113.9 | 116.6 | 112.8 |
| 2 M–2 h–1:5 |
5.4 | 123.6 | 112.5 | 117.1 | 112.9 | 115.4 | 111.8 |
4. Conclusions
We have proposed and experimentally demonstrated the feasibility of an integrated CO2 capture process for CO2 abatement in the iron and steel industry. In such a process, highly effective CaO-based CO2 sorbents were easily prepared via acid extraction of waste steel slag using acetic acid. Encouragingly, the recycling efficiency of iron from the steel slag residues remaining after acid extraction increased significantly due to improvement in both the recovery of iron-rich materials and the iron content in the materials recovered, allowing for recycling of high-quality iron ore with iron content as high as 55.1–70.6%. The steel slag-derived, CaO-based CO2 sorbents 1 M–0.5 h–1:10, 1 M–2 h–1:10, 2 M–0.5 h–1:5, and 2 M–2 h–1:5 could achieve CaO content as high as ∼90 wt%. The well-defined, porous morphology ensured these materials superior CO2 reactivity, and importantly, significantly slower linear deactivation with CaL cycles, compared to commercial CaO. Although costing more than naturally derived CaO in the purchase of feedstock, the final cost of the steel slag-derived, CaO-based CO2 sorbent developed here was compensated by the byproducts recovered, i.e., the high-purity CaO, high-quality iron ore, and acetone. Materials 1 M–0.5 h–1:10 and 1 M–2 h–1:10 were much more cost-effective than naturally derived CaO, making the integrated CO2 capture process proposed in this study potentially very attractive for CO2 abatement in the iron and steel industry.
Acknowledgements
The authors gratefully acknowledge the Tsinghua University Initiative Scientific Research Program (Grant No. 2014z22075) and the National Natural Science Foundation of China (Grant No. 21576156) for financial support. Also, the authors would like to acknowledge the financial support of the UK CCS Research Centre (http://www.ukccsrc.ac.uk) in carrying out this work. The UKCCSRC is funded by the EPSRC as part of the RCUK Energy Programme.References
- IPCC, Industry, in Climate Change 2014: Mitigation of Climate Change. Contribution of Working Group III to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA, 2014 Search PubMed.
- T. A. Napp, A. Gambhir, T. P. Hills, N. Florin and P. S. Fennell, A review of the technologies, economics and policy instruments for decarbonising energy-intensive manufacturing industries, Renewable Sustainable Energy Rev., 2014, 30, 616–640 CrossRef CAS.
- U.S. Energy Information Administration (U.S. EIA), International Energy Outlook 2013: With Projections to 2040, Washington, DC 20585; 2013. To be found at: http://www.eia.gov/forecasts/ieo/pdf/0484(2013).pdf.
- International Energy Agency (IEA), Energy technology perspectives 2010: scenarios & strategies to 2050, OECD/IEA, Paris and 2010 Search PubMed.
- IPCC, Climate Change 2007: Synthesis Report. Contribution of Working Groups I, II and III to the Fourth Assessment Report of the Intergovernmental Panel on Climate Change, Geneva, Switzerland, 2007.
- International Energy Agency (IEA), Executive Summary for Energy Technology Perspectives 2012: Pathways to a Clean Energy System, OECD/IEA, Paris, 2012. To be found at: http://www.iea.org/textbase/npsum/ETP2012SUM.pdf Search PubMed.
- International Energy Agency (IEA), Tracking Clean Energy Progress 2014, OECD/IEA, Paris, 2014. To be found at: http://www.iea.org/publications/freepublications/publication/Tracking_clean_energy_progress_2014.pdf Search PubMed.
- S. Tian, J. Jiang, F. Yan, K. Li and X. Chen, Synthesis of highly efficient CaO-based, self-stabilizing CO2 sorbents via structure-reforming of steel slag, Environ. Sci. Technol., 2015, 49, 7464–7472 CrossRef CAS PubMed.
- D. P. Hanak, E. J. Anthony and V. Manovic, A review of developments in pilot-plant testing and modelling of calcium looping process for CO2 capture from power generation systems, Energy Environ. Sci., 2015, 8, 2199–2249 CAS.
- B. Arias, M. E. Diego, J. C. Abanades, M. Lorenzo, L. Diaz, D. Martínez, J. Alvarez and A. Sánchez-Biezma, Demonstration of steady state CO2 capture in a 1.7 MWth calcium looping pilot, Int. J. Greenhouse Gas Control, 2013, 18, 237–245 CrossRef CAS.
- J. Blamey, E. J. Anthony, J. Wang and P. S. Fennell, The calcium looping cycle for large-scale CO2 capture, Prog. Energy Combust. Sci., 2010, 36, 260–279 CrossRef CAS.
- M. Alonso, M. E. Diego, C. Perez, J. R. Chamberlain and J. C. Abanades, Biomass combustion with in situ CO2 capture by CaO in a 300 kWth circulating fluidized bed facility, Int. J. Greenhouse Gas Control, 2014, 29, 142–152 CrossRef CAS.
- M. Zhao, A. I. Minett and A. T. Harris, A review of techno-economic models for the retrofitting of conventional pulverised-coal power plants for post-combustion capture (PCC) of CO2, Energy Environ. Sci., 2013, 6, 25–40 CAS.
- D. P. Hanak, C. Biliyok and V. Manovic, Calcium looping with inherent energy storage for decarbonisation of coal-fired power plant, Energy Environ. Sci., 2016, 9, 971–983 CAS.
- D. C. Ozcan, B. H. Shanks and T. D. Wheelock, Improving the stability of a CaO-based sorbent for CO2 by thermal pretreatment, Ind. Eng. Chem. Res., 2011, 50, 6933–6942 CrossRef CAS.
- V. Manovic and E. J. Anthony, Thermal activation of CaO-based sorbent and self-reactivation during CO2 capture looping cycles, Environ. Sci. Technol., 2008, 42, 4170–4174 CrossRef CAS PubMed.
- Z. Chen, H. S. Song, M. Portillo, C. J. Lim, J. R. Grace and E. J. Anthony, Long-term calcination/carbonation and thermal pretreatment for CO2 capture by limestone and dolomite, Energy Fuels, 2009, 23, 1437–1444 CrossRef CAS.
- V. Manovic and E. J. Anthony, Steam reactivation of spent CaO-based sorbent for multiple capture cycles, Environ. Sci. Technol., 2007, 41, 1420–1425 CrossRef CAS PubMed.
- B. V. Materić, C. Sheppard and S. I. Smedley, Effect of repeated steam hydration reactivation on CaO-based sorbents for CO2 capture, Environ. Sci. Technol., 2010, 44, 9496–9501 CrossRef PubMed.
- F. Donat, N. H. Florin, E. J. Anthony and P. S. Fennell, Influence of high-temperature steam on the reactivity of CaO sorbent for CO2 capture, Environ. Sci. Technol., 2012, 46, 1262–1269 CrossRef CAS PubMed.
- N. Phalak, N. Deshpande and L. S. Fan, Investigation of high-temperature steam hydration of naturally derived calcium oxide for improved carbon dioxide capture capacity over multiple cycles, Energy Fuels, 2012, 26, 3903–3909 CrossRef CAS.
- B. Arias, G. S. Grasa, M. Alonso and J. C. Abanades, Post-combustion calcium looping process with a highly stable sorbent activity by recarbonation, Energy Environ. Sci., 2012, 5, 7353–7359 CAS.
- W. Q. Liu, B. Feng, Y. Q. Wu, G. X. Wang, J. Barry and J. C. D. da Costa, Synthesis of sintering-resistant sorbents for CO2 capture, Environ. Sci. Technol., 2010, 44, 3093–3097 CrossRef CAS PubMed.
- M. Broda and C. R. Müller, Synthesis of highly efficient, Ca-based, Al2O3-stabilized, carbon gel-templated CO2 sorbents, Adv. Mater., 2012, 24, 3059–3064 CrossRef CAS PubMed.
- S. Wang, L. Fan, C. Li, Y. Zhao and X. Ma, Porous spherical CaO-based sorbents via PSS-assisted fast precipitation for CO2 capture, ACS Appl. Mater. Interfaces, 2014, 6, 18072–18077 CAS.
- R. Filitz, A. M. Kierzkowska, M. Broda and C. R. Müller, Highly efficient CO2 sorbents: development of synthetic, calcium-rich dolomites, Environ. Sci. Technol., 2012, 46, 559–565 CrossRef CAS PubMed.
- F. N. Ridha, V. Manovic, A. Macchi and E. J. Anthony, High-temperature CO2 capture cycles for CaO-based pellets with kaolin-based binders, Int. J. Greenhouse Gas Control, 2012, 6, 164–170 CrossRef CAS.
- M. Zhao, J. Shi, X. Zhong, S. Tian, J. Blamey, J. Jiang and P. S. Fennell, A novel calcium looping absorbent incorporated with polymorphic spacers for hydrogen production and CO2 capture, Energy Environ. Sci., 2014, 7, 3291–3295 CAS.
- M. Broda, A. M. Kierzkowska and C. R. Müller, Development of highly effective CaO-based, MgO-stabilized CO2 sorbents via a scalable “one-pot” recrystallization technique, Adv. Funct. Mater., 2014, 24, 5753–5761 CrossRef CAS.
- F. N. Ridha, V. Manovic, Y. Wu, A. Macchi and E. J. Anthony, Pelletized CaO-based sorbents treated with organic acids for enhanced CO2 capture in Ca-looping cycles, Int. J. Greenhouse Gas Control, 2013, 17, 357–365 CrossRef CAS.
- F. N. Ridha, V. Manovic, A. Macchi, M. A. Anthony and E. J. Anthony, Assessment of limestone treatment with organic acids for CO2 capture in Ca-looping cycles, Fuel Process. Technol., 2013, 116, 284–291 CrossRef CAS.
- M. T. Ho, A. Bustamante and D. E. Wiley, Comparison of CO2 capture economics for iron and steel mills, Int. J. Greenhouse Gas Control, 2013, 19, 145–159 CrossRef CAS.
- J. Oda, K. Akimoto, T. Tomoda, M. Nagashima, K. Wada and F. Sano, International comparisons of energy efficiency in power, steel, and cement industries, Energy Policy, 2012, 44, 118–129 CrossRef CAS.
- P. Renforth, C. L. Washbourne, J. Taylder and D. A. C. Manning, Silicate production and availability for mineral carbonation, Environ. Sci. Technol., 2011, 45, 2035–2041 CrossRef CAS PubMed.
- A. Telesca, D. Calabrese, M. Marroccoli, M. Tomasulo, G. L. Valenti, G. Duelli and F. Montagnaro, Spent limestone sorbent from calcium looping cycle as a raw material for the cement industry, Fuel, 2014, 118, 202–205 CrossRef CAS.
- C. C. Dean, D. Dugwell and P. S. Fennell, Investigation into potential synergy between power generation, cement manufacture and CO2 abatement using the calcium looping cycle, Energy Environ. Sci., 2011, 4, 2050–2053 CAS.
- N. Rodríguez, R. Murillo and J. C. Abanades, CO2 capture from cement plants using oxyfired precalcination and/or calcium looping, Environ. Sci. Technol., 2012, 46, 2460–2466 CrossRef PubMed.
- K. Atsonios, P. Grammelis, S. K. Antiohos, N. Nikolopoulos and E. Kakaras, Integration of calcium looping technology in existing cement plant for CO2 capture: process modeling and technical considerations, Fuel, 2015, 153, 210–223 CrossRef CAS.
- Y. Li, C. Zhao, L. Duan, C. Liang, Q. Li, W. Zhou and H. Chen, Cyclic calcination/carbonation looping of dolomite modified with acetic acid for CO2 capture, Fuel Process. Technol., 2008, 89, 1461–1469 CrossRef CAS.
- Y. Li, C. Zhao, H. Chen, C. Liang, L. Duan and W. Zhou, Modified CaO-based sorbent looping cycle for CO2 mitigation, Fuel, 2009, 88, 697–704 CrossRef CAS.
- J. C. Abanades, E. J. Anthony, D. Y. Lu, C. Salvador and D. Alvarez, Capture of CO2 from combustion gases in a fluidized bed of CaO, AIChE J., 2004, 50, 1614–1622 CrossRef CAS.
- D. Alvarez and J. C. Abanades, Determination of the critical product layer thickness in the reaction of CaO with CO2, Ind. Eng. Chem. Res., 2005, 44, 5608–5615 CrossRef CAS.
- Z. Li, H. Sun and N. Cai, Rate equation theory for the carbonation reaction of CaO with CO2, Energy Fuels, 2012, 26, 4607–4616 CrossRef CAS.
- E. R. Sacia, S. Ramkumar, N. Phalak and L. S. Fan, Synthesis and regeneration of sustainable CaO sorbents from chicken eggshells for enhanced carbon dioxide capture, ACS Sustainable Chem. Eng., 2013, 1, 903–909 CrossRef CAS.
- A. MacKenzie, D. L. Granatstein, E. J. Anthony and J. C. Abanades, Economics of CO2 capture using the calcium cycle with a pressurized fluidized bed combustor, Energy Fuels, 2007, 21, 920–926 CrossRef CAS.
- L. M. Romeo, Y. Lara, P. Lisbona and A. Martínez, Economical assessment of competitive enhanced limestones for CO2 capture cycles in power plants, Fuel Process. Technol., 2009, 90, 803–811 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6gc00400h |
| This journal is © The Royal Society of Chemistry 2016 |