
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
C–H carboxylation of heteroarenes with ambient CO2†
Sabine
Fenner
and
Lutz
Ackermann
*
Institut fuer Organische und Biomolekulare Chemie, Georg-August-Universitaet, Tammannstrasse 2, 37077 Goettingen, Germany. E-mail: Lutz.Ackermann@chemie.uni-goettingen.de; Fax: +49 551 396777
First published on 4th April 2016
Abstract
The C–H carboxylation of heteroarenes was achieved under transition metal-free reaction conditions with naturally abundant CO2 as the C1 source at relatively low temperature. The C–H carboxylation was mediated by KOt-Bu at atmospheric pressure of CO2, and thereby provided atom- and step-economical access to various heteroaromatic carboxylic acid derivatives.
Introduction
Strategies for the fixation of carbon dioxide (CO2) as an easily accessible, inexpensive, naturally abundant, and renewable C1 source towards valuable commodity chemicals1,2 have attracted major topical interest.3 While significant progress has been witnessed in the chemical use of CO2 during the recent decade,4–14 the vast majority of these procedures require pre-functionalized substrates, such as aryl halides or aryl boronic acids.1 The synthesis of the prerequisite pre-oxidized arenes calls for a number of reaction steps, which contradicts the principles of green chemistry.15 In contrast, the direct functionalization of otherwise inert C–H bonds represents a considerably more atom- and step-economical strategy,16 with important advances in direct carboxylations17,18 accomplished by Iwasawa,19–21 Nolan,22,23 Hou,24 Hu,25 Klankermayer/Leitner,11 and Beller,26,27 among others.28 Within our program on catalytic C–H activation,29,30 we became attracted by devising reaction conditions for sustainable C–H carboxylation with ambient CO2 under mild conditions.31 As a result of our efforts, we have developed a highly effective protocol for step-economical C–H carboxylations of heteroarenes with ambient CO2 under transition metal-free reaction conditions, on which we now31 wish to report herein. In contrast to previously reported methods,22–24 our C–H carboxylation protocol is operative in the absence of transition metals at relatively low temperatures of only 80–100 °C.Results and discussion
At the outset of our studies, we chose reaction conditions similar to the ones previously described for the carboxylations of organoboronic esters with CO2 (Table 1).32 Thus, when reacting benzo[d]oxazole (1a) in the presence of 10 mol% of the well-defined N-heterocyclic carbene copper(I) complex [Cu(IPr)Cl] in DMF at 80 °C under an atmosphere of CO2, 82% isolated yield of methylbenzo[d]oxazole-2-carboxylate (3a) were obtained upon treatment with methyl iodide (2a) (Table 1, entry 1). In order to establish a more economical and user-friendly method, the reaction was conducted with simple CuCl as the catalyst under ligand-free reaction conditions, which provided product 3a in a comparable yield (entry 2). Interestingly, when conducting a test reaction in the absence of a transition metal catalyst solely with the base KOt-Bu33 in DMF at 100 °C,34 the desired product 3a was isolated in 80% yield (entry 3).‡ An elevated reaction temperature of 125 °C failed to afford an improvement (entry 4), whilst a reaction conducted at 80 °C proceeded efficiently (entries 4 and 5), clearly highlighting the beneficial features of KOt-Bu as compared to Cs2CO3 that required 125 °C (vide infra).25 Polar solvents other than DMF, such as NMP, 1,4-dioxane, THF and DMSO, provided less satisfactory results (entries 7–10), as did apolar toluene (entry 11). However, the encouraging result obtained with NMP as the solvent (entry 7) indicates the potential of our strategy for the use of greener solvents, such as 1-butylpyrrolidinone or Cyrene.35 On the contrary, the C–H functionalization performed in DMA furnished carboxylic acid ester 3a with a high efficacy (entry 12). Interestingly, bases other than KOt-Bu, including Cs2CO3 or Rb2CO3, proved to be considerably less effective under otherwise identical reaction conditions (entries 13 and 14), illustrating the unique power of KOt-Bu as the base, particularly at a reaction temperature of 80 °C (entry 15 versus 5).![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a
a
|
|
|||||
|---|---|---|---|---|---|
| Entry | Base | Solvent | T 1 [°C] | T 2 [°C] | 3a [%] |
| a Reaction conditions: 1a (1.0 mmol), base (1.2 mmol), solvent (5.0 mL), CO2 (1 atm), T1, 18 h; 2a (3.0 mmol), T2, 2 h; yields of isolated products; GC-conversion in parentheses. b With [Cu(IPr)Cl] (10 mol%). c With CuCl (10 mol%). | |||||
| 1 | KOt-Bu | DMF | 80 | 65 | 82b |
| 2 | KOt-Bu | DMF | 100 | 65 | 76c |
| 3 | KOt-Bu | DMF | 100 | 65 | 80 |
| 4 | KOt-Bu | DMF | 125 | 65 | 80 |
| 5 | KOt-Bu | DMF | 80 | 65 | 71 |
| 6 | KOt-Bu | DMF | 40 | 40 | (10) |
| 7 | KOt-Bu | NMP | 100 | 65 | 69 |
| 8 | KOt-Bu | 1,4-Dioxane | 100 | 65 | — |
| 9 | KOt-Bu | THF | 65 | 65 | (4) |
| 10 | KOt-Bu | DMSO | 100 | 65 | 52 |
| 11 | KOt-Bu | PhMe | 100 | 65 | (11) |
| 12 | KOt-Bu | DMA | 100 | 65 | 77 |
| 13 | Cs2CO3 | DMF | 100 | 65 | 69 |
| 14 | Rb2CO3 | DMF | 100 | 65 | 8 |
| 15 | Cs2CO3 | DMF | 80 | 65 | 23 |

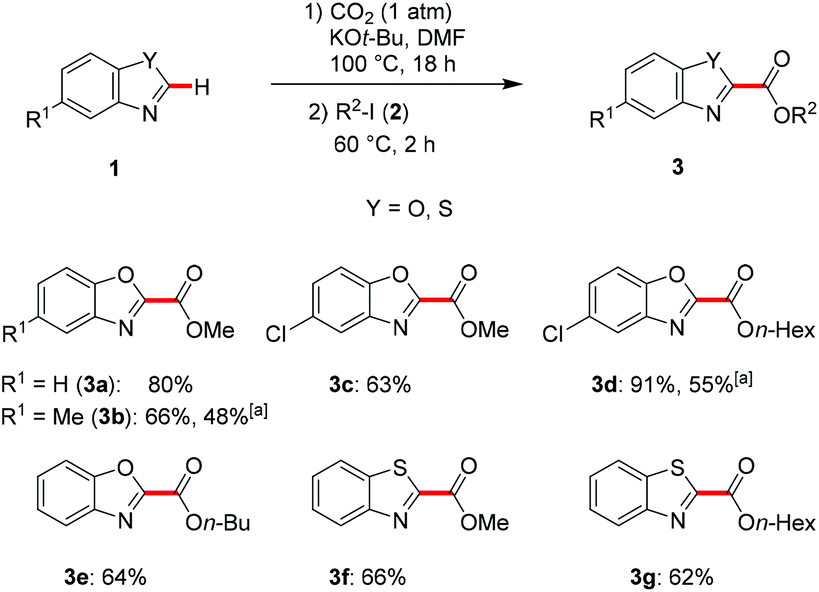
With the optimized reaction conditions in hand, the scope of the C–H carboxylation was explored next (Scheme 1).36 A series of representative heteroarenes 1 was successfully converted into the desired carboxylic acid esters 3 under transition metal-free reaction conditions with atmospheric CO2.
 | ||
| Scheme 1 C–H carboxylation with ambient CO2. aCs2CO3 as the base. | ||
Various alkyl carboxylates 3 were obtained upon subsequent esterification with different alkyl iodides 2 under rather mild reaction conditions. Methyl- as well as chloro-substituted benzo[d]oxazoles 1b and 1c were site-selectively functionalized, affording the 2-substituted carboxylic acid esters 3b–d in high yields after treatment with the corresponding alkyl iodide 2. Notably, the use of Cs2CO3 as the base under otherwise identical reaction conditions resulted in an inferior yield of only 48% for product 3b. Likewise, it is noteworthy that chloro-substituted azole 1c provided the corresponding product 3d in an excellent yield of 91%, whereas Cs2CO3 delivered only 55% of the desired ester 3d. As showcased in a representative set of C–H functionalizations, our sustainable approach was not restricted to the use of methyl iodide as the electrophile, but also allowed esterification with a variety of alkyl iodides 2. Moreover, our protocol set the stage for the C–H carboxylation of benzothiazole in a step-economical fashion. Indeed, the corresponding methyl ester 3f and hexyl ester 3g were isolated in 66% and 62% yield, respectively.
Furthermore, oxazoles 4 served as viable substrates for the C–H carboxylation, delivering the corresponding carboxylic acid derivatives 5a–d in a step- and atom-economical manner (Scheme 2).37 Intriguingly, valuable chlorine substituents on the heteroarenes were well tolerated under the optimized reaction conditions, which should prove instrumental for further late-stage diversification by inter alia cross-coupling technology.
 | ||
| Scheme 2 C–H carboxylation of oxazoles 4. aGC-conversion. | ||
Finally, we were pleased to observe that 1,3,4-oxadiazoles 6 proved to be viable substrates for the C–H carboxylation under an ambient CO2 atmosphere as well, providing the desired carboxylic acid esters 7a–c with high levels of selectivity control (Scheme 3).
 | ||
| Scheme 3 C–H carboxylation of oxadiazoles 6. | ||
Based on the literature precedents,25,38 we propose the reaction to proceed by initial reversible C–H cleavage (Scheme 4), along with subsequent C–C formation by the action of ambient CO2.
 | ||
| Scheme 4 Proposed mechanism for the C–H carboxylation. | ||
Conclusions
In summary, we have reported on the use of CO2 as an easily accessible, inexpensive, and renewable C1 source for green C–H carboxylations under transition metal-free reaction conditions. Hence, KOt-Bu enabled efficient C–H functionalizations on heteroarenes with an ample substrate scope under mild39 reaction conditions, namely at a rather low reaction temperature and ambient pressure of CO2.Acknowledgements
Support by the CaSuS (Catalysis for Sustainable Synthesis) PhD program is gratefully acknowledged.Notes and references
- Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933 CrossRef PubMed.
- T. E. Mueller and W. Leitner, Beilstein J. Org. Chem., 2015, 11, 675–677 CrossRef CAS PubMed.
- For selected recent reviews, see: (a) L. Zhang and Z. Hou, Chem. Sci., 2013, 4, 3395–3403 RSC; (b) Y. Tsuji and T. Fujiharaa, Chem. Commun., 2012, 48, 9956–9964 RSC; (c) M. Cokoja, C. Bruckmeier, B. Rieger, W. A. Herrmann and F. E. Kühn, Angew. Chem., Int. Ed., 2011, 50, 8510–8537 CrossRef CAS PubMed; (d) M. Beller and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2014, 53, 4527–4528 CrossRef CAS PubMed; (e) K. Huang, C.-L. Sun and Z.-J. Shi, Chem. Soc. Rev., 2011, 40, 2435–2452 RSC; (f) T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS PubMed , and references cited therein.
- I. I. F. Boogaerts and S. P. Nolan, Chem. Commun., 2011, 47, 3021–3024 RSC.
- L. Yang and H. Wang, ChemSusChem, 2014, 7, 962–998 CrossRef CAS PubMed.
- R. Martin and A. W. Kleij, ChemSusChem, 2011, 4, 1259–1263 CrossRef CAS PubMed.
- T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS PubMed.
- P. Braunstein, D. Matt and D. Nobel, Chem. Rev., 1988, 88, 747–764 CrossRef CAS.
- A. Boddien, F. Gaertner, C. Federsel, P. Sponholz, D. Mellmann, R. Jackstell, H. Junge and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 6411–6414 CrossRef CAS PubMed.
- K. Beydoun, G. Ghattas, K. Thenert, J. Klankermayer and W. Leitner, Angew. Chem., Int. Ed., 2014, 53, 11010–11014 CrossRef CAS PubMed.
- K. Beydoun, T. vom Stein, J. Klankermayer and W. Leitner, Angew. Chem., Int. Ed., 2013, 52, 9554–9557 CrossRef CAS PubMed.
- X. Wang, M. Nakajima and R. Martin, J. Am. Chem. Soc., 2015, 137, 8924–8927 CrossRef CAS PubMed.
- X. Wang, Y. Liu and R. Martin, J. Am. Chem. Soc., 2015, 137, 6476–6479 CrossRef CAS PubMed.
- A. Correa and R. Martin, J. Am. Chem. Soc., 2009, 131, 15974–15975 CrossRef CAS PubMed.
- L. Ackermann, A. R. Kapdi, H. K. Potukuchi and S. I. Kozhushkov, in Handbook of Green Chemistry, ed. C.-J. Li, Wiley-VCH, Weinheim, 2012, pp. 259–305 Search PubMed.
- Representative reviews on C–H activation: (a) J. G. Kim, K. Shin and S. Chang, Top. Organomet. Chem., 2016, 55, 29–51 CrossRef; (b) M. Moselage, J. Li and L. Ackermann, ACS Catal., 2016, 6, 498–525 CrossRef CAS; (c) O. Daugulis, J. Roane and L. D. Tran, Acc. Chem. Res., 2015, 48, 1053–1064 CrossRef CAS PubMed; (d) Y. Segawa, T. Maekawa and K. Itami, Angew. Chem., Int. Ed., 2015, 54, 66–81 CrossRef CAS PubMed; (e) N. Kuhl, N. Schroeder and F. Glorius, Adv. Synth. Catal., 2014, 356, 1443–1460 CrossRef CAS; (f) S. A. Girard, T. Knauber and C.-J. Li, Angew. Chem., Int. Ed., 2014, 53, 74–100 CrossRef CAS PubMed; (g) J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369–375 CrossRef CAS PubMed; (h) S. R. Neufeldt and M. S. Sanford, Acc. Chem. Res., 2012, 45, 936–946 CrossRef CAS PubMed; (i) T. Satoh and M. Miura, Chem. – Eur. J., 2010, 16, 11212–11222 CrossRef CAS PubMed; (j) L. Ackermann, R. Vicente and A. Kapdi, Angew. Chem., Int. Ed., 2009, 48, 9792–9826 CrossRef CAS PubMed; (k) R. G. Bergman, Nature, 2007, 446, 391–393 CrossRef CAS PubMed , and references cited therein.
- A. Uhe, M. Hoelscher and W. Leitner, Chem. – Eur. J., 2012, 18, 170–177 CrossRef CAS PubMed.
- S. D. Stoychev, C. M. Conifer, A. Uhe, M. Hoelscher and W. Leitner, Dalton Trans., 2014, 43, 11180–11189 RSC.
- H. Mizuno, J. Takaya and N. Iwasawa, J. Am. Chem. Soc., 2011, 133, 1251–1253 CrossRef CAS PubMed.
- T. Suga, H. Mizuno, J. Takaya and N. Iwasawa, Chem. Commun., 2014, 50, 14360–14363 RSC.
- K. Sasano, J. Takaya and N. Iwasawa, J. Am. Chem. Soc., 2013, 135, 10954–10957 CrossRef CAS PubMed.
- I. I. F. Boogaerts, G. C. Fortman, M. R. L. Furst, C. S. J. Cazin and S. P. Nolan, Angew. Chem., Int. Ed., 2010, 49, 8674–8677 CrossRef CAS PubMed.
- I. I. F. Boogaerts and S. P. Nolan, J. Am. Chem. Soc., 2010, 132, 8858–8859 CrossRef CAS PubMed.
- L. Zhang, J. Cheng, T. Ohishi and Z. Hou, Angew. Chem., Int. Ed., 2010, 49, 8670–8673 CrossRef CAS PubMed.
- O. Vechorkin, N. Hirt and X. Hu, Org. Lett., 2010, 12, 3567–3569 CrossRef CAS PubMed.
- Y. Li, T. Yan, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2014, 53, 10476–10480 CrossRef CAS PubMed.
- Y. Li, I. Sorribes, T. Yan, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 12156–12160 CrossRef CAS PubMed.
- L. Ackermann, Angew. Chem., Int. Ed., 2011, 50, 3842–3844 CrossRef CAS PubMed.
- L. Ackermann, Synlett, 2007, 507–526 CrossRef CAS.
- L. Ackermann, Acc. Chem. Res., 2014, 47, 281–295 CrossRef CAS PubMed.
- S. Fenner, PhD thesis, Georg-August-University, 2012.
- L. Zhang, J. Cheng, T. Ohishi and Z. Hou, Angew. Chem., Int. Ed., 2010, 49, 8670–8673 CrossRef CAS PubMed.
- KOt-Bu was purchased from Sigma-Aldrich, where it was sublimed and analyzed by ICP-MS, which revealed only trace amounts of transition metals (inter alia <0.5 ppm Fe; <0.1 ppm Cu, Pd, Ni).
- (a) For representative pKa values, see http://www.chem.wisc.edu/areas/reich/pkatable/index.htm; (b) For calculated pKa values, see: K. Shen, Y. Fu, J.-N. Li, L. Liu and Q.-X. Guo, Tetrahedron, 2007, 63, 1568–1576 CrossRef CAS, and references cited therein. (c) For DFT studies on carboxylations of heteroarenes by copper(I) complexes, see: A. Ariafard, F. Zarkoob, H. Batebi, R. Stranger and B. F. Yates, Organometallics, 2011, 30, 6218–6224 CrossRef CAS.
- J. Sherwood, M. D. Bruyn, A. Constantinou, L. Moity, C. R. McElroy, T. J. Farmer, T. Duncan, W. Raverty, A. J. Hunta and J. H. Clark, Chem. Commun., 2014, 50, 9650–9652 RSC.
- All direct carboxylation reactions were performed in new glassware using novel stirring bars.
- For detailed information see the ESI.†.
- A. Banerjee, G. R. Dick, T. Yoshino and M. W. Kanan, Nature, 2016, 531, 215–219 CrossRef CAS PubMed.
- J. Wencel-Delord, T. Droege, F. Liu and F. Glorius, Chem. Soc. Rev., 2011, 40, 4740–4761 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data, and 1H and 13C NMR spectra for products. See DOI: 10.1039/c6gc00200e |
| ‡ Representative procedure for the C–H carboxylation using CO2: synthesis of methylbenzo[d]oxazole-2-carboxylate (3a): a mixture of 1a (118 mg, 0.99 mmol), KOt-Bu (135 mg, 1.20 mmol) and DMF (5.0 mL) was degassed in a Schlenk-tube. The Schlenk-tube was then flushed with CO2via a balloon and CO2 was bubbled through the reaction mixture for 10–20 minutes. After removal of the balloon, the reaction mixture was heated to 100 °C for 18 h. After cooling to 65 °C, 2a (3.00 equiv.) was added and the reaction mixture was stirred at 65 °C for 2 h. At ambient temperature, the reaction mixture was diluted with H2O (25 mL) and Et2O (25 mL). The aqueous layer was extracted with Et2O (3 × 25 mL) and the combined organic layers were dried over Na2SO4. Purification by column chromatography (n-pentane/Et2O = 20/1 → 10/1 → 7/1 → 5/1) yielded 3a (141 mg, 80%) as a colorless solid. |
| This journal is © The Royal Society of Chemistry 2016 |