
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Application of mild autohydrolysis to facilitate the dissolution of wood chips in direct-dissolution solvents†
Somdatta
Deb
a,
Sara R.
Labafzadeh
a,
Unna
Liimatainen
a,
Arno
Parviainen
a,
Lauri K. J.
Hauru
b,
Shoaib
Azhar
c,
Martin
Lawoko
c,
Tuomas
Kulomaa
a,
Tia
Kakko
a,
Juha
Fiskari
a,
Marc
Borrega
b,
Herbert
Sixta
b,
Ilkka
Kilpeläinen
*a and
Alistair W. T.
King
*a
aLaboratory of Organic Chemistry, Department of Chemistry, University of Helsinki A.I. Virtasen aukio 1 (Chemicum), P.O. Box 55, 00014 Helsinki, Finland. E-mail: alistair.king@helsinki.fi; ilkka.kilpeläinen@helsinki.fi; Tel: +358505279446
bDepartment of Forest Products Technology, School of Chemical Technology, Aalto University, Finland
cWallenberg Wood Science Center (WWSC), Department of Fiber and Polymer Technology, Royal Institute of Technology (KTH), Stockholm, Sweden
First published on 11th February 2016
Abstract
Wood is not fully soluble in current non-derivatising direct-dissolution solvents, contrary to the many reports in the literature quoting wood ‘dissolution’ in ionic liquids. Herein, we demonstrate that the application of autohydrolysis, as a green and economical wood pre-treatment method, allows for a massive increase in solubility compared to untreated wood. This is demonstrated by the application of two derivitising methods (phosphitylation and acetylation), followed by NMR analysis, in the cellulose-dissolving ionic liquids 1-allyl-3-methylimidazolium chloride ([amim]Cl) and 1,5-diazabicyclo[4.3.0]non-5-enium acetate ([DBNH][OAc]). In addition, the non-derivitising tetrabutylphosphonium acetate ([P4444][OAc])![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DMSO-d6 electrolyte also allowed for dissolution of the autohydrolysed wood samples. By combination of different particle sizes and P-factors (autohydrolysis intensity), it has been clearly demonstrated that the solubility of even wood chips can be drastically increased by application of autohydrolysis. The physiochemical factors affecting wood solubility after autohydrolysis are also discussed.
:DMSO-d6 electrolyte also allowed for dissolution of the autohydrolysed wood samples. By combination of different particle sizes and P-factors (autohydrolysis intensity), it has been clearly demonstrated that the solubility of even wood chips can be drastically increased by application of autohydrolysis. The physiochemical factors affecting wood solubility after autohydrolysis are also discussed.
Introduction
Wood is often reported to be the most abundant renewable feedstock for future production of materials, chemicals and energy. Wood is known to be ‘recalcitrant’ towards chemical conversion but the well-documented cellulose dissolution capability of basic ionic liquids1–4 and electrolytes5–7 is commonly used to justify the ability to dissolve wood in similar solvents. Kilpeläinen et al.8 showed that wood can be dissolved and chemically modified from chloride ionic liquids, such as [amim]Cl, but the degree of degradation caused during these dissolution and modification steps was not apparent. At least a highly acetylated and chloroform-soluble polymeric material was identified by NMR analysis. However, Kyllönen et al.9 have subsequently shown that wood is simply not soluble in non-derivatising direct-dissolution solvents, such as ionic liquids, without some kind of mechanical or chemical pre-treatment. It is suggested that this is in part due to the lignin–carbohydrate complexes (LCCs).10–13 These are often regarded to be covalent linkages between mainly hemicelluloses and lignin and render the LC matrix resistant to dissolution. However, the presence of LCCs, as covalent linkages, in native (untreated) wood is disputed.In any case, in order to maintain the molecular weight (MW) of the constituent polymers during homogenisation, methods for selective deconstruction of the wood matrix are required. This would allow for the application of these high MW polymers in a wide range of potential material applications, i.e. preparation of synthetic regenerated fibres, films, thermoplastics, compositing and formation of polymeric blends, before or after further chemical modification.
Previously Hauru et al.14 showed that a better solubilisation of wood and more efficient fractionation was possible after initial autohydrolysis pre-treatment, followed by ionic liquid fractionation, compared to the untreated birch wood. This was demonstrated for particle sizes similar to sawdust-size. Autohydrolysis is the treatment of wood under hydrothermal conditions, by which some components of wood are extracted. Acetic acid released during the initial hydrolysis of acetate groups mainly on hemicelluloses, further catalyse hydrolysis and release of the wood components.15,16 There are several synonyms or related processes, such as hot water extraction, pressurised hot water extraction, steam pre-treatment, steam explosion, etc. All result in extraction of hemicelluloses and some lignin, to different degrees. Autohydrolysis is also implemented in industry, as a pre-treatment prior to kraft pulping, to produce dissolving-grade pulp (high purity and molecular weight cellulose designed for polymeric chemical or textile uses). The resulting pulp is typically called pre-hydrolysis kraft (PHK) pulp. As a stand-alone process there are several advantages to the use of autohydrolysis: (i) the process is relatively ‘green’ as water is used, (ii) hemicelluloses may be recovered and converted to a wide range of useful chemicals, such as xylitol, prebiotics, antioxidants, pharmaceutical and cosmetic ingredients, etc.17 Most PHK mills however simply combust the extract for its calorific value. (iii) Solid wood residue enriched in cellulose and lignin can be further utilised in alternative applications, such as in biocomposites.17
As the aim of this study is to assess the effect of autohydrolysis on the solubility of wood in non-derivatising direct-dissolution solvents, 3 experimental elements were combined: (1) preparation of wood samples of different particle sizes to assess how wider particle size ranges affect solubility, (2) autohydrolysis of wood samples to varying degrees to assess how the intensity of autohydrolysis affects solubility and (3) correct choice of analytical procedures to quantify wood solubility. This last point was satisfied by using ionic liquids to dissolve/swell the wood samples, prior to NMR analysis. Two of these methods also involved chemical derivatisation and one did not, to demonstrate that dissolution was not an artefact of the chemical modification steps. Therefore, the goals of the present manuscript concern the investigation of autohydrolysis, as a sustainable wood pre-treatment method, to allow for much more thorough dissolution and chemical modification of wood chips, as the most suitable low-cost feedstock for bulk industrial application.
Experimental
Various wood samples of different particle sizes were prepared by combinations of Wiley milling, sieving and planetary milling steps (Fig. 1). | ||
| Fig. 1 Methods for the production of the particle-sizes used in this study. | ||
Wood samples: ‘Sawdusts’
Silver birch (Betula pendula) chips and autohydrolysed birch chips were Wiley milled to pass a 22-mesh (1 mm) sieve.Wood samples: planetary milled wood
Soxhlet-extracted (acetone, 24 h) Norway spruce (Picea abies) and birch wood chips were Wiley milled to pass a 40-mesh (0.5 mm) screen. These samples were then planetary milled using a PM400 Retsch Planetary Ball Mill, to the required degree (Fig. 1). The following conditions were used: 36 g of extractive-free wood samples were planetary ball milled at 3000 rpm from 0–24 h.Wood samples: larger particle size-ranges
First, the unextracted wood chips and autohydrolysed wood chips were Wiley milled without sieving to collect the bulk effluent (Fig. 1). This was then sized using a sieve stack (<0.16 mm, 0.16–0.4 mm, 0.4–1.0 mm, 1–3 mm, 3–6 mm and >6 mm, Fig. 2). | ||
| Fig. 2 Large particle sizes produced during Wiley milling and sieving. | ||
Wood samples: autohydrolysed wood chips
Autohydrolysed birch wood was prepared by extracting the wood chips with hot water at various intensities (by varying the extraction temperature and isothermal extraction time), according to Borrega et al.18 The intensity of hot water extractions is typically described using the P-factor, in units of hours.19 This is a measure of cumulative conversion of hemicellulose (based on hydrolysis kinetics) under the given temperature conditions and over time. It is derived by the time-dependent integration of the rate constant from a modified Arrhenius equation, using the activation energy for xylan hydrolysis (125.6 kJ mol−1 (ref. 19)). In Borrega et al.,18 pressurised hot water was re-circulated through the chip bed (1 kg of chips, 3 kg of deionised water) in a batch reactor (10 L). After the treatment, the water extract was drained and the wood residue was recovered and stored in a freezer. These were then used in the present publication after the various mechanical treatments and sieving steps. The conditions used to prepare the samples analysed in this study are presented in Table 1 and are based on Borrega et al.18| Sample | Reactor set temp. (°C) | Isothermal reaction timea (min) | Reactor heat-up time (min) | P-factor (h) |
|---|---|---|---|---|
| a This is the reaction time at the set temperature, after the reactor heat up time. | ||||
| 180:0 |
180 | 0 | 18 | 51 |
| 200:0 |
200 | 0 | 21 | 167 |
| 180:10 |
180 | 10 | 27 | 262 |
| 180:30 |
180 | 30 | 47 | 766 |
| 200:10 |
200 | 10 | 29 | 1057 |
| 220:2 |
220 | 2 | 25 | 1322 |
| 200:30 |
200 | 30 | 49 | 3600 |
| 220:25 |
220 | 25 | 47 | 8166 |
31P NMR procedure
Preparation of [amim]Cl and the 31P NMR analysis were performed according to Kyllönen et al. (ESI).9 Briefly, wood samples were heated at various times and temperatures in dry [amim]Cl. This allows for significant swelling of the biomass and complete dissolution in some cases. Hydroxyl groups were then functionalized as phosphite esters (Fig. 3) and the whole spectral region (151.5–133 ppm) was then integrated against the internal standard to give a value of ‘available hydroxyls’ (mmol g−1). This is essentially a measure of the solubility of the wood samples as those hydroxyl groups that are still in the solid and gel states are not visible using this solution-state NMR technique. For the >3 mm particle size range the particles may have required splitting to fit into an 8 ml sample vial for the derivatisation reaction. | ||
| Fig. 3 31P NMR analysis procedure for quantifying wood hydroxyls. | ||
Ionic liquid acetylation and NMR analysis
Acetylation was performed by dissolving the wood samples in [DBNH][OAc] followed by acetylation with isopropenyl acetate (Fig. 4). [DBNH][OAc] was prepared according to Parviainen et al. (1:1 addition of DBN and acetic acid in the absence of solvent).3 Freshly prepared [DBNH][OAc] (1.90 g) was then added to the wood samples (100 mg) in a screw-top glass vial and heated, with stirring at 130 °C, for 1 h under argon. The mixture was allowed to cool to 80 °C and isopropenyl acetate (670 μl) was added. The reaction mixture was then stirred vigorously with a spatula and after a few minutes the vial was left in an oil bath at 80 °C for 30 min. Water (50 ml) was added and the mixture was refluxed for 1 h to remove traces of the ionic liquid. The product was filtered and dried under vacuum. All samples had changed from the original dark brown to a light brown colour. The product was extracted with chloroform (20 ml) with gentle heating and filtered through a G3 glass sinter. Chloroform was removed by rotary evaporation. The dried filtrate and the undissolved material were weighed and yields were determined.
 | ||
| Fig. 4 Mild acetylation procedure applied to the acetylation of autohydrolysed birch samples. | ||
The ATR-IR and 1H NMR spectra (chloroform-d1) were recorded for all samples. DEPT-edited HSQC were also recorded for some samples.
Ionic liquid electrolyte dissolution and NMR analysis
1H NMR analysis of dissolved wood was possible, under non-derivatising conditions, through the use of [P4444][OAc] (Fig. 5) as the DMSO-d6 electrolyte. [P4444][OAc]:DMSO-d6 stock solution (200 μl, 50:50 wt%) was added to the wood samples (33.8 mg) in an 8 ml sealable vial. Further DMSO-d6 (400 μl) was added and the samples were heated, with stirring, for 3 h at 100 °C. The solutions (600 μl) were transferred to a 5 mm NMR tube while hot, via a needle and syringe. 1H NMR spectra (at 65 °C) were recorded for all solutions. [P4444][OAc] was prepared according to Holding et al. (ESI).7
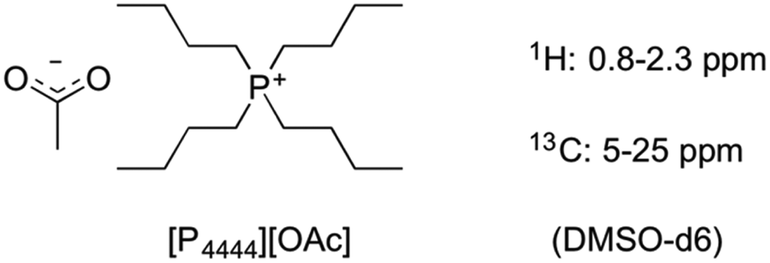 | ||
| Fig. 5 Structure of [P4444][OAc] used for NMR analysis of wood samples as the DMSO-d6 electrolyte under non-derivatising conditions. | ||
Results and discussion
Solubility of unextracted wood (low particle size-range)
Both spruce and birch were planetary milled from the sawdust and subjected to the 31P NMR analysis conditions. The results show that only after extended milling times are the samples fully soluble (Fig. 6), with ∼12 mmol g−1 representing the total number of hydroxyl groups in wood. This is consistent with what was published previously for Norway spruce by Kyllönen et al.9 The results show that for both hard and softwood species mechanical degradation is required to reach full solubility under mild dissolution conditions (90 °C, 3 h) in [amim]Cl. The solubility was also investigated as a function of temperature and time (Fig. 7). If the samples are heated at 100 °C or below over an extended period there is no increase in the available hydroxyls as a function of time.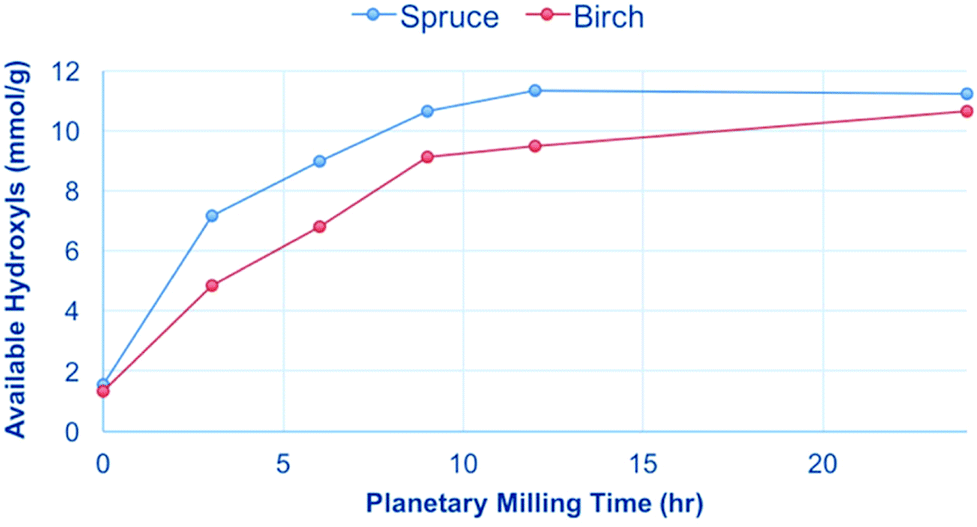 | ||
| Fig. 6 Increase in solubility of spruce and birch after extended planetary milling, as determined by [amim]Cl dissolution and 31P NMR analysis.9 Solubility is represented by the ‘available hydroxyls’ corresponding to the number of measurable phosphite esters. | ||
 | ||
| Fig. 7 Solubility of birch wood sawdust after heating in [amim]Cl at different temperatures over 3 h heating time (left) and at 120 °C for extended heating times (right). | ||
However, if the pre-dissolution temperature is increased from 90 °C to 140 °C, for a dissolution time of 3 h the solubility increases. If the dissolution time at 120 °C is increased from 1–24 h the solubility also increases. As this increase in solubility is not observed at <100 °C this is likely due to degradation occurring at the elevated temperature.
This is quite consistent with early reports of the depolymerisation of cellulose or lignin in chloride ionic liquids20,21 and confirms the need for lower temperatures (100 °C or lower) to preserve polymer molecular weights. This is particularly relevant for analytical procedures using [amim]Cl and other chloride anion analogues. Other analogues, in particular containing the acetate anion, are also shown to be reactive towards cellulose. This topic has been thoroughly discussed in previous reports.22,23
Solubility of autohydrolysed birch (sawdust)
To measure the effect of autohydrolysis intensity on wood solubility, autohydrolysis was then applied to birch chips according to Borrega et al. (Table 1).18 The chips were then Wiley milled to pass a 1 mm sieve (sawdust size) and were then subjected to 31P NMR analysis (Fig. 8). For comparison, the theoretical OH content was also calculated based on the chemical composition of the different biomass samples,18 before and after autohydrolysis (cellulose content, xylan content, lignin content and acetyl content). The values were determined using eqn (1) and (2):| OH(Th) = OH(Cell) + OH(Xyl) + OH(Lig) − OH(Ac) | (1) |
 | (2) |
 | ||
| Fig. 8 Solubility (available OH) of autohydrolysed sawdust samples, with increasing P-factor, compared to the theoretical available OH values from sugar, lignin and acetyl analyses.18 | ||
where OH(X) (mmol g−1) are the different theoretical hydroxyl contributions to eqn (1), %(X) are the different mass percentages of those species in the wood samples and XOH (mmol g−1) are the available hydroxyls for the different species: OH(Th) is the theoretical hydroxyl content of the whole biomass, OH(Cell) is the theoretical cellulose hydroxyl content, OH(Xyl) is the theoretical xylan hydroxyl content without acetyl functionalities and OH(Ac) is the theoretical hydroxyl content consumed by residual acetate groups. In the second equation, CellOH (18.5 mmol g−1) is the available hydroxyls per unit mass of cellulose (3/162). XylOH is the available hydroxyls per unit mass of xylan (14.2 mmol g−1) and was determined by King et al.24 based on data published by Timell concerning the sugar composition of xylan.25 LigOH is the available hydroxyls per unit mass of lignin (ESI) and are mostly predicted values based on 31P NMR analysis of milled wood lignin, after the autohydrolysis of birch wood at increasing P-factors.26 Only lower P-factors were analysed but LigOH values for higher P-factors are estimated based on the fact that higher intensity treatments cause significant condensation of lignin. AcOH is the unavailable hydroxyls per unit mass of acetyl (23.3 mmol g−1) incorporated into the biomass, as acetate esters. %(Cell) is the percentage by mass of cellulose in the samples, %(Xyl) is the percentage by mass of xylan in the samples, %(Lig) is the percentage by mass of lignin in the samples (based on Klason and acid-soluble lignin), %(Ac) is the percentage by mass of acetyl species in the biomass samples. All values are given in the ESI.†
The results in Fig. 8 clearly show that application of a P-factor close to 200 h allows for practically complete solubilisation of the sawdust samples, as opposed to the almost completely insoluble samples for the 0 h P-factor (untreated samples). P-factors of around 800–1000 h are typically those which are applied during pre-hydrolysis for PHK pulp production. Clearly mild autohydrolysis is a good economic substitute for costly planetary milling when one wishes to achieve a more complete chemical modification of wood.
Dissolution vs. ‘reactive dissolution’
The reagent used in the 31P NMR phosphitylation procedure is rather reactive and some degree of ‘reactive dissolution’ may be expected. Reactive dissolution has been observed previously for cellulose modification where cellulose can be dissolved, in the absence of direct-dissolution solvents, particularly using acid chlorides as acylation reagents.27,28 However, in this context we wish also to avoid other possible degradation effects produced by the high reactivity or the phosphitylation reagent. Therefore, two additional mild procedures were tested: (1) acetylation in [DBNH][OAc] with isopropenyl acetate, and (2) direct-dissolution into [P4444][OAc]:DMSO-d6. NMR analysis was performed on the acetylated materials after extraction with chloroform and also on the biomass dissolved in the [P4444][OAc]:DMSO-d6 electrolyte solutions. [DBNH][OAc] has been used previously for the acetylation of hemicelluloses, with acetic anhydride as the reagent.29 An even milder procedure has however been developed using the lower reactivity acetate ester, isopropenyl acetate (Fig. 9).30 Acetylation using these reagents is expected to avoid any issues with reactive dissolution. The sawdust samples (5 wt%) were first dissolved over 1 h at 130 °C to pre-swell the biomass. Some degradation is expected under these conditions but is not expected to be significant due to the shorter heating times and degradation seems to occur to a much lesser extent in [DBNH][OAc], compared to chloride ionic liquids. However, no accurate study has been done on this to date. Isopropenyl acetate was then introduced and the reaction quenched. The filtered and dried products were then extracted with chloroform to give an additional measure of the solubility of the samples. The yields after acetylation and chloroform extraction are given in Fig. 9. Here we can clearly see that there is a dramatic increase in the chloroform solubility of the acetylated autohydrolysed samples, compared to the untreated samples. The weight percent gains (WPGs) compared to the theoretical WPGs are all above 75%, even for the untreated sample. This nicely corresponds with the ATR-IR analysis (ESI†) showing high degrees of acetylation for all samples. At intermediate P-factors the WPGs are almost 100% of the theoretical WPGs indicating more thorough acetylation. At higher P-factors (>3000 h) the yields slightly decrease again indicating that further degradation of the samples may reduce yields, after aqueous precipitation. It is commonly known that low molecular weight samples may not fully precipitate upon the addition of a non-solvent. However, as the samples were only acetylated on a 100 mg scale, there may also be error due to unavoidable losses during filtration and transfer of samples. Above a P-factor of around 260 h the chloroform-soluble portion of the recovered acetylated material reaches 93%, compared to 14% for the untreated wood sample. As with the 31P NMR analysis this indicates a very significant increase in the solubility of wood after an autohydrolysis P-factor of around 200 h. All samples after extraction with chloroform and drying in a rotary evaporator are film-forming (ESI†). For direct comparison of the [amim]Cl-31P NMR results with the [DBNH][OAc]-acetylation results, the 31P NMR results can be plotted against the acetylation results (ESI†). While some error exists, clearly there is a strong correlation between the two analytical procedures, indicating that reactive dissolution during the phosphitylation procedure does not have a significant effect when dissolving the minimally treated (autohydrolysed) samples. However, the yields from acetylation are not close to 100%, as in the case of the 31P NMR procedure indicating that there is some effect. Importantly the increase in solubility on going from untreated up to a P-factor of around 200 h is significant using both procedures. NMR analysis was performed on the acetylated materials (Fig. 10). This included 1H NMR on all acetylated samples and DEPT-edited HSQC on the untreated sample (0 h P-factor) and the sample autohydrolysed to a P-factor of 766 h. From Fig. 10 we can see that as the P-factor increases the cellulose in the samples (1H: C3 – 5.07 ppm, C2 – 4.80 ppm, C1 – 4.42 ppm, gem-C6 – 4.38 & 4.06 ppm, C4 – 3.71 ppm, C5 – 3.54 ppm) clearly becomes soluble. A relatively significant peak which may correspond to the lignin methoxy (1H/13C: 3.76/56.25 ppm) is present. Some unidentified peaks upfield of the cellulose backbone (1H: 3.5 ppm) are also present. At this point in the 766 h P-factor sample these are thought to be lignin resonances which are possibly methylene species (CH2), alpha to both ketone and oxygen. This is however speculative without much more detailed analyses. For the untreated sample (0 h P-factor) there are more unknown resonances that may be related to extractives, saccharides and lignin. While there are peaks that roughly correspond to acetylated cellulose, these are rather weak, relative to the unknown resonances, indicating minimal solubilisation of cellulose during the acetylation and chloroform extraction. There are also several unidentified saccharide resonances, consistent with the solubilisation of mainly the lower molecular weight fractions and not the cellulose.
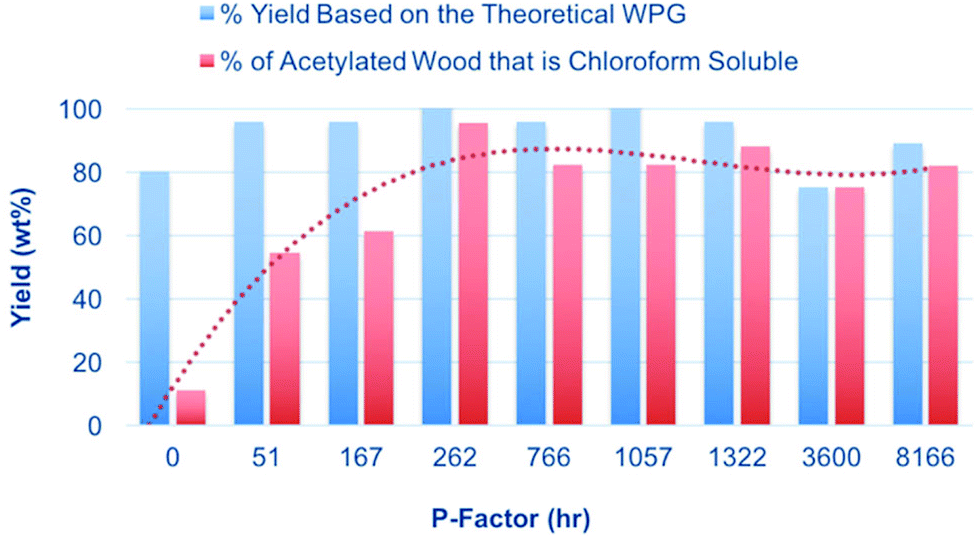 | ||
| Fig. 9 Yields after [DBNH][OAc] dissolution, acetylation (Fig. 5) and solubilisation of autohydrolysed sawdust samples into chloroform. | ||
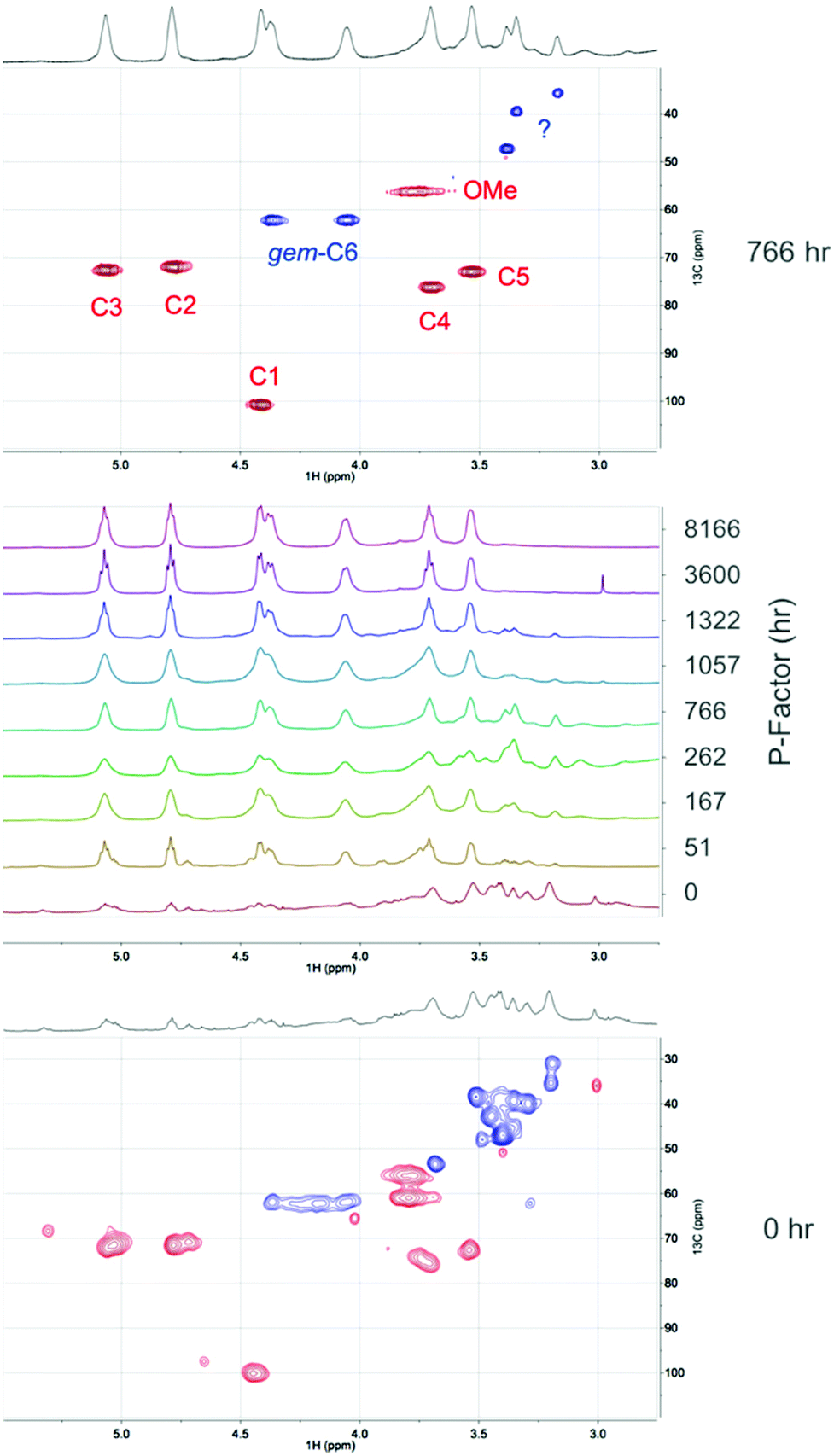 | ||
| Fig. 10 1H NMR saccharide backbone region for the [DBNH][OAc]-acetylated CDCl3-soluble fractions for the different P-factors of autohydrolysed birch wood (middle). The 2D spectra are DEPT-edited (CH & CH3 in red, CH2 in blue) HSQC spectra (above and below). Assignments in the 766 h P-factor HSQC are for the cellulose backbone peaks. | ||
Dissolution under ‘non-derivatising’ conditions
As a further demonstration of how reactive dissolution does not effect solubility at lower P-factors, the samples were dissolved in the ionic liquid electrolyte [P4444][OAc]:DMSO-d6. Similar electrolytes have been used for the analysis of high molecular weight pulp samples, containing mainly cellulose.7,31 All components of wood are soluble in these electrolytes, as demonstrated previously by Holding et al.7 Therefore, if it is truly possible to solubilize autohydrolysed wood, this medium should allow for this. 5 wt% samples were dissolved into the electrolyte solutions and 1H NMR analysis was performed (Fig. 11). Clearly for the extended P-factors the cellulose in the samples is dissolving, indicating a much more complete solubilisation of the samples. For the 0 and 51 h P-factors little cellulose is visible indicating poor solubility of the whole biomass sample. This is entirely consistent with the 31P and acetylation results.
 | ||
| Fig. 11
1H NMR saccharide backbone region for the different P-factors of autohydrolysed birch wood dissolved in [P4444][OAc]:DMSO-d6. The concentrations are the same for all spectra and normalised against the [P4444][OAc] signals, i.e. solubility is represented by cellulose backbone signal intensity. The assignments are for the different cellulose backbone 1H signals, according to Holding et al.7,31 | ||
Solubility of autohydrolysed birch (chips)
From a techno-economic standpoint, sawdust is not a suitable feedstock to use for bulk application. While it is readily available as a low-cost by-product from saw mills, it is only available on a limited scale industrially. Industrial chips are much more common and at a much lower cost compared to the additional milling steps required to reach lower particle sizes, such as sawdust. As such, we wished to investigate larger particle-size ranges to see if they could also be solubilised after autohydrolysis. To achieve this, the starting untreated and autohydrolysed wood chips (1057 h P-factor) were Wiley milled and sieved to give different particle size ranges (Fig. 2). These were then subjected to [amim]Cl dissolution, phosphite ester derivatisation and the 31P NMR analysis procedure (Fig. 12). From the results presented in Fig. 12 it is clear that decreasing particle size slightly improves the solubility of the untreated chips. However, after autohydrolysis to a P-factor of 1057 h, all particle size ranges are equally soluble. Some error exists but the results clearly show that autohydrolysis can be used to drastically enhance the solubility of wood chips over the untreated chips, with the untreated chips being practically insoluble.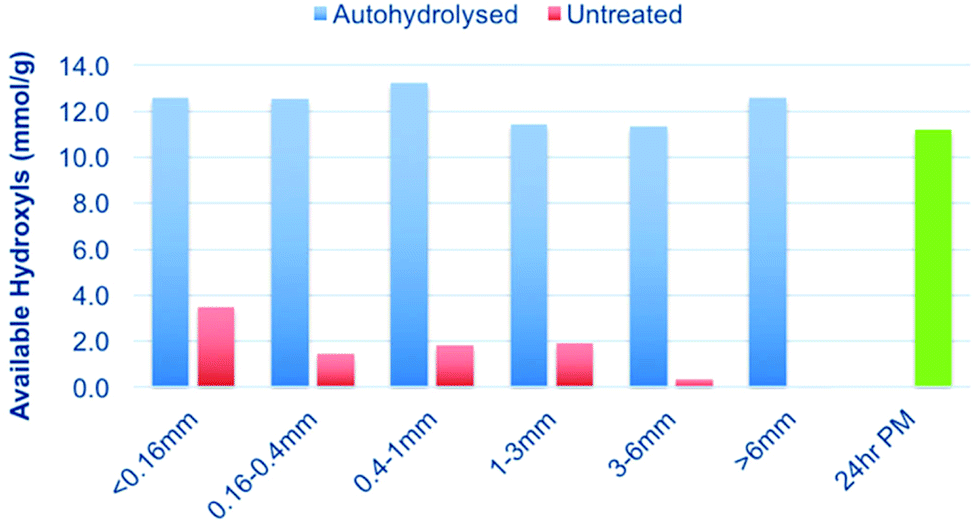 | ||
| Fig. 12 Solubility (available OH) of increasing particle size ranges, up to chip-size, after autohydrolysis of the birch wood chips at a P-factor of 1057 h. 24 h PM refers to the experimental value for the untreated birch sample which was fully soluble, after 24 h of planetary milling. | ||
Mechanism causing increased solubility
In Kyllönen et al.9 it was argued that the insolubility of wood was due to the recalcitrant ‘lignin–carbohydrate matrix (LCM)’. LCCs are of course implicated in this but it was not specifically argued that these were actual branching covalent linkages, between polysaccharide and lignin. Extensive covalent LCCs would bind the wood matrix components to rapidly approach a molecular weight equivalent of Avogadro's number, for that molar quantity of material (much too high MW to be soluble). As mentioned in the previous article the scepticism about the occurrence of covalent LCCs in native (unprocessed) woods is mainly due to the inability to isolate LCC fractions for high resolution analyses, without the need for chemically or mechanically harsh treatments. This is the Schrödinger's cat of wood chemistry. As a consequence, we would like to present several options for discussion concerning the solubility of autohydrolysed wood:(1) Increased porosity – it has been known for some time that during autohydrolysis of wood, hemicelluloses and lignin are extracted. Smiljanski and Stanković32 have shown that there is an increase in the sub-microscopic porosity of beech wood (Fagus moesica) as glucuronoxylans are extracted during pre-hydrolysis. This increase in porosity would clearly allow for much more rapid penetration of the wood matrix with reagents.
(2) Increased fibrillation – after autohydrolysis the wood chips become much softer and easier to break apart, parallel with the fibre orientation. In addition, the chips fibrillate quite rapidly in ionic liquid ([amim]Cl and [DBNH][OAc]) as opposed to unextracted chips, which essentially do not fibrillate at all. This effect clearly also rapidly increases surface area, similar to increased porosity. This is mainly a result of the removal of acid-soluble lignin and pectins from the middle lamella, which acts as a cement keeping the adjacent fibres together. It is not clear if this, combined with removal of hemicellulose (binder), is enough to allow for complete dissolution of the remaining wood matrix.
(3) LCC breakage – autohydrolysis, by definition, is an acid-catalysed process for the degradation and extraction of hemicelluloses. Therefore, acid-labile linkages are expected to be degraded during this process. The proposed LCC structures (benzyl ethers, benzyl esters and phenyl glycosides)10–13 will have variable stabilities under pH conditions. If they are actually present in native wood then particular types are expected to be cleaved, potentially diminishing the overall molecular weight of the LCM low enough to be rendered into solution.
(4) Lignin condensation – at higher P-factors lignin starts to condense.26 This may have the opposite effect of LCC breakage, i.e. leading to reduced solubility of the matrix, if the condensed fragments become large enough in size. Therefore, keeping to low P-factors will likely be beneficial for many reasons.
Aside from the structural changes in wood, ionic liquids themselves are often the culprits when it comes to enhanced ‘solubility’ of wood in seemingly non-derivatising ionic liquids. There is now quite much evidence demonstrating that certain ionic liquids react with wood functionalities or catalyse reactions. This reactivity often goes hand-in-hand with the cellulose dissolution capability as both depend on anion basicity.22 A good example of this is the conjugation of 1-ethyl-3-methylimidazolium acetate ([emim][OAc]) with reducing ends of cellulose.33,34 This has recently been developed by Clough et al.23 where it has been shown that [emim][OAc] allows for conversion of the carbon in the glucopyranose ring into an imidazolium C1–formaldehyde adduct. 1-Butyl-3-methylimidazolium chloride ([bmim]Cl) was shown to be an alternative structure that did not form this adduct, presumably due to the reduced basicity of the anion.22 Similar reports can also be found for reaction with lignin.
Potential applications and sustainability
Autohydrolysis in itself is a pre-treatment method performed in water on a 2nd generation bio-based feedstock. It is both green and sustainable as it is already implemented for the production of PHK. It yields an extractive fraction which can be combusted for its calorific value (steam or electricity generation) or used for chemicals production. Under mild autohydrolysis conditions this fraction typically contains acetic acid and hemicelluloses, both valuable chemicals. In terms of chemistry, the hemicellulose (xylan) fraction may be chemically modified to polymeric chemicals or it is also already industrially converted to xylose (for xylitol production) and furfural. Polymeric hemicelluloses also already have wide application in the food industry. Regarding the sustainability of the subsequent direct-dissolution step on the residual chips, the most obvious applications are in further chemical modification of the wood matrix or regeneration to films and fibres, e.g. a lyocell using the ionic liquid as the direct-dissolution solvent.35,36 Provided low toxicity ionic liquids and reagents are used, the main stumbling block to sustainability is the removal of water from ionic liquids during the recycling stage, as typically water is used to regenerate the biomass. Therefore, the value of the product must be high enough to offset this cost or advances made that either minimise the use or energy consumption of water use.Conclusions
In this paper, we demonstrate that mild autohydrolysis can be applied to allow for almost complete solubilisation of wood chips in ionic liquid. Without the autohydrolysis pre-treatment both wood chips and particle sizes much lower than sawdust (<0.16 mm) are mostly insoluble, until you get down to the very small particle sizes that extended planetary milling affords. As autohydrolysis is an existing green and economical pre-treatment method, typically applied prior to kraft pulping, this could now open the door to much more thorough chemical modification of wood chips, as a low cost feedstock. This could most obviously lead to new applications in thermoplastics, films, polymer blends, composite materials etc. It could also lead to novel dissolution/regeneration applications, such as a wood-based lyocell process where high tensile strength wood fibres are regenerated. The factors affecting the solubility of autohydrolysed chips over unextracted chips are also discussed, with no significant conclusions other than to further clarify the different potential physiochemical factors involved. Understanding all of these processes is required to make predictions about wood recalcitrance. However, clearly there will be interesting opportunities in the future to start dissecting the different contributions to the recalcitrance of wood. New economical pre-treatments, e.g. mild acid/base, ammonolysis, electron beam, redox processes, etc., may also prove to be more effective or economical than autohydrolysis and offer different application areas.Acknowledgements
We would like to acknowledge FIBIC (The Finnish Bioeconomy Cluster) Oy, now known as CLIC Innovation, for funding under the FuBio (Future Biorefinery) program. We would also like to thank the WWSC (Wallenberg Wood Science Center) for additional financial support.References
- R. P. Swatloski, S. K. Spear, J. D. Holbrey and R. D. Rogers, J. Am. Chem. Soc., 2002, 124, 4974–4975 CrossRef CAS PubMed.
- Y. Fukaya, K. Hayashi, M. Wada and H. Ohno, Green Chem., 2008, 10, 44–46 RSC.
- A. Parviainen, A. W. T. King, I. Mutikainen, M. Hummel, C. Selg, L. K. J. Hauru, H. Sixta and I. Kilpeläinen, ChemSusChem, 2013, 6, 2161–2169 CrossRef CAS PubMed.
- A. W. T. King, J. Asikkala, I. Mutikainen, P. Järvi and I. Kilpeläinen, Angew. Chem., Int. Ed., 2011, 50, 6301–6305 CrossRef CAS PubMed.
- S. Spange, A. Reuter, E. Vilsmeier, T. Heinze, D. Keutel and W. Linert, Polym. Chem., 1998, 36, 1945 CrossRef CAS.
- R. Rinaldi, Chem. Commun., 2011, 47, 511–513 RSC.
- A. J. Holding, M. Heikkilä, I. Kilpeläinen and A. W. T. King, ChemSusChem, 2014, 7, 1422–1434 CrossRef CAS PubMed.
- I. Kilpeläinen, H. Xie, A. W. T. King, M. Granström, S. Heikkinen and D. S. Argyropoulos, J. Agric. Food Chem., 2007, 55, 9142–9148 CrossRef PubMed.
- L. Kyllönen, A. Parviainen, S. Deb, M. Lawoko, M. Gorlov, I. Kilpeläinen and A. W. T. King, Green Chem., 2013, 15, 2374–2378 RSC.
- D. Fengel and G. Wegener, Lignin-polysaccharide complexes, in Wood chemistry, ultrastructure and reactions, W. De Gruyter, Berlin, 1984, pp. 167–174 Search PubMed.
- M. Lawoko, G. Henriksson and G. Gellerstedt, Biomacromolecules, 2005, 6, 3467 CrossRef CAS PubMed.
- M. Balakshin, E. Capanema, H. Gracz, H.-M. Chang and H. Jameel, Planta, 2011, 233, 1097–1110 CrossRef CAS PubMed.
- M. Y. Balakshin, E. Capanema and H.-M. Chang, Holzforschung, 2007, 61, 1–7 CrossRef CAS.
- L. K. J. Hauru, Y. Ma, M. Hummel, M. Alekhina, A. W. T. King, I. Kilpeläinen, P. A. Penttila, R. Serimaa and H. Sixta, RSC Adv., 2013, 3, 16365–16373 RSC.
- G. Garrote, H. Domiguez and J. C. Parajo, Holz Roh– Werkst., 1999, 57, 191–202 CrossRef CAS.
- G. Garrote, H. Domiguez and J. C. Parajo, J. Chem. Technol. Biotechnol., 1999, 74, 1101–1109 CrossRef CAS.
- P. Gullon, A. Romani, C. Vila, G. Garrote and J. C. Parajo, Biofuels, Bioprod. Biorefin., 2012, 6, 219–232 CrossRef CAS.
- M. Borrega, L. K. Tolonen, F. Bardot, L. Testova and H. Sixta, Bioresour. Technol., 2013, 135, 665–671 CrossRef CAS PubMed.
- H. Sixta, A. Potthast and A. W. Krotschek, in Chemical Pulping Processes: Handbook of Pulp, ed. H. Sixta, Wiley-VCH, Weinheim, 2006, vol. 1, pp. 343–345 Search PubMed.
- G. Bentivoglio, T. Röder, M. Fasching, M. Buchberger, H. Schottenberger and H. Sixta, Lenzinger Ber., 2006, 86, 154–161 CAS.
- G. Laus, G. Bentivoglio, H. Schottenberger, V. Kahlenberg, H. Kopacka, T. Röder and H. Sixta, Lenzinger Ber., 2005, 84, 71–85 CAS.
- A. W. T. King, A. Parviainen, P. Karhunen, J. Matikainen, L. K. J. Hauru, H. Sixta and I. Kilpelainen, RSC Adv., 2012, 2, 8020–8026 RSC.
- M. T. Clough, K. Geyer, P. A. Hunt, S. Son, U. Vagt and T. Welton, Green Chem., 2015, 17, 231–243 RSC.
- A. W. T. King, L. Zoia, I. Filpponen, A. Olszewska, H. Xie, I. Kilpeläinen and D. S. Argyropoulos, J. Agric. Food Chem., 2009, 57, 8236–8243 CrossRef CAS PubMed.
- T. E. Timell, Wood Sci. Technol., 1967, 1, 45–70 CrossRef CAS.
- T. Rauhala, A. W. T. King, G. Zuckerstatter, S. Suuronen and H. Sixta, Nord. Pulp Pap. Res. J., 2011, 26, 386–391 CAS.
- S. R. Labafzadeh, J. Kavakka, K. Sievänen, J. Asikkala and I. Kilpeläinen, Cellulose, 2012, 19, 1295–1304 CrossRef CAS.
- T. Kulomaa, J. Matikainen, P. Karhunen, M. Heikkilä, J. Fiskari and I. Kilpeläinen, RSC Adv., 2015, 5, 80702–80708 RSC.
- A. M. Stepan, A. W. T. King, T. Kakko, G. Toriz, I. Kilpeläinen and P. Gatenholm, Cellulose, 2013, 20, 2813–2824 CrossRef CAS.
- T. Kakko, S. Asaadi, A. W. T. King, M. Hummel, H. Sixta and I. Kilpeläinen, ISWFPC 2015 (Extended Abstract Proceedings – P8), 22–25.
- A. J. Holding, V. Mäkelä, L. Tolonen, H. Sixta, I. Kilpeläinen and A. W. T. King, ChemSusChem, 2016 DOI:10.1002/cssc.201501511.
- S. Smiljanski and S. Stanković, Cellul. Chem. Technol., 1974, 8, 283–294 CAS.
- T. Liebert and T. Heinze, Bioresources, 2008, 3, 576–601 Search PubMed.
- G. Ebner, S. Schiehser, A. Potthast and T. Rosenau, Tetrahedron Lett., 2008, 49, 7322–7324 CrossRef CAS.
- H. Sixta, A. Michud, L. Hauru, S. Asaadi, Y. Ma, A. W. T. King, I. Kilpeläinen and M. Hummel, Nord. Pulp Pap. Res. J., 2015, 30, 43–57 CAS.
- Y. Ma, S. Asaadi, L.-S. Johansson, P. Ahvenainen, M. Reza, M. Alekhina, L. Rautkari, A. Michud, L. Hauru, M. Hummel and H. Sixta, ChemSusChem, 2015, 8, 4030–4039 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6gc00183a |
| This journal is © The Royal Society of Chemistry 2016 |