
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of pyrrolidinone derivatives from aniline, an aldehyde and diethyl acetylenedicarboxylate in an ethanolic citric acid solution under ultrasound irradiation†
Hamideh
Ahankar
a,
Ali
Ramazani
*a,
Katarzyna
Ślepokura
b,
Tadeusz
Lis
b and
Sang Woo
Joo
*c
aDepartment of Chemistry, University of Zanjan, P O Box 45195-313, Zanjan, Iran. E-mail: aliramazani@gmail.com
bFaculty of Chemistry, University of Wrocław, 14 Joliot-Curie St., 50-383 Wrocław, Poland
cSchool of Mechanical Engineering, Yeungnam University, Gyeongsan 712-749, Republic of Korea. E-mail: swjoo@yu.ac.kr
First published on 11th March 2016
Abstract
The ultrasound-promoted one-pot multicomponent synthesis of substituted 3-pyrrolin-2-ones using citric acid as a green additive in a green solvent is reported. Citric acid catalyzed the reaction efficiently without the need for any other harmful organic reagents. Clean reaction profile, easy work-up procedure, excellent yields and short reaction times are some remarkable features of this method. The utilization of ultrasound irradiation makes this method potentially very useful, fast, clean and convenient.
Introduction
In recent years, there has been increasing interest in the applications of multicomponent reactions (MCRs) in modern synthetic organic chemistry. Many research studies into the synthesis of the most abundant biologically active compounds for organic, medicinal and combinatorial chemistry, and in drug discovery have been enabled by one-pot multicomponent reactions. These reactions have a highly important position in comparison with other reactions.1,2MCRs based on the synthesis of heterocyclic compounds containing a 2-pyrrolidinone skeleton have gained much importance in organic synthesis. These are famous compounds because of their vast number involved in biological activities, their huge potential for applications in drug development and their existence in natural products and the agricultural sector. 2-Pyrrolidinones are a class of 5-membered lactams with a four-carbon heterocyclic ring structure with biological interest.3 The simplest 2-pyrrolidinone is named 2-pyrrolidone; it is a common component of larger natural products and is sometimes referred to as simply pyrrolone (Fig. 1).
 | ||
| Fig. 1 Molecular structure of 2-pyrrolidinone. | ||
2-Pyrrolidinones are important compounds that are found in many pharmaceuticals (Fig. 2) and in active natural products (Fig. 3). One common biochemically important 2-pyrrolidinone is cotinine (1), an alkaloid found in tobacco that is the predominant metabolite of nicotine.4 Doxapram (2) to form doxapram hydrochloride is a respiratory stimulant; administered intravenously, doxapram stimulates an increase in tidal volume and respiratory rate.5 Ethosuximide (3) is a succinimide anticonvulsant, used mainly in the absence of seizures.6
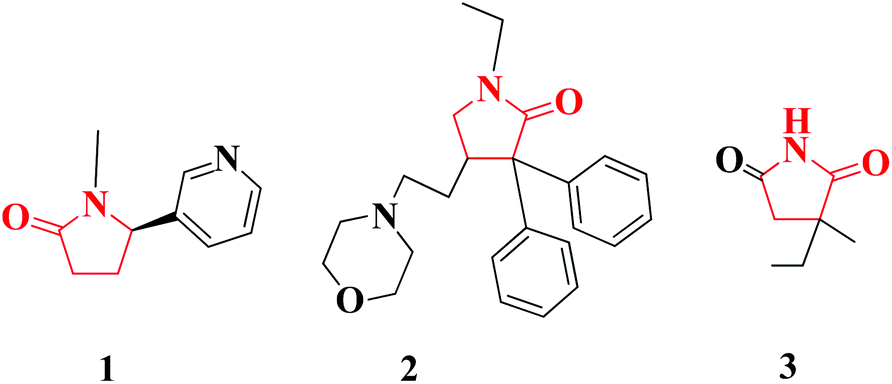 | ||
| Fig. 2 Selected drugs with a 2-pyrrolidinone moiety. | ||
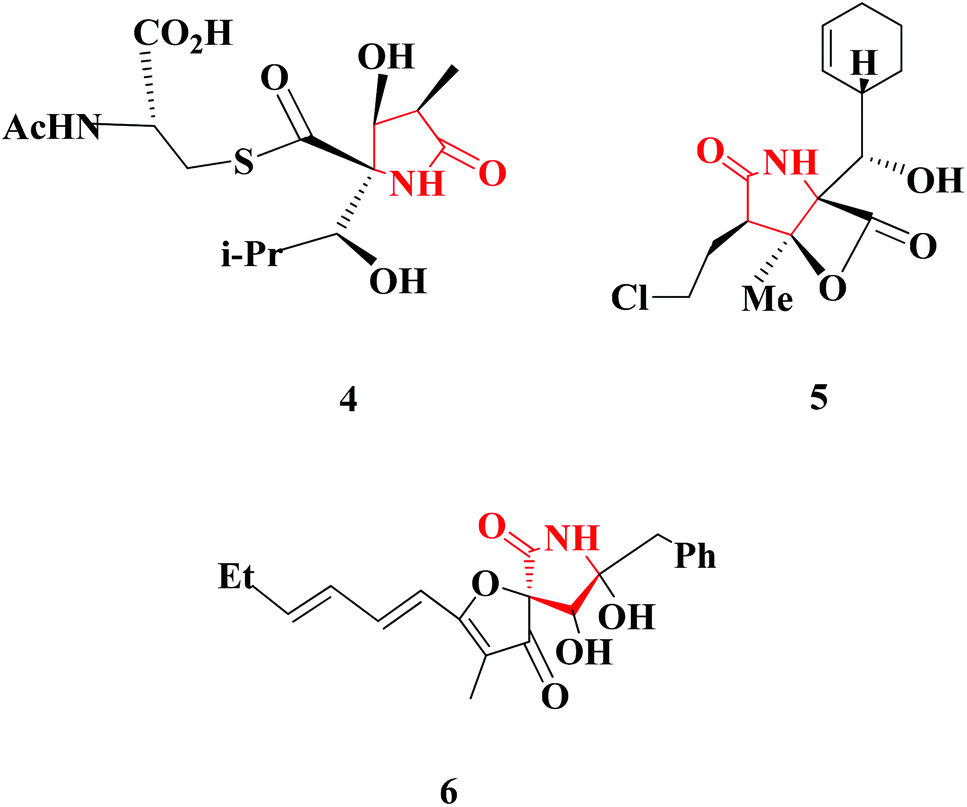 | ||
| Fig. 3 Selected natural products with a 2-pyrrolidinone moiety. | ||
Several notable examples of natural products have the 2-pyrrolidinone moiety. 2-Pyrrolidinone-containing natural products display extremely interesting biological activities. Lactacystin (4), for example, is an organic compound naturally synthesized by bacteria of the genus Streptomyces, as was first described in 1991.7 Another attractive natural product is named Salinosporamide A (5); this marine natural product is produced by the obligate marine bacteria Salinispora tropica and Salinispora arenicola, which are found in ocean sediment.8 (−)-Azaspirene (6) is a novel angiogenesis inhibitor that is isolated from the fungus Neosartorya sp.9
Substituted 3-pyrrolin-2-ones with a 2-pyrrolidinone moiety are also of use in medicinal chemistry as many derivatives have shown significant pharmacological and biological activities, as, e.g., anti-cancer agents,10 antitumours,11 HIV-1 integrase inhibitors,12 anti-microbial,13 antibacterial14 and anti-inflammatory.15 In view of the importance of substituted pyrrolidinones, various synthetic methods have been reported.16–24
Ultrasound-assisted organic synthesis is used as a modern and eco-friendly technique to accelerate organic synthesis. The chemical effects of ultrasound were first reported by Richards and Loomis in 1917.25 The use of ultrasound to accelerate reactions has proven to be a useful tool for meeting the green chemistry goals of minimization of the production of waste and the reduction of energy requirements. Ultrasound may also support cleaner reactions by improving yields and selectivities, particularly those involving free radical intermediates. Sonochemistry is widely used for improving reactions that use expensive reagents, high temperatures and prolonged reaction conditions.26 An acoustic cavitation phenomenon happens in an irradiated liquid during ultrasound-assisted reactions, which involves the formation, growth and collapse of bubbles in the liquid. This effect enforces high local temperatures and pressures to increase the rate of reactions.27
Citric acid (2-hydroxy-1,2,3-propanetricarboxylic acid) is a weak organic acid of huge industrial importance. It is a natural preservative present in citrus fruits. This acid is a nearly universal intermediate product of metabolism and its traces are found in virtually all plants and animals. It is known to be harmless to the environment and commercially available with stability toward humidity.28 Despite its great importance, only a few papers have reported on its catalytic application in organic synthesis.29–36
Green chemistry is an efficient technology that minimizes or preferably eliminates the formation of waste, avoids the use of toxic solvents and reagents and utilizes renewable raw materials as far as possible.37 The introduction of combinatorial chemistry, which has become almost universally linked with lead compound development, appears in many applications to be contrary to the principles of green chemistry as, once again, the focus is on the desired product with little concern for the overall efficiency of the process with regards to transforming reactants into products with the least possible production of waste.
Considering the importance of clean chemistry, in this study we have developed a new methodology for the synthesis of substituted 3-pyrrolin-2-ones in the presence of citric acid as a green additive in a green solvent under ultrasound irradiation (Scheme 1). However, in past reports, the existence of some drawbacks, such as long reaction times, harsh conditions, a toxic organic solvent, high temperature, high cost and low product yield have been mentioned. Ultimately, expensive, difficult to reuse, and unecofriendly additives limit the use of the reported methods. Whereas, a cleaner reaction profile, easy work-up, excellent yields and short reaction times are some of remarkable features of the proposed method.
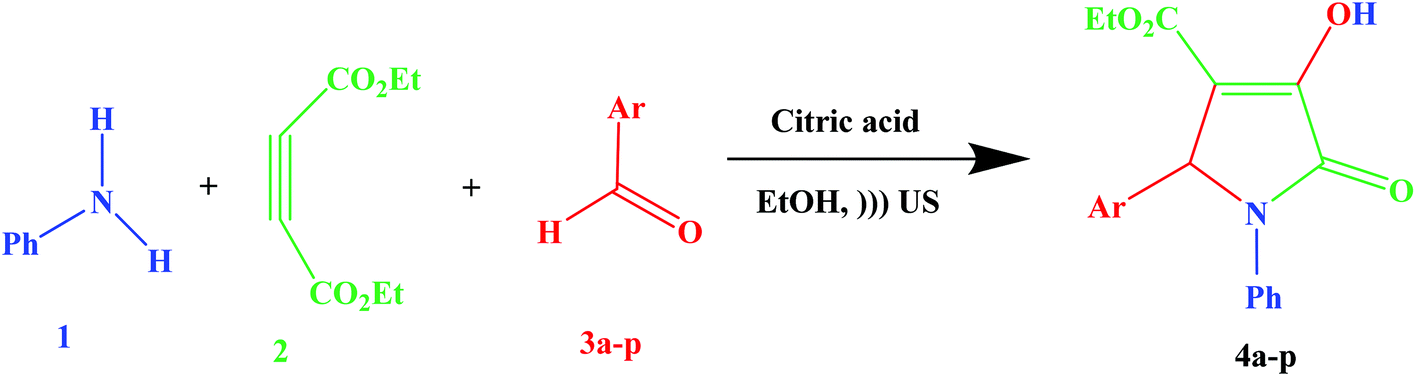 | ||
| Scheme 1 Synthesis of substituted 3-pyrrolin-2-ones under ultrasound irradiation. | ||
Results and discussion
Our research was focused on the verification of pyrrolidinones synthesis via a convenient and easy method. The effect of ultrasound irradiation in accelerating the reactions attracted our attention. Ultrasound irradiation has been established as a significant technique in synthetic organic chemistry, where it has been applied as an efficient energy and heating source for organic reactions.16,25–27 To achieve suitable conditions for the synthesis of substituted 3-pyrrolin-2-ones (4a–p), various reaction conditions were investigated for the reaction of aniline, diethyl acetylenedicarboxylate, 4-chlorobenzaldehyde and citric acid as a green additive in ethanol solvent as a model reaction. We first optimized the reaction conditions, such as the effects of solvents, additive amount and ultrasound irradiation power.First, to optimize the reaction conditions, different solvents, such as H2O, C2H5OH, C2H5OH–H2O, CH3OH, CH3CN and CH2Cl2, were tested in the synthesis of 4d as a model compound. The best result was obtained by the reaction of aniline, diethyl acetylenedicarboxylate, 4-chlorobenzaldehyde and citric acid as a green additive in ethanol solvent under ultrasound irradiation (100 W) for the preparation of 4d, which occurred in 15 min with 89% yield (Table 1, entry 2). Water, water-ethanol, methanol, dichloromethane and acetonitrile afforded moderate yields of desired products but took comparatively longer reaction time (Table 1, entries 1 and 3–6). So, ethanol was selected as the best solvent for all our further reactions.
| Entry | Solvent | Time (min) | Yieldb (%) |
|---|---|---|---|
| a Reaction conditions: aniline 1 (1 mmol), diethyl acetylenedicarboxylate 2 (1 mmol), 4-chlorobenzaldehyde 3d (1 mmol) and citric acid monohydrate (2 mmol) as a green additive in various solvents under ultrasound irradiation (100 W). b Isolated yield. | |||
| 1 | H2O | 80 | 70 |
| 2 | C2H5OH | 15 | 89 |
| 3 | H2O-C2H5OH | 60 | 78 |
| 4 | CH3OH | 35 | 72 |
| 5 | CH2Cl2 | 40 | 62 |
| 6 | CH3CN | 35 | 68 |
Then, to find the optimum amount of the citric acid additive, the model reaction of aniline 1 (1 mmol), diethyl acetylenedicarboxylate 2 (1 mmol) and 4-chlorobenzaldehyde 3d (1 mmol) for the synthesis of compound 4d (Table 5, entry 4) was carried out by varying the quantity of additive, as shown (Table 2, entries 2–4). The maximum yield of the target product was obtained when 2 mmol of additive was used. The results are summarized in Table 2.
| Entry | Additive (mmol) | Time (min) | Yieldb (%) |
|---|---|---|---|
| a Reaction conditions: aniline 1 (1 mmol), diethyl acetylenedicarboxylate 2 (1 mmol), 4-chlorobenzaldehyde 3d (1 mmol) and citric acid as a green additive in ethanol solvent under ultrasound irradiation (100 W). b Isolated yield. | |||
| 1 | — | 60 | — |
| 2 | 1.0 | 60 | 68 |
| 3 | 1.5 | 40 | 78 |
| 4 | 2.0 | 15 | 89 |
In order to verify the effect of ultrasound irradiation, we performed the reaction of aniline 1 (1 mmol), diethyl acetylenedicarboxylate 2 (1 mmol), 4-chlorobenzaldehyde 3d (1 mmol) and citric acid monohydrate (2 mmol) as a green additive in 4 ml ethanol solvent under ultrasound irradiation of different powers. After sonication at 100 W, the solution temperature was ca. 40 °C. The reaction temperature was monitored with a thermometer and the reaction was completed within 15 min (as indicated by TLC) in an 89% yield (Table 3, entry 2).
| Entry | Ultrasonic power (W) | Time (min) | Yieldb (%) |
|---|---|---|---|
| a Reaction conditions: aniline 1 (1 mmol), diethyl acetylenedicarboxylate 2 (1 mmol), 4-chlorobenzaldehyde 3d (1 mmol) and citric acid monohydrate (2 mmol) as a green additive in different ultrasonic power. b Isolated yield. | |||
| 1 | 50 | 25 | 80 |
| 2 | 100 | 15 | 89 |
| 3 | 150 | 15 | 85 |
As shown in Table 3, also a survey of the influence of ultrasonic power inputs from 50 to 150 W in the synthesis of compound 4d (Table 5, entry 4) as a model reaction was evaluated. The product yield was seen to be affected by the ultrasonic power. The best product yield was obtained when the ultrasonic power was 100 W. The reaction yield increased with the ultrasonic power at 100 W in comparison to 50 W. The reaction time of 4d did not change from 100 to 150 W, but the product yield decreased slightly. Therefore, 100 W of ultrasound irradiation was sufficient to push the reaction forward.
To delineate the role of ultrasound and the effect of cavitation on the acceleration or the thermal effect, the synthesis of compound 4d (Table 5, entry 4) was studied under different conditions, such as at room temperature for 10 h, with conventional heating at 40 °C (15, 30, 60 min) and by refluxing for different times (15, 30, 90, 180, 360 min) without ultrasonic waves irradiation. The conversions and isolated yields were calculated with respect to the starting substrate under a plot, in order to show a comparison of the various reaction times and yields of the reactions with or without sonication under different reaction conditions. The desired conditions for the reactions mixture are listed in Table 4. The residue (crude product) was used for preparation of the corresponding 1H NMR spectrum (see pages S54–S63 of ESI†). When the reaction was performed under ultrasound irradiation for 15 min, it gave 100% conversion and an 89% isolated yield (Table 4, entry 1). In addition, the conversion and isolated yield were 97% and 86% at room temperature for 10 h, respectively (Table 4, entry 2). As shown in Table 4, the residue (crude product 4d) of the reaction mixture at 40 °C (Table 4, entries 3–5) gave relatively good conversions, which could be a result of the formation of multiple intermediates, but without the final product 4d. Moreover, our investigation was performed under reflux conditions in different times in ethanol solvent (Table 4, entries 6–10). The reactions (Table 4, entries 6–8) resulted in multiple intermediates (without the final product 4d), too. To prolong the reaction time, the conversion and isolated yields were increased, albeit these were lower in comparison to in ultrasonic-induced synthesis. Thus, it is clear from the presented data (Table 4, entry 1) that ultrasound irradiation could accelerate the reaction and could afford a higher yield in comparison to room temperature, conventional heating and under reflux conditions.
| Entry | Conditions | Time (min) | Conversion (%) | Isolated yield (%) |
|---|---|---|---|---|
| a Reaction conditions: aniline 1 (1 mmol), diethyl acetylenedicarboxylate 2 (1 mmol), 4-chlorobenzaldehyde 3d (1 mmol) and citric acid monohydrate (2 mmol) as a green additive in ethanol solvent (4 ml) under different conditions and times. After the mentioned reaction time, the solvent was removed under reduced pressure and the residue was mixed with 20 ml CH2Cl2. The insoluble solid additive (citric acid monohydrate crystals) was removed from the reaction mixture by simple filtration. The solvent was removed under reduced pressure and the residue (crude product 4d) was used for the preparation of the corresponding 1H NMR spectrum (see pages S54–S63 in ESI). | ||||
| 1 | U. S. | 15 | 100 | 89 |
| 2 | R. T. | 600 | 97 | 86 |
| 3 | 40 °C | 15 | 81 | 0 |
| 4 | 40 °C | 30 | 84 | 0 |
| 5 | 40 °C | 60 | 90 | 0 |
| 6 | Reflux | 15 | 88 | 0 |
| 7 | Reflux | 30 | 94 | 0 |
| 8 | Reflux | 90 | 99 | 0 |
| 9 | Reflux | 180 | 100 | 69 |
| 10 | Reflux | 360 | 100 | 78 |
| Entry | Ar | Product | With sonicationa | Without sonicationb | ||
|---|---|---|---|---|---|---|
| Time (min) | Yieldc (%) | Time (min) | Yieldc (%) | |||
| a Reaction conditions: aniline 1 (1 mmol), diethyl acetylenedicarboxylate 2 (1 mmol), aldehyde 3a–p (1 mmol) and citric acid monohydrate (2 mmol) as a green additive in ethanol solvent (4 ml) under ultrasound irradiation (100 W). b Reaction conditions: aniline 1 (1 mmol), diethyl acetylenedicarboxylate 2 (1 mmol), aldehyde 3a–p (1 mmol) and citric acid monohydrate (2 mmol) as a green additive in ethanol solvent (4 ml) at room temperature under stirring conditions. c Isolated yield. | ||||||
| 1 | Ph |
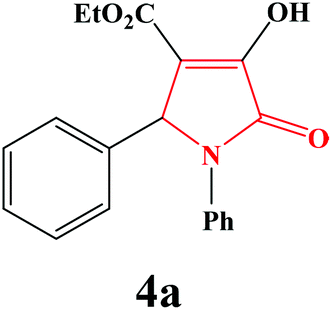
|
20 | 90 | 600 | 85 |
| 2 | 4-Me-C6H4 |
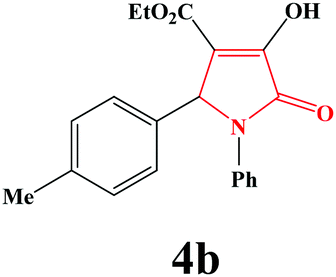
|
35 | 91 | 600 | 85 |
| 3 | 2-Naphthal |
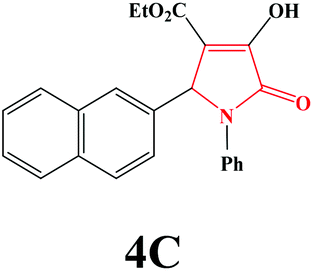
|
20 | 92 | 600 | 83 |
| 4 | 4-Cl-C6H4 |
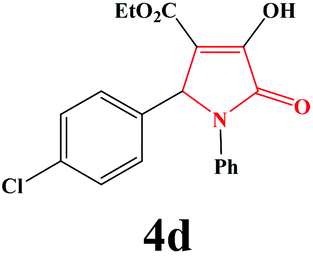
|
15 | 89 | 600 | 86 |
| 5 | 3-Cl-C6H4 |
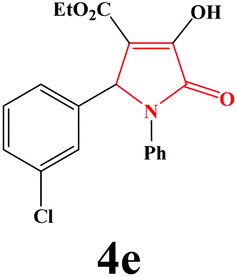
|
15 | 92 | 600 | 87 |
| 6 | 2-Cl-C6H4 |
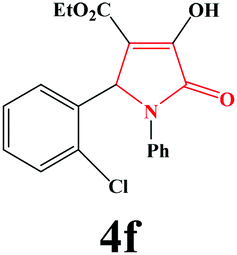
|
10 | 91 | 600 | 86 |
| 7 | 4-F-C6H4 |
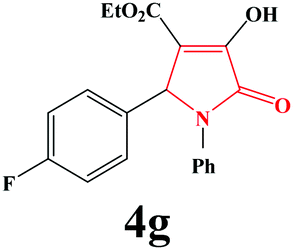
|
15 | 90 | 600 | 85 |
| 8 | 4-Br-C6H |

|
15 | 92 | 600 | 86 |
| 9 | 3-Br-C6H4 |
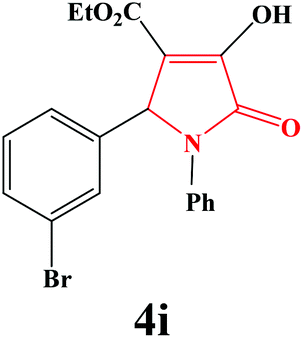
|
10 | 91 | 600 | 82 |
| 10 | 4-NO2-C6H4 |
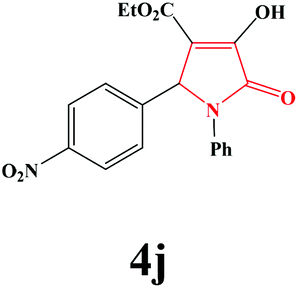
|
30 | 83 | 600 | 82 |
| 11 | 3-NO2-C6H4 |
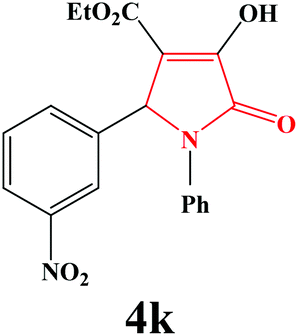
|
20 | 88 | 600 | 85 |
| 12 | 2-NO2-C6H4 |
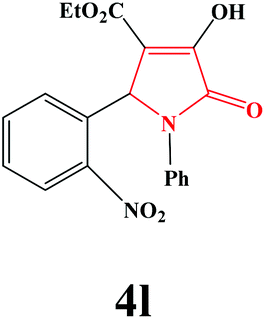
|
25 | 82 | 600 | 79 |
| 13 | 4-Ph-C6H4 |
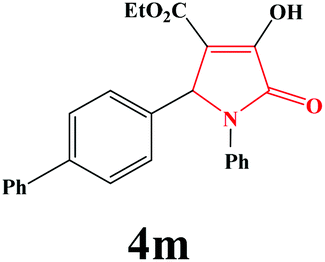
|
20 | 86 | 600 | 81 |
| 14 | 4-OH-C6H4 |
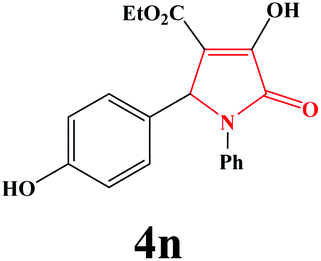
|
30 | 80 | 600 | 80 |
| 15 | 4-MeS-C6H4 |
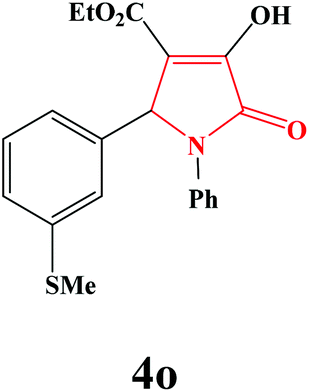
|
40 | 85 | 600 | 78 |
| 16 | 4-MeO-C6H4 |
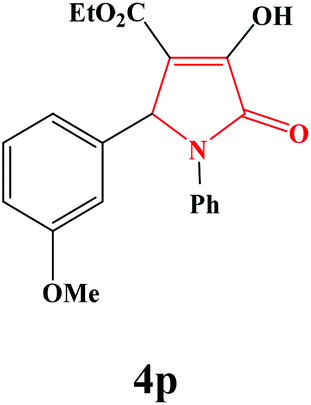
|
50 | 82 | 600 | 80 |
After finding a suitable and green solvent (C2H5OH) and power (100 W) to determine the important role of ultrasound, this method was examined with the reaction of aniline, diethyl acetylenedicarboxylate and several substituted aldehyde 3a–p in the presence of citric acid as an additive in ethanol solvent under ultrasound irradiation and without ultrasound irradiation at room temperature (Table 5).
As shown in Table 5, when the reactions were carried out with the conventional method, these took a comparatively longer time and resulted in lower yields; whereas when the same reactions were performed under the influence of ultrasonic conditions, they gave higher yields in shorter reaction times. Generally, a similar effect was seen in all reactions, and it was thus apparent that ultrasound irradiation can accelerate the reaction significantly to reduce the reaction times but giving higher yields. We found that ultrasonic irradiation was very effective and useful in our work, because the products could be synthesized in a short time with excellent yields.
In the last few years, there has been no report on the synthesis of substituted 3-pyrrolin-2-ones by ultrasound irradiation as an efficient procedure using citric acid as a green organoadditive. Therefore, this fact prompted us to apply a new method under eco-friendly conditions for the synthesis of substituted 3-pyrrolin-2-ones with a facile and appropriate ultrasound-promoted one-pot three-component approach. Then, various aromatic aldehydes carrying electron-donating and electron-withdrawing groups on the aromatic ring in the ortho, meta and para positions were evaluated. Yields of all the reactions were good to excellent. It was found that the aldehydes with electron-donating groups reacted longer than the aldehydes with electron-withdrawing groups (Table 5). Ultimately, the synthesis of substituted 3-pyrrolin-2-ones under the mentioned conditions were preferred due to the shorter reaction times and higher yields (Table 5). The structures of the products 4 were supported by FTIR, 1H- and 13C-NMR spectroscopic data. The structure of the product 4g was also confirmed by single-crystal X-ray analysis.
In the following part of our research work, various substrates, such as aliphatic, unsaturated, aromatic and heteroaromatic aldehydes, were examined. The corresponding product 4 could not be generated when we used acetaldehyde, isobutyraldehyde, 3-phenylpropionaldehyde, trans-cinnamaldehyde, salicylaldehyde, 2-hydroxy-1-naphthaldehyde, 4-(dimethylamino)benzaldehyde, furfural or glyoxal under the reaction conditions with or without sonication, and in all the cases, a mixture of several products and starting materials was observed (based on TLC investigation). Formaldehyde, pyrrole-2-carboxaldehyde and thiophene-2-carbaldehyde did not work well under the reaction conditions, and gave very low yields. Prolonging the reaction time did not lead to an increased yield for the corresponding product 4. In all the cases, a mixture of several products and starting materials was also observed (based on TLC investigation).
The suggested mechanism for the synthesis of substituted 3-pyrrolin-2-ones (4a–p) is illustrated in Scheme 2. The reaction proceeds very fast and cleanly under ultrasound irradiation and no side reactions were observed. On the basis of the chemistry of substituted 3-pyrrolin-2-ones under ultrasonic irradiation conditions, it is reasonable to assume that the first step in the reaction may proceed via the acid-catalyzed condensation of aniline and aromatic aldehyde in ethanol solvent in the presence of citric acid to produce imine. Then, diethyl acetylenedicarboxylate with water undergoes a nucleophilic addition to give 1,3-dipolar intermediate. In the following step, the addition of 1,3-dipolar intermediate to imine happens. Subsequently, the intramolecular attack of an amino group on one of the asters results in the formation of the nitrogen-containing five-membered ring. The elimination of ethanol and citric acid molecules gives the target product as indicated in Scheme 2.
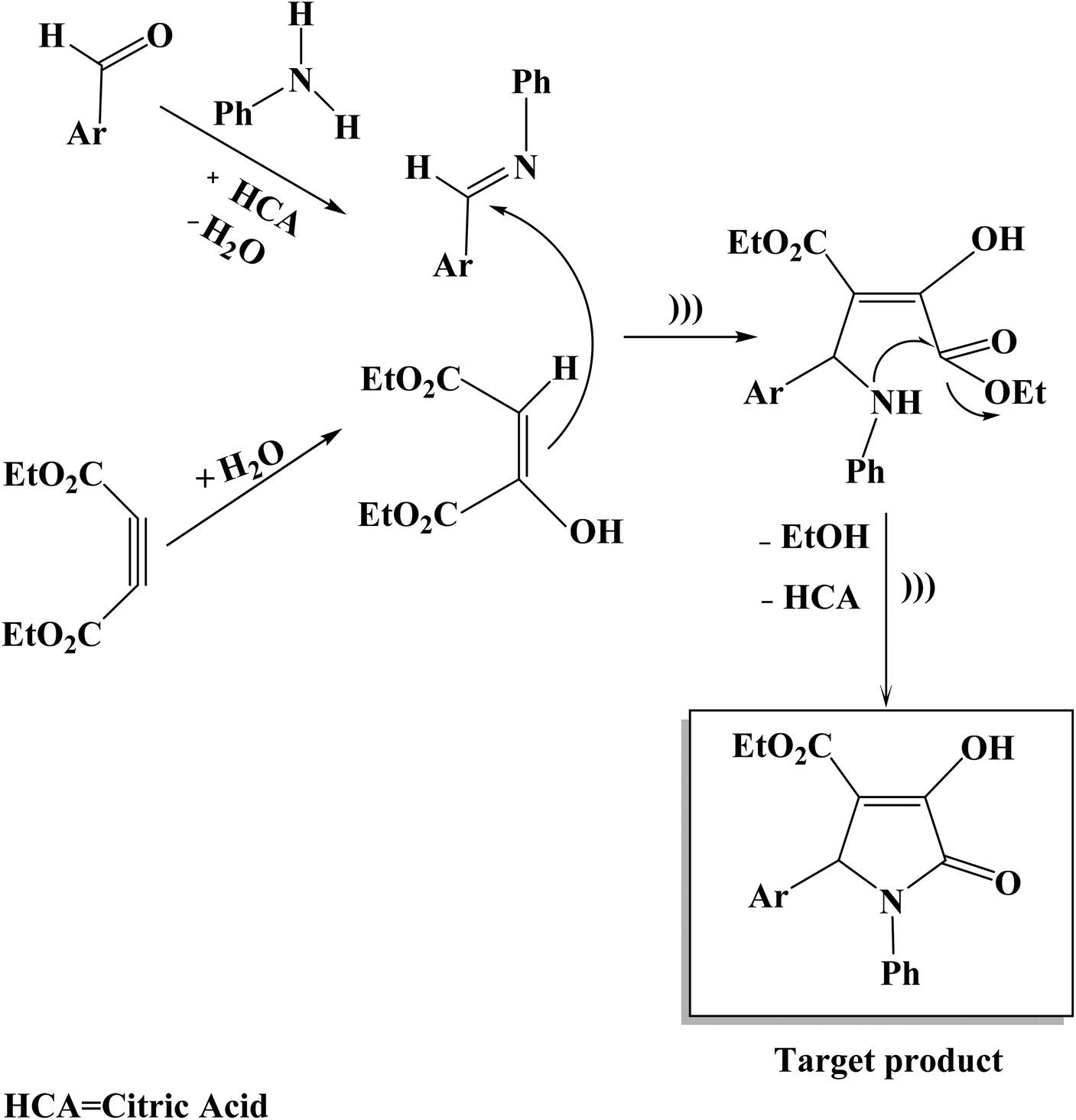 | ||
| Scheme 2 Proposed mechanistic path for the synthesis of substituted 3-pyrrolin-2-ones (4a–p). | ||
The crystal structure of one of the products, namely compound 4g, was confirmed by single-crystal X-ray analysis. As shown in Fig. 4a, there are two different molecules of the compound in the asymmetric unit of the crystal; however, their conformations are almost identical (Fig. 4b). Since the crystal is centrosymmetric, it contains a racemic compound 4g. The molecular structure of the (R)-enantiomer is shown in Fig. 4. The selected geometrical parameters are given in Table S1 (see page S67 in the ESI†). The overall conformation of 4g is almost identical to that observed in the isomorphous 2-Ph analogue.23 The C(O)OEt groups are planar and are almost coplanar with the planes of the pyrroline rings, and the molecules have a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Oester, C–OH syn-periplanar arrangement (which is different to 2-(4-Cl-Ph)–3-C(O)OMe derivative).23 As shown in Fig. 4a, the mutual orientation of the carbonyl O atoms from 3-C(O)OEt and 4-hydroxyl groups is accompanied by the intramolecular O–H⋯O hydrogen bonds (see Table 6 for the geometry). In the crystal lattice of 4g, two molecules of the same chirality are joined to each other by the bifurcated O–H⋯O hydrogen bonds, which gives rise to homochiral dimers, as shown in Fig. 5, within which the [R22(4)] ring motif is formed (Table 6). Between the adjacent dimers, again of the same chirality, F⋯π contacts are observed (with the F⋯centroid distance amounting to 2.923(2) Å, the perpendicular distance to 2.827 Å, and the C36–F2⋯centroid angle is 137.3(2)°), resulting in infinite chains (Fig. 5). The inter-chain contacts are provided by the extensive network of centrosymmetric C–H⋯π interactions (Table 6).
Oester, C–OH syn-periplanar arrangement (which is different to 2-(4-Cl-Ph)–3-C(O)OMe derivative).23 As shown in Fig. 4a, the mutual orientation of the carbonyl O atoms from 3-C(O)OEt and 4-hydroxyl groups is accompanied by the intramolecular O–H⋯O hydrogen bonds (see Table 6 for the geometry). In the crystal lattice of 4g, two molecules of the same chirality are joined to each other by the bifurcated O–H⋯O hydrogen bonds, which gives rise to homochiral dimers, as shown in Fig. 5, within which the [R22(4)] ring motif is formed (Table 6). Between the adjacent dimers, again of the same chirality, F⋯π contacts are observed (with the F⋯centroid distance amounting to 2.923(2) Å, the perpendicular distance to 2.827 Å, and the C36–F2⋯centroid angle is 137.3(2)°), resulting in infinite chains (Fig. 5). The inter-chain contacts are provided by the extensive network of centrosymmetric C–H⋯π interactions (Table 6).
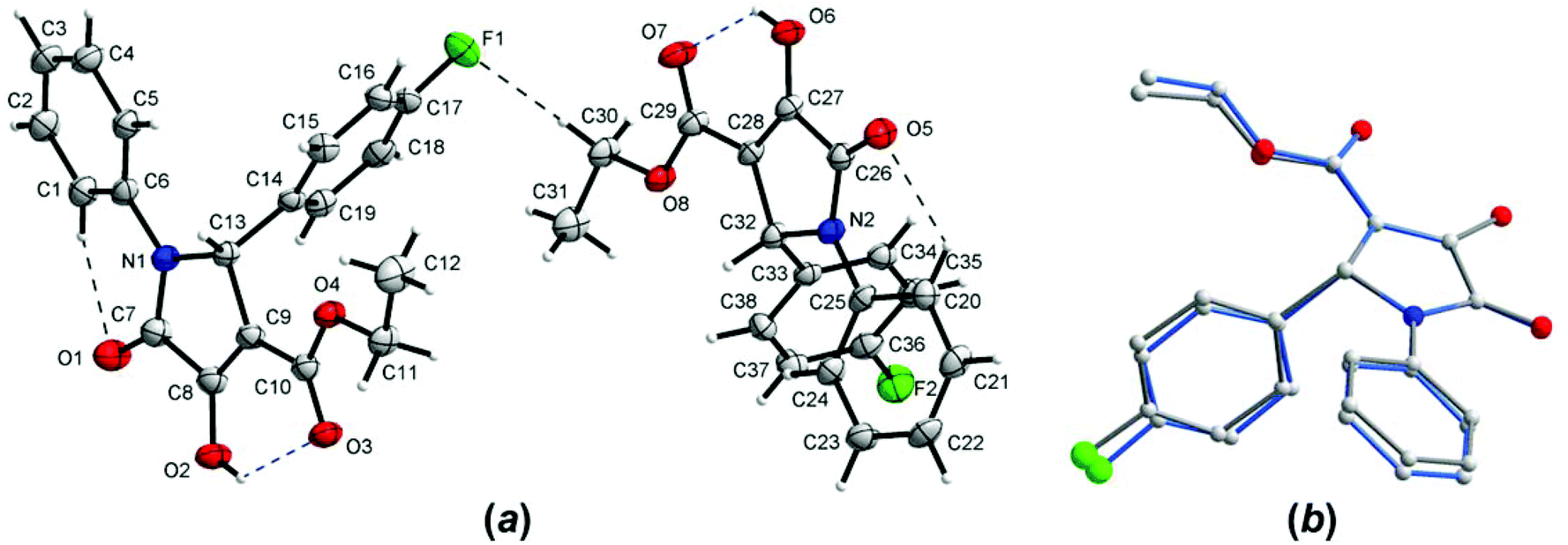 | ||
| Fig. 4 (a) Two crystallographically independent molecules of 4g present in the asymmetric unit of its crystal, showing the atom-numbering scheme and the symmetry-independent hydrogen bonds (dashed lines). The R enantiomers are shown. Displacement ellipsoids are drawn at the 50% probability level. (b) Comparison of the conformations of both molecules. H atoms are omitted for clarity. The common reference points are pyrroline atoms. | ||
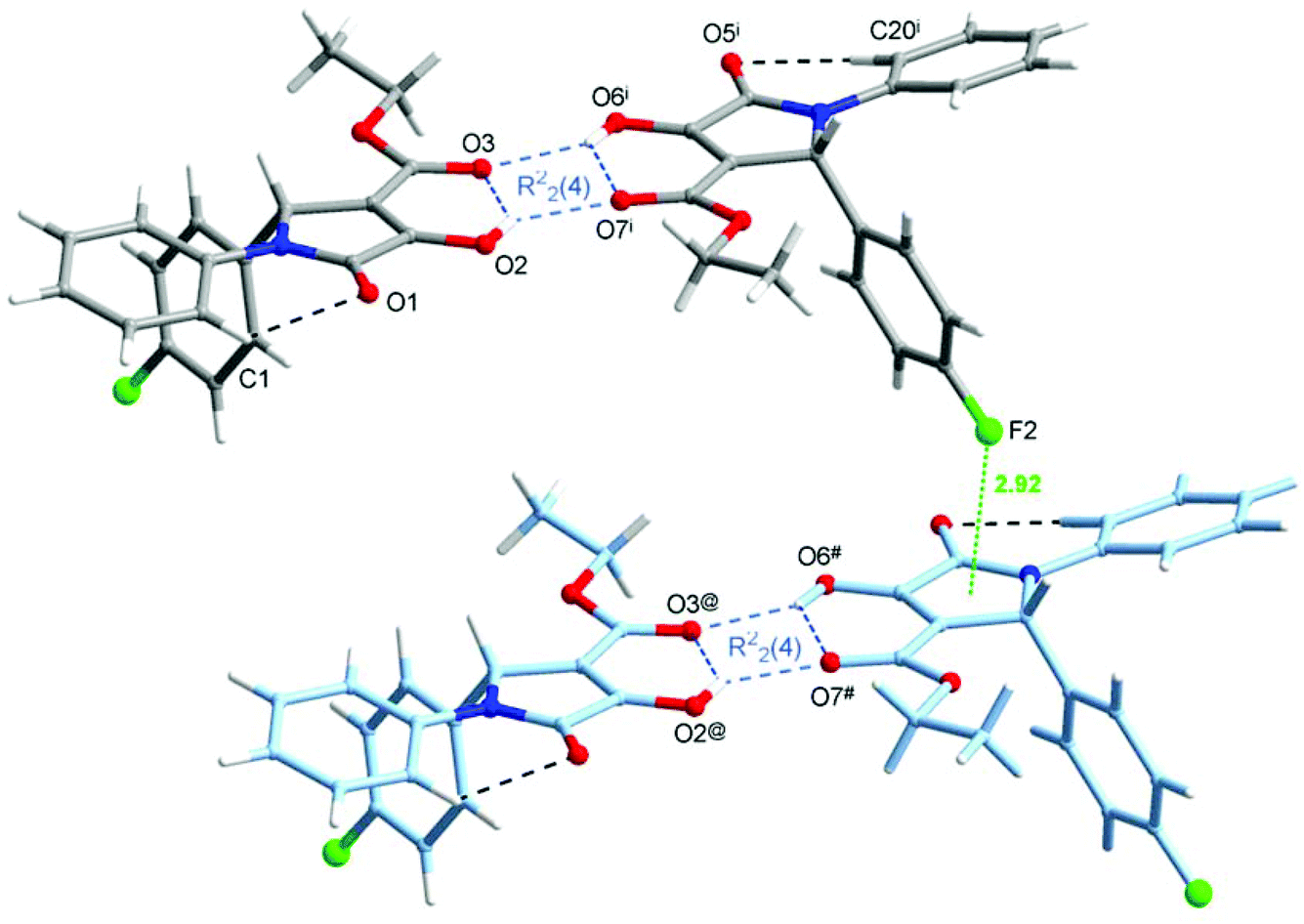 | ||
| Fig. 5 Molecular dimers built up from two molecules of 4g of the same chirality, joined by the bifurcated O–H⋯O hydrogen bonds (blue dashed lines), giving rise to [R22(4)] ring motifs, and the F⋯π contact (green dotted line) realized between the adjacent dimers. Symmetry code (i) is given in Table 6; (@) x − 1, y, z; (#) x − 1, y + 1, z. | ||
| D–H⋯A | D–H | H⋯A | D⋯A | D–H⋯A |
|---|---|---|---|---|
| O2–H2O⋯O3 | 0.84 | 2.25 | 2.894(3) | 134 |
| O2–H2O⋯O7i | 0.84 | 2.07 | 2.804(3) | 146 |
| C1–H1⋯O1 | 0.95 | 2.29 | 2.904(3) | 121 |
| O6–H6O⋯O7 | 0.84 | 2.26 | 2.902(3) | 133 |
| O6–H6O⋯O3ii | 0.84 | 2.05 | 2.797(2) | 147 |
| C20–H20⋯O5 | 0.95 | 2.30 | 2.902(3) | 121 |
| C21–H21⋯O1iii | 0.95 | 2.59 | 3.511(3) | 164 |
| C30–H30B⋯F1 | 0.99 | 2.46 | 3.449(4) | 176 |
| C–H⋯π | H⋯π | H⋯π⊥ | C⋯π | C–H⋯π |
|---|---|---|---|---|
| Symmetry codes: (i) x, y + 1, z; (ii) x, y − 1, z; (iii) x, y − 1, z + 1; (iv) −x + 1, −y + 1, −z; (v) −x + 1, −y + 1, −z + 1; (vi) −x + 2, −y + 2, −z; Cg1, Cg2 Cg3, Cg4, Cg5 and Cg6 are the centroids of C33–C38, C20–C25, N1–C13, C14–C19, C1–C6 and N2–C32 rings, respectively. | ||||
| C3–H3⋯Cg1iv | 2.89 | 2.75 | 3.714(4) | 146 |
| C11–H11A⋯Cg2v | 2.88 | 2.87 | 3.770(3) | 150 |
| C13–H13⋯Cg3vi | 2.89 | 2.84 | 3.728(3) | 142 |
| C19–H19⋯Cg3 | 2.71 | 2.57 | 2.993(3) | 98 |
| C22–H22⋯Cg4v | 2.82 | 2.72 | 3.646(3) | 146 |
| C30–H30A⋯Cg5iv | 2.80 | 2.79 | 3.706(3) | 152 |
| C34–H34⋯Cg6 | 2.83 | 2.61 | 3.079(3) | 96 |
Generally, when ultrasound is passed through a liquid system, bubble cavitation causes a series of unique physical phenomena that can affect the materials in the solution system. High energetic shockwaves are produced by cavitation. During the collapse of a cavity, high local temperatures and pressures arise, which result in a pressure shockwave. We believe that the ultrasonic irradiation played an important role in this reaction and enabled the reaction to proceed smoothly under mild conditions in a short time.
To summarize, this new procedure provides the first example of an efficient and ultrasound-promoted approach for the synthesis of target products with high yields and excellent purities. This method is the most simple and convenient and would be applicable for the synthesis of different substituted 3-pyrrolin-2-ones. The structures of all the synthesized compounds were established by IR, 1H NMR and 13C NMR. The molecular structure of product 4g was also established by single-crystal X-ray analysis.
Conclusions
In this work, the reported method offers a new, simple and efficient route for the one-pot sonochemical synthesis of substituted 3-pyrrolin-2-ones by citric acid as a green additive. Some important superiorities of this method are its short reaction time, use of green solvent, easy work-up, non-chromatographic purification technique, high yields and high purity. Furthermore, the application of a green, inexpensive, eco-friendly and commercially available additive makes it a useful procedure in modern synthetic methodologies.Experimental
General remarks
Starting materials were obtained from Merck (Germany), Fluka (Switzerland) and Sigma-Aldrich (USA) and were used without further purification. The methods used to follow the reactions were TLC. Sonication was performed in a Bandelin SONOPULS ultrasonic homogenizer (made in Germany) with 20 kHz processing frequency, a nominal power of 250 W and uniform sonic waves. Melting points were measured on an Electrothermal 9100 apparatus (LABEQUIP LTD, Markham, Ontario, Canada) and are uncorrected. 1H and 13C NMR spectra (CDCl3 and DMSO-d6) were recorded on a Bruker DRX-250 Avance spectrometer at 250.13 and 62.90 MHz, respectively. IR spectra were measured on a Jasco 6300 FTIR spectrometer. Elemental analyses were performed using a Heraeus CHN-O-Rapid analyzer.General procedure for the synthesis of substituted 3-pyrrolin-2-ones
A solution of aniline 1 (1 mmol), diethyl acetylenedicarboxylate 2 (1 mmol) and ethanol (4 ml) was magnetically stirred at room temperature. To the mixture, aldehyde 3a–p (1 mmol) and citric acid monohydrate (2 mmol) were added and the content was stirred at room temperature. The progress of the reaction was checked by TLC (n-hexane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EtOAc, 10:7). After completion of the reaction, the solid product was filtered and the pure product was obtained by recrystallization from hot ethanol.
:EtOAc, 10:7). After completion of the reaction, the solid product was filtered and the pure product was obtained by recrystallization from hot ethanol.
Ultrasound-promoted synthesis of substituted 3-pyrrolin-2-ones
A solution of aniline 1 (1 mmol), diethyl acetylenedicarboxylate 2 (1 mmol) in ethanol (4 ml) was magnetically stirred at room temperature. To the mixture, aldehyde 3a–p (1 mmol) and citric acid monohydrate (2 mmol) were added and the content was sonicated under ultrasound irradiation. The progress of the reaction was checked by TLC (n-hexane:EtOAc, 10:7). After completion of the reaction, the solid product was filtered and the pure product was obtained by recrystallization from hot ethanol.
Representative procedure for synthesis of 4d
A solution of aniline (0.091 ml, 1 mmol) and diethyl acetylenedicarboxylate (0.160 ml, 1 mmol) in ethanol (4 ml) was magnetically stirred at room temperature. To the mixture, 4-chlorobenzaldehyde (0.141 g, 1 mmol) and citric acid monohydrate (0.42 g, 2 mmol) were added and the content was sonicated under ultrasound irradiation. The same reaction was also conducted without sonication at room temperature. The progress of the reactions was checked by TLC (n-hexane:EtOAc, 10:7). After completion of the reactions, the solid product was filtered and the pure product was obtained by recrystallization from hot ethanol.
4a. Yield 0.291 g (90%). M.p. 174–177 °C. IR: νmax/cm−1 3250, 3077, 2970, 1710, 1458, 1090 cm−1; 1H NMR (250.13 MHz, CDCl3) δH 1.16 (t, 3H, J = 7.00 Hz, 3H), 4.17 (q, J = 7.00 Hz, 2H), 5.74 (s, 1H), 7.05–7.50 (m, 9H), 9.14 (br s, 1H); 13C NMR (62.90 MHz, CDCl3) δC 13.91, 61.23, 61.56, 113.19, 122.27, 125.81, 127.53, 128.50, 128.57, 128.94, 135.07, 136.24, 156.36, 162.94, 165.05. Anal. Calc. for C19H17NO4 (323.35): C 70.58, H 5.30, N 4.33; Found: C 70.62, H 5.32, N 4.29.
4b. Yield 0.307 g (91%). M.p. 202–205 °C. IR: νmax/cm−1 3250, 3077, 2970, 1710, 1458, 1090 cm−1; 1H NMR (250.13 MHz, CDCl3) δH 1.19 (t, J = 7.00 Hz, 3H), 2.25 (s, 3H), 4.18 (q, J = 7.00 Hz, 2H), 5.71 (s, 1H), 7.02–7.50 (m, 9H), 9.00 (br s, 1H); 13C NMR (62.90 MHz, CDCl3) δC 13.93, 21.11, 61.21, 61.32, 113.24, 122.24, 125.73, 127.36, 128.90, 128.27, 131.93, 136.33, 138.24, 156.30, 162.90, 165.11. Anal. Calc. for C20H19NO4 (337.37): C 71.20, H 5.68, N 4.15; Found: C 70.26, H 5.70, N 4.18.
4c. Yield 0.343 g (92%). M.p. 225–228 °C. IR: νmax/cm−1 3250, 3077, 2970, 1710, 1458, 1090 cm−1; 1H NMR (250.13 MHz, CDCl3) δH 1.13 (t, 3H, J = 7.00 Hz, 3H), 4.14 (q, 2H, J = 7.00 Hz, 2H), 5.91 (s, 1H), 7.02–7.47 (m, 12H), 9.18 (br s, 1H). 13C NMR (62.90 MHz, CDCl3) δC 13.89, 61.26, 61.69, 113.13, 122.24, 123.83, 125.87, 126.38, 127.71, 127.77, 127.89, 128.67, 128.98, 132.46, 133.06, 133.26, 136.27, 156.59, 162.93, 164.11. Anal. Calc. for C23H19NO4 (373.41): C 73.98, H 5.13, N 3.75; Found: C 73.96, H 5.10, N 3.71.
4d. Yield 0.318 g (89%). M.p. 195–198 °C. IR: νmax/cm−1 3307, 3067, 2982, 1733, 1499, 1015 cm−1; 1H NMR (250.13 MHz, CDCl3) δH 1.19 (t, 3H, J = 7.00 Hz, 2H), 4.20 (q, J = 7.00 Hz, 2H), 5.72 (s, 1H), 7.08–7.74 (m, 9H), 9.07 (br s, 1H). 13C NMR (62.9 MHz, CDCl3) δC 13.97, 60.83, 61.38, 112.77, 122.26, 126.05, 128.87, 129.08, 133.74, 134.29, 135.93, 156.40, 162.76, 164.81. Anal. Calc. for C19H16ClNO4 (357.79): C 63.78, H 4.51, N 3.91; Found: C 63.75, H 4.53, N 3.94.
4e. Yield 0.329 g (92%). M.p. 184–187 °C. IR: νmax/cm−1 3302, 3050, 2979, 1725, 1475, 1021 cm−1; 1H NMR (250.13 MHz, CDCl3) δH 1.20 (t, 3H, J = 7 Hz, 3H), 4.20 (q, 2H, J = 7 Hz, 2H), 5.71 (s, 1H), 7.10–7.48 (m, 9H), 9.17 (br s, 1H). 13C NMR (62.9 MHz, CDCl3) δC 13.90, 60.87, 61.39, 112.72, 122.14, 125.49, 126.03, 127.88, 128.77, 129.10, 129.93, 134.37, 135.99, 137.35, 156.56, 162.75, 164.83. Anal. Calc. for C19H16ClNO4 (357.79): C 63.78, H 4.51, N 3.91; Found: C 63.76, H 4.55, N 3.93.
4f. Yield 0.325 g (91%). M.p. 204–207 °C. IR: νmax/cm−1 3300, 3064, 2986, 1728, 1499, 1076 cm−1; 1H NMR (250.13 MHz, CDCl3) δH 1.16 (t, J = 7.15 Hz, 3H), 4.16 (q, J = 7.15 Hz, 2H), 6.43 (s, 1H), 6.93–7.54 (m, 9H), 9.24 (br s, 1H); 13C NMR (62.90 MHz, CDCl3) δC 13.73, 56.45, 61.34, 112.5, 119.2, 121.62, 125.82, 126.88, 127.49, 129.03, 129.64, 132.77, 136.05, 157.33, 162.72, 165.22. Anal. Calc. for C19H16ClNO4 (357.79): C 63.78, H 4.51, N 3.91; Found: C 63.79, H 4.54, N 3.92.
4g. Yield 0.307 g (90%). M.p. 193–196 °C. IR: νmax/cm−1 3295, 3066, 2984, 1717, 1499, 1027 cm−1; 1H NMR (250.13 MHz, CDCl3) δH 1.19 (t, J = 7.00 Hz, 3H), 4.19 (q, J = 7.00 Hz, 2H), 5.72 (s, 1H), 6.90–7.45 (m, 9H), 9.06 (br s, 1H); 13C NMR (62.90 MHz, CDCl3) δC 13.93, 60.86, 61.28, 112.94, 115.63 (d, 2JCF = 21.89 Hz), 122.39, 125.99, 129.07, 129.29, 129.08 (d, 3JCF = 9.37 Hz), 130.89 (d, 4JCF = 3.08 Hz), 136.03, 156.38, 160.58, 162.72, 164.70 (d, 1JCF = 23.40 Hz). Anal. Calc. for C19H16FNO4 (341.34): C 66.86, H 4.72, N 4.10; Found: C 66.90, H 4.74, N 4.06.
4h. Yield 0.370 g (92%). M.p. 190–193 °C. IR: νmax/cm−1 3297, 3052, 2981, 1717, 1499, 1027 cm−1; 1H NMR (250.13 MHz, DMSO-d6) δH 1.06 (t, J = 7.00 Hz, 3H), 3.64 (br s, 1H), 4.01 (q, J = 7.00 Hz, 2H), 6.05 (s, 1H), 7.07–7.60 (m, 9H); 13C NMR (62.90 MHz, DMSO-d6) δC 14.41, 60.23, 60.42, 112.20, 121.46, 122.93, 125.99, 126.70, 129.15, 131.60, 136.03, 142.07, 153.27, 162.36, 164.32. Anal. Calc. for C19H16BrNO4 (402.24): C 58.73, H 4.01, N 3.48; Found: C 58.76, H 4.04, N 3.45.
4i. Yield 0.366 g (91%). M.p. 192–194 °C. IR: νmax/cm−1 3304, 3048, 2980, 1727, 1499, 1023 cm−1; 1H NMR (250.13 MHz, CDCl3) δH 1.22 (t, J = 7.13 Hz, 3H), 4.21 (q, J = 7.13 Hz, 2H), 5.68 (s, 1H), 7.09–7.48 (m, 9H), 9.16 (br s, 1H); 13C NMR (62.90 MHz, CDCl3) δC 13.92, 60.80, 61.41, 112.73, 122.13, 122.44, 125.90, 126.04, 129.12, 130.22, 130.80, 131.68, 135.97, 137.58, 156.57, 162.74, 164.84. Anal. Calc. for C19H16BrNO4 (402.24): C 58.73, H 4.01, N 3.48; Found: C 58.75, H 4.02, N 3.47.
4j. Yield 0.305 g (83%). M.p. 181–183 °C. IR: νmax/cm−1 3299, 3073, 2983, 1732, 1498, 1025 cm−1; 1H NMR (250.13 MHz, DMSO-d6) δH 7.06–8.05 (m, 9H), 6.25 (s, 1H), 4.00 (q, J = 7.00 Hz, 2H), 3.97 (br s, 1H), 1.05 (t, J = 7.00 Hz, 3H); 13C NMR (62.90 MHz, DMSO-d6) δC 14.39, 60.17, 60.26, 111.53, 122.85, 123.77, 126.03, 129.48, 130.66, 136.42, 145.21, 147.53, 153.98, 162.31, 164.45. Anal. Calc. for C19H16N2O6 (368.35): C 61.96, H 4.38, N 7.61; Found: C 62.00, H 4.36, N 7.58.
4k. Yield 0.324 g (88%). M.p. 225–227 °C. IR: νmax/cm−1 3319, 3087, 2982, 1736, 1498, 1022 cm−1; 1H NMR (250.13 MHz, CDCl3) δH 1.46 (t, J = 7.25 Hz, 3H), 4.21 (q, J = 7.08 Hz, 2H), 5.87 (s, 1H), 7.10–8.24 (m, 9H), 9.09 (br s, 1H); 13C NMR (62.90 MHz, DMSO-d6) δC 14.30, 60.05, 60.28, 111.72, 122.92, 123.48, 123.63, 126.03, 129.28, 130.34, 134.36, 136.32, 139.78, 147.93, 153.81, 162.25, 164.34. Anal. Calc. for C19H16N2O6 (368.35): C 61.96, H 4.38, N 7.61; Found: C 61.99, H 4.39, N 7.57.
4l. Yield 0.302 g (82%). M.p. 203–205 °C. IR: νmax/cm−1 3294, 3080, 2982, 1732, 1499, 1021 cm−1; 1H NMR (250.13 MHz, CDCl3) δH 1.20 (t, J = 7.13 Hz, 3H), 4.18 (q, J = 7.00 Hz, 2H), 5.65 (s, 1H), 7.08–7.68 (m, 9H), 9.10 (br s, 1H); 13C NMR (62.90 MHz, DMSO-d6) δC 14.34, 54.98, 60.39, 112.19, 122.58, 125.18, 126.11, 127.84, 129.54, 131.64, 134.21, 136.65, 150.51, 153.85, 160.13, 162.26, 164.83. Anal. Calc. for C19H16N2O6 (368.35): C 61.96, H 4.38, N 7.61; Found: C 61.97, H 4.37, N 7.58.
4m. Yield 0.343 g (86%). M.p. 222–225 °C. IR: νmax/cm−1 3238, 3030, 2976, 1702, 1485, 1018 cm−1; 1H NMR (250.13 MHz, CDCl3) δH 1.20 (t, J = 7.13 Hz, 3H), 4.21 (q, J = 7.17 Hz, 2H), 5.79 (s, 1H), 7.08–7.5 (m, 12H), 9.10 (br s, 1H); 13C NMR (62.90 MHz, CDCl3) δC 14.0, 61.23, 61.31, 113.12, 122.23, 125.85, 126.21, 126.94, 127.38, 127.92, 128.76, 129.01, 1334.04, 136.30, 140.18, 141.28, 156.46, 162.88, 165.09. Anal. Calc. for C25H21NO4 (399.45): C 75.17, H 5.30, N 3.51; Found: C 75.14, H 5.33, N 3.48.
4n. Yield 0.271 g (80%). M.p. 243–245 °C (dec.). IR: νmax/cm−1 3311, 3027, 2990, 1698, 1451, 1071 cm−1; 1H NMR (250.13 MHz, DMSO-d6) δH 1.10 (t, J = 7.00 Hz, 3H), 3.55 (br s, 1H), 3.99 (q, J = 7.00 Hz, 2H), 5.93 (s, 1H), 6.55–7.69 (m, 9H), 9.34 (br s, 1H); 13C NMR (62.90 MHz, DMSO-d6) δC 14.44, 60.08, 60.78, 112.94, 115.41, 123.03, 125.67, 126.66, 129.03, 129.24, 136.79, 152.57, 157.24, 162.48, 164.27. Anal. Calc. for C19H17NO5 (339.35): C 67.25, H 5.05, N 4.13; Found: C 67.28, H 5.08, N 4.09.
4o. Yield 0.314 g (85%). M.p. 159–161 °C. IR: νmax/cm−1 3250, 3077, 2970, 1710, 1458, 1090 cm−1; 1H NMR (250.13 MHz, CDCl3) δH 1.18 (t, J = 7.00 Hz, 3H), 2.38 (s, 3H), 4.17 (q, J = 7.00 Hz, 2H), 5.68 (s, 1H), 7.10–7.47 (m, 9H), 9.15 (br s, 1H); 13C NMR (62.90 MHz, CDCl3) δC 13.98, 15.26, 61.12, 61.27, 113.07, 122.24, 125.84, 126.13, 127.95, 128.96, 131.58, 136.14, 139.00, 156.00, 163.00, 164.81. Anal. Calc. for C20H19NO4S (369.43): C 65.02, H 5.18, N 3.79; Found: C 65.05, H 5.15, N 3.80.
4p. Yield 0.290 g (82%). M.p. 150–153 °C. IR: νmax/cm−1 3250, 3029, 2929, 1706, 1458, 1107, 1032 cm−1; 1H NMR (250.13 MHz, CDCl3) δH 1.20 (t, J = 7.25 Hz, 3H), 3.73 (s, 3H), 4.19 (q, J = 7.08 Hz, 2H), 5.69 (s, 1H), 6.74–7.46 (m, 9H), 9.15 (br s, 1H); 13C NMR (62.90 MHz, CDCl3) δC 13.98, 55.16, 61.03, 61.25, 113.21, 113.97, 122.37, 125.80, 126.71, 128.66, 128.94, 136.23, 156.38, 159.52, 162.00, 165.00. Anal. Calc. for C20H19NO4S (353.37): C 67.98, H 5.42, N 3.96; Found: C 65.68.00, H 5.39, N 3.97.
X-Ray crystallography
After recrystallization from hot ethanol, the pure powder of 4g was dissolved in hot ethanol. X-ray quality crystals of 4g were obtained in excellent yield after slow evaporation of the mother liquor at room temperature.The crystallographic measurement of 4g was performed on a Kuma KM4-CCD κ-geometry automated four-circle diffractometer equipped with a CCD camera Sapphire2 and graphite-monochromatized Mo Kα radiation (λ = 0.71073 Å). The data were collected at 150(2) K by using the Oxford-Cryosystems cooler. Data were corrected for the Lorentz and polarization effects. Data collection, cell refinement, data reduction and analysis were carried out with KM4-CCD software, CrysAlisCCD and CrysAlisRED, respectively.384g is isomorphous with the Ph derivative reported in the literature,23 and was deposited at the Cambridge Structural Database (CSD, Version 5.35),39 with the CSD refcode LIFBEJ. Therefore, the refinement of its structure was started by using the coordinates of non-H atoms taken from LIFBEJ. Fluorine atoms were added from Fourier maps in the next step. The refinement was carried out by a full-matrix least-squares technique with SHELXL201440 and with the anisotropic thermal parameters for non-H atoms. All the H atoms were found in difference Fourier maps and were refined isotropically. In the final refinement cycles, the C-bonded H atoms were repositioned in their calculated positions and refined using a riding model, with C–H = 0.95–1.00 Å, and with Uiso(H) = 1.2Ueq(C) for CH and CH2, and 1.5Ueq(C) for CH3. The hydroxyl H atom was refined with the O–H distance restrained to 0.840(2) Å, and with Uiso(H) = 1.5Ueq(O), and then it was constrained to ride on its parent atom (AFIX 3 instruction in SHELXL2014). Figures were made with the Diamond program.41 The crystallographic information file (CIF) was deposited with The Cambridge Crystallographic Data Centre (http://www.ccdc.cam.ac.uk/; deposition number CCDC 1429951) and provided as ESI.†
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 8.747(4), b = 11.406(5), c = 16.580(6) Å, α = 80.72(3)°, β = 85.21(3)°, γ = 85.46(4)°, V = 1623.2(12) Å3, T = 150(2) K, Z = 4, μ = 0.11 mm−1 (for Mo Kα, λ = 0.71073 Å), multi-scan absorption correction, Tmin = 0.846, Tmax = 1.000, 13925 reflections measured, 8006 unique (Rint = 0.053), 4465 observed (I > 2σ(I)), (sinθ/λ)max 0.838 Å−1, 453 parameters, 2 restraints, R = 0.068, wR = 0.171 (observed refl.), GOOF = S = 1.00, (Δρmax) = 0.35 and (Δρmin) = −0.34 e Å−3.
, a = 8.747(4), b = 11.406(5), c = 16.580(6) Å, α = 80.72(3)°, β = 85.21(3)°, γ = 85.46(4)°, V = 1623.2(12) Å3, T = 150(2) K, Z = 4, μ = 0.11 mm−1 (for Mo Kα, λ = 0.71073 Å), multi-scan absorption correction, Tmin = 0.846, Tmax = 1.000, 13925 reflections measured, 8006 unique (Rint = 0.053), 4465 observed (I > 2σ(I)), (sinθ/λ)max 0.838 Å−1, 453 parameters, 2 restraints, R = 0.068, wR = 0.171 (observed refl.), GOOF = S = 1.00, (Δρmax) = 0.35 and (Δρmin) = −0.34 e Å−3.
Acknowledgements
This work is funded by the grant NRF-2015-002423 of the National Research Foundation of Korea.Notes and references
- (a) C. O. Kappe, Acc. Chem. Res., 2000, 33, 879–888 CrossRef CAS PubMed; (b) A. Dömling and I. Ugi, Angew. Chem., Int. Ed., 2000, 39, 3168–3210 CrossRef; (c) J. Zhu, Eur. J. Org. Chem., 2003, 1133–1144 CrossRef CAS; (d) G. Balme, E. Bossharth and N. Monteiro, Eur. J. Org. Chem., 2003, 4101–4111 CrossRef CAS; (e) A. Ramazani, M. Rouhani, E. Mirhadi, M. Sheikhi, K. Ślepokura and T. Lis, Nano. Chem. Res., 2016, 1, 87–107 Search PubMed; (f) A. Ramazani and A. R. Kazemizadeh, Curr. Org. Chem., 2011, 15, 3986–4020 CrossRef CAS.
- (a) M. Sh. Singh and S. Chowdhury, RSC Adv., 2012, 2, 4547–4592 RSC; (b) A. R. Kazemizadeh and A. Ramazani, Curr. Org. Chem., 2012, 16, 418–450 CrossRef CAS; (c) I. Ugi, B. Werner and A. Dömling, Molecules, 2003, 8, 53–66 CrossRef; (d) H. Ahankar, A. Ramazani and S. W. Joo, Res. Chem. Intermed., 2016, 42, 2487–2500 CrossRef CAS; (e) A. Ramazani, A. T. Mahyari, M. Rouhani and A. Rezaei, Tetrahedron Lett., 2009, 50, 5625–5627 CrossRef CAS; (f) A. Ramazani, M. Khoobi, A. Torkaman, F. Zeinali Nasrabadi, H. Forootanfar, M. Shakibaie, M. Jafari, A. Ameri, S. Emami, M. A. Faramarzi, A. Foroumadi and A. Shafiee, Eur. J. Med. Chem., 2014, 78, 151–156 CrossRef CAS PubMed; (g) B. L. Li, P. H. Li, X. N. Fang, C. X. Li, J. L. Sun, L. P. Mo and Z. H. Zhang, Tetrahedron, 2013, 69, 7011–7018 CrossRef CAS; (h) J. Yang, J.-N. Tan and Y. Gu, Green Chem., 2012, 14, 3304–3317 RSC.
- (a) D. L. Priebbenow and C. Bolm, RSC Adv., 2013, 3, 10318–10322 RSC; (b) J. Liu, H.-R. Zhang, X.-R. Lin, Sh.-J. Yan and J. Lin, RSC Adv., 2013, 3, 27582–27590 Search PubMed; (c) J. A. Joule and K. Mills, Heterocyclic Chemistry, Blackwell Science Publishing, Oxford, UK, 4th edn, 2000 Search PubMed.
- L. P. Dwoskin, L. Teng, S. T. Buxton and P. A. Crooks, J. Pharmacol. Exp. Ther., 1999, 288, 905–911 CAS.
- P. Singh, V. Dimitriou, R. P. Mahajan and A. W. Crossley, Br. J. Anaesth., 1993, 71, 685–688 CrossRef CAS PubMed.
- P. N. Patsalos, Epilepsia, 2005, 46, 140–144 CrossRef CAS PubMed.
- S. Omura, T. Fujimoto, K. Otoguro, K. Matsuzaki, R. Moriguchi, H. Tanaka and Y. Sasaki, J. Antibiot., 1991, 44, 113–116 CrossRef CAS PubMed.
- R. H. Feling, G. O. Buchanan, T. J. Mincer, C. A. Kauffman, P. R. Jensen and W. Fenical, Angew. Chem., Int. Ed., 2003, 42, 355–357 CrossRef CAS PubMed.
- Y. Asami, H. Kakeya, R. Onose, A. Yoshida, H. Matsuzaki and H. Osada, Org. Lett., 2002, 4, 2845–2848 CrossRef CAS PubMed.
- (a) T. Michael, A. Michael, T. Andreas, H. Ulrich, B. Mirko and N. A. Johannes, Patent WO2008055945(A1), 2008 Search PubMed; (b) V. O. Koz'minykh, N. M. Igidov, S. S. Zykova, V. E. Kolla, N. S. Shuklina and T. Odegova, Pharm. Chem. J., 2002, 36, 188–191 CrossRef.
- Y. Geng, X. Wang, L. Yang, H. Sun, Y. Wang, Y. Zhao, R. She, M.-X. Wang, D.-X. Wang and J. Tang, PLoS One, 2015, 10, 1–15 Search PubMed.
- (a) K. Ma, P. Wang, W. Fu, X. Wan, L. Zhou, Y. Chu and D. Ye, Bioorg. Med. Chem. Lett., 2011, 21, 6724–6727 CrossRef CAS PubMed; (b) A. Pendri, T. L. Troyer, M. J. Sofia, M. A. Walker, B. N. Naidu, J. Banville, N. A. Meanwell, I. Dicker, Z. Lin, M. Krystal and S. W. Gerritz, J. Comb. Chem., 2010, 12, 84–90 CrossRef CAS PubMed.
- V. L. Gein, M. N. Armisheva, N. A. Rassudikhina, M. I. Vakhrin and E. V. Voronina, Pharm. Chem. J., 2011, 45, 162–164 CrossRef CAS.
- V. L. Gein, V. A. Mihalev, N. N. Kasimova, E. V. Voronina, M. I. Vakhrin and E. B. Babushkina, Pharm. Chem. J., 2007, 41, 208–210 CrossRef CAS.
- V. L. Gein, V. V. Yushkov, N. N. Kasimova, N. S. Shuklina, Y. M. Vasil'eva and M. V. Gubanova, Pharm. Chem. J., 2005, 39, 484–487 CrossRef CAS.
- M. S. F. Franco, G. A. Casagrande, C. Raminelli, S. Moura, M. Rossatto, F. H. Quina, C. M. P. Pereira, A. F. C. Flores and L. Pizzuti, Synth. Commun., 2015, 45, 692–701 CrossRef CAS.
- M. Astada and Sh.-I. Hashimoto, Tetrahedron Lett., 1998, 39, 79–82 CrossRef.
- D.-R. Choi, K.-Y. Lee, Y.-S. Chung, J.-E. Joo, Y.-H. Kim, Ch.-Y. Oh, Y.-S. Lee and W.-H. Ham, Arch. Pharm. Res., 2005, 28, 151–158 CrossRef CAS PubMed.
- L. E. Burgess and A. I. Meyers, J. Org. Chem., 1992, 57, 1656–1662 CrossRef CAS.
- L. E. Overman and T. P. Remarchuk, J. Am. Chem. Soc., 2002, 124, 12–13 CrossRef CAS PubMed.
- V. Singh, R. Saxena and S. Batra, J. Org. Chem., 2005, 70, 353–356 CrossRef CAS PubMed.
- R. Sarkar and Ch. Mukhopadhyay, Tetrahedron Lett., 2013, 54, 3706–3711 CrossRef CAS.
- J. Sun, Q. Wu, E.-Y. Xia and Ch.-G. Yan, Eur. J. Org. Chem., 2011, 2981–2986 CrossRef CAS.
- Q. Zhu, H. Jiang, J. Li, Sh. Liu, Ch. Xia and M. Zhang, J. Comb. Chem., 2009, 11, 685–696 CrossRef CAS PubMed.
- (a) G. A. Shvekhgeimer, Chem. Heterocycl. Compd., 1994, 30, 633–660 CrossRef; (b) R. Cella and H. A. Stefani, Tetrahedron, 2013, 65, 2619–2641 CrossRef; (c) W. T. Richards and A. L. Loomis, J. Am. Chem. Soc., 1927, 49, 3086–3100 CrossRef CAS.
- (a) L. Ji-Tai, W. Shu-Xiang, C. Guo-Fengi and L. Tong-Shuang, Curr. Org. Synth., 2005, 2, 415–419 CrossRef; (b) M. H. Mosslemin and M. R. Nateghi, Ultrason. Sonochem., 2010, 17, 162–167 CrossRef CAS PubMed; (c) J. L. Luche and C. Allavena, Tetrahedron, 1988, 29, 5369–5372 CrossRef CAS; (d) A. G. Nladhari and F. Saki, Ultrason. Sonochem., 2013, 20, 571–579 CrossRef PubMed; (e) A. Ramazani, M. Rouhani and S. W. Joo, Ultrason. Sonochem., 2016, 28, 393–399 CrossRef CAS PubMed; (f) M. Rouhani, A. Ramazani and S. W. Joo, Ultrason. Sonochem., 2015, 22, 391–396 CrossRef CAS PubMed; (g) M. Rouhani, A. Ramazani and S. W. Joo, Ultrason. Sonochem., 2014, 21, 262–267 CrossRef CAS PubMed; (h) R. Moosavi, A. R. Abbasi, M. Yousefi, A. Ramazani and A. Morsali, Ultrason. Sonochem., 2012, 19, 1221–1226 CrossRef CAS PubMed; (i) A. R. Abbasi, H. Kalantary, M. Yousefi, A. Ramazani and A. Morsali, Ultrason. Sonochem., 2012, 19, 853–857 CrossRef CAS PubMed; (j) M. Rouhani, A. Ramazani, S. W. Joo and Y. Hanifehpour, Bull. Korean Chem. Soc., 2012, 33, 4127–4130 CrossRef CAS; (k) A. Ramazani, M. Rouhani, F. Zeinali Nasrabadi and F. Gouranlou, Phosphorus, Sulfur Silicon, 2015, 190, 20–28 CrossRef CAS.
- (a) D. Nagargoje, P. Mandhane, S. Shingote, P. Badadhe and C. Gill, Ultrason. Sonochem., 2012, 19, 94–96 CrossRef CAS PubMed; (b) P. V. Shinde, B. B. Shingate and M. S. Shingare, Bull. Korean Chem. Soc., 2011, 32, 1179–1182 CrossRef CAS; (c) F. Dang, N. Y. Enomoto, J. C. Hojo and K. J. Enpuku, Ultrason. Sonochem., 2009, 16, 649–654 CrossRef CAS PubMed.
- M. Papagianni, Biotechnol. Adv., 2007, 25, 244–263 CrossRef CAS PubMed.
- M. A. Zolfigol, M. Mokhlesi and Sh. Farahmand, J. Iran. Chem. Soc., 2013, 10, 577–581 CrossRef CAS.
- P. B. Pawar, S. D. Jadhav, M. B. Deshmukh and S. Patil, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 2014, 53, 1185–1193 Search PubMed.
- K. A. Shaikh, U. N. Chaudhar and V. B. Ningdale, IOSR J. Appl. Chem., 2014, 7, 90–93 CrossRef.
- A. de Vasconselos, P. S. Oliveira, M. Ritter, R. A. Freitag, R. L. Romano, F. H. Quina, L. Pizzuti, C. M. P. Pereira, F. M. Stefanello and A. G. Barshak, J. Biochem. Mol. Toxicol., 2012, 26, 155–161 CrossRef PubMed.
- R. Enugala, S. Nuvvula, V. Kotra, R. Varala and S. R. Adapa, Heterocycles, 2008, 75, 2523–2533 CrossRef CAS.
- S. R. Thopate, S. R. Kote, S. V. Rohokale and N. M. Thorat, J. Chem. Res., 2011, 35, 124 CrossRef CAS.
- A. D. Carbaugh, W. Vosburg, T. J. Scherer, C. E. Castillo, M. A. Christianson, J. Kostarellas, S. J. Gosai and M. S. Leonard, ARKIVOC, 2007, 12, 43–54 Search PubMed.
- A. Dondoni, J. Orduna and J. P. Merino, Synthesis, 1992, 201–208 CrossRef CAS.
- (a) R. A. Sheldon, Chem. Ind., 1992, 903–906 CAS; (b) R. A. Sheldon, Green Chem., 2007, 9, 1273–1283 RSC.
- CrysAlisCCD and CrysAlisRED in KM4-CCD software, Oxford Diffraction Ltd., Abingdon, England, 2009 Search PubMed.
- C. R. Groom and F. H. Allen, Angew. Chem., Int. Ed., 2014, 53, 662–671 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- K. Brandenburg, DIAMOND Version 3.2k, Crystal Impact GbR, Bonn, Germany, 2014 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: FT-IR, 1H NMR, 13C NMR spectra of the products, the X-ray structure analysis and 1H NMR spectra of the crude product. CCDC 1429951. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6gc00157b |
| This journal is © The Royal Society of Chemistry 2016 |