
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of monosubstituted thioureas by vapour digestion and mechanochemical amination of thiocarbamoyl benzotriazoles†
Mateja
Đud
a,
Oxana V.
Magdysyuk
b,
Davor
Margetić
a and
Vjekoslav
Štrukil
*a
aDivision of Organic Chemistry and Biochemistry, Ruđer Bošković Institute, Bijenička cesta 54, 10 000 Zagreb, Croatia. E-mail: vstrukil@irb.hr
bDiamond Light Source Ltd, Harwell Science and Innovation Campus, OX11 0DE Didcot, UK
First published on 3rd March 2016
Abstract
Thiocarbamoyl benzotriazoles, as safe and easy-to-handle isothiocyanate equivalents, were quantitatively converted to N-monosubstituted thioureas by vapour digestion synthesis under an ammonia atmosphere. This simple, but timely process provided a synthetic platform that enabled the “slow” amination reaction to be successfully transformed into a rapid one aided by mechanochemical milling. The ammonium chloride/sodium carbonate equimolar mixture allowed in situ formation of ammonia under ball-milling conditions. This novel and green approach yielded aromatic and aliphatic primary thioureas in near-quantitative isolated yields with workup entirely based on using only water. In addition, the molecular and crystal structures of selected polyaromatic primary thioureas were determined from the synchrotron powder diffraction data.
Introduction
Solid-state synthesis promoted by simple aging of reaction mixtures,1a manual treatment in mortar using a pestle, high-intensity mechanochemical milling in shaker and planetary ball mills1b or mechanical agitation during twin screw extrusion,1c,d has gained much popularity among synthetic chemists as a viable alternative to the conventional solution-based approach to chemicals. The green chemistry aspect of these methods, as well as ease of preparation and isolation of compounds, have contributed to their wide-spread utility in research laboratories, but also for educational purposes.2 The examples of successful application of mechanosynthesis in different fields of chemistry research are numerous, ranging from materials science,3a pharmaceuticals,3b organic3c and organometallic chemistry,3d catalysts,3e,f polymers3g to novel use of ball milling in treatment of lignin as a renewable source of valuable products.3h Not only does the mechanochemistry enable more efficient, cleaner, faster and environmentally-friendly approach to known chemicals, but it also allows for the discovery of their unknown phases or polymorphs by exploitation of the unique solid-state reactivity through liquid-assisted grinding (LAG)4 and ion- and liquid-assisted grinding (ILAG) methods.5 The role of these catalytic additives and their effect on the solid-state reactivity are still obscured, but the recent advancements made by in situ real-time monitoring of mechanochemical reactions6 using synchrotron radiation,7 Raman spectroscopy8 or the combination of both9 have greatly helped in bringing the knowledge of the solid-state mechanisms to a higher level.The benzotriazole chemistry, developed by Katritzky, has been established as a valuable tool to access a variety of organic compounds under conventional solution synthesis conditions.10 Led by a hypothesis that mechanochemistry could offer new opportunities for the development of benzotriazole reagents, we recently reported the first successful isolation and characterisation of aromatic thiocarbamoyl benzotriazoles as bench-stable synthetic equivalents of isothiocyanates.11 This has been achieved through mechanochemical milling synthesis under LAG conditions coupled with solid-state characterisation methods to obviate their solution sensitivity. These compounds have been shown to undergo a quantitative conversion to N,N′-disubstituted aromatic thioureas in reaction with different anilines. Following this path, we envisaged that the milling approach might also be utilised for the synthesis of N-monosubstituted (primary) thioureas. Thioureas are generally regarded as an important structural motif in biologically active compounds,12a–c as well as synthetic intermediates to amidines, guanidines and many heterocycles.12d,e Typically, the solution synthesis of primary thioureas mostly relies on the reaction of primary amines with alkali metal or ammonium thiocyanate in the presence of an acid (or without if amine hydrochloride salts are used),13a base hydrolysis of benzoyl thioureas,13b thermal rearrangement of ammonium thiocyanate in the presence of a primary amine13c or ammonia addition to isothiocyanates.13d In a previous report by Kaupp, gas–solid amination reactions using ammonia or N-methylamine under pressure and a low temperature (0.4–1 bar, −30 to 0 °C) with isothiocyanates were studied, and were found to proceed in quantitative yields after overnight exposure of the solid reactant to the amine gas (Scheme 1).14
 | ||
| Scheme 1 Common methods for the synthesis of primary thioureas. | ||
In this paper, the scope of mechanochemical synthesis of thiocarbamoyl benzotriazoles in terms of an amine reagent has been extended, including disubstituted anilines, aliphatic amines, as well as polyaromatic hydrocarbon (PAH) amines (naphthalene, anthracenes, phenanthrene, chrysene and pyrene). We also report on the behaviour of thiocarbamoyl benzotriazoles, as bench-stable and easy-to-handle masked isothiocyanates, in reaction with ammonia under experimental conditions that would not require working with hazardous reagents, pressurized gases or special low temperature apparatus. In what appears to be the first demonstration of an amination reaction under milling conditions, primary thioureas have been accessed from amines in a rapid two-step procedure. Its advantages as an environmentally-friendly alternative to existing methods are mirrored in complete reduction of toxic solvent usage, quantitative conversions, simple isolation based on aqueous work-up, and excellent isolated yields.
Results and discussion
Synthesis of thiocarbamoyl benzotriazoles 2
The scope of mechanochemical thiocarbamoylation of aromatic amines synthesised and structurally characterised previously (p-bromo 2a, chloro 2b, methyl 2d, methoxy 2e and amino 2j derivatives)11 has now been extended to several other m- and p-substituted anilines, polyaromatic hydrocarbon amines, and a few alkyl amines, e.g. benzyl and 4-methoxybenzyl (Scheme 2). In this respect, the reactions were found to proceed in near-quantitative isolated yields (≥95%) not only with solid amines, but liquid ones as well, e.g. 4-fluoroaniline (2c) and benzylamine (2l). However, it was found that highly deactivated anilines, such as 4-nitro and 4-cyano are not suitable substrates for this type of transformation under the investigated experimental conditions. This was not the case with 3-nitro and 3-cyano derivatives which reacted smoothly to give thiocarbamoylated products 2h and 2i in quantitative yield. Next, the synthesis of bis-thiocarbamoylated benzidine derivative 2n, incorporating a rigid biphenyl spacer, also proceeded smoothly as well as the thiocarbamoylation reaction of mono-Boc protected benzidine, leading to two orthogonal functionalities on protected/masked amino groups in 2o, allowing further derivatisation at these two sites. We also attempted to transform PAH amines to thiocarbamoyl benzotriazoles 2p–v through milling synthesis, and gratifyingly found that the near-quantitative isolated yields were not affected by condensation of aromatic units. The only exception was 9-aminoanthracene which failed to afford the desired product 2s, presumably due to steric hindrance around the amino group.15 | ||
| Scheme 2 Mechanochemical synthesis of thiocarbamoyl benzotriazoles 2a–v followed by amination to thioureas 3a–v using the NH4Cl/Na2CO3 system (4 eq.) under milling conditions. Typical milling conditions were: a 10 mm stainless steel ball, a 10 mL jar, 30 Hz, dry acetonitrile as the grinding liquid (η = 0.25 μL mg−1), 10 minutes (1st step) and 60 minutes (2nd step). In the cases of 2j and 2n, two equivalents of 1 were used. Amination of 2l and 2m was achieved using 5 eq. of Na2CO3 under LAG conditions. | ||
All of the products have been characterised by 1H/13C NMR and FTIR-ATR spectroscopy. In the IR spectra, a characteristic N–H stretching vibration of the thiocarbamoyl NH group is observed in the range from 3200 to 3250 cm−1. In cases where the solution incompatibility of aromatic thiocarbamoyl benzotriazoles still allowed the NMR spectra to be acquired, characteristic chemical shift values in chloroform at 10.6–11.1 ppm for NH protons, along with benzotriazole doublets at ca. 8.95 and 8.15 ppm, and triplets at ca. 7.65 and 7.55 ppm, respectively, undoubtedly confirmed the structural identity of thiocarbamoylated benzotriazoles 2a–v. All these results testify the wide applicability of mechanochemistry to the synthesis of thiocarbamoyl benzotriazoles, particularly aromatic ones as useful and practical synthetic intermediates.
Amination of thiocarbamoyl benzotriazoles – synthesis of primary thioureas 3
Amination of 4-bromophenyl thiocarbamoyl benzotriazole (2a) was selected as a model reaction to investigate 2a reactivity in ammonia vapours. A sample of 2a was exposed to NH3(g) in a closed vial containing aqueous ammonia solution at room temperature and the progress of the reaction was monitored by an ex situ FTIR-ATR analysis. Recently, FTIR has been utilized as a fast and reliable spectroscopic technique for probing the course of mechanochemical reactions such as hydroamination of alkynes16 and amide synthesis.17 The absorption bands positioned at 1617 and 509 cm−1 characteristic for N-(4-bromophenyl)thiourea (3a) appeared after 90 minutes, whereas 6 hours into aging resulted in significant transformation to 3a as evidenced by reduction of 2a band intensities at 1588, 1520, 1157, 1143, 968, 924 and 494 cm−1.Simultaneously, thiourea 3a bands at 1009 and 800 cm−1 alongside absorption at 1617 and 509 cm−1, increased in the intensity. Notably, a very broad absorption band of low intensity positioned between 2200 and 2000 cm−1 could be detected during the reaction, suggesting the ammonia-induced decomposition of 2a to 4-bromophenyl isothiocyanate as the reactive species.10,11 Finally, overnight exposure to NH3(g) led to disappearance of 2a bands, quantitatively yielding thiourea 3a (Fig. 1a–c).
 | ||
| Fig. 1 Up: FTIR-ATR analysis of 4-bromophenyl thiocarbamoyl benzotriazole 2a amination by aqueous ammonia solution. Aging of 2a in ammonia vapours at room temperature in a closed vial after: (a) 90 minutes, (b) 6 hours and (c) overnight. (d) LAG reaction using NH3(aq), (e) LAG using water with in situ ammonia formation from NH4Cl and Na2CO3, (f) 2a and (g) thiourea 3a. Middle: Visual changes during aging of 2a in ammonia vapours in a closed vial. Initially the light yellow sample of 2a (25 mg) gradually becomes off-white on exposure to ammonia gas (1 mL of conc. NH3(aq) was placed in the small test tube). Bottom: Colour change after mechanochemical synthesis of 2a (1st step) and the amination reaction to produce thiourea 3a (2nd step). | ||
Visual inspection of the aging reaction also suggested a chemical transformation as the colour of the sample changed from pale yellow to off-white (Fig. 1). Next, several aryl-substituted thiocarbamoyl benzotriazoles 2b, 2d, 2h and 2i were subjected to overnight ammonia vapour digestion, again affording thioureas 3b, 3d, 3h and 3i in quantitative yields, based on FTIR-ATR analyses (Fig. S62–S65, ESI†).
While aging has recently been introduced as an attractive methodology to convert metal oxides, sulfides and other salts into useful metal–organic frameworks (MOFs),18a–c the results in the present work establish aging or vapour digestion as a viable approach in the field of organic synthesis as well. Indeed, previous reports on syntheses of Schiff bases under different humidity or solvent vapour levels,18d and of organometallic palladium complexes18e support the paradigm of aging being an operationally simple, low-energy and environmentally-friendly way of practicing “slow chemistry”.1a
Intrigued by the possibility to convert thiocarbamoyl benzotriazoles to primary thioureas by ammonia vapour digestion, we speculated on a liquid-assisted grinding experiment in a ball mill, where aqueous ammonia solution NH3(aq) would be used as the grinding liquid. In this case, NH3(aq) could provide the source of the amination reagent (NH3) but also of a liquid medium necessary to conduct the LAG experiment (water). Rather disappointingly, FTIR-ATR analysis revealed a small amount of thiourea 3a in the crude product while the bromo-derivative 2a remained unchanged (conversion 23.6%, Fig. 1d). The obvious discrepancy in the extent of the amination reaction carried out by milling (low conversion) or aging (quant. conversion), led us to a conclusion that water during LAG using NH3(aq) played a key role in failure of the milling approach. For this reason, we resorted to other sources of ammonia.
Most conveniently for solid-state mechanochemical synthesis, we found that ammonia gas can easily be generated in situ by milling equimolar amounts of ammonium chloride and sodium carbonate.19 To corroborate the above hypothesis of “LAG inhibition” in 2a amination by NH3(aq), intermediate 2a was subjected to 60 minutes of LAG using water, in the presence of NH4Cl/Na2CO3 (4 equivalents) as the NH3 source. FTIR-ATR analysis showed only traces of thiourea 3a (weak signals at 1617 and 509 cm−1) in a mixture with unreacted starting material 2a (Fig. 1e), despite ammonia whose presence was indicated by strong smell on opening the jar. The conversion was estimated to 3.1%, based on the ratio of peak areas at 509 cm−1 (for 3a) and 494 cm−1 (for 2a).
As a tentative explanation for the observed inhibitory effect of water in this type of LAG reaction, we propose strong hydrogen-bonding solvation of NH3 molecules in water as opposed to alcohols and acetonitrile (vide infra) which have a lower tendency to form hydrogen bonds.20 Ammonia molecules are likely to form clusters with water molecules resulting in at least partial protonation of NH3, leading to solvated ammonia cation species NH4+(H2O)n.21 The nucleophilicity of the nitrogen atom in such molecular assemblies would be poor, thus explaining its very low reactivity towards thiocarbamoylated intermediate 2a. We also tested the ability of ammonium chloride alone to induce the transformation of intermediate 2a to thiourea 3a, either by aging at room temperature or by neat milling for 60 minutes. In both cases, FTIR-ATR analyses confirmed that no reaction had taken place under these conditions (Fig. S66, ESI†), which is in line with our expectations. Finally, when a mixture of 2a and four equivalents of Na2CO3 in the absence of NH4Cl was milled in the presence of water or ethanol, decomposition to 4-bromophenyl isothiocyanate occurred with some intermediate still unreacted. On the other hand, when acetonitrile was employed, or under solvent-free conditions, the degree of decomposition was small based on IR analysis, showing that water and ethanol as protic solvents in LAG do not interfere with the base-promoted decomposition of thiocarbamoyl intermediate 2a (Fig. S67†). When NH4Cl (pKa 9.24)22 is present, sodium carbonate (pKa 10.25) is likely to deprotonate it first as the most acidic species in the typical mixture,23 which was suggested by the lack of isothiocyanate bands in IR spectra of the crude mixture and worked-up samples from the water-assisted grinding reaction (only traces of thiourea product 3a and unreacted 2a, Fig. 1e). In the case of addition of an extra equivalent of sodium carbonate, IR analysis showed only slightly better conversion of 2a to thiourea 3a (13.1% vs. 3.1%, Fig. S68†). However, the evolved ammonia gas itself can act as the base, but for the above mentioned solvation effects in water, its proton accepting ability would be disturbed, thus leaving 2a virtually intact. With acetonitrile, the reactivity of gaseous ammonia in terms of basicity and nucleophilicity would not be affected and the reaction is proposed to proceed by the NH3-induced decomposition of 2a into 4-bromophenyl isothiocyanate, which is then rapidly consumed by ammonia to quantitatively yield the thiourea product 3a, reminiscent of aging amination (Scheme 3).
 | ||
| Scheme 3 Proposed mechanism for thiocarbamoyl benzotriazole amination under neat or liquid-assisted grinding and aging conditions. | ||
The effect of water on the stoichiometric composition of zinc and copper MOFs in a stepwise LAG synthesis has been studied and tracked down to mole fractions and activity of water.24 Inspired by these findings we conducted a series of LAG experiments where water–acetonitrile and water–ethanol binary mixtures of different compositions (20, 40, 60, 80 and 90% v/v) were used as the grinding liquid (η = 0.25 μL mg−1), in order to evaluate the influence of the water content in an organic solvent on the conversion of thiocarbamoyl benzotriazole 2a to thiourea 3a. The progress of the LAG reactions was monitored by FTIR-ATR.25 As the diagram in Fig. 2 shows, no linear relationship between the conversion and the water content in a solvent (expressed as volume percentage v/v% or mole fraction x(H2O)) was observed. Instead, we found that the quantitative formation of 3a in LAG is not affected by the presence of water over a broad range of volume percentage (up to 60% v/v) and mole fraction values (up to x(H2O) 0.7). A significant negative effect occurred when the water content reached threshold values of v/v% > 60 and x(H2O) > 0.8.
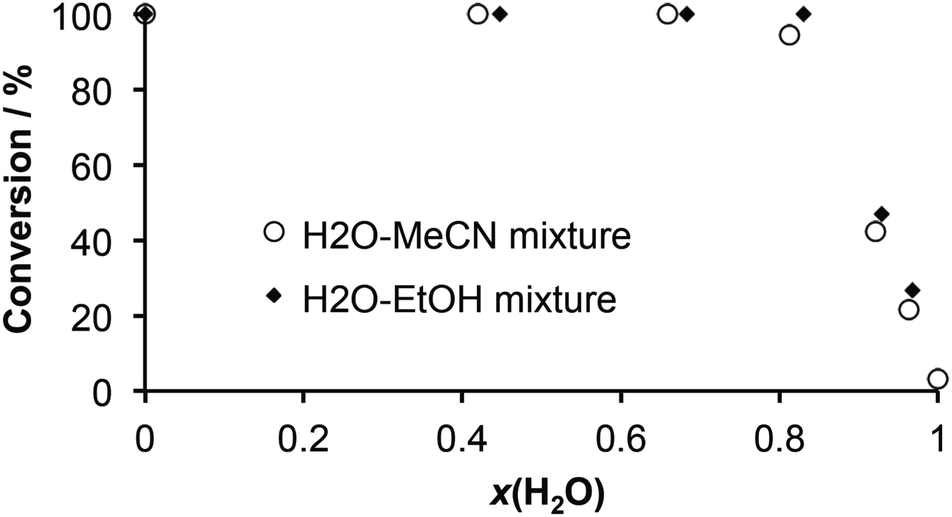 | ||
| Fig. 2 A correlation between the mole fraction of water in binary solvent mixtures used in LAG and the conversion of thiocarbamoyl benzotriazole 2a to primary thiourea 3a. | ||
In comparison with acetonitrile mixtures, water–ethanol mixtures consistently yielded higher conversions. We explain this observation by higher water activity in an aprotic solvent (such as acetonitrile), resulting from differences in hydrogen bond basicities of acetonitrile (pKHB 0.91) and ethanol (pKHB 1.02).20 As a poor hydrogen bond acceptor, acetonitrile is less likely to disrupt the hydrogen bond network in ammonia–water clusters at higher water content levels.
On the other hand, ethanol as a protic solvent competes with ammonia for water molecules, thus increasing the effective concentration of unsolvated ammonia molecules available for the nucleophilic addition reaction. At lower mole fractions, the apparent insensitivity of the conversion in acetonitrile and ethanol mixtures is ascribed simply to the dilution effect of the organic solvent where differences in hydrogen bond basicities do not play a major role. We also note that this behaviour in LAG using aqueous organic solvents is the first case of such observation in mechanochemical organic synthesis, thus making it a contribution to better understanding of LAG reactions and related solvent effects.26
Switching from water to dry tert-butanol, ethanol and acetonitrile as the grinding liquids afforded the desired thiourea 3a in an excellent 90% isolated yield over two steps in 60 minutes.27 An ex situ FTIR-ATR analysis showed that LAG reaction using acetonitrile reached completion within only 5–10 minutes whereas neat grinding, which also worked well with quantitative conversions and the same isolated yield, took up to 20 minutes (Fig. S69 and S70, ESI†). Despite LAG being faster with 2a as the substrate, but bearing in mind the green chemistry aspect of the reaction and the maximum reduction in solvent usage, we opted for the solvent-free approach in a reasonably short reaction time (60 min) to ensure completion of amination with a range of thiocarbamoyl benzotriazoles 2a–v differing in reactivity (Scheme 2).
In general, simple aromatic thioureas 3a–k were prepared and isolated in ≥90% yield, as well as mono- (3o) and bis-thioureas (3n) derived from benzidine. Interestingly, the alkyl derivatives 3l and 3m required an additional equivalent of the base under LAG conditions for complete conversion to thioureas. We interpret this result through increased stability of alkyl thiocarbamoyl benzotriazole intermediates 2l and 2m which are stable enough to be prepared and even isolated by conventional solution synthesis.10b Next, PAH thiocarbamoyl benzotriazoles 2p–v were successfully transformed to thioureas 3p–v. Extending the aromatic system in naphthalene derivative 3p to 1-, 2- and 9-anthracene thioureas 3q–s and phenanthrene thiourea 3t had no effect on the efficiency of the amination reaction, keeping the isolated yields of pure products over 94%. In the case of thiourea 3s, the amination was carried out by reacting the corresponding anthracene-9-isothiocyanate for 90 minutes using acetonitrile as the grinding liquid in LAG, demonstrating that isothiocyanates can also be utilised as reactants when the respective thiocarbamoyl benzotriazoles are not available. Thiocarbamoyl benzotriazoles 2u and 2v, derived from 6-aminocrysene and 1-aminopyrene as representatives of PAHs with four fused aromatic rings, also cleanly underwent the solvent-free amination to afford thioureas 3u and 3v in near-quantitative yield. To demonstrate the superiority of the herein presented green mechanochemical approach to primary thioureas, we also performed the conventional solution synthesis of N-(2-anthracenyl)thiourea (3r) following the protocol by Douglass and Dains.13b Starting from 2-aminoanthracene, N-(2-anthracenyl)-N′-benzoylthiourea was synthesised in the first step in 55% yield (after purification by recrystallisation from toluene). This intermediate was next hydrolysed with sodium hydroxide solution, affording thiourea 3r in 95% yield, and 52% based on 2-aminoanthracene (for details, see the ESI†). For comparison, mechanosynthesis afforded pure 3r in total yield of 94% in just over an hour, with entirely aqueous workup.28
Exploiting the stability differences between aromatic (less stable) and aliphatic (more stable) thiocarbamoyl benzotriazoles as mentioned above, enabled chemoselective preparation of mono-thiourea 6 and bis-thiourea 7 (Scheme 4). LAG treatment of 4-aminobenzyl amine (4) with two equivalents of 1 for 10 minutes quantitatively yielded bis-thiocarbamoylated benzylic derivative 5. Further neat grinding of 5 with four equivalents of an equimolar NH4Cl/Na2CO3 mixture resulted in a regioselective amination to afford a mixed thiocarbamoyl benzotriazole thiourea 6.
 | ||
| Scheme 4 Directing the reactivity of a mixed aromatic-aliphatic bis-thiocarbamoyl benzotriazole 5 by switching from neat (NG) to liquid-assisted grinding (LAG) and adding an extra equivalent of the base. The A and B reaction pathways reflect different stabilities of aromatic vs. aliphatic thiocarbamoyl benzotriazoles. | ||
As expected, the benzotriazole moiety was displaced on the aromatic side due to higher reactivity of aromatic thiocarbamoyl benzotriazoles.11 However, continuing the amination of 6 under LAG conditions using an extra Na2CO3 equivalent results in the formation of bis-thiourea 7 in 82% isolated yield. The bis-thiourea 7 can also be accessed directly from 5 by milling with a NH4Cl (8 eq.)/Na2CO3 (9 eq.) mixture in 70% isolated yield.
Crystallography
The crystal structures of simple aromatic primary thioureas have already been determined for several compounds shown in Scheme 2 (e.g. p-substituted thioureas 3a–e),29 thus we focused our attention on structural features of PAH derivatives 3p–v with fused aromatic rings. The only known structure from this class is of 1-naphthylthiourea 3p, which consists of individual 3p molecules connected to a chain and held by N–H⋯S hydrogen bonds in a typical R22(8) thiourea supramolecular synthon.30 Powder X-ray diffraction (PXRD) analysis of PAH thioureas 3p–v showed that all of the products were crystalline solids. The measured diffractogram of 1-naphthylthiourea 3p perfectly matches the simulated one from the reported crystal structure (ref. code MOJDAS), confirming that mechanosynthesis provides the same polymorph as solution crystallisation (Fig. S72, ESI†). Despite all our attempts to crystallise PAH-thioureas 3q–v, we did not succeed in obtaining single crystals suitable for diffraction analysis. However, enormous progress made over the years in the field of crystal structure determination from PXRD data31 enabled us to unravel the molecular and crystal structures for some of these compounds. Specifically, the structures of 1-anthracenylthiourea (3q) and 1-pyrenylthiourea (3v) were successfully elucidated by employing this approach.The crystal structures of 3q and 3v were determined from synchrotron PXRD data collected at the Diamond Light Source, beamline I12-JEEP (λ = 0.23307 Å).32 Indexing of both powder patterns revealed monoclinic unit cells with Z = 4 and subsequent structure solution using TOPAS33 revealed Z′ = 1. In both molecules, 3q and 3v, thiourea units are essentially planar and almost perpendicular to the aromatic ring.
In the crystal structure, each 3q molecule is hydrogen-bonded to the neighboring molecule via R22(8) homosynthon forming a centrosymmetric dimer, while in 3p (as an analogue with two fused aromatic rings) each molecule forms two dimers via R22(8) homosynthons. The intermolecular N–H⋯S bonds in these assemblies are equal to 3.31(7) Å. The dimers are further linked by N–H⋯S hydrogen bonds (d = 3.20(4) Å) in R24(8) synthon forming an infinite ladder-type ribbon stretching along the a-axis. Notably, single chains within these ribbons are aligned in an antiparallel fashion as a result of head-to-tail stacking of 3q molecules. Both hydrogen atoms of the NH2 group are involved in the formation of N–H⋯S hydrogen bonds, while the free hydrogen atom of the NH group points towards the aromatic ring of the nearest molecule (Fig. 3a).
 | ||
| Fig. 3 (a) Supramolecular organization of dimers in the crystal structure of 1-anthracenylthiourea 3q, (b) a chain of hydrogen-bonded 1-pyrenylthiourea 3v, (c) and (d) Rietveld refinement for 3q and 3v, respectively. | ||
The pyrene derivative 3v displays a structural motif very similar to those found in 1-naphthylthiourea 3p. The molecules of 3v are associated with N–H⋯S interactions (3.23(2) Å and 3.47(2) Å) in distorted R22(8) homosynthons and organized into ribbons along the c-axis. While the hydrogen-bonded thiourea moieties in 3p are almost aligned in a single plane, the geometry of intermolecular N–H⋯S bonds in 3v deviates significantly from the planar arrangement of hydrogen bond donors and acceptors, resulting in weaker N–H⋯S interactions than in 3p or 3q. The hydrogen bond N–H⋯S distances for 3q and 3v are typical for organic phenylthiourea structures.34 An interesting feature of the 3v crystal structure is the weak Caromatic–H⋯S interactions (3.80(7) Å) between the thiourea unit and the aromatic ring of the neighboring molecule inserted into the ribbon (Fig. 3b). This interaction, together with the weak π⋯π stacking (3.76 Å) between pyrene subunits leads to a supramolecular arrangement where the parallel ribbons are organised into two-dimensional staggered sheets (Fig. S75 and S76, ESI†).
Conclusions
In summary, the synthesis of novel thiocarbamoyl benzotriazoles and their reactivity as electrophiles in the solid–gas aging reaction with ammonia vapours is described. In the next step, we have utilised ammonia vapour digestion as a platform to develop an environmentally-friendly mechanochemically-based strategy that allowed facile and solvent-free construction of N-monosubstituted thioureas directly from amines. The effectiveness of the presented synthetic method is reflected in near-quantitative isolated yields, high purity of the products, and simple and straightforward aqueous workup. Neat and liquid-assisted grinding gave similar results, however, the mechanochemical amination seems to be incompatible with water as the grinding liquid. The screening of different water levels in acetonitrile and ethanol aqueous mixtures revealed that the presence of water at x(H2O) > 0.8 led to lower conversions in both solvent systems. This negative effect was more pronounced in mixtures with acetonitrile due to its aprotic character and higher water activity, which supports the hypothesis that water retards the amination process by strong solvation of ammonia molecules through hydrogen bonding. Keeping in mind all the benefits of solvent-free research laboratory offered by mechanochemistry,35 the organic solvent-free synthesis of thioureas was coupled with structure determination from synchrotron X-ray diffraction data to gain insight into the molecular arrangement of polyaromatic primary thioureas in the solid-state.Acknowledgements
This work has been supported by the Croatian Science Foundation grant no. 9310. We gratefully acknowledge Dr Ernest Meštrović (Pliva TAPI) for collecting the powder X-ray diffraction patterns of intermediates and thioureas. We also thank Diamond Light Source for access to beamline I12-JEEP that contributed to the results presented here.Notes and references
- (a) X. Lim, Nat. News, 2015, 524, 20–21 CrossRef CAS PubMed; (b) S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friščić, F. Grepioni, K. D. M. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steed and D. C. Waddell, Chem. Soc. Rev., 2012, 41, 413–447 RSC; (c) D. Daurio, K. Nagapudi, L. Li, P. Quana and F.-A. Nunez, Faraday Discuss., 2014, 170, 235–249 RSC; (d) D. Crawford, J. Casaban, R. Haydon, N. Giri, T. McNally and S. L. James, Chem. Sci., 2015, 6, 1645–1649 RSC.
- (a) S. H. Leung and S. A. Angel, J. Chem. Educ., 2004, 81, 1492–1493 CrossRef CAS; (b) A. Wixtrom, J. Buhler and T. Abdel-Fattah, J. Chem. Educ., 2014, 91, 1232–1235 CrossRef CAS.
- (a) G. Kaupp, CrystEngComm, 2009, 11, 388–403 RSC; (b) W. Jones and M. D. Eddleston, Faraday Discuss., 2014, 170, 9–34 RSC; (c) G.-W. Wang, Chem. Soc. Rev., 2013, 42, 7668–7700 RSC; (d) J. G. Hernández, I. S. Butler and T. Friščić, Chem. Sci., 2014, 5, 3576–3582 RSC; (e) V. Štrukil, M. D. Igrc, M. Eckert-Maksić and T. Friščić, Chem. – Eur. J., 2012, 18, 8464–8473 CrossRef PubMed; (f) J. G. Hernández and C. Bolm, Chem. Commun., 2015, 51, 12582–12584 RSC; (g) D. A. Davis, A. Hamilton, J. Yang, L. D. Cremar, D. Van Gough, S. L. Potisek, M. T. Ong, P. V. Braun, T. J. Martínez, S. R. White, J. S. Moore and N. R. Sottos, Nature, 2009, 459, 68–72 CrossRef CAS PubMed; (h) T. Kleine, J. Buendia and C. Bolm, Green Chem., 2013, 15, 160–166 RSC.
- (a) T. Friščić, A. V. Trask, W. Jones and W. D. S. Motherwell, Angew. Chem., Int. Ed., 2006, 45, 7546–7550 CrossRef PubMed; (b) T. Friščić, S. L. Childs, S. A. A. Rizvi and W. Jones, CrystEngComm, 2009, 11, 418–426 RSC.
- T. Friščić, D. G. Reid, I. Halasz, R. S. Stein, R. E. Dinnebier and M. J. Duer, Angew. Chem., Int. Ed., 2010, 49, 712–715 CrossRef PubMed.
- (a) I. Halasz, T. Friščić, S. A. J. Kimber, K. Užarević, A. Puškarić, C. Mottillo, P. Julien, V. Štrukil, V. Honkimaki and R. E. Dinnebier, Faraday Discuss., 2014, 170, 203–221 RSC; (b) K. Užarević, I. Halasz and T. Friščić, J. Phys. Chem. Lett., 2015, 6, 4129–4140 CrossRef PubMed.
- (a) T. Friščić, I. Halasz, P. J. Beldon, A. M. Belenguer, F. Adams, S. A. J. Kimber, V. Honkimäki and R. E. Dinnebier, Nat. Chem., 2013, 5, 66–73 CrossRef PubMed; (b) I. Halasz, A. Puškarić, S. A. J. Kimber, P. J. Beldon, A. M. Belenguer, F. Adams, V. Honkimäki, R. E. Dinnebier, B. Patel, W. Jones, V. Štrukil and T. Friščić, Angew. Chem., Int. Ed., 2013, 52, 11538–11541 CrossRef CAS PubMed; (c) A. Katsenis, A. Puškarić, V. Štrukil, C. Mottillo, P. A. Julien, K. Užarević, M.-H. Pham, T.-O. Do, S. A. J. Kimber, P. Lazić, O. Magdysyuk, R. E. Dinnebier, I. Halasz and T. Friščić, Nat. Commun., 2015, 6, 6662 CrossRef CAS PubMed.
- D. Gracin, V. Štrukil, T. Friščić, I. Halasz and K. Užarević, Angew. Chem., Int. Ed., 2014, 53, 6193–6197 CrossRef CAS PubMed.
- L. Batzdorf, F. Fischer, M. Wilke, K.-J. Wenzel and F. Emmerling, Angew. Chem., Int. Ed., 2015, 54, 1799–1802 CrossRef CAS PubMed.
- (a) A. R. Katritzky, R. M. Witek, V. Rodriguez-Garcia, P. P. Mohapatra, J. W. Rogers, J. Cusido, A. A. A. Abdel-Fattah and P. J. Steel, J. Org. Chem., 2005, 70, 7866–7881 CrossRef CAS PubMed; (b) A. R. Katritzky, S. Ledoux, R. M. Witek and S. K. Nair, J. Org. Chem., 2004, 69, 2976–2982 CrossRef CAS PubMed; (c) A. R. Katritzky and B. V. Rogovoy, Chem. – Eur. J., 2003, 9, 4586–4593 CrossRef CAS PubMed.
- V. Štrukil, D. Gracin, O. Magdysyuk, R. E. Dinnebier and T. Friščić, Angew. Chem., Int. Ed., 2015, 54, 8440–8443 CrossRef PubMed.
- (a) M. Bukvić Krajačić, M. Perić, K. S. Smith, Z. Ivezić Schönfeld, D. Žiher, A. Fajdetić, N. Kujundžić, W. Schönfeld, G. Landek, J. Padovan, D. Jelić, A. Ager, W. K. Milhous, W. Ellis, R. Spaventi and C. Ohrt, J. Med. Chem., 2011, 54, 3595–3605 CrossRef PubMed; (b) J. Stefanska, D. Szulczyk, A. E. Koziol, B. Miroslaw, E. Kedzierska, S. Fidecka, B. Busonera, G. Sanna, G. Giliberti, P. La Colla and M. Struga, Eur. J. Med. Chem., 2012, 55, 205–213 CrossRef CAS PubMed; (c) P. A. Yonova and G. M. Stoilkova, J. Plant Growth Regul., 2005, 23, 280–291 CrossRef; (d) G. V. Boyd, in Amidines and Imidates, ed. S. Patai and Z. Rappoport, Wiley, Chichester, 1991, vol. 2, ch. 7, pp. 339–366 Search PubMed; (e) Y. Yamamoto and S. Kojima, in Amidines and Imidates, ed. S. Patai and Z. Rappoport, Wiley, Chichester, 1991, vol. 2, ch. 10, pp. 485–526 Search PubMed.
- (a) R. J. Herr, J. L. Kuhler, H. Meckler and C. J. Opalka, Synthesis, 2000, 1569–1574 CrossRef CAS; (b) I. B. Douglass and F. B. Dains, J. Am. Chem. Soc., 1934, 56, 1408–1409 CrossRef CAS; (c) D. C. Schroeder, Chem. Rev., 1955, 55, 181–228 CrossRef CAS; (d) M. L. Moore and F. S. Crossley, Org. Synth., 1955, 617 Search PubMed , Collect. vol. 3.
- G. Kaupp, J. Schmeyers and J. Boy, Tetrahedron, 2000, 56, 6899–6911 CrossRef CAS.
- Thiocarbamoyl benzotriazoles 2p, 2t and 2u, derived from 1-aminonaphthalene, 9-aminophenanthrene and 6-aminocrysene were found by IR and 1H NMR analysis to be partially decomposed to isothiocyanates, whereas the other synthesised derivatives were stable and pure after aqueous workup.
- M. Weiße, M. Zille, K. Jacob, R. Schmidt and A. Stolle, Chem. – Eur. J., 2015, 21, 6511–6522 CrossRef PubMed.
- T.-X. Métro, J. Bonnamour, T. Reidon, A. Duprez, J. Sarpoulet, J. Martinez and F. Lamaty, Chem. – Eur. J., 2015, 21, 12787–12796 CrossRef PubMed.
- (a) M. J. Cliffe, C. Mottillo, R. S. Stein, D.-K. Bučar and T. Friščić, Chem. Sci., 2012, 3, 2495–2500 RSC; (b) F. Qi, R. S. Stein and T. Friščić, Green Chem., 2014, 16, 121–132 RSC; (c) C. Jia, J. Wang, X. Feng, Q. Lin and W. Yuan, CrystEngComm, 2014, 16, 6552–6555 RSC; (d) D. Cinčić, I. Brekalo and B. Kaitner, Chem. Commun., 2012, 48, 11683–11685 RSC; (e) M. Juribašić, I. Halasz, D. Babić, D. Cinčić, J. Plavec and M. Ćurić, Organometallics, 2014, 33, 1227–1234 CrossRef.
- In a paper by Konnert et al. formation of ammonia during milling, as one of the by-products of potassium cyanate and trimethylsilyl isocyanate hydrolysis, was reported. However, the evolved gas did not participate in the reaction, either as a base or a nucleophile. For details, see L. Konnert, B. Reneaud, R. M. de Figueiredo, J.-M. Campagne, F. Lamaty, J. Martinez and E. Colacino, J. Org. Chem., 2014, 79, 10132–10142 CrossRef CAS PubMed.
- C. Laurence and J.-F. Gal, Lewis Basicity and Affinity Scales: Data and Measurement, Wiley, Chichester, 2010 Search PubMed.
- (a) D. E. Bacelo, J. Phys. Chem. A, 2002, 106, 11190–11196 CrossRef CAS; (b) S. Karthikeyan, N. J. Singh and K. S. Kim, J. Phys. Chem. A, 2008, 112, 6527–6532 CrossRef CAS PubMed; (c) P. Hvelplund, T. Kurtén, K. Støchkel, M. J. Ryding, S. Brøndsted Nielsen and E. Uggerud, J. Phys. Chem. A, 2010, 114, 7301–7310 CrossRef CAS PubMed.
- G. Busca, Chem. Rev., 2010, 110, 2217–2249 CrossRef CAS PubMed.
- Gas phase basicity (GB) of 2a, calculated at the M06-2X/6-311++G(2df,2pd)//M06-2X/6-31+G(d,p) level, is 322.8 kcal mol−1, while of ammonia the GB value is 200.0 kcal mol−1. The calculated pKa values of 2a and ammonia in water are 9.01 and 4.02, respectively, with the SMD/M06-2X/6-31+G(d,p)//M06-2X/6-31+G(d,p) approach. While the theoretical prediction of the pKa value of ammonia in water is often compromised due to its small size and high charge density in the cation (in this case, underestimated by more than 5 pKa units relative to the experimental value of 9.24), we note the correct and expected trend in GB values, suggesting ammonia as the most acidic species in the reaction mixture.
- F. C. Strobridge, N. Judaš and T. Friščić, CrystEngComm, 2010, 12, 2409–2418 RSC.
- Standard powder mixtures of 2a and 3a simulating different conversion values were prepared by manual grinding and the quantitative analysis was based on the ratio of peak areas at 509 cm−1 (for 3a) and 494 cm−1 (for 2a). After each milling experiment, the IR spectrum of the crude mixture was recorded, followed by aqueous workup and IR quantitative analysis.
- M. Tireli, M. Juribašić Kulcsar, N. Cindro, D. Gracin, N. Biliškov, M. Borovina, M. Ćurić, I. Halasz and K. Užarević, Chem. Commun., 2015, 51, 8058–8061 RSC.
- FTIR-ATR analysis of the crude reaction mixtures revealed the quantitative conversion of the bromo-intermediate 2a to thiourea 3a.
- For more examples of aqueous work-up in mechanosynthesis, see: (a) V. Štrukil, B. Bartolec, T. Portada, I. Đilović, I. Halasz and D. Margetić, Chem. Commun., 2012, 48, 12100–12102 RSC; (b) D. Tan, C. Mottillo, A. D. Katsenis, V. Štrukil and T. Friščić, Angew. Chem., Int. Ed., 2014, 53, 9321–9324 CrossRef CAS PubMed.
- Cambridge Structural Database, accessed July 2015.
- M. A. AlDamen and M. Sinnokrot, J. Struct. Chem., 2014, 55, 53–60 CrossRef CAS.
- K. D. M. Harris, Top. Curr. Chem., 2012, 315, 133–178 CrossRef CAS PubMed.
- M. Drakopoulos, T. Connolley, C. Reinhard, R. Atwood, O. Magdysyuk, N. Vo, M. Hart, L. Connor, B. Humphreys, G. Howell, S. Davies, T. Hill, G. Wilkin, U. Pedersen, A. Foster, N. De Maio, M. Basham, F. Yuan and K. Wanelik, J. Synchrotron Radiat., 2015, 22, 828–838 CrossRef CAS PubMed.
- A. A. Coelho, TOPAS, Version 4.1, 2007, Coelho Software, Brisbane, Australia Search PubMed.
- (a) B. M. Yamin, H. F. Salem and S. F. M. Yusoff, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2013, 69, o1468 CAS; (b) M. M. Rosli, M. S. Karthikeyan, H.-K. Fun, I. A. Razak and P. S. Patil, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, o67 CAS; (c) Y.-F. Lin, G.-X. Zhong, F. Xu and W.-X. Hu, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, o3549 CAS; (d) M. M. Rosli, M. S. Karthikeyan, H.-K. Fun, I. A. Razak, P. S. Patil, B. S. Holla and S. M. Dharmaprakash, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2006, 62, o5692 CAS; (e) A. Saeed, U. Shaheen and U. Floerke, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2010, 66, o1558 CAS; (f) Y.-H. Shen and D.-J. Xu, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2004, 60, o1193 CAS; (g) T. Steiner, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1998, 54, 1121 Search PubMed; (h) L. Xian, Y. Wang and B. Zhang, Z. Kristallogr. - New Cryst. Struct., 2008, 223, 411–412 CAS; (i) H.-K. Fun, C. K. Quah, P. S. Nayak, B. Narayana and B. K. Sarojini, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2012, 68, o2460 CAS; (j) H.-K. Fun, C. K. Quah, P. S. Nayak, B. Narayana and B. K. Sarojini, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2012, 68, o2462 CAS; (k) H.-K. Fun, C. K. Quah, P. S. Nayak, B. Narayana and B. K. Sarojini, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2012, 68, o2423 CAS; (l) H. F. Saleem and B. M. Yamin, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2010, 66, o789 CAS; (m) H.-B. Shi, W.-X. Hu and Y.-F. Lin, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 65, o2401 CAS; (n) B. K. Sarojini, B. Narayana, K. Sunil, H. S. Yathirajan and M. Bolte, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, o3754 CAS.
- V. Štrukil, M. D. Igrc, L. Fábián, M. Eckert-Maksić, S. L. Childs, D. G. Reid, M. J. Duer, I. Halasz, C. Mottillo and T. Friščić, Green Chem., 2012, 14, 2462–2473 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures for the synthesis of thiocarbamoyl benzotriazoles and monosubstituted thioureas, FTIR-ATR and NMR spectra, and crystallographic information for thioureas 3q and 3v. CCDC 1445153 and 1445154. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6gc00089d |
| This journal is © The Royal Society of Chemistry 2016 |