
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enhancing the stability of ionic liquid media for cellulose processing: acetal protection or carbene suppression?†
Matthew T.
Clough
 ab,
Jeraime A.
Griffith
a,
Olga
Kuzmina
a and
Tom
Welton
*a
ab,
Jeraime A.
Griffith
a,
Olga
Kuzmina
a and
Tom
Welton
*a
aDepartment of Chemistry, Imperial College London, London, SW7 2AZ, UK. E-mail: t.welton@imperial.ac.uk
bMax-Planck-Institut für Kohlenforschung, Kaiser-Wilhelm-Platz 1, 45470 Mülheim-an-der-Ruhr, Germany
First published on 4th April 2016
Abstract
Although excellent candidate solvents for cellulose, capable of dissolving ≥20 wt% of the carbohydrate for electrospinning processes, dialkylimidazolium carboxylate ionic liquids undergo undesirable side reactions with the reducing end of saccharides, terminating in an equilibrium concentration of a 2-(hydroxymethyl)-substituted imidazolium ‘adduct’. The addition of small molar quantities of a benign, non-toxic and inexpensive co-solvent, e.g. glycerol, reduces the rate of adduct accumulation, thereby enhancing the long-term thermal stability and recyclability of the expensive ionic liquid component. NMR, UV-vis and mass spectrometry experiments reveal that the improved stability is likely attributable to suppression of the transient dialkylimidazol-2-ylidene carbene, via hydrogen-donation by the protic co-solvent, rather than by cyclic acetal protection of the carbohydrate. The incorporation of (up to) 10 wt% of glycerol into the solvent mixture does not exacerbate the rate of cellulose depolymerisation compared to in the neat ionic liquid, and high solubility of cellulose is maintained. Furthermore, a colourimetric comparison of the recovered solvents, following cellulose re-precipitation, demonstrates that glycerol does not increase the concentration of contaminant reducing sugars in the organic electrolyte.
Introduction
In view of the rapidly depleting planetary fossil fuel reserves, accompanied by a fast-growing global population, there is a mounting imperative to identify and to exploit renewable resources in order to meet heightening energy and material demands. Lignocellulosic biomass is one excellent candidate renewable resource; the separation and subsequent deconstruction/reprocessing of the three major biopolymer components (cellulose, hemicellulose and lignin) affords a broad spectrum of textiles, fuels and commodity chemicals, potentially alleviating the reliance on petrochemical sources.1–3Ionic Liquids (ILs), salts that are molten at (or just above) room temperature, are increasingly finding favour as solvents for cellulose, which accounts for 35–50% of the weight of dry biomass and is the most abundant bio-renewable resource on earth.4–7 It is possible to prepare high-quality crystalline cellulose fibres by initially dissolving crude cellulose in an IL, and then ‘electrospinning’ the viscous solution into an anti-solvent pool (e.g. water).8–12
However, there is the need for an informed selection of the incorporated IL ions; both the anion and the cation4 play a critical role in dismantling the extensive inter-/intra-molecular hydrogen-bonding framework of crystalline cellulose. Accordingly, the pairing of strongly hydrogen-bond basic13,14 anions (high β,15e.g. Cl−, [OAc]− or [(CH3)2PO4]−) with cations that offer a hydrogen-bond acidity within a suitable range16 and that contribute to thermodynamically-favourable dissolution17 (e.g. 1-ethyl-3-methylimidazolium, ‘[C2C1im]+’) affords ILs that are effective cellulose-dissolving solvents. Furthermore, a certain degree of research attention has been devoted to the so-called ‘organic electrolyte solutions’, mixtures of an ionic species with a polar aprotic organic solvent (for example DMSO,18–20 (CH3)2CO21). LiCl in dimethylacetamide (DMA) is the archetypal organic electrolyte for cellulose dissolution, described first by Spange et al.22 The application of LiCl/DMA for solvation of cellulose is restricted by the toxicity of DMA; nevertheless, this report22 was the first to identify that basicity is a primary criterion for effective cellulose dissolution. Contemporary organic electrolyte solvent systems have the potential to maintain (or even to improve on18–20) moderate-to-high cellulose solubility, whilst simultaneously minimising difficulties associated with handling highly viscous solutions of cellulose in neat ILs.
Employing ILs for the dissolution of cellulose has the potential to circumvent profound limitations associated with contemporary industrial technologies; the Lyocell process is restricted by the limited thermal stability of the solvent, N-methylmorpholine-N-oxide (NMMO)23 and the modern Viscose process employs toxic, volatile, highly flammable CS2.24 ILs are not intrinsically expensive.25 However, when using comparatively costly cellulose-dissolving ILs of the form [CnC1im][X] (where n is typically 2 or 4 and [X]− is commonly Cl−, [OAc]− or [(CH3)2PO4]−), there is a necessity for ready and continuous recycling of the IL component. Recycling of the pure IL, following one cellulose reprocessing cycle, may be hindered via several pathways: (i) inherent thermal degradation of the IL;26,27 (ii) volatilisation of the intact IL ions28 (expected to be a minimal contribution for dialkylimidazolium carboxylate ILs on account of their high vaporisation enthalpies29,30), and; (iii) direct reaction between the IL and the cellulose solute.
Previously, Liebert and Heinze31 and Ebner et al.32 demonstrated the ambient-temperature reaction of the cellulose-dissolving IL [C4C1im][OAc], at the imidazolium C2 position, with the reducing (aldehyde) end of either D-(+)-glucose or cellulose itself. Reaction occurs via a transient N-Heterocyclic Carbene (NHC) intermediate, facilitated by the basicity of the IL anion, to yield a hydroxyalkylated adduct species.31–33
Recently, we further examined this reactivity by exposing mixtures of the IL [C2C1im][OAc] with carbohydrates to temperatures analogous to those of industrial cellulose reprocessing protocols (100–120 °C), for extended periods of time (up to 24 h); the mixtures underwent a sequence of decomposition reactions, terminating in an equilibrium concentration of the ‘C1 adduct’ compound, 1-ethyl-2-(hydroxymethyl)-3-methylimidazolium acetate, [C2C1(HO)C12im][OAc].34 Evaluation of the equilibrium concentration of this C1 adduct, and the rate at which maximum adduct concentration is reached, represents a useful proxy for assessing cellulose (or carbohydrate model compound) degradation when dissolved in ILs.
This observed sequence of degradation steps represents an undesired side-reaction between the IL and cellulose, hindering recycling of the solvent and, furthermore, damaging the carbohydrate fibres. Considering these reactive dialkylimidazolium carboxylate ILs, on initial inspection, blocking the C2–H substituent (e.g. methylation) will prevent the formation of an NHC intermediate and therefore suppress reactivity with cellulose. However, this substitution yields a salt with higher melting point and a smaller stable liquid range.35 Moreover, it is increasingly recognised that the acidic ring C2–H substituent participates in hydrogen-bonding interactions with hydroxyl residues along the cellulosic backbone, facilitating the actual dissolution process.36 The parent IL cation can be potentially recovered from the C1 adduct via high-temperature treatment (180 °C).34 However, temperatures of this magnitude are problematic regarding the inherent thermal instability of dialkylimidazolium carboxylate ILs.35 Therefore for a dialkylimidazolium carboxylate IL capable of dissolving a significant wt% of cellulose, strategies for enhancing the stability of the mixture via a structural modification of the IL ions appear unfeasible; this observation motivated our research group to investigate alternative approaches.
We proposed that it might be possible, instead, to suppress the sequence of IL-carbohydrate degradation mechanisms by inclusion of a covalent protecting group at the reducing end of cellulose, thereby leaving the cellulose backbone unchanged and retaining the beneficial high biopolymer solubility in the IL. The conversion of an aldehyde (e.g., the open form of the cellulose reducing end) or ketone into a hemi-acetal or acetal is one of the best-known organic protection strategies. In the circumstance that the aldehyde functionality can be protected in situ, in the form of a cyclic acetal, undesirable side reaction of the IL with cellulose could be suppressed. Investigations were undertaken to evaluate the feasibility of this protecting group strategy for cellulose dissolved in carboxylate ILs. The structures of all ILs (1, 2), carbohydrates (3–5) and co-solvents (6–12) employed in this investigation are displayed in Fig. 1. Syntheses of 1 and 2 are described in the ESI,† and experimental procedures are outlined below.
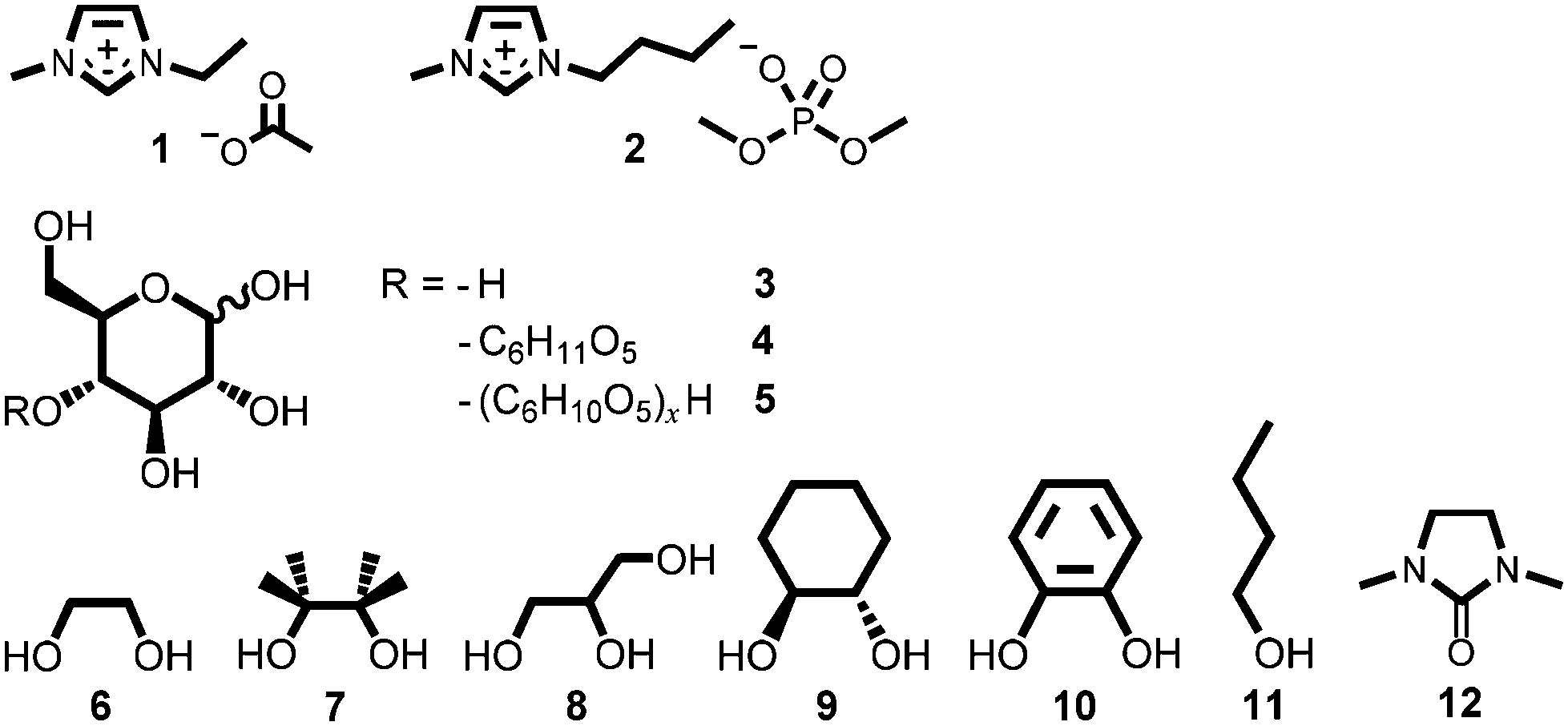 | ||
| Fig. 1 Structures of the ionic liquids (1, 2), carbohydrates (3–5) and co-solvents/additives (6–12) employed in this investigation. | ||
Experimental
Solvent–carbohydrate mixtures: preparation & analysis
A sample of carbohydrate (D-(+)-glucose 3, D-(+)-cellobiose 4, or cellulose 5) (0.050 ± 0.001 g) was carefully weighed into a 5 ml single-necked round-bottomed flask, fitted with an adapter for Schlenk apparatus and a magnetic stirrer bar. Where employed, a co-solvent/additive (ethane-1,2-diol, 6, 2,3-dimethylbutane-2,3-diol (pinacol), 7, glycerol, 8, trans-cyclohexane-1,2-diol, 9, benzene-1,2-diol, 10, 1-butanol, 11, or 1,3-dimethylimidazolidin-2-one, 12) and acetic acid (one drop, 11 ± 3 mg) were added to the reaction flask. A sample of the IL ([C2C1im][OAc] 1, or [C4C1im][(CH3)2PO4], 2) (0.50 ± 0.01 g), was then measured into the flask (water content of each ionic liquid sample <3 wt% for 3, 4, <0.5 wt% for 5). Subsequently, the flask was suspended in an oil bath, and the mixture was maintained at a temperature of 60 °C and at ∼0.1 mbar pressure for a pre-determined period of time (the ‘protection time’), with gentle stirring at a rate of 250 rpm. The flask was then raised from the oil bath and the temperature of the bath was rapidly increased to 120 °C. The vacuum in the flask was replaced by an ambient pressure of dry nitrogen gas. The flask was again suspended in the oil bath, and the mixture was maintained at 120 °C for a further minimum of four hours, with stirring at 250 rpm under a gentle flow of dry nitrogen. Small aliquots of the mixture (0.02 ± 0.01 g) were extracted at pre-determined intervals, ‘tx’ (e.g. x = 0, 0.5, 1, 2, 4 hours, with additional points for cellulose 5), from which 1H NMR spectra were obtained (in DMSO-d6 solvent). In addition, for the samples incorporating 5, the cellulose ‘degree of polymerisation’ was determined at t1 and t4 time points by Gel Permeation Chromatography (GPC, see experimental description below). The oil bath temperature fluctuated by no more than ±2 °C from the desired value during the course of the experiment. Compositional and experimental data, for all investigated IL-carbohydrate–(co-solvent) mixtures, is tabulated in the ESI (Table E1†). For each time point, tx, the ‘% Conversion’, representing the proportion of the parent IL cation ([CnC1im]+) that had undergone decomposition (conversion) with the carbohydrate to form the C1 adduct, [CnC1(HO)C12im]+, was evaluated according to the following equation:where: ‘
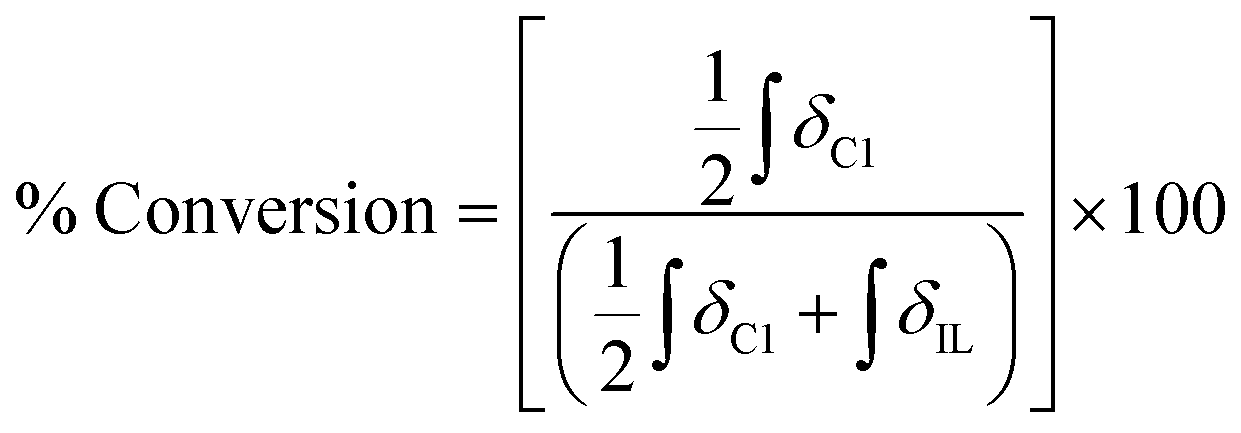
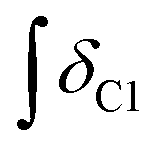 ’ is the integral of the hydroxymethyl methylene (CH2OH) peak of the C1 adduct cation, [C2C1(HO)C12im]+ or [C4C1(HO)C12im]+, centred at δ = ∼4.80 or ∼4.73 ppm, respectively (1H NMR, DMSO-d6 solvent); ‘
’ is the integral of the hydroxymethyl methylene (CH2OH) peak of the C1 adduct cation, [C2C1(HO)C12im]+ or [C4C1(HO)C12im]+, centred at δ = ∼4.80 or ∼4.73 ppm, respectively (1H NMR, DMSO-d6 solvent); ‘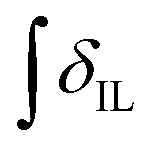 ’ is the integral of the aromatic imidazolium NCHCHN peak of [C2C1im]+ or [C4C1im]+, centred at δ = ∼7.83 or ∼7.81 ppm, respectively (1H NMR, DMSO-d6 solvent). These integral values are represented in graphical form in the ESI (Fig. E2†). It was observed that selecting different δ cut-off values, when integrating 1H NMR peaks, yielded marginally different integral values (
’ is the integral of the aromatic imidazolium NCHCHN peak of [C2C1im]+ or [C4C1im]+, centred at δ = ∼7.83 or ∼7.81 ppm, respectively (1H NMR, DMSO-d6 solvent). These integral values are represented in graphical form in the ESI (Fig. E2†). It was observed that selecting different δ cut-off values, when integrating 1H NMR peaks, yielded marginally different integral values (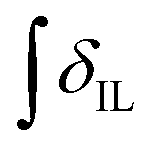 and
and 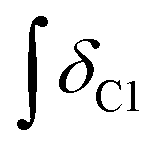 ). For example, for a C1 adduct singlet peak centred at δ = 4.80 ppm, a different value of
). For example, for a C1 adduct singlet peak centred at δ = 4.80 ppm, a different value of  is obtained by integrating across the range of 4.83–4.77 ppm instead of 4.82–4.78 ppm. Caution was therefore taken to integrate NMR peaks in a careful and consistent manner; nevertheless, error values arising from integration were observed to be smaller in magnitude than those of repeat experiments on the same mixture composition (e.g. experiments i–iii in Table E1, ESI†).
is obtained by integrating across the range of 4.83–4.77 ppm instead of 4.82–4.78 ppm. Caution was therefore taken to integrate NMR peaks in a careful and consistent manner; nevertheless, error values arising from integration were observed to be smaller in magnitude than those of repeat experiments on the same mixture composition (e.g. experiments i–iii in Table E1, ESI†).
Determination of cellulose degree of polymerisation
The degrees of polymerisation (DP) and polydispersity (PDI) of cellulose samples were determined for the cellulose (5) solutions in solvents (neat IL 1, 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 wt/wt 1:8, and 90:10 wt/wt 1:8) at time points t0 and t4. DP values were measured by Gel Permeation Chromatography (GPC) in a lithium chloride/dimethylacetamide (LiCl/DMA) organic electrolyte mobile phase, as described by Potthast et al.37 DP values of cellulose for the three solvent systems at t0 and t4 are listed in Table 1, compared against the DP of untreated Sigmacell cellulose.
:5 wt/wt 1:8, and 90:10 wt/wt 1:8) at time points t0 and t4. DP values were measured by Gel Permeation Chromatography (GPC) in a lithium chloride/dimethylacetamide (LiCl/DMA) organic electrolyte mobile phase, as described by Potthast et al.37 DP values of cellulose for the three solvent systems at t0 and t4 are listed in Table 1, compared against the DP of untreated Sigmacell cellulose.
| Solvent | Time point (h) | DP (±5) | PDI |
|---|---|---|---|
| IL 1 | 0 | 271 | 2.10 |
| 4 | 257 | 2.09 | |
| IL 1:glycerol 8 (95:5 wt/wt) + cat. AcOH |
0 | 255 | 2.09 |
| 4 | 249 | 2.05 | |
| IL 1:glycerol 8 (90:10 wt/wt) + cat. AcOH |
0 | 270 | 1.87 |
| 4 | 255 | 2.00 | |
| Untreated Sigmacell cellulose | — | 263 | 2.19 |
Quantification of reducing sugars in recovered solvents
1 wt% 3,5-dinitrosalicylic acid (DNS) solution was prepared by carefully mixing 3,5-dinitrosalicylic acid (2.5 g), sodium sulfite (0.1 g) and sodium hydroxide (2.0 g) into a flask, and subsequently diluting the mixture with deionized water up to a volume of 0.25 L.38Following pretreatment at 60 °C under reduced pressure and consequent reaction for 4 hours at 120 °C, 0.5 ml samples of the neat IL-/organic electrolyte–cellulose solutions were treated with deionized water (20 ml) in order to precipitate cellulose, were filtered, and were dried under high vacuum to recover the solvents. Samples (0.3 ml) of these recovered solvents were treated with an aliquot of the 1 wt% DNS solution (0.3 ml), and were heated to 90 °C for 15 minutes in order to develop the red-brown colouration. 40% Potassium sodium tartrate solution (‘Rochelle salt’, one drop) was added to the mixture, which was subsequently allowed to cool to room temperature. The UV-vis absorbance, A, for each sample was then measured at 537 nm. Finally, the reducing sugar concentration in each sample was determined by interpolation of an appropriate calibration curve (it was necessary to account for the possible influence of the IL and/or glycerol on the absorption behaviour of the DNS dye). Initially, one calibration curve was obtained from four aqueous glucose standard solutions (0.1, 1, 2 and 4 g L−1, curve displayed in ESI†). This calibration curve, based on aqueous glucose solutions, was validated to be suitable for determining reducing sugar concentration in the recovered neat IL, because analysis of a freshly-prepared 2 g L−1 glucose in IL (1) sample yielded a result of 2.16 g L−1 using the calibration curve. However, for determination of the reducing sugar concentration in the 90:10 wt/wt IL (1):glycerol (8) recovered solvent, it was necessary to create a second calibration curve derived from three 90:10 wt/wt 1:8 standard solutions of known glucose concentration (2, 3 and 5 g L−1, curve displayed in ESI†); the incorporation of glycerol was found to give a negative bias for the DNS assay (i.e. impression of lower reducing sugar concentration) when comparing against fully aqueous standards, as has similarly been observed with the inclusion of moderate-high concentrations of ethanol.39 Although in our investigation the DNS assay affords valuable comparative data between the different IL/organic electrolyte solvent systems, it must be recognised that measured reducing sugar concentrations obtained via the DNS method commonly differ significantly from those obtained using HPLC techniques.40
Results and discussion
Initially, control mixtures were prepared incorporating 0.5 g IL ([C2C1im][OAc], 1) with 10 wt% of D-(+)-glucose, 3 (as a model compound for cellulose). Alongside these ‘unprotected’ control mixtures, analogous ‘protected’ mixtures were prepared with the addition of a diol/triol co-solvent, 6–10, and one drop glacial acetic acid (11 ± 3 mg) to catalyse formation of a cyclic acetal. Mixtures were first treated at a low temperature of 60 °C under high vacuum for two hours (denoted the ‘protection time’) to encourage formation of the acetal and to remove any water evolved from any protective addition (or remnant from the starting mixture). Mixtures were then heated at the higher temperature of 120 °C for four hours, under a gentle positive pressure of nitrogen gas. At regular time intervals, small aliquots of the mixtures were carefully extracted and were analysed by 1H NMR spectroscopy, to assess the quantity of C1 adduct [C2C1(HO)C12im]+ that had formed. A graph highlighting relative beneficial stabilising effects of co-solvents 6–10 is shown below (Fig. 2).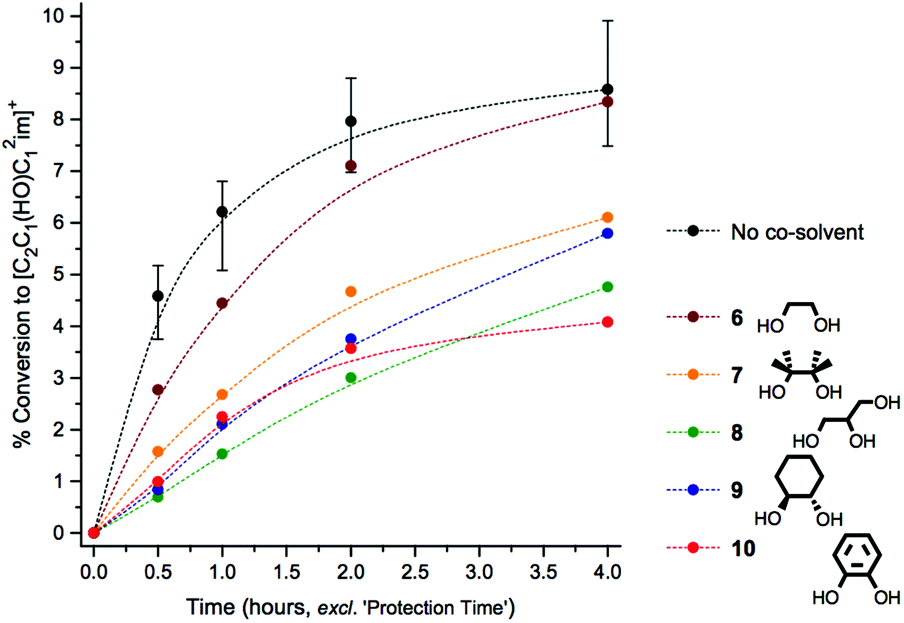 | ||
| Fig. 2 Comparison of the stabilising ability of diol/triol compounds 6–10, when incorporated in mixtures of [C2C1im][OAc] 1 + 10 wt% D-(+)-glucose 3, heated to 120 °C for four hours. Two molar equivalents of the diol/triol is added (relative to 3), and one drop acetic acid is included. Black data points represent median averages of triplicate ‘unprotected’ experiments, in the absence of diol/triol or acid. | ||
Comparison of the rates of C1 adduct evolution for the control mixture, against that incorporating the simplest diol co-solvent 6, revealed that only a modest stabilising effect had been conferred on D-(+)-glucose, 3, by inclusion of ethane-1,2-diol (at concentration 2:1 mol/mol of 6:3) and acetic acid. Any suppression of degradation was most pronounced in the early time points (t0.5 and t1). By contrast additives 7–10 each noticeably outperformed 6, bringing about a significant reduction in the rate of C1 formation which persisted over the course of the four-hour experiment. Moreover, the visible and intense discolouration that characterised heating of the unprotected mixture was substantially slower with addition of 7–10. Of the investigated diols/triols, glycerol (8) and benzene-1,2-diol (10) yielded the most profound enhancements to glucose stability (Fig. 2). Reactions performed with 1-butanol (11, data in Fig. E3d†), a simple mono-alcohol species, yielded no significant stabilising effect persisting for the four hour experiment, indicating that cyclic acetal formation is a possible preliminary explanation for the stabilising influence of 7–10.
Subsequently, the diol concentration and the identity of the acid were independently modified. The simplest co-solvent, 6, was employed in each experiment, and the rate of C1 adduct formation was monitored as before. The data is represented in the ESI (Fig. E3b and c†). As expected, doubling the concentration of 6 (with respect to D-(+)-glucose, 3, 4:1 mol/mol) yielded a pronounced improvement in the stabilising effect (Fig. E3b†). By contrast, stability was largely insensitive to subtle changes in acid identity/concentration, yet exclusion of acid altogether dramatically reduced the stabilising effect (Fig. E3c†).
Previously, it has been demonstrated that substituting IL 1 with [C4C1im][(CH3)2PO4], 2, dramatically reduces the rate of C1 adduct formation.34 In this study, employing the same strategy, for analogous mixtures with 2, appeared to confer no observable protecting effect; similar darkening of ‘protected’ and ‘unprotected’ mixtures was observed, although the minor % conversion to C1 adduct within the experimental duration allows for only a tentative conclusion. Hereafter, all discussion is for mixtures employing [C2C1im][OAc], 1.
Following the experiments employing D-(+)-glucose 3, the investigation was extended to D-(+)-cellobiose (4), and to the carbohydrate of interest cellulose (5); the (partially) optimised experimental conditions were employed, as before (i.e., a 4:1 glycerol:carbohydrate mole ratio, 1 h ‘protection time’ and one drop of AcOH). Glycerol (8) was selected as the co-solvent/additive of choice on the basis of its low cost41 and toxicity, its environmentally-benign nature and the promising stabilising effect it exhibits for IL–glucose mixtures (Fig. 2).
For the experiments employing D-(+)-cellobiose, 4, only a moderate suppression of the rate of C1 adduct formation was observed (Fig. E4†). It was postulated that residual water in the mixture may restrict the stabilising capability of glycerol, 8, via hydrolytic cleavage of the β-1,4-glycosidic bond of 4,42–44 thereby exposing another (reactive) reducing end.
Therefore for the experiments on cellulose, 5, a more strict restriction on water content (≤0.5 wt% instead of 3 wt%) was selected. Three mixtures were investigated and compared: the neat IL 1, 95:5 wt/wt 1:8, and 90:10 wt/wt 1:8 mixtures, each with 10 wt% of cellulose (5) added (relative to the quantity of IL), and one drop of AcOH added to the latter two mixtures. The solutions were heated at 120 °C for 30 hours, and the rate of C1 adduct evolution was monitored as before. Experimental details are described in the Experimental section. Relative rates of C1 adduct formation for the three mixtures are displayed in Fig. 3.
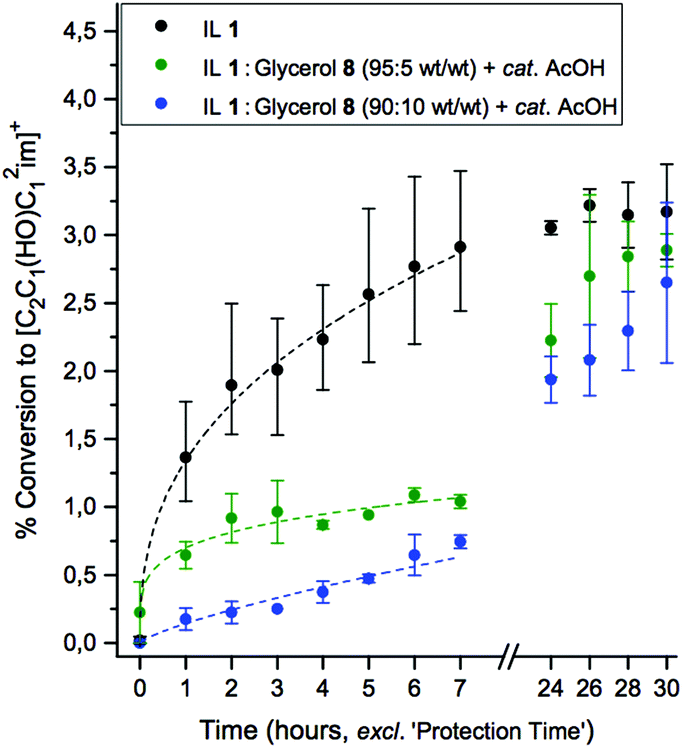 | ||
| Fig. 3 A comparison of thermal decomposition of cellulose (10 wt% relative to IL 1) dissolved in the neat IL vs. IL:glycerol (+ one drop AcOH) systems, assessed by the rate of evolution of the C1 adduct, 1-ethyl-2-(hydroxymethyl)-3-methylimidazolium acetate. | ||
The 95:5 wt/wt 1:8 mixture afforded a marked improvement in stability over the neat IL mixture over the first seven hours of the experiment (Fig. 3), represented by a >two-fold reduction in the quantity of the C1 adduct at each time point. Therefore, an encouraging reduction in cellulose (5) degradation is achieved by addition of minor quantities of inexpensive and environmentally-benign glycerol and acetic acid. Increasing the added quantity of glycerol, 8, in the case of the 90:10 wt/wt 1:8 experiments, further enhanced the protecting ability, exhibiting a >four-fold reduction in C1 adduct concentration at each time point within the first six hours (relative to the unprotected mixture). All stabilising ability appears restricted to the earlier time points; after 24 hours of heating, equilibrium concentrations of the C1 adduct were equivalent (within errors) for the mixtures with or without the addition of a co-solvent. Nevertheless the application of ILs (or organic electrolytes) to electrospinning of cellulose fibres requires that cellulose only be dissolved for relatively short durations of time, ≪24 hours. Therefore, the 90:10 wt/wt mixture of IL 1 with glycerol (8), and one drop AcOH, is an attractive alternative to the pure IL system, maintaining good cellulose solubility (≥10 wt% at 60 °C), and significantly enhancing the number of feasible cellulose dissolution–extrusion cycles whilst minimising undesired adduct formation/IL degradation.
NMR analysis alone of the IL/organic electrolyte–cellulose mixtures, as described above (Fig. 3), demonstrates formation of the C1 adduct species derived from the dialkylimidazolium IL cation. However, it is important also to understand the precise behaviour and fate of the cellulose, both in terms of depolymerisation and the quantity of carbohydrate residue in the recovered solvent following one heating cycle.
For each of the three investigated IL/organic electrolyte–cellulose mixtures (Fig. 3), at time points t0 and t4, small aliquots of the mixtures were investigated using Gel Permeation Chromatography (GPC) with LiCl/DMA mobile phase, to determine cellulose ‘degree of polymerisation’ (DP) and ‘polydispersity’ (PDI) parameters. GPC traces for the ‘t4’ time point are shown in Fig. 4, traces for the ‘t0’ time point are shown in the ESI (Fig. E5†). DP and PDI values are tabulated (Table 1). The DP of untreated Sigmacell cellulose was measured as 263 ± 5.
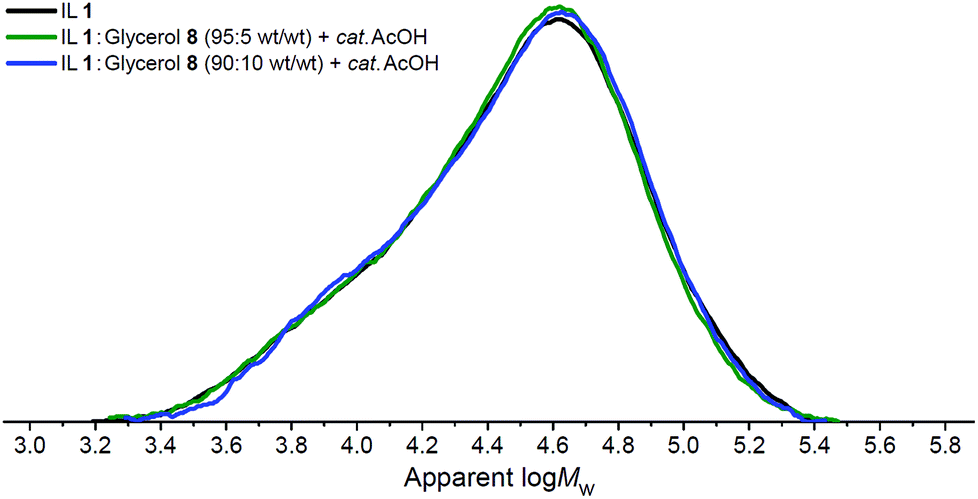 | ||
| Fig. 4 Gel Permeation Chromatography (‘GPC’) traces for cellulose samples recovered from IL and organic electrolyte solvents at the ‘t4’ time point (2 h ‘protection time’ at 60 °C then 4 h at 120 °C) (Fig. 3). | ||
A comparison of the DP values at t0 and t4 for each of the three solvent systems revealed that depolymerisation had occurred to a minor extent in all cases during pretreatment and reaction. The small magnitudes of ΔDP are consistent with the mechanism proposed previously,34 whereby reaction occurs solely at the reducing end of the cellulose polymer and glucopyranose residues are consumed in a sequential manner. Interestingly, the incorporation of up to 10 wt% glycerol, 8, in the solvent mixture appears to confer no change to the depolymerisation behaviour of the cellulose; the DP values for the neat IL 1 system and the 90:10 wt/wt IL 1:glycerol 8 blend at t0 (271 and 270, respectively) and t4 (257 and 255, respectively) are equivalent within the experimental error. Similarly, in all circumstances (Table 1), the DP and PDI values of the extracted samples are (close or) equal in magnitude to the measured values of untreated Sigmacell cellulose, within the error. GPC traces of samples recovered at t0 (Fig. E5†) and t4 (Fig. 4) time points are almost identical.
Subsequently, recyclabilities of the neat IL-/90:10 (wt/wt) IL:glycerol–cellulose blends were compared by measuring the quantities of residual reducing sugar in the recovered solvents after high-temperature treatment. The colourimetic method employing 3,5-dinitrosalicylic acid is detailed fully in the Experimental section, above.
The results demonstrate approximately equal concentrations of residual reducing sugar for the neat IL (1) (3.6 g L−1) and IL 1:glycerol 8 (90:10 wt/wt) (3.2 g L−1) recovered solvents. These similar measured reducing concentrations appear sensible on the basis of the observed similarity in depolymerisation behaviour (ΔDP) for the three solvent systems (Table 1). Therefore, the accumulation of reactive/contaminant reducing sugars in recovered solvents is neither significantly enhanced or inhibited with the inclusion of 10 wt% 8 in the solvent.
Rationalisation of improved stability with co-solvent
The above investigations (Fig. 2 and 3†), monitoring formation of the IL-derived C1 adduct species, 1-ethyl-2-(hydroxymethyl)-3-methylimidazolium acetate, provide no clear evidence of an exact explanation (or mechanism) for the improved stability of carbohydrates in IL (1) media. It had been suggested that the incorporation of a diol (or triol) with catalytic acid may enable the formation of a cyclic acetal protecting group at the carbohydrate reducing end.To examine this hypothesis, experiments were performed employing the UV-active diol additive 2,3-naphthalenediol. A mixture of [C2C1im][OAc] 1 + 10 wt% D-(+)-cellobiose (4), 2,3-naphthalenediol (2.4 molar equivalents relative to 4) and 1 drop AcOH was heated to 60 °C under vacuum conditions for 1 h, analogous to the above-described experiments. The solution was subsequently diluted with distilled water, and excess diol was extracted in EtOAc until no further diol could be detected by TLC. The aqueous and organic (EtOAc) phases were characterized using Electrospray mass spectrometry (procedures/spectra shown in ESI, Fig. E6†) and UV-vis spectroscopy.
The obtained mass spectra offered no clear evidence of the protection-group pathway; the anticipated signal at m/z 484 – corresponding to a naphthalenediol-capped cellobiose species – was absent from both organic and aqueous phase spectra. Instead, the major peak at m/z 453 in the aqueous phase (Fig. E6a†) spectrum could be attributed to the ‘C12’ decomposition adduct formed from reaction of a [C2C1im]+ cation with open-form cellobiose, as described previously.34 The major peak of the organic phase, at m/z 623, was tentatively ascribed also to the C12 adduct, clustered together with a [C2C1im][OAc] ion pair (Fig. E6b†).
In addition, the UV-vis experiments were unable to detect the formation of any novel naphthalene-containing species besides the un-coordinated 2,3-naphthalenediol itself.
Furthermore, the treatment of a mixture of [C2C1im][OAc] 1 + 10 wt% D-(+)-glucose, one drop of AcOH and one molar equivalent of 1,3-13C2-glycerol (relative to 3), heated at 60 °C under vacuum conditions in an NMR tube, afforded no new peaks in the 13C NMR spectrum. This observation lends clear evidence against the formation of a new covalent bond (for a protecting group) between the glycerol and glucose, in addition to the lack of a signal for the anticipated naphthalenediol-cellobiose adduct in electrospray mass spectrometry and UV-vis investigations.
An alternative explanation for improved stability conferred by the co-solvent may be the increased dilution of the reactive IL ions (or a change in the ‘ionicity’ due to lower solvent viscosity45). To test this hypothesis, investigations employing IL 1 + 10 wt% D-(+)-glucose (3) were undertaken, substituting glycerol, 8, for 1,3-dimethylimidazolidin-2-one (‘DMI’, 12), a previously-investigated organic electrolyte co-solvent18 with a molecular weight approximately equivalent to glycerol (yet by contrast an aprotic solvent). The experiment (Fig. E3d†) revealed that 12 confers no significant persisting stabilising effect on stability (over the four hour period) relative to the neat IL system, as had also been observed with 1-butanol, 11. This therefore suggests that dilution effects are unlikely to be the primary contributing factor towards the observed stabilisation phenomenon; rather, the protic functional groups of the diol/triol co-solvent (and acetic acid) likely play a specific role.
Subsequently, further investigation into reactivity was undertaken using isotopically-labelled co-solvents – perdeuterated acetic acid (‘AcOH-d4’) and glycer(ol-d3) (both ≥99 atom% D). Mixtures were prepared incorporating IL 1 + 10 wt% carbohydrate (3/5) + four molar equivalents (relative to the carbohydrate) of either AcOH-d4 or glycer(ol-d3). The mixtures were prepared in sealed NMR tubes, and were heated at 60 °C for a period of two hours and subsequently at 120 °C for four hours, recording 1H NMR spectra at 30 min intervals (Fig. 5 and E7†).
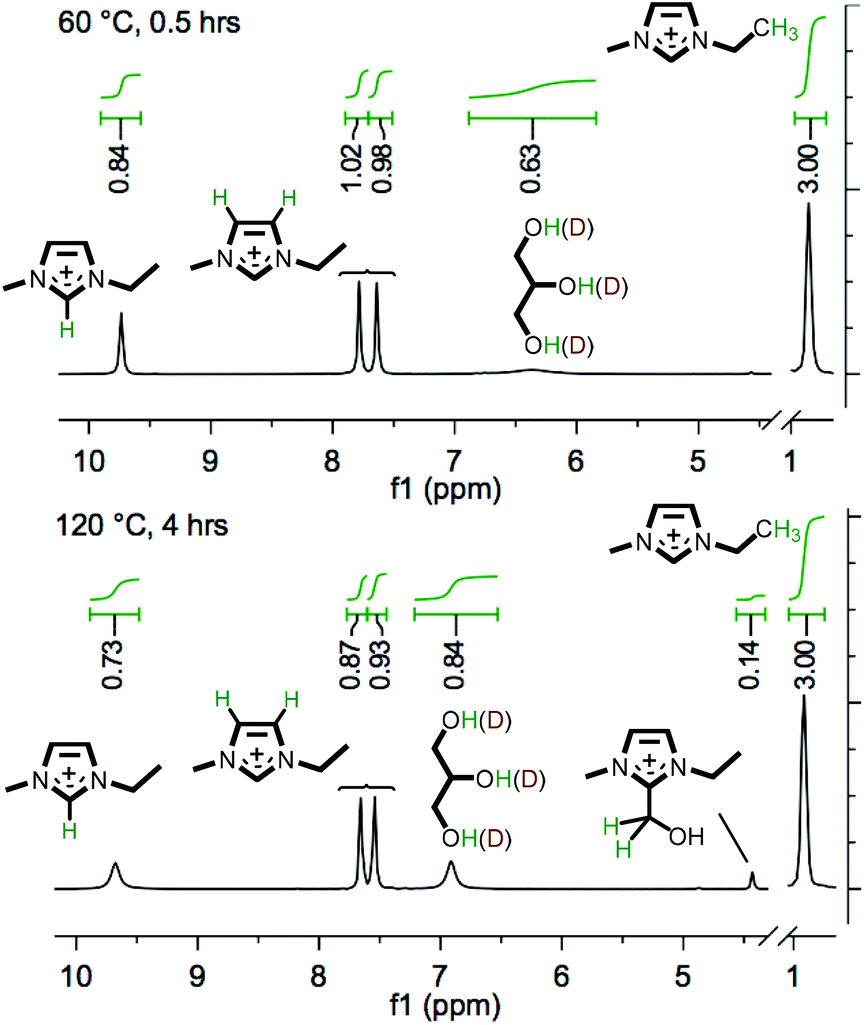 | ||
| Fig. 5 The in situ H/D exchange at the imidazolium ring C2 (60 °C) and C4/C5 (120 °C) positions, for the system [C2C1im][OAc] (1) + 10 wt% D-(+)-glucose (3) + four molar equivalents (relative to 3) of glycer(ol-d3). The 1H NMR integrations are determined relative to the NCH2CH3 peak set at 3.00 (assumed not to exchange with D). Heating times (0.5 and 4 hours, top and bottom panels, respectively) refer to the time heated at the specified temperature (same sample). | ||
For the inclusion of either AcOH-d4 or glycer(ol-d3) in the solution, the first recorded time point (at 60 °C) demonstrated that rapid exchange/scrambling of the co-solvent deuterium atoms with the imidazolium C2 ring proton had occurred (Fig. 5 and E7†), indicated by the lower integral of the C2 proton (δ = ∼9.7 ppm, e.g. 0.84 relative to 3.00 for the NCH2CH3 peak, Fig. 5). The decrease in integration for the C2 imidazolium signal was accompanied by the emergence of a broad signal for glycerol O–H, or acetic acid AcO–H. Interestingly, upon increasing the temperature to 120 °C, H/D exchange was also observed with the ‘back’ ring protons, indicated by a decrease in intensity of C4/C5 peaks (e.g. to 0.90 ± 0.03, Fig. 5). Furthermore, LSIMS mass spectra, obtained for the mixture following the heating experiment, demonstrated evolution of a peak at m/z 112 (ca. 25% the intensity of m/z 111, undeuterated [C2C1im]+) and a very small peak for m/z 113, indicating the formation of deuterated imidazolium cations and confirming the H/D exchange. It has long been recognised that dialkylimidazolium ILs may undergo ready H/D exchange at the C2 ring position in D2O,46 and that a transient imidazol-2-ylidene NHC may be trapped in the presence of a suitable reactive species (e.g. CO2,47,48 S849 or a transition metal complex50,51). Moreover, the formation of ‘abnormal carbenes’ at the C4/C5 position has previously been described for 1,3-disubstituted imidazolium species.52,53
Replacing the acetate IL (1) with [C4C1im][NTf2] quenched all H/D exchange, indicating that the IL anion plays a crucial role in the mechanism (and suggesting that a sufficiently basic anion is required for formation of the NHC intermediate). By contrast identical H/D exchange behaviour was observed with or without the inclusion of the carbohydrate (3/5), suggesting that the glucose/cellulose itself does not play a key role in the exchange mechanism (Fig. E7†). Recently, Zanatta et al. highlighted a plausible mechanism for H/D exchange at the C2–Me substituent of [C4C1C12im]-derived ILs incorporating [HCO3]− or prolinate anions,54 whereby exchange is associated with the presence of intimate contact ion pairs (not anion basicity), and the IL behaves as a neutral base for the transfer of D from the deuterated solvent (e.g. CDCl3) to the IL cation. However, this mechanism is not plausible for acetate IL 1 in our investigation, which lacks a labile proton on the anion.
Therefore on the basis of the collective mass spectrometry, UV-vis and NMR investigations with 2,3-naphthalenediol and isotopically-labelled co-solvents, the enhanced thermal stability of [C2C1im][OAc]-glycerol(-acetic acid) organic electrolyte solvent systems (Fig. 3) cannot sensibly be ascribed to the formation of cyclic acetal protection group moieties as initially had been hypothesised. Instead, this stabilisation is tentatively explained on the basis of suppression of the transient imidazol-2-ylidene NHC intermediate, via proton-donation from the protic neutral co-solvent species, regenerating the 1-ethyl-3-methylimidazolium cation. The protic co-solvent (e.g. glycerol, acetic acid) may therefore ‘compete’ with the cellulose reducing end for reaction with the NHC. Stabilisation on the basis of suppression of the NHC may also be corroborated by the pattern of the kinetics of C1 adduct evolution (Fig. 3), whereby inclusion of glycerol, 8, strongly impedes the initial rate of adduct formation, yet each of the three IL-/organic electrolyte–cellulose mixtures equilibrates to an identical (within the error) concentration of [C2C1(HO)C12im]+ after 30 hours of reaction.
The approach outlined in this investigation is a preventative measure focusing on in situ suppression of undesirable adduct formation. Reductive pre-treatment of the cellulose pulp (e.g. using 1% NaBH4, at 15 °C for 24 h (ref. 55)) – commonly employed to inhibit oxidative peeling of cellulose during paper bleaching – could serve to deactivate the reactive (aldehyde) reducing ends prior to mixing with the IL/organic electrolyte solvent. This approach adds complexity by introduction of further reaction stages/separation, but is nonetheless worthy of future investigation. We continue to explore alternative methods for preventing undesirable side reaction between ILs and cellulose.
Conclusions
Overall, it has been demonstrated that the incorporation of small mole fractions of a protic co-solvent (e.g. χglycerol ≤ 0.17) substantially reduces the rate of adduct accumulation when dissolving cellulose in carboxylate ILs (1). It is likely that this phenomenon is attributable to the suppression of the in situ N-Heterocyclic Carbene (NHC) species (derived from the IL cation), via proton donation from the co-solvent, thereby minimising reactivity between the carbohydrate aldehyde reducing end and the NHC.Considering these insights in the context of cellulose electrospinning from IL media (and, furthermore, other applications of ILs with carbohydrates), several significant beneficial implications arise: (i) IL adduct formation is suppressed, minimising changes to the physicochemical properties of the mixture, maintaining favourable solvating interaction between the IL and carbohydrate, and maximising recyclability of the expensive IL component; (ii) inclusion of glycerol does not increase the accumulation of contaminant reducing sugars in the recovered solvent; (iii) depolymerisation of cellulose strands is not exacerbated in the presence of (up to) 10 wt% glycerol in the solvent mixture, and; (iv) the relatively high cellulose solubility (≥10 wt%, relative to IL) is maintained with the incorporation of up to 10 wt% glycerol into the solvent system. Changes in the viscosities of the (glycerol) organic electrolyte–cellulose mixtures, though not formally quantified in this investigation, did not impede rapid dissolution of 10 wt% cellulose at 60 °C with trivial magnetic stirring (i.e. without input of mechanical stirring).
Glycerol and acetic acid are both inexpensive41 and non-hazardous, therefore this strategy is remarkably benign. Experimental conditions are as yet far from optimised, and it is not wholly certain that NHC suppression via proton donation is the precise explanation for improved stability. Nevertheless, these observations highlight one feasible strategy towards overcoming a recurring barrier against industrial-scale implementation of ILs for cellulose solvation/reprocessing – that they are prohibitively expensive on account of their instability, contamination and, therefore, poor recyclability.
Acknowledgements
The authors are grateful to Dr Lisa Haigh of Imperial College London, for assistance with mass spectrometry experiments. The authors wish to acknowledge the EPSRC (EP/L017679/1) for generous sponsorship. Supporting data may be obtained from Prof. T. Welton (t.welton@imperial.ac.uk). The authors acknowledge Dr Antje Potthast and Dr Kyujin Ahn of the Department of Chemistry, Division of Chemistry of Renewables, University of Natural Resources and Life Sciences, Vienna, Austria, for GPC analysis of cellulose samples.References
- R. D. Perlack, L. L. Wright, A. F. Turhollow, R. L. Graham, B. J. Stokes and D. C. Erbach, Biomass as feedstock for a bioenergy and bioproducts industry: the technical feasibility of a billion-ton annual supply, DTIC Document, 2005 Search PubMed.
- R. Rinaldi, Angew. Chem., Int. Ed., 2014, 53, 8559–8560 CrossRef CAS PubMed.
- R. Rinaldi, R. Jastrzebski, M. T. Clough, J. Ralph, M. Kennema, P. C. A. Bruijnincx and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2016, 55 DOI:10.1002/anie.201510351.
- A. Brandt, J. Gräsvik, J. P. Hallett and T. Welton, Green Chem., 2013, 15, 550–583 RSC.
- A. Pinkert, K. N. Marsh, S. Pang and M. P. Staiger, Chem. Rev., 2009, 109, 6712–6728 CrossRef CAS PubMed.
- H. Wang, G. Gurau and R. D. Rogers, Chem. Soc. Rev., 2012, 41, 1519–1537 RSC.
- X. Yuan and G. Cheng, Phys. Chem. Chem. Phys., 2015, 17, 31592–31607 RSC.
- L. Meli, J. Miao, J. S. Dordick and R. J. Linhardt, Green Chem., 2010, 12, 1883–1892 RSC.
- L. J. Hauru, M. Hummel, A. Michud and H. Sixta, Cellulose, 2014, 21, 4471–4481 CrossRef CAS.
- M. Hummel, A. Michud, M. Tanttu, S. Asaadi, Y. Ma, L. J. Hauru, A. Parviainen, A. T. King, I. Kilpeläinen and H. Sixta, in Ionic Liquids for the Production of Man-Made Cellulosic Fibers: Opportunities and Challenges, Springer, Berlin, Heidelberg, 2015, ch. 307, pp. 1–36 Search PubMed.
- Y. Ma, M. Hummel, M. Maattanen, A. Sarkilahti, A. Harlin and H. Sixta, Green Chem., 2016, 18, 858–866 RSC.
- L. K. J. Hauru, M. Hummel, K. Nieminen, A. Michud and H. Sixta, Soft Matter, 2016, 12, 1487–1495 RSC.
- R. P. Swatloski, S. K. Spear, J. D. Holbrey and R. D. Rogers, J. Am. Chem. Soc., 2002, 124, 4974–4975 CrossRef CAS PubMed.
- M. A. Ab Rani, A. Brandt, L. Crowhurst, A. Dolan, M. Lui, N. H. Hassan, J. P. Hallett, P. A. Hunt, H. Niedermeyer, J. M. Perez-Arlandis, M. Schrems, T. Welton and R. Wilding, Phys. Chem. Chem. Phys., 2011, 13, 16831–16840 RSC.
- M. J. Kamlet and R. W. Taft, J. Am. Chem. Soc., 1976, 98, 377–383 CrossRef CAS.
- A. Parviainen, A. W. T. King, I. Mutikainen, M. Hummel, C. Selg, L. K. J. Hauru, H. Sixta and I. Kilpelainen, ChemSusChem, 2013, 6, 2161–2169 CrossRef CAS PubMed.
- H. F. N. de Oliveira and R. Rinaldi, ChemSusChem, 2015, 8, 1577–1584 CrossRef PubMed.
- R. Rinaldi, Chem. Commun., 2011, 47, 511–513 RSC.
- S. Xu, J. Zhang, A. He, J. Li, H. Zhang and C. C. Han, Polymer, 2008, 49, 2911–2917 CrossRef CAS.
- S.-L. Quan, S.-G. Kang and I.-J. Chin, Cellulose, 2010, 17, 223–230 CrossRef CAS.
- M. Kostag, T. Liebert and T. Heinze, Macromol. Rapid Commun., 2014, 35, 1419–1422 CrossRef CAS PubMed.
- S. Spange, A. Reuter, E. Vilsmeier, T. Heinze, D. Keutel and W. Linert, J. Polym. Sci., Part A: Polym. Chem., 1998, 36, 1945–1955 CrossRef CAS.
- S. Dorn, F. Wendler, F. Meister and T. Heinze, Macromol. Mater. Eng., 2008, 293, 907–913 CrossRef CAS.
- T. Liebert, Cellulose Solvents: For Analysis, Shaping and Chemical Modification, American Chemical Society, 2010 Search PubMed.
- L. Chen, M. Sharifzadeh, N. Mac Dowell, T. Welton, N. Shah and J. P. Hallett, Green Chem., 2014, 16, 3098–3106 RSC.
- A. W. T. King, A. Parviainen, P. Karhunen, J. Matikainen, L. K. J. Hauru, H. Sixta and I. Kilpelainen, RSC Adv., 2012, 2, 8020–8026 RSC.
- C. Maton, N. De Vos and C. V. Stevens, Chem. Soc. Rev., 2013, 42, 5963–5977 RSC.
- M. J. Earle, J. Esperanca, M. A. Gilea, J. N. C. Lopes, L. P. N. Rebelo, J. W. Magee, K. R. Seddon and J. A. Widegren, Nature, 2006, 439, 831–834 CrossRef CAS PubMed.
- A. Chandran, K. Prakash and S. Senapati, Chem. Phys., 2010, 374, 46–54 CrossRef CAS.
- K. Shimizu, M. Tariq, M. F. C. Gomes, L. S. P. N. Rebelo and J. N. C. Lopes, J. Phys. Chem. B, 2010, 114, 5831–5834 CrossRef CAS PubMed.
- T. Liebert and T. Heinze, BioResources, 2008, 3, 576–601 Search PubMed.
- G. Ebner, S. Schiehser, A. Potthast and T. Rosenau, Tetrahedron Lett., 2008, 49, 7322–7324 CrossRef CAS.
- X. Wei, Z. Han and D. Zhang, Carbohydr. Res., 2013, 374, 40–44 CrossRef CAS PubMed.
- M. T. Clough, K. Geyer, P. A. Hunt, S. Son, U. Vagt and T. Welton, Green Chem., 2015, 17, 231–243 RSC.
- M. T. Clough, K. Geyer, P. A. Hunt, J. Mertes and T. Welton, Phys. Chem. Chem. Phys., 2013, 47, 20480–20495 RSC.
- R. S. Payal and S. Balasubramanian, Phys. Chem. Chem. Phys., 2014, 16, 17458–17465 RSC.
- A. Potthast, S. Radosta, B. Saake, S. Lebioda, T. Heinze, U. Henniges, A. Isogai, A. Koschella, P. Kosma, T. Rosenau, S. Schiehser, H. Sixta, M. Strlic, G. Strobin, W. Vorwerg and H. Wetzel, Cellulose, 2015, 22, 1591–1613 CrossRef CAS.
- G. L. Miller, Anal. Chem., 1959, 31, 426–428 CrossRef CAS.
- N. Z. Numan and J. C. Ford, Res. Rev.: J. Chem., 2015, 4, 31–34 Search PubMed.
- D. B. Rivers, S. J. Gracheck, L. C. Woodford and G. H. Emert, Biotechnol. Bioeng., 1984, 26, 800–802 CrossRef CAS PubMed.
- For example, sigmaaldrich.com: ‘Glycerol ACS reagent ≥99.5%’; Sigma Aldrich G7893, £3215 for 200 litres, correct as of January 2016.
- R. Rinaldi, R. Palkovits and F. Schüth, Angew. Chem., Int. Ed., 2008, 47, 8047–8050 CrossRef CAS PubMed.
- C. Li, Q. Wang and Z. K. Zhao, Green Chem., 2008, 10, 177–182 RSC.
- H. F. N. de Oliveira, C. Farès and R. Rinaldi, Chem. Sci., 2015, 6, 5215–5224 RSC.
- O. Holloczki, F. Malberg, T. Welton and B. Kirchner, Phys. Chem. Chem. Phys., 2014, 16, 16880–16890 RSC.
- R. A. Olofson, W. R. Thompson and J. S. Michelman, J. Am. Chem. Soc., 1964, 86, 1865–1866 CrossRef CAS.
- G. Gurau, H. Rodríguez, S. P. Kelley, P. Janiczek, R. S. Kalb and R. D. Rogers, Angew. Chem., Int. Ed., 2011, 50, 12024–12026 CrossRef CAS PubMed.
- O. Hollóczki, D. S. Firaha, J. Friedrich, M. Brehm, R. Cybik, M. Wild, A. Stark and B. Kirchner, J. Phys. Chem. B, 2013, 117, 5898–5907 CrossRef PubMed.
- H. Rodriguez, G. Gurau, J. D. Holbrey and R. D. Rogers, Chem. Commun., 2011, 47, 3222–3224 RSC.
- J. Dupont and J. Spencer, Angew. Chem., Int. Ed., 2004, 43, 5296–5297 CrossRef CAS PubMed.
- M. E. M. Berger, D. Assenbaum, N. Taccardi, E. Spiecker and P. Wasserscheid, Green Chem., 2011, 13, 1411–1415 RSC.
- H. Lebel, M. K. Janes, A. B. Charette and S. P. Nolan, J. Am. Chem. Soc., 2004, 126, 5046–5047 CrossRef CAS PubMed.
- R. H. Crabtree, Coord. Chem. Rev., 2013, 257, 755–766 CrossRef CAS.
- M. Zanatta, A. L. Girard, N. M. Simon, G. Ebeling, H. K. Stassen, P. R. Livotto, F. P. dos Santos and J. Dupont, Angew. Chem., Int. Ed., 2014, 126, 13031–13035 CrossRef.
- H. Sixta, Handbook of pulp, Wiley-vch, 2006 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6gc00027d |
| This journal is © The Royal Society of Chemistry 2016 |