
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A facile route for rubber breakdown via cross metathesis reactions†
R. F.
Smith
,
S. C.
Boothroyd
,
R. L.
Thompson
* and
E.
Khosravi
*
Department of Chemistry, Durham University, Mountjoy Site, Durham, DH1 3LE, UK. E-mail: r.l.thompson@durham.ac.uk; ezat.khosravi@durham.ac.uk
First published on 3rd March 2016
Abstract
A new approach towards reprocessing cross-linked rubbery materials by catalytic disassembly of polymer chains, which eliminates the need for energy intensive mechanical processes, is demonstrated. First and second generation (G1 and G2) Grubbs’ ruthenium catalysts break down polybutadiene (PBd) networks at their double bonds via cross-metathesis (CM) reactions to produce readily soluble molecules. A dramatic reduction in molecular weight to around 2000 g mol−1 was observed by size exclusion chromatography and the breakdown of cross-linked networks was confirmed by rheometry. This process was repeated with a styrene-butadiene rubber sheet, a common component of vehicle tyres, with a G2 catalyst and a diester to accelerate the breakdown. A sufficient amount of G2 catalyst and a diester were found to diffuse into the styrene-butadiene rubber sheet, to catalyse its breakdown into rubber crumb. This reaction can be achieved at room temperature within 2.5 h. Increasing the reaction time and temperature increased the extent of the breakdown and under these conditions some breakdown of rubber occurred with the addition of only the G2 catalyst, without the need for a diester. We speculate that, when present, pendant ethylene groups in the PBd chain structure can participate in CM reactions, enabling break-down of the cross-linked network into individual molecules with lasso-like structures.
Introduction
The production of cross-linked rubber goods such as vehicle tyres, latex gloves etc., is a global industry responsible for the consumption of millions of tons of rubbery polymers such as polybutadiene (PBd), polyisoprene (PI) and natural rubber every year. All of these materials comprise long chain hydrocarbon molecules with unsaturated carbons in their backbone. While cross-linking, or vulcanisation is essential to provide the necessary mechanical properties, this process also renders the tyre material insoluble; therefore making it extremely difficult to recycle or reprocess tyre components at the end of their life.1 Traditionally, rubber is reprocessed either by ambient temperature milling or cryofracturing, both of which are energy intensive processes, leading to a crumb product, which can be mixed with a fresh elastomer to produce a new material, typically with somewhat diminished properties.2,3 The loss of properties has significant implications for the reuse of rubber reclaimed in this way, and it is generally restricted to low performance materials. Applications requiring high performance such as vehicle tyres typically do not incorporate more than a small fraction of reclaimed rubber.4 Since many rubber materials contain a variety of fillers, a further complication of a rubber reclaiming process that does not break down the rubber on a molecular level is that the filler type and content cannot easily be exchanged for the next intended application. Given these factors, there remains a significant need for a new approach to reprocessing rubber materials, and preferably one that could allow the polymer to be extracted and separated from the filler.Very recently, Zhang et al.5 have demonstrated a process, which uses CuCl2 as a catalyst incorporated into vulcanised rubber to enable PBd recovery by disulfide metathesis at the vulcanization points. This approach led to a recyclable rubber that also had self-healing properties, but currently has the limitation that the CuCl2 catalyst had to be incorporated into the rubber at the point when it was prepared. There still remains a significant challenge to recycling vulcanised materials that have not been prepared with this foresight for future recycling.
Ring-opening metathesis polymerization (ROMP) is now a well-established and versatile chain-growth polymerization technique in which mono or polycyclic olefins undergo ring opening, in the presence of alkylidene catalysts, thereby forming a linear polymer chain.6,7 Ruthenium-based catalysts, due to their functional group tolerance and non-sensitivity to air and moisture, have been widely used to extend the scope of ROMP.7,8 Cross metathesis (CM), exchange of double bond fragments between two olefins, in the presence of alkylidene complexes has emerged as a powerful tool in the synthesis of organic compounds.9–11 This has been due to the development of catalysts with variable activities and functional group tolerance, such as Grubbs 1st (“G1”) and 2nd (“G2”) generation ruthenium catalysts, Fig. 1.12,13
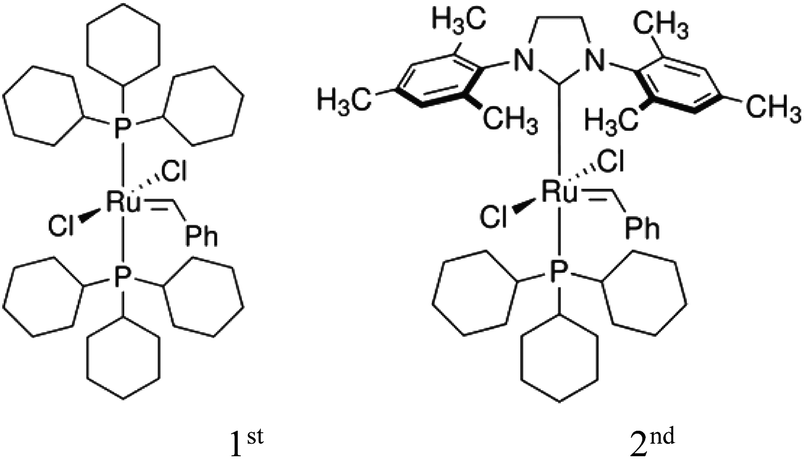 | ||
| Fig. 1 Sketch of 1st and 2nd generation Grubbs’ catalysts used in cross-metathesis reactions. | ||
The use of metathesis reactions to break and reform polymer chains has emerged as a potent technique to achieve restructuring of materials. Otsuka et al.14 carried out cross-metathesis reactions between 1,4-polybutadiene and unsaturated polyester prepared from polycondensation of cis-2-butene-diol and adipoyl chloride. They reported the formation of a polymer hybrid containing polybutadiene, unsaturated polyester and polymer segments containing both butadiene and ester groups.
Two reports by Guan et al.15,16 have demonstrated the ability of small amounts of Grubbs catalysts to achieve dramatic new effects in cross-linked polybutadienes. In both articles, low loadings of the catalyst were shown to facilitate the breaking and reformation of double bonds within a (99% 1,4) PBd network. Perhaps most relevant to our present work is the utilisation of Ru catalysis to render an insoluble cross-linked PBd network malleable. By enabling bonds to break and reform, these networks can relax the applied stress at a rate that increases with increasing catalyst loading.16 Similarly, the incorporation of low loadings of the Ru catalyst has been shown to confer self-healing properties at a fractured PBd network surface.15 In this example, the dynamic breaking and reformation of double bonds allow a network to reform across an interface, provided that at least one side of the fractured material contains some catalyst. The idea of incorporating active, reversibly reactable groups into polymers appears to be quite general, and has also been exploited to generate self-healing thermosetting polymers.17
Here, we demonstrate for the first time the effectiveness of CM reactions to break down rubbery polymers into viscous liquids, which then have the potential to be reprocessed. This article is set out as follows: first the validity of the CM approach is tested on a chemical level with simple, linear PBd and unsaturated diester molecules. The diesters were added in order to introduce new chain ends to the PBd via CM reactions; therefore breaking down the chains. Next the impact of this reaction mechanism on the molar mass distribution and mechanical properties of a model cross-linked PBd is explored. Finally the effectiveness of this approach is described for commercial sheet rubber samples. The influence of CM reactions is monitored on a molecular level by SEC, which is sensitive to the molar mass distribution of soluble polymers and rheology, which can verify the breakdown of cross-linked polymers into discrete fragments. NMR and ion beam analysis experiments, described in the following section, enable the analysis of chemical processes and the quantification of the residual catalyst in the test materials to be determined.
Experimental
Materials
Poly(butadiene), (cis-1,4 36%, trans-1,4 56%, vinyl 8%) nominal molecular weight ∼420![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 (measured Mw = 280000 g mol−1), benzoyl peroxide (“BPO”), 1st and 2nd generation Grubbs’ catalysts, dimethylmaleate (“DM”) and dimethylfumarate (“DF”) were supplied by Sigma-Aldrich (UK) and used as received. Rubber was obtained from ARTIS Independent Rubber Consultants, UK, in 2 mm thick styrene butadiene rubber sheets (BR/SBR, T00931A, VSL 5228-2).
000 g mol−1 PBd was dissolved in dichloromethane (DCM), and BPO (1% relative to the molarity of double bonds in the PBd) was added and the solution was stirred. There is exactly one double bond per C4H6 repeat unit of PBd; therefore the molarity of double bonds is given by the PBd mass per litre divided by the molar mass of C4H6, 54 g mol−1. The solution was poured into a Teflon® mould. The mould was left in the fume hood to allow the DCM to evaporate, leaving behind a film containing PBd and BPO. The film was placed in a vacuum oven (to prevent oxidation) at 100 °C for 16 h to crosslink the sample. The film was removed from the mould and swelled and then washed in DCM to remove any unreacted BPO. The film was dried as before and then weighed. The cross-linking reaction was immediately evident from the elastic feel of the film upon removal from the mould and this was confirmed by rheometry.
000 g mol−1 (measured Mw = 280000 g mol−1), benzoyl peroxide (“BPO”), 1st and 2nd generation Grubbs’ catalysts, dimethylmaleate (“DM”) and dimethylfumarate (“DF”) were supplied by Sigma-Aldrich (UK) and used as received. Rubber was obtained from ARTIS Independent Rubber Consultants, UK, in 2 mm thick styrene butadiene rubber sheets (BR/SBR, T00931A, VSL 5228-2).
000 g mol−1 PBd was dissolved in dichloromethane (DCM), and BPO (1% relative to the molarity of double bonds in the PBd) was added and the solution was stirred. There is exactly one double bond per C4H6 repeat unit of PBd; therefore the molarity of double bonds is given by the PBd mass per litre divided by the molar mass of C4H6, 54 g mol−1. The solution was poured into a Teflon® mould. The mould was left in the fume hood to allow the DCM to evaporate, leaving behind a film containing PBd and BPO. The film was placed in a vacuum oven (to prevent oxidation) at 100 °C for 16 h to crosslink the sample. The film was removed from the mould and swelled and then washed in DCM to remove any unreacted BPO. The film was dried as before and then weighed. The cross-linking reaction was immediately evident from the elastic feel of the film upon removal from the mould and this was confirmed by rheometry.
Methods
Results and discussion
CM reactions on linear PBd
Cross-metathesis reactions were carried out on the linear PBd with the Grubbs G1 and G2 catalyst using the DF diester. Initial experiments used 1 mole of DF for every 3 moles of PBd double bonds. According to the reaction scheme in Fig. 2, this would be sufficient to break down the polymer into fragments of the order of 3 repeat units, each with a monoester terminal group. Despite the possible steric hindrance in the CM reaction with DF, our results were consistent with this calculated activity insofar as the PBd was broken down into fragments that were completely soluble in methanol, which is a very poor solvent for PBd. No solid product remained that could be recovered by precipitation, which shows that no PBd of molecular weight greater than a few kg mol−1 remained after the CM reaction. In order to explore the remarkable efficiency of the breakdown further, experiments were repeated with a much lower (1:100) loading of diester:PBd double bonds. In this case the expected number-average molecular weight of CM products would be at least Mn ∼ 5400 g mol−1 corresponding to 100 repeat units.
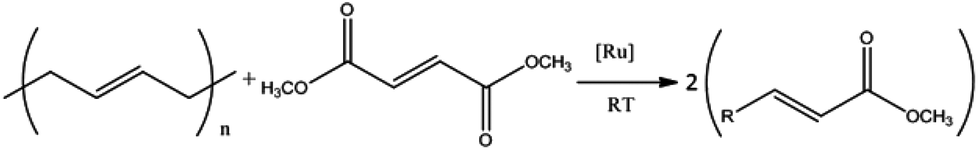 | ||
| Fig. 2 Scheme for cross-metathesis reaction of diester (DF) to break down PBd chain. | ||
The low molecular weight PBd product would be expected to be only sparingly soluble in methanol, and indeed, a small amount of precipitate was found in the cross-metathesis reaction products. The 1H NMR spectra of these products as well as DF and PBd are shown in Fig. 3. It is clear that the recovered reaction products (3c) and (3d) are essentially similar to the original PBd polymer (3b), with peaks at 2.0 and 5.4 ppm due to allyl and olefin groups, respectively. In other words the CM reaction preserves the basic chemistry of the PBd, which is important for the downstream re-use of recovered products. The resonances at 3.75 or 6.8 ppm, due to the methyl and olefin groups, respectively, for DF are absent in either reaction product (3c) or (3d). This is perhaps surprising, but may simply indicate that this fraction of the recovered material is relatively high in molecular weight; therefore ester end-groups form only a small fraction of the total material in the sample.
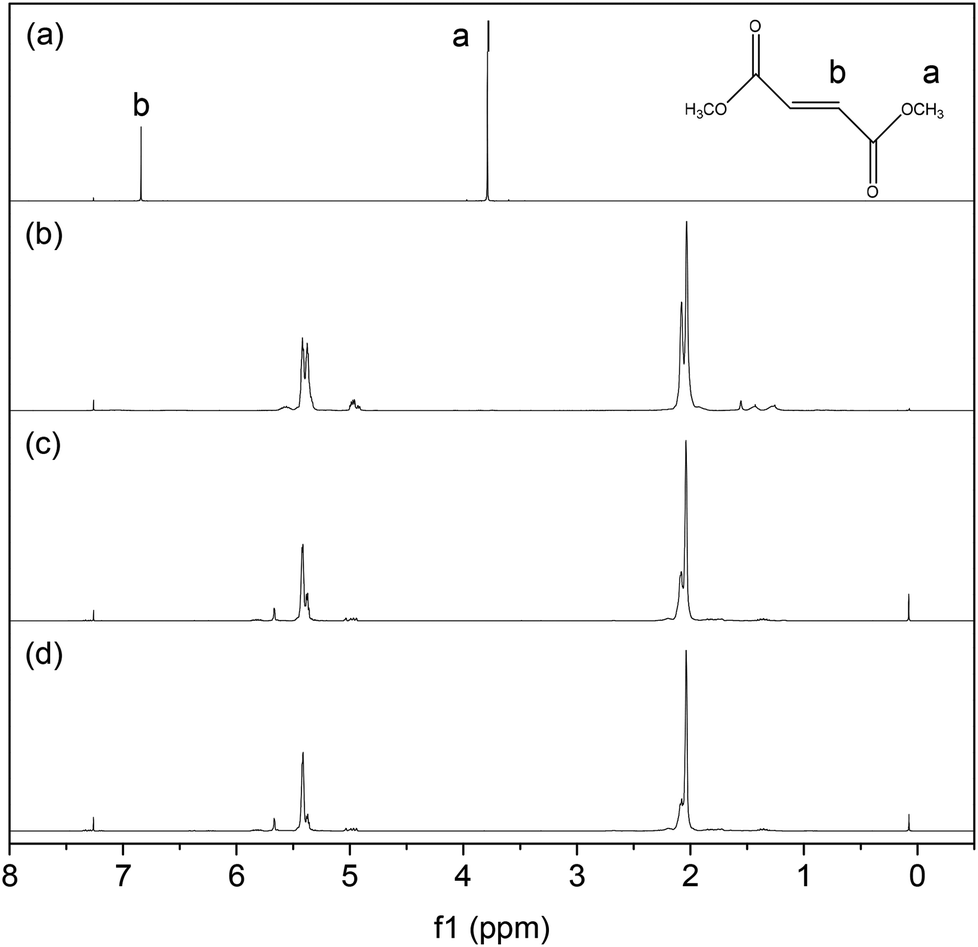 | ||
| Fig. 3 1H NMR spectra of (a) DF, (b) linear PBd, and the solid precipitates recovered from reaction using 1 mol% DF in PBd and (c) G1 catalyst or (d) G2 catalyst. | ||
Finally it is interesting to note that the cis:trans ratio, defined by the relative peak intensities has shifted significantly with respect to the starting material (3b), as detailed in Table 1. The cis, trans and vinyl contents were assigned as shown in Fig. 4. The linear PBd contains 36.3% cis and 56.0% trans conformation. Following reaction with G1 (3c) this changes to 25.6% cis and 68.3% trans, while with G2 (3d) the change is even greater, to 18.0% cis and 75.4% trans. The vinyl content drops by ∼1.5% with the G1 catalyst, and 1% with G2. This provides additional strong evidence for the catalysts acting on the polymer chain, giving rise to an increase in the favoured trans configuration at the expense of the more reactive cis repeat units, and further that there may be a greater degree of activity for the G2 catalyst than the G1. Ultimately, the increase in trans-content will have some impact on the final properties of PBd reprocessed by this route, so it may be important to control the extent of reaction at the breakdown stage in order to control the chain microstructure in recycled materials.
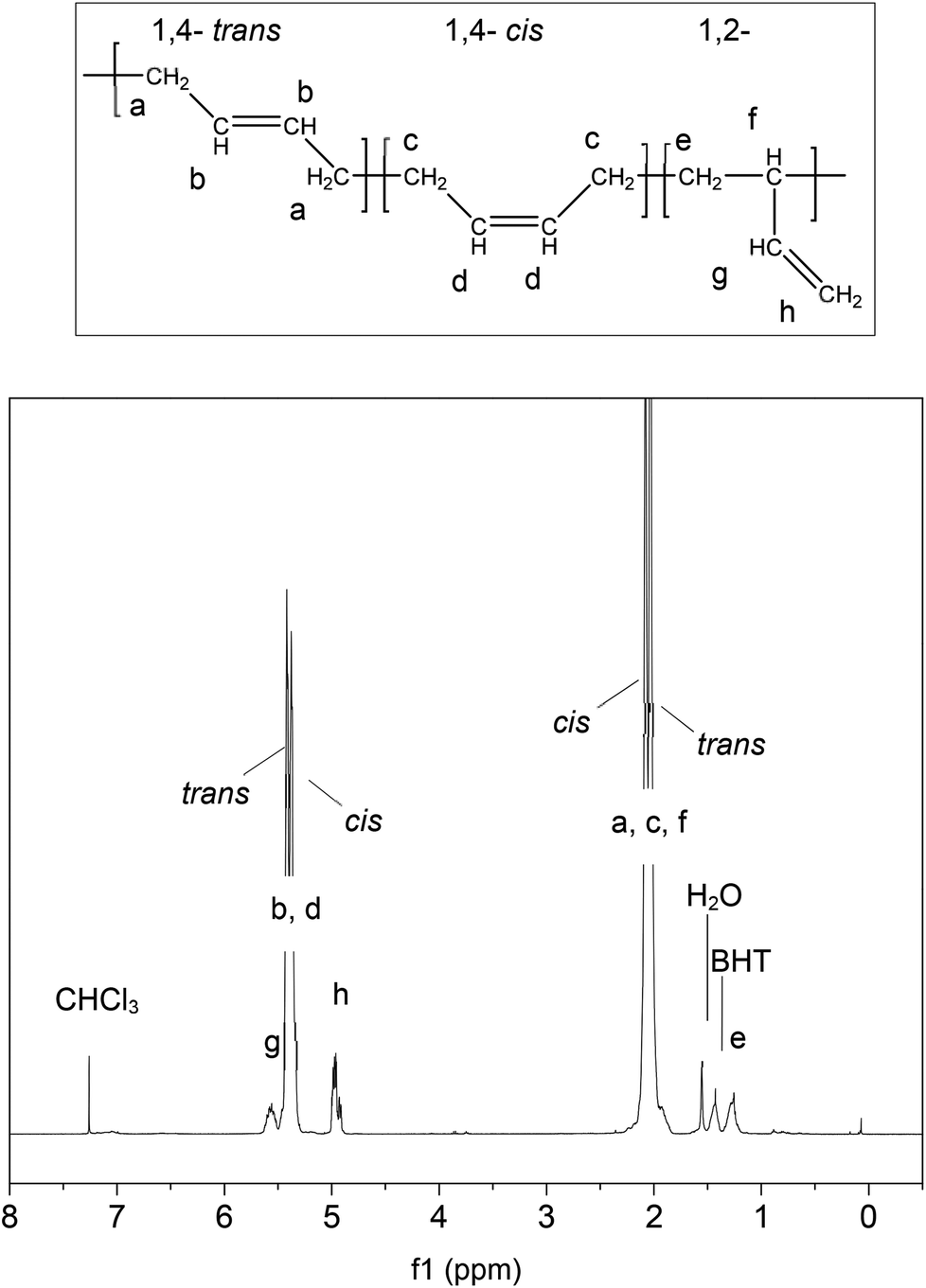 | ||
| Fig. 4 1H NMR assignments21 for PBd cis, trans and vinyl groups, showing the NMR of the linear PBd starting material. | ||
| Description | % | ||
|---|---|---|---|
| vinyl | cis | trans | |
| (a) DF | — | — | — |
| (b) Linear PBd | 7.7 | 36.3 | 56.0 |
| (c) Precipitates after PBd reaction with 1 mol% DF and G1 | 6.1 | 25.6 | 68.3 |
| (d) Precipitates after PBd reaction with 1 mol% DF and G2 | 6.6 | 18.0 | 75.4 |
CM reactions on cross-linked PBd
Having established the ability of the Grubbs’ catalysts to break down linear PBd, the approach was next tested on a model cross-linked PBd. The same approach to the CM reaction was used as before, except that this time, because the BPO cross-linked PBd was insoluble in DMF, small sections of this material were cut and put in the catalyst solution, whereupon they became swollen. Again, major breakdown of the PBd was evident, and it appeared that the cross-linking did little to inhibit the reaction. In this case a further control experiment was carried out in which the diester was entirely absent. Quite unexpectedly, even this reaction with the G1 catalyst resulted in breakdown of the cross-linked PBd, and this was quantified more rigorously by SEC (Fig. 5). Clearly even the catalyst acting in the absence of diester does more than simply break the BPO cross-links, because this would lead to reaction products of similar molecular weight to the original linear PBd starting material. In both cases the PBd (original Mw = 280000 g mol−1) which had been cross-linked to form a continuous network, was broken down to form oligomers of approximately 2000 g mol−1. The influence of the diester, initially thought to be essential for the reaction to proceed is almost negligible.
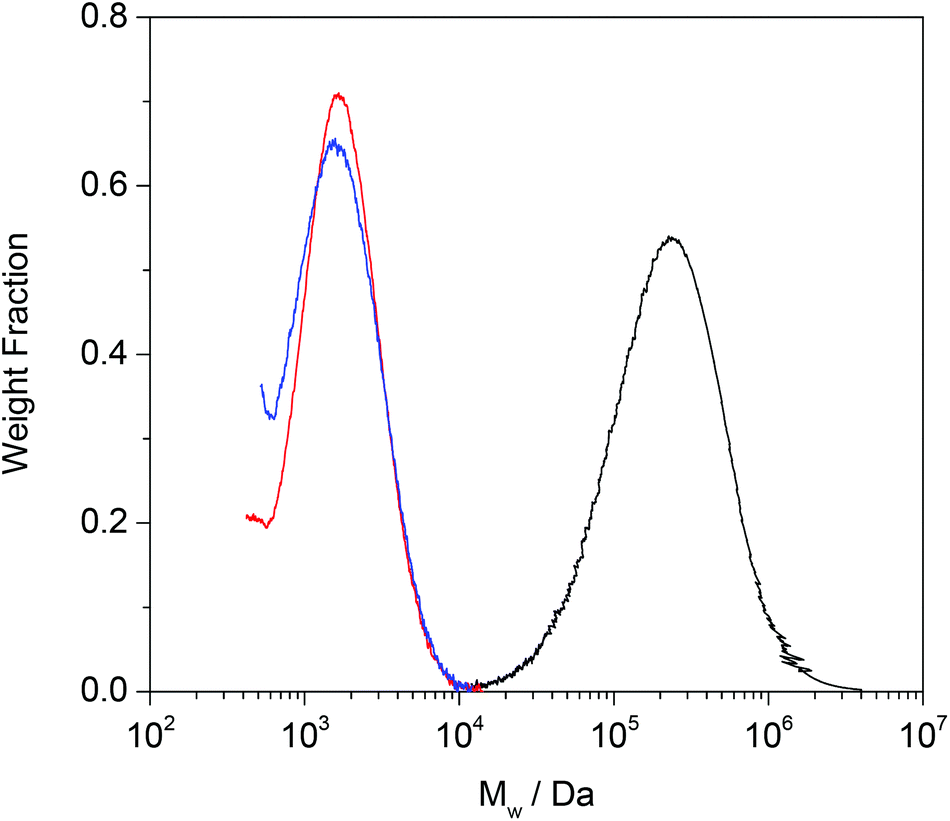 | ||
| Fig. 5 SEC traces showing the Grubbs’ G1 catalysed breakdown of cross-linked PBd. Traces shown are (black) linear PBd (prior to cross-linking), (red) breakdown product for G1 catalyst + 0.01 mole fraction DM diester, (blue) G1 catalyst but no diester. | ||
Rheometry provides further evidence for the breakdown of cross-linked PBd, and importantly gives some insight into the properties such as viscosity and modulus relevant to processing. For the PBd materials indicated by SEC before cross-linking and following breakdown of the cross-linked products, we have calculated the frequency dependent rheology explicitly.
Fig. 6 demonstrates this approach using the polydisperse double reptation model embodied in Reptate software.22–27 This well-established model allows the quantitative calculation of rheology data from the molecular weight distribution of a polymer, by approximating the molecular weight distribution measured by SEC as a multimodal blend of ∼10 monodisperse fractions. For the starting linear PBd material, an excellent fit to the experimental data is obtained in which the Rouse relaxation time, τe (= 1.14 × 10–6 s), of a single entanglement, the plateau modulus, Ge (= 1.54 × 106 Pa), and entanglement molecular weight, Me (= 2600 g mol−1), were varied. The data correspond to the rubbery plateau region of the viscoelastic spectrum in which G′ exceeds G′′, Ge is well defined and the fitted result is very close to the expected literature value.28 The fitted values for τe and Me are also in reasonable agreement within the expected range for polybutadienes, and it is possible to predict that the crossover between viscous and elastic behaviour occurs at 0.28 ± 0.02 rad s−1, which is equivalent to a terminal relaxation time of approximately 3.5 s. The rheology results for the breakdown products are consistent with the very low molecular weights determined by SEC. After breakdown, the material is liquid-like (note that G′′ now exceeds G′ over a broad range of angular frequency, ω), even on much shorter timescales and the moduli drop by orders of magnitude. In fact, the elastic modulus is so low that it is not well characterised by the rheometer due to inertial effects in the instrument. Since at low frequencies, rheology is very sensitive to small quantities of high molecular weight materials, this test convincingly demonstrates the quantitative conversion of high molecular weight polymer networks to oligomers. Both SEC and rheometry clearly demonstrated that even with the less active first generation Grubbs’ catalyst, a massive change in the mechanical properties of the cross-linked PBd is achieved, and the cross linked elastic materials can readily be converted into viscous liquids.
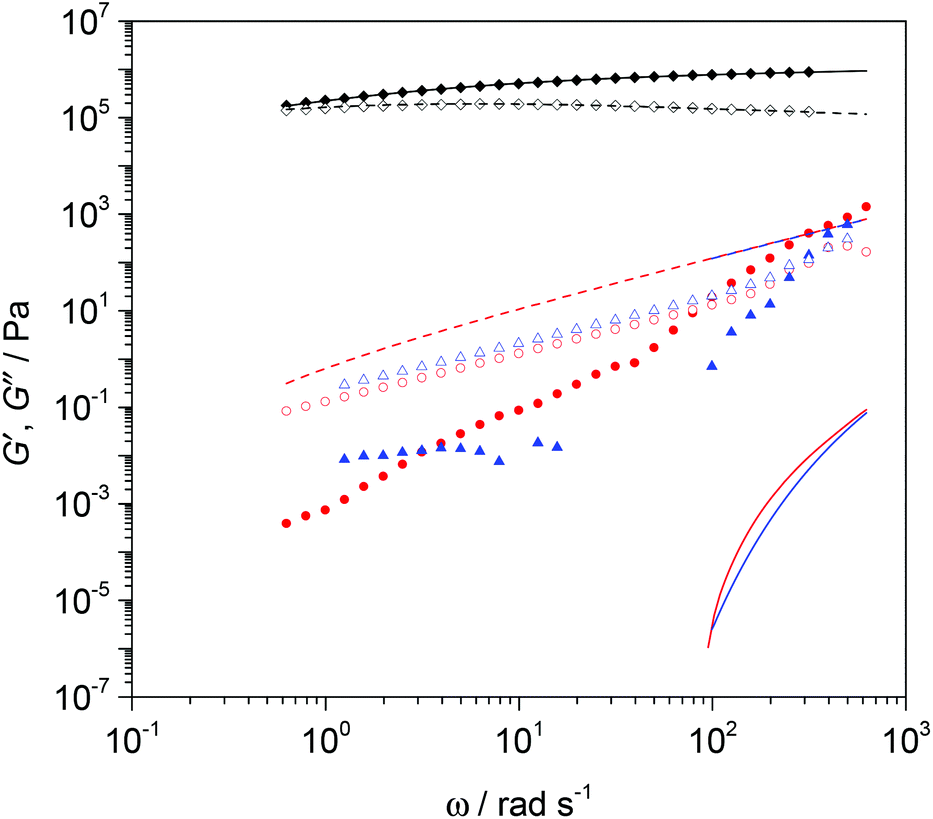 | ||
| Fig. 6 Complex modulus frequency sweep data showing impact of G1 catalyst on cross-linked PBd. G′ and G′′ are denoted by solid and open symbols respectively. Calculated values using Reptate for G′ and G′′ are shown as solid and dotted curves respectively. Data are shown for (black) linear PBd prior to cross-linking, (red) breakdown product for G1 catalyst + 0.01 mole fraction DM diester, (blue) G1 catalyst but no diester. | ||
Prior to this investigation, it was anticipated that the only means by which CM reactions could break down PBd was by the introduction of new chain ends using small molecules, such as DM or DF, as illustrated in Fig. 7(a). However, the remarkable ability of Grubbs’ catalysts to break down PBd, even in the absence of a low molecular weight diester such as DF, proves that breakdown of PBd is also possible by some internal CM reaction within the PBd. We postulate that two further mechanisms could be responsible for this result, which are shown in the schematic form in Fig. 7(b). Firstly, a CM reaction between the backbone double bonds in a single polymer chain between crosslink points would lead to the formation of two smaller chains, one of which would be cyclic. Although this mechanism could not completely break down a network, it could convert a continuous network into a more densely cross-linked network plus some free ring polymers. This mechanism is illustrated in the sketch in Fig. 7(bi). However, this reaction is likely to be quite limited in its extent, since the formation of tight ring polymers and very dense networks would be sterically hindered. We also note that such a mechanism could equally apply to the materials in the studies of Guan et al.16 but this behaviour was not reported. Instead, the pendant –CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 groups resulting from 1,2 enchainment appear to be the more likely candidates for participation in a CM reaction with a backbone –CHCH– resulting in a lasso topography and a linear polymer. This process is sketched in Fig. 7(bii), and significantly, this mechanism could account for the complete breakdown of a network without leaving an insoluble residue. Furthermore, the virtual absence of 1,2 enchainment in the materials studied by Guan et al., would preclude this mechanism from having a significant role in their materials. Noting that our original PBd comprised 7.7% 1,2 (vinyl) groups, there are more than sufficient numbers of these groups present to account for the levels of breakdown indicated by SEC analysis.
CH2 groups resulting from 1,2 enchainment appear to be the more likely candidates for participation in a CM reaction with a backbone –CHCH– resulting in a lasso topography and a linear polymer. This process is sketched in Fig. 7(bii), and significantly, this mechanism could account for the complete breakdown of a network without leaving an insoluble residue. Furthermore, the virtual absence of 1,2 enchainment in the materials studied by Guan et al., would preclude this mechanism from having a significant role in their materials. Noting that our original PBd comprised 7.7% 1,2 (vinyl) groups, there are more than sufficient numbers of these groups present to account for the levels of breakdown indicated by SEC analysis.
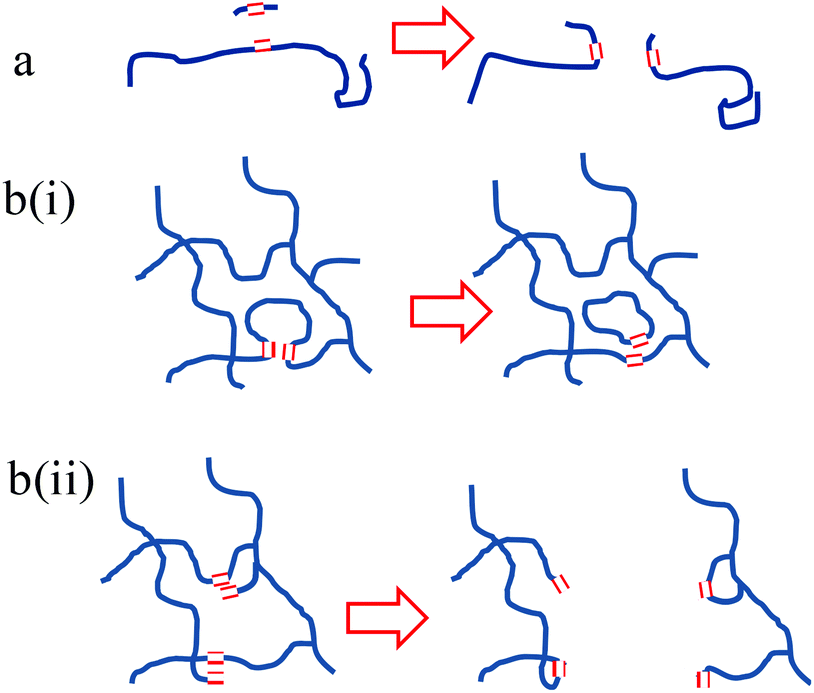 | ||
| Fig. 7 Sketch of network topographical arrangements made possible by CM reactions (a) facilitated by an additional unsaturated diester or (b) by internal rearrangement: (i) between two 1,4 PBd units and (ii) between 1,2 PBd units and 1,4 PBd units. | ||
Breaking down styrene butadiene rubber (SBR) with CM reactions
The reaction protocol used for the cross-linked PBd was repeated on 8 mm diameter, 2 mm thick disc samples of SBR sheets. When immersed in DMF with the G2 catalyst and 0.1 mole fraction DM, the discs were broken down to a crumb-like consistency within 24 h at room temperature, Fig. 8. When this procedure was repeated for shorter times, the discs were found to be intact after 1 h, but completely broken down within 2.5 h. Swelling tests in heptane revealed that 13 ± 1% of the crumb product was soluble after 2.5 h reaction, indicating that a significant quantity of low molecular weight PBd chains had been liberated by the reaction. Although it was found that longer reaction times led to somewhat larger soluble fractions, we note that the styrene component of the SBR would not be expected to be dissolved by heptane, hence complete dissolution is not expected. | ||
| Fig. 8 Photograph of SBR 8 mm diameter disc samples before and after breakdown with G2 catalyst at room temperature over 24 h. | ||
Further evidence for the participation of the catalyst in the breakdown of SBR can be obtained by examining the insoluble residue of the breakdown products for bound Grubbs’ catalyst. Particle-induced X-ray emission (PIXE) analysis is a convenient method for quantifying the presence of heavy metals in organic materials, and our results of this analysis confirm the presence of Ru from the Grubbs’ catalyst in all of our materials following immersion in a DMF solution of the catalyst. Results of PIXE analysis shown in Fig. 9 are typical, and confirm the presence of Grubbs’ catalyst in solid rubber samples. The filled squares correspond to the surface of the sheet material and the open circles correspond to the PIXE spectrum of the solid recovered following the CM reaction with the G2 catalyst and the DM diester at 10 mol% with respect to PBd double bonds in the SBR sheet sample. The catalyst is most clearly and uniquely identified by the Ru Kα X-rays at approximately 19 keV, and corroborated by the peaks around 2.5 keV due to the Ru L X-rays. For the sample exposed to the catalyst, both peaks were analysed using Gupixwin software18 to quantify the amount of Ru present, yielding a consistent value of 3 ± 1 parts per thousand by mass (pptm). This loading of catalyst equates to approximately 1 mole of catalyst per 700 moles of PBd repeat units, and so underlines the efficiency of this catalyst in transforming the state of the cross-linked rubber. The breakdown product also shows the presence of some other elements, notably Br (3 pptm) and K, (0.3 pptm) which were not apparent in the original sheet sample. Here, it is important to note that the X-ray yield due to the ion beam used in these experiments is dominated by the uppermost 10 μm of the sample. At greater depths, the 4He beam energy falls below 3.0 MeV and the X-ray yield is minimal. It appears most likely that these additional elements are due to fillers and bromobutyl rubber residues which were not initially present at the surface of the SBR sheet, but were exposed by the G2 catalysed breakdown.
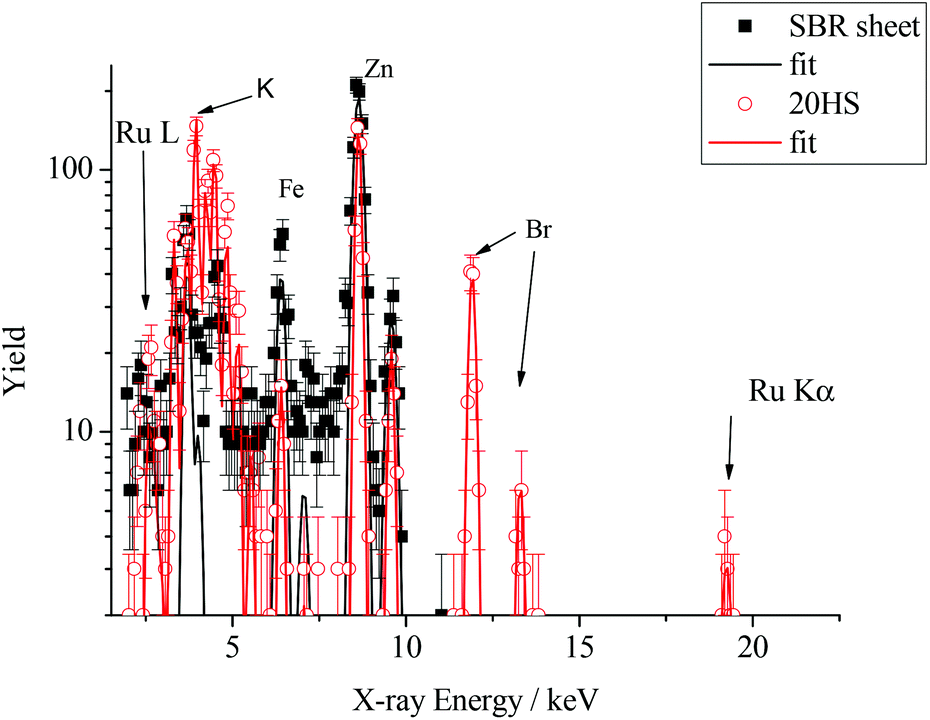 | ||
| Fig. 9 PIXE spectra for SBR sheet before (filled squares) and after (open circles) exposure to 2nd generation Grubbs’ catalyst. The solid curves are fits to the data obtained using Gupixwin analysis software. | ||
Since the CM reactions of the SBR sheet mediated by DM were successful, a reaction with DF was carried out to observe whether this diester, having a more sterically hindered olefin, produced a less favourable reaction, and a further control reaction with no diester present was also attempted. Remarkably, these reactions also reduced the SBR sheet to rubber crumb products, indicating that significant breakdown of the rubber was possible.
NMR spectra of the products obtained from the filtrate shown in Fig. 10 provide insight into the dependence of the CM reaction on the species present in the soluble products generated. The cis, trans and vinyl contents of the PBd were also calculated, as detailed in Table 2. In the first experiment (Fig. 10a) an SBR sheet was heated to 40 °C in DCM without a diester or a catalyst. In this control experiment, the rubber sheet became swollen, but there was no evidence of breakdown. NMR analysis of the soluble filtrate revealed traces of cis and trans PBd, which were apparent from the small peaks at 2.0 and 5.4 ppm. A large peak at 1.26 ppm shows a predominantly vinyl structure (∼96%) however. This indicates that a small fraction of the SBR starting material was not cross-linked in the network; the sol fraction. The spectrum from the CM reaction, G2 + 10 mol% DM with respect to PBd, at 40 °C, Fig. 10(b), indicates much larger peaks for the PBd among the breakdown products than were found in the control experiment, Fig. 10(a). There is a significant change in the microstructure of the PBd, with a reduction to 49% vinyl content. The cis and trans contents increase to 12 and 39% respectively. This is the same reaction mixture for which the visual evidence (Fig. 8) shows breakdown to crumb and the PIXE spectrum (Fig. 9) shows incorporation of G2 in the crumb residue. Ultimately the change in the microstructure may be a consequence of the fact that in Fig. 10(a) the rubber had not been broken down, so was not representative of the true sample. These results indicate that the CM reactions have succeeded in breaking down some of the SBR such that it can now dissolve in methanol. In Fig. 10(c), the reaction was reattempted with the less reactive DF instead of the DM diester. Although the material was broken down to a crumb product, the soluble filtrate analysed by NMR showed large peaks for unreacted DF and only small PBd peaks. The strong DF peak indicates that DF is unlikely to have participated in the CM reaction at room temperature. From these results it can be concluded that using the less sterically hindered diester, DM, results in a more effective CM reaction and a greater yield of soluble PBd. The final NMR spectrum, Fig. 10(d), shows that when the reaction with the G2 catalyst is attempted without any diester, but at 40 °C, a significant yield of PBd is obtained in the filtrate. We therefore conclude that the G2 catalyst is also able to cause the breakdown of the SBR sheet rubber via a similar mechanism to that was postulated for the in-house cross-linked PBd, Fig. 7b(ii).
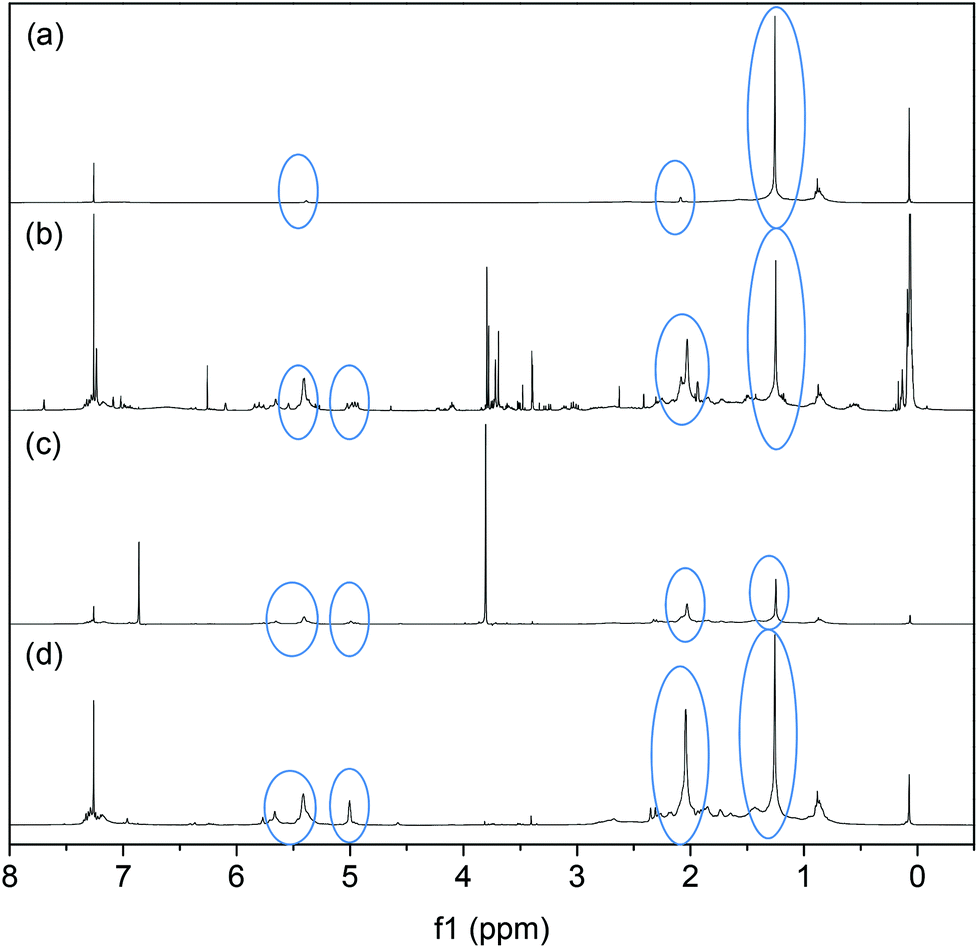 | ||
| Fig. 10 1H NMR spectra of soluble products recovered from SBR sheet (a) heated to 40 °C without catalyst or diester, (b) 40 °C with G2 and 10 mol% DM, (c) room temperature G2 with DF diester, (d) 40 °C with G2 without diester. The positions of the PBd peaks are circled. | ||
| Reaction | %vinyl | %cis | %trans |
|---|---|---|---|
| (a) SBR heated to 40 °C, no catalyst or diester | 95.6 | 3.6 | 0.8 |
| (b) 40 °C with G2 and 10 mol% DM | 49 | 12 | 39 |
| (c) Room temperature, G2, DF | 44.7 | 14.8 | 40.5 |
| (d) 40 °C, G2, no diester | 50.9 | 8.4 | 40.6 |
Scope for development as a ‘green’ process
Thus far, we have tested and demonstrated the efficacy of this catalytic approach to the breakdown of cross-linked rubber on a small scale. Although the use of chlorinated solvents would not be suitable for larger scale processing, it should be noted that the catalysts used here are now established as being effective in a wide variety of solvents, including more environmentally benign solvents13 such as pentane, and even some imidazolium ionic liquids.29 The choice of solvent would then be dictated by balance between the effectiveness of the catalyst in that solvent and the ability of the catalyst solution to penetrate the rubber network in order to minimise the need to cut the rubber sample. The next stage to exploit these breakdown products will be to re-polymerise them into a rubbery material. Our analysis clearly shows that as well as being readily soluble, the breakdown products retain their PBd-like structure. We have also shown that the presence of double bonds in this structure can readily enable cross-linking reactions with BPO, and we anticipate that this and other reaction mechanisms can readily be applied to the breakdown products. Although beyond the scope of the present work, there is considerable flexibility for future development of this methodology both in terms of utilising greener and more scalable approaches, and a final reprocessing step to regenerate a rubber product.Conclusions
We have shown that polybutadiene (PBd) and the rubber used in tyres can be broken down using cross metathesis (CM) reactions. These react together with two double carbon bonds in the presence of a Grubbs catalyst, leading to the rearrangement and scission of PBd double bonds in the backbone chain. It has been discovered that CM reactions with PBd and another olefin, such as a diester (e.g. dimethyl maleate), accelerate the breakdown of the polymer chain.By using CM reactions PBd chains can be broken down to a fraction of their original size to give readily soluble products. This occurred regardless of crosslinking. The effects of changing the diester used to one with a less sterically hindered double carbon bond and varying the quantity added were investigated, but in fact breakdown was significant regardless of the diester structure. Both Grubbs’ 1st and 2nd generation ruthenium catalysts were used in reactions to produce similar outcomes, although it was seen that the G2 catalyst was marginally more active, resulting in a greater proportion of trans-linkages in the backbone of the breakdown products. It has been observed that adding the catalyst without the diester caused the breakdown of the PBd, even when it was cross-linked, suggesting that pendant ethyl groups can also be utilised in the breakdown process.
Styrene-butadiene rubber (SBR) was tested in the sheet form and so the breakdown of this material via the CM reaction was evident by the formation of a crumb-like product. Running the reaction for 24 h at 40 °C and using a diester resulted in extensive breakdown of the SBR discs. However, the discs could also be broken down into crumb within 2.5 h at room temperature.
CM reactions could be used as an efficient way of making rubber crumb since only a very small amount of catalyst is added and the extent to which it is broken down can be determined by the reaction time. These reactions for linear PBd and SBR show that the higher the crosslinking, the longer the reaction time needed. Nevertheless, our results show that CM is an effective method for the breakdown of PBd and this reaction has potential to be used in the breakdown of rubber crumb and hence might be developed as a method for chemically reprocessing waste vehicle tyres. We anticipate that other polymers which contain carbon double bonds (e.g. polyisoprenes, natural rubber) in the chain backbone could also be broken down using this method. If the full breakdown of tyres via a CM reaction can be achieved it could redefine the way rubber is recycled.
Acknowledgements
We are grateful to Dr Alan Kenwright (NMR analysis service), Prof. Lian Hutchings (SEC analysis service) and Stephen Millington (ARTIS) for providing the rubber sheet.References
- B. Adhikari, D. De and S. Maiti, Prog. Polym. Sci., 2000, 25, 909–948 CrossRef CAS.
- T. P. Utomo, U. Hasanudin and E. Suroso, in Proc. of the World Congress on Engineering, 2010 Search PubMed.
- M. Myhre and D. A. MacKillop, Rubber Chem. Technol., 2002, 75, 429–474 CrossRef CAS.
- M. Myhre, S. Saiwari, W. Dierkes and J. Noordermeer, Rubber Chem. Technol., 2012, 85, 408–449 CrossRef CAS.
- H. P. Xiang, H. J. Qian, Z. Y. Lu, M. Z. Rong and M. Q. Zhang, Green Chem., 2015, 17, 4315–4325 RSC.
- C. W. Bielawski and R. H. Grubbs, Prog. Polym. Sci., 2007, 32, 1–29 CrossRef CAS.
- Handbook of Metathesis - Polymer Synthesis, ed. R. H. Grubbs and E. Khosravi, Wiley VCH, 2nd edn, 2015 Search PubMed.
- U. Frenzel and O. Nuyken, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 2895–2916 CrossRef CAS.
- S. J. Connon and S. Blechert, Angew. Chem., Int. Ed., 2003, 42, 1900–1923 CrossRef CAS PubMed.
- Handbook of Metathesis: Applications in Organic Synthesis, ed. R. H. Grubbs and D. J. O'Leary, 2nd edn, Wiley-VCH, 2015 Search PubMed.
- A. K. Chatterjee, T. L. Choi, D. P. Sanders and R. H. Grubbs, J. Am. Chem. Soc., 2003, 125, 11360–11370 CrossRef CAS PubMed.
- P. Schwab, R. H. Grubbs and J. W. Ziller, J. Am. Chem. Soc., 1996, 118, 100–110 CrossRef CAS.
- M. S. Sanford, J. A. Love and R. H. Grubbs, J. Am. Chem. Soc., 2001, 123, 6543–6554 CrossRef CAS PubMed.
- H. Otsuka, T. Muta, M. Sakada, T. Maeda and A. Takahara, Chem. Commun., 2009, 1073–1075 RSC.
- Y. X. Lu and Z. B. Guan, J. Am. Chem. Soc., 2012, 134, 14226–14231 CrossRef CAS PubMed.
- Y. X. Lu, F. Tournilhac, L. Leibler and Z. B. Guan, J. Am. Chem. Soc., 2012, 134, 8424–8427 CrossRef CAS PubMed.
- M. Capelot, D. Montarnal, F. Tournilhac and L. Leibler, J. Am. Chem. Soc., 2012, 134, 7664–7667 CrossRef CAS PubMed.
- T. L. Hopman, Z. Nejedly, J. A. Maxwell and J. L. Campbell, Nucl. Instrum. Methods Phys., Res. Sect. B, 2002, 189, 138–142 CrossRef CAS.
- T. B. Johansson, K. R. Akselsson and S. A. E. Johansson, Nucl. Instrum. Methods, 1970, 84, 141 CrossRef CAS.
- J. L. Campbell, T. L. Hopman, J. A. Maxwell and Z. Nejedly, Nucl. Instrum. Methods Phys. Res., Sect. B, 2000, 170, 193–204 CrossRef CAS.
- M. A. Hillmyer and F. S. Bates, Macromolecules, 1996, 29, 6994–7002 CrossRef CAS.
- J. Des Cloizeaux, Macromolecules, 1990, 23, 3992–4006 CrossRef CAS.
- A. E. Likhtman and T. C. B. McLeish, Macromolecules, 2002, 35, 6332–6343 CrossRef CAS.
- J. Ramirez and A. E. Likhtman, RepTate, http://www.reptate.com.
- C. Tsenoglou, Macromolecules, 1991, 24, 1762–1767 CrossRef CAS.
- W. H. Tuminello, Polym. Eng. Sci., 1986, 26, 1339–1347 CAS.
- J. L. Viovy, M. Rubinstein and R. H. Colby, Macromolecules, 1991, 24, 3587–3596 CrossRef CAS.
- S. J. Park, P. S. Desai, X. Chen and R. G. Larson, Macromolecules, 2015, 48, 4122–4131 CrossRef CAS.
- Y. S. Vygodskii, A. S. Shaplov, E. I. Lozinskaya, O. A. Filippov, E. S. Shubina, R. Bandari and M. R. Buchmeiser, Macromolecules, 2006, 39, 7821–7830 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Data for SEC, NMR, rheology and PIXE. See DOI: 10.1039/c5gc03075g |
| This journal is © The Royal Society of Chemistry 2016 |