
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Transketolase catalysed upgrading of L-arabinose: the one-step stereoselective synthesis of L-gluco-heptulose†
Fabiana
Subrizi
a,
Max
Cárdenas-Fernández
b,
Gary J.
Lye
b,
John M.
Ward
b,
Paul A.
Dalby
b,
Tom D.
Sheppard
a and
Helen C.
Hailes
*a
aDepartment of Chemistry, University College London, 20 Gordon Street, London, WC1H 0AJ, UK. E-mail: h.c.hailes@ucl.ac.uk
bThe Advanced Centre for Biochemical Engineering, Department of Biochemical Engineering, University College London, Bernard Katz Building, London, WC1H 0AH, UK
First published on 16th February 2016
Abstract
Conversion of biomass using biocatalysis is likely to become a technology that contributes significantly to the future production of chemical building blocks, materials and transport fuels. Here the synthesis of a value-added chemical from L-arabinose, a major component of the carbohydrates in sugar beet pulp (SBP), in a concise and sustainable manner has been investigated. Biocatalytic conversions using transketolase variants have been developed for the efficient, scalable synthesis of a rare naturally occurring ketoheptose, L-gluco-heptulose, from L-arabinose. New active E. coli TK mutants that readily accept L-arabinose were identified using a versatile colorimetric screening assay and the reaction was performed on a preparative scale.
Introduction
Sustainability issues related to the use of crude oil are progressively directing the chemical and pharmaceutical industries towards renewable sources for the production of organic synthons. Indeed, biomass represents an attractive alternative as a renewable source for chemical building blocks, materials and biofuels.1 In a bio-based economy, biocatalysis has been recognised as a green, environmentally friendly technology with significant potential due to the mild reaction conditions used and high chemo- and enantioselectivities that can be achieved.2Transketolase (TK), a key enzyme in metabolic regulation providing a link between the pentose phosphate pathway and glycolysis, is a thiamine diphosphate (ThDP) dependent enzyme that has been used in stereospecific carbon–carbon bond formation.3In vivo, TK catalyses the reversible transfer of a two-carbon fragment from a five-carbon ketose (D-xylulose-5-phosphate) to a five-carbon aldose (D-ribose-5-phosphate) to form a seven-carbon ketose (D-sedoheptulose-7-phosphate). The ability of TK to perform the reaction irreversibly with the loss of carbon dioxide, when using β-hydroxypyruvate (HPA) as the ketol donor, makes it an attractive biocatalyst in industrial synthesis.4,5 To achieve commercial viability in large-scale processes with non-phosphorylated substrates, variants of TK with greater specific activity are required. The TK reaction with lithium hydroxypyruvate (Li-HPA 1) as a donor has been studied with a range of aldehyde acceptors (Scheme 1) and TK variants exhibiting enhanced activity towards non-phosphorylated aliphatic and aromatic substrates have been developed.6
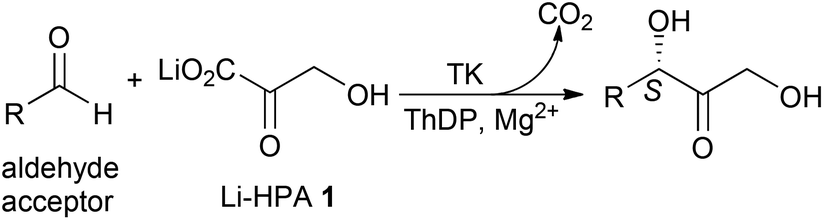 | ||
| Scheme 1 TK reaction with lithium β-hydroxypyruvate (Li-HPA) 1, highlighting the WT-TK S-stereochemistry in the α,α′-dihydroxyketone product. | ||
Ketoheptoses are rare seven-carbon sugars found in nature, that have significant pharmacological potential as inhibitors of sugar metabolism. For example, in the 1960s D-manno-heptulose was isolated from avocados and reported to inhibit glucose metabolism and insulin secretion in vivo and in vitro.7 Following the identification of D-manno-heptulose as an inhibitor of glucokinase, interest in ketoheptoses for biomedical applications has increased and concise synthetic routes to these compounds are required.8 Multistep syntheses of D-manno-heptulose and other ketoheptoses have been described, including the use of rearrangements and chain extensions resulting in the formation of diastereomeric mixtures and poor overall yields.9 For example, the most recent approach reported the stereoselective preparation of D-manno-heptulose and D-gluco-heptulose in eight synthetic steps and 56% overall yield, using toxic reagents.10
In a current multidisciplinary project, we are investigating the use of the waste biomass sugar beet pulp (SBP) for the synthesis of value-added chemicals. L-Arabinose 2, is one of the major sugars in this feedstock, and can be obtained from the hydrolysis of pectin.11 The acceptance of L-arabinose as a substrate with TK would generate L-gluco-heptulose 3, a rare naturally-occurring ketoheptose with potential therapeutic applications in hypoglycaemia and cancer (Scheme 2).8 Here we report for the first time the one-step biocatalytic preparation and characterisation of L-gluco-heptulose 3. TK libraries were screened through a medium-throughput assay and a colorimetric screening assay was also developed for high-throughput applications and successfully applied to identify productive enzymes. The use of a high-performing TK variant with L-arabinose was then used for the efficient stereoselective, one-step synthesis of L-gluco-heptulose 3 on a preparative laboratory scale.
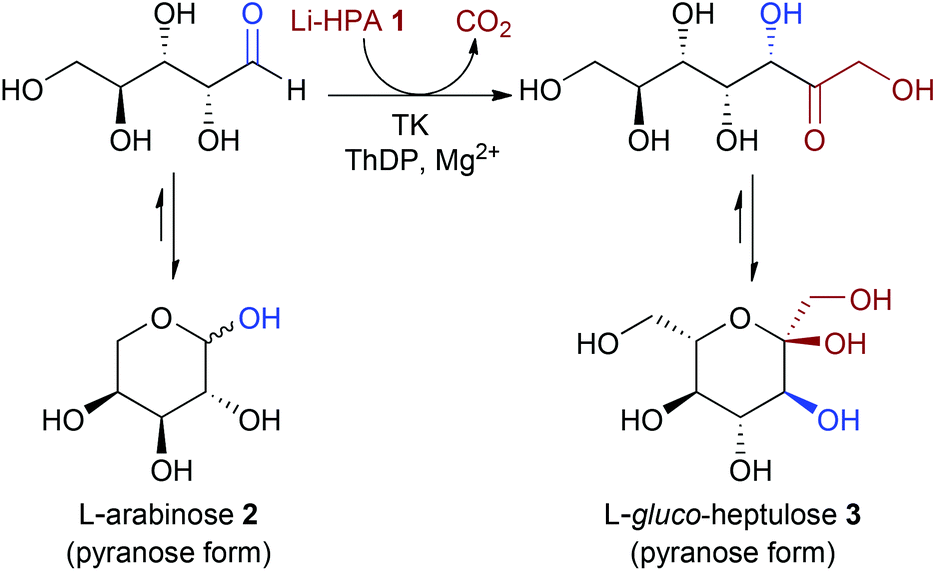 | ||
| Scheme 2 TK mediated synthesis of L-gluco-heptulose 3 from Li-HPA 1 and L-arabinose 2. | ||
Results and discussion
Identifying an improved transketolase for L-arabinose acceptance
Compared to the natural aldehyde acceptors D-ribose-5-phosphate and erythrose-4-phosphate, TK in general has low affinity towards non-phosphorylated sugars.12 In 1971, Axelrod et al. reported that L-arabinose 2 was accepted by wild-type (WT) Saccharomyces cerevisiae TK, with a KM > 200-fold higher than the natural phosphorylated substrate D-ribose-5-phosphate (KM 120 mM and 0.4 mM for L-arabinose and D-ribose-5-phosphate respectively).13 More recently, WT S. cerevisiae and E. coli TK specific activities of approximately 0.045 U mg−1 were noted, more than 400-fold less than that for glycolaldehyde, using a pH based assay.14 Studies to evolve S. cerevisiae TK for improved activities against D-ribose and D-glucose led to a double mutant R526Q/S252T that gave some of the highest improvements in specific activities (2.6-fold for D-ribose and 2.1-fold for D-glucose).15 The pentose sugars D-ribose and L-arabinose 2 share the same stereochemistry at C-2, but have the opposite stereochemistries at C-3 and C-4, and to the best of our knowledge no attempt to evolve TK towards the acceptor L-arabinose has been reported to date.In our preliminary investigation of over 20 E. coli TK variants from our UCL-TK libraries, selected on the basis of previous studies using polyhydroxylated substrates, several single and double active-site mutants were identified that were able to accept L-arabinose 2, including WT (as previously reported), F434A and A29E/H461S (Table 1). For this smaller library a GC-MS protocol for identification of the heptulose product 3 was used (see Experimental).16 GC-MS is particularly valuable in terms of sensitivity and product identification, as well as giving access to relative stereoselectivity data. The gas chromatogram for all the active mutants identified revealed a single product peak, suggesting a fully stereoselective reaction. The new stereocentre in the ketose product was assigned as the S-configuration (D-threo product), on the basis of the established WT-stereoselectivity for hydroxylated aldehyde substrates and a comparison of the characterization data for 3 to the enantiomer reported in the literature.3,10,12 (see below and ESI†).
| TKE.coli variant | Heptulose 3 | TKE.coli variant | Heptulose 3 |
|---|---|---|---|
| a Reaction conditions: Li-HPA 1 (33 mM), 2 (33 mM), ThDP (1.7 mM) and MgCl2 (6 mM) in Tris-HCl (50 mM) at pH 7.0, with 33% v/v TK lysate for 24 h. b Traces corresponds to <5% conversion; ✓ corresponds to ∼6–20% conversion; ✗ corresponds to no conversion. | |||
| WT | ✓ | F434A/R520Q | Traces |
| D469X (X = T, Y, E) | ✗ | A29E/H461S | ✓ |
| F434A | ✓ | A29E/R520V | Traces |
| H461S | ✗ | A29E/D469Y | ✗ |
| A29E | ✗ | H461S/R520Q | Traces |
| R520V | Traces | R358P/D469T | ✗ |
| D469X/R520Q (X = T, Y, E) | ✗ | S385X/D469 T/R520Q (X = T, Y, E) | ✗ |
Although the variants showing activity towards 2 did not lead to a significant increase in activity compared to WT, it was possible to rationalise the substrate specificity observed. Notably, none of the D469X variants possessing either polar ionic (E) or uncharged (T and Y) residues led to active mutants, suggesting a crucial role for D469 in binding of the sugar 2. Computational docking analysis of L-arabinose 2 in the most abundant cyclic α-pyranose (63% at equilibrium in water)17 form into the active site of the E. coli wild-type TK (PDB ID: 1QGD) containing the ThDP-enamine intermediate (Fig. 1) highlighted a key interaction between the D469 residue and C-2 hydroxyl group of the acceptor.18
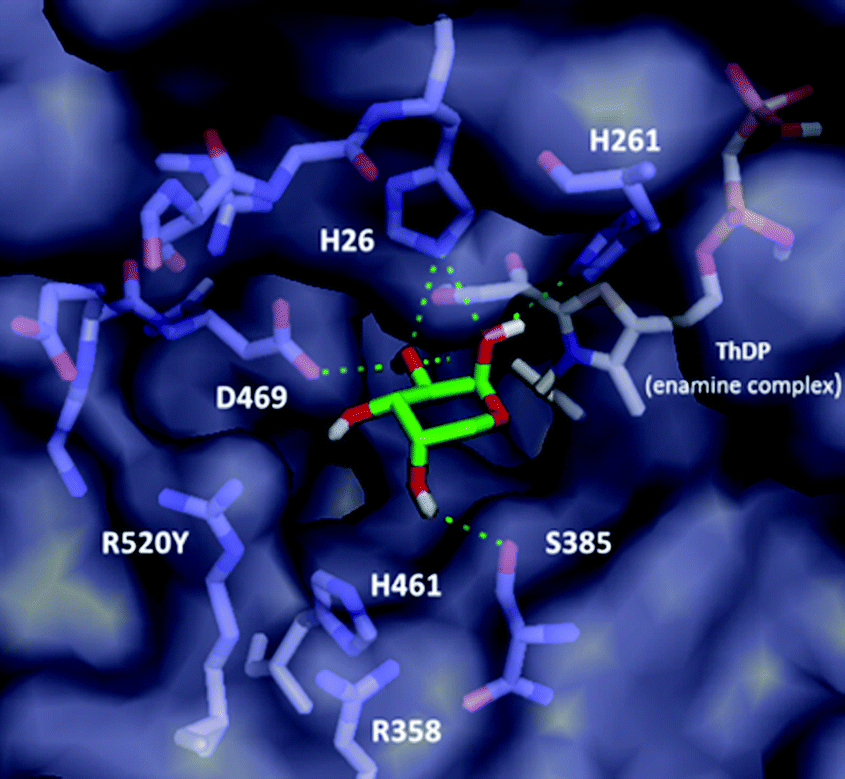 | ||
| Fig. 1 Computational docking of L-arabinose 2 in the cyclic pyranose form into the active site of E. coli WT-TK (PDB ID: 1QGD) containing the ThDP-enamine intermediate. | ||
Docking analysis also provided further insights: the inner part of the substrate channel contains several His residues, and the middle section a Ser residue. These residues (H26, H261 and S385) interact with the C-1 and C-2 hydroxyl groups of the L-arabinose 2via polar interactions, similar to those in the crystal structure with D-ribose-5-phosphate (R5P) in its cyclic five-membered ring form (cR5P) in the WT-TK active site.14,18,19 They can also contribute to maintaining the close proximity of the acceptor substrate to the nucleophilic centre on the ThDP-enamine intermediate in the correct orientation. In addition, the two Arg residues and the His residues in the outer part of the active site (R358, H461 and R520), interact with the phosphate moiety of cR5P via a mixture of hydrogen bonds and electrostatic interactions. These residues represent interesting sites for mutation to enhance the activity of TK toward non-phosphorylated long chain polyhydroxylated compounds, and have previously been highlighted in other mutational studies.6,14 The same set of interactions (except for residue S385) was also identified in the docking analysis of L-arabinose in its acyclic form (see ESI†), which represent less than 0.1% at equilibrium in water.
On the basis of the docking calculation and preliminary data, high-throughput screening with three libraries of mutants previously prepared by saturation mutagenesis of the conserved residues (R358, H461, and R520) was performed.6 Each library contained 90 randomly picked variants and three WT controls. Screening of the reaction plates, containing cell free lysate in each well, co-factors, Li-HPA 1 and L-arabinose 2 was performed using a new colorimetric assay.
Development of a high-throughput colorimetric assay
A crucial component in enzyme library screening is a rapid and reliable high-throughput method for identifying variants that catalyse the reaction. To date, colorimetric, fluorogenic and a colorimetric pH-based assay have been used to screen TK mutant libraries.14,20,21 However these methods either cannot be used with α-hydroxy aldehyde acceptors, are limited to a restricted substrate, or are based on the consumption of Li-HPA. Our aim was to develop an assay that could monitor the formation of ketose products in the TK reaction from L-arabinose 2 or other aldose sugars. Seliwanoff's test distinguishes between ketoses and aldoses in a reaction using 6 M HCl and resorcinol.22 Ketose dehydration under acidic conditions is followed by condensation with resorcinol to give a red xanthenone product. Aldoses, such as L-arabinose, however react more slowly than ketoses giving a yellow to green coloration. The reaction was therefore developed as a colorimetric assay to detect L-gluco-heptulose 3 formation in the presence of L-arabinose 2via production of a red xanthenone. An assay for L-gluco-heptulose formation was performed (Scheme 3) using the Seliwanoff's reagent and solutions containing different concentrations of 3 (Scheme 3, line A). After heating, a red coloration was evident in wells containing 3, and in addition a correlation could be observed between the amount of 3 present and intensity of the coloration generated. No coloration was observed in control reactions (also see experimental) containing L-arabinose alone or in combination with the cofactors (Scheme 3, line B). This qualitative assay was then used to rapidly identify mutants from the TKs libraries that accepted L-arabinose 2 to give 3. After 24 h the TK reactions in 96-well plates were treated with Seliwanoff's reagent, and heated at 100 °C. Colour formation due to the presence of the ketosugar 3 (and conversion to a xanthenone) was observed within 15 min (see ESI†). | ||
| Scheme 3 Assay for L-gluco-heptulose 3. Wells (W1–W4) line A correspond to the following concentrations of 3: W1, 50 mM; W2, 30 mM; W3, 15 mM; W4, 5 mM; line B: W1, L-arabinose 2 (33 mM), ThDP (1.6 mM) and MgCl2 (6 mM) and W2 L-arabinose 2 (33 mM). | ||
Visual analysis of the colorimetric assay plates (performed in duplicate) indicated that the E. coli TK libraries for R358, H461 and R520 contained active variants. In total, 31 active variants (17 from the R520 library, 11 from R358 library and 3 from the H461 library) were selected and sequenced. Among the R520 variants, two were revealed to be WT at a higher expression level while two mutations were identified for the other variants: R520Y and R520P. Similarly, three different mutants were identified from the 11 R358 variants sequenced: R358S, R358P and R358I. Finally, from the H461 library the single point mutant H461Y only was identified. In parallel a reaction plate for each library was also analysed by HPLC to validate the colorimetric screening method and confirmed the formation of 3. The positive hits from the colorimetric assay and HPLC screening were furthermore repeated and analysed by GC-MS, which corroborated the formation of 3.16 The new TK assay is versatile and potentially applicable to a variety of sugar-like acceptor substrates, including hexoses.
Determination of TK specific activities towards L-arabinose
All variants identified with the colorimetric assay were expressed and activities compared to E. coli WT-TK (Fig. 2). Reactions were monitored using high-performance anion-exchange chromatography with pulsed amperometric detection (HPAC-PAD). The specific activities of the selected mutants R358P, R358S, H461Y, R520P and R520Y were greater than or comparable to WT-TK. The greatest improvements relative to WT were a 2.1-fold increase for R520Y, and 1.5-fold for R520P. In studies to evolve yeast TK for improved activities against D-ribose and D-glucose, R526Y (residue R526 in yeast TK is equivalent to R520 in E. coli TK) gave specific activities compared to WT of 1.5-fold and 0.9-fold, respectively. This highlights the variation in activities with different aldose stereochemistries and chain lengths.15 The conversion of 2 to 3 was also performed for all 6 variants, and expression levels noted (Table 2).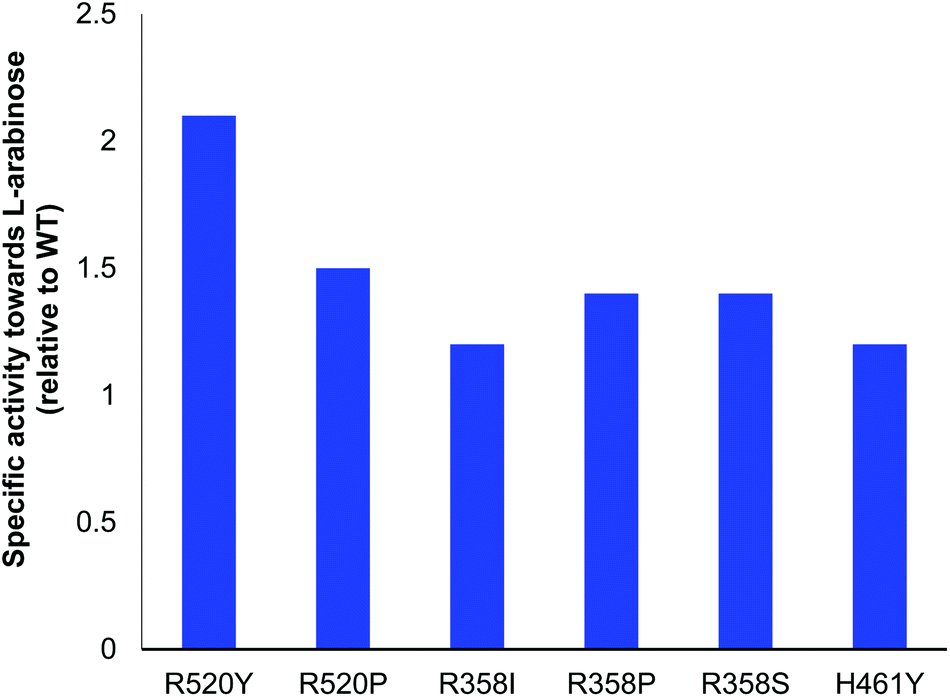 | ||
| Fig. 2 E. coli TK specific activities (μmol min−1 mg−1) towards L-arabinose 1, relative to WT (clarified lysates) using Li-HPA 1 (33 mM), 2 (33 mM), ThDP (1.7 mM) and MgCl2 (6 mM) in Tris-HCl (50 mM) at pH 7.0. The specific activity of the WT-TK was 0.021 μmol min−1 mg−1. | ||
| TK | Conversiona/% | mg mL−1 TK | Initial rateb (mM h−1) |
|---|---|---|---|
| a Conversions were determined based on L-arabinose consumption after 24 h by high-performance anion-exchange chromatography with pulsed amperometric detection (HPAC-PAD). b Initial rates were determined by monitoring the production of 3 using HPAC-PAD. Reactions were performed using Li-HPA 1 (33 mM), 2 (33 mM), ThDP (1.67 mM) and MgCl2 (6 mM) in Tris-buffer (50 mM) at pH 7.0 and using clarified lysate (v/v 33%) in a total reaction volume of 900 μL. | |||
| WT | 48 | 0.67 | 0.84 |
| R520Y | 54 | 0.48 | 1.27 |
| R520P | 31 | 0.28 | 0.54 |
| R358I | 22 | 0.23 | 0.36 |
| R358P | 35 | 0.48 | 0.84 |
| R358S | 48 | 0.75 | 1.31 |
| H461Y | 54 | 0.73 | 1.10 |
The higher conversion yields for R520Y and H461Y reflected the generally higher reaction rates compared to the other mutants, although a higher amount of H461Y is needed with respect to R520Y. Replacement of the positively charged residues (R and H) with a hydrophobic and uncharged tyrosine residue has previously been reported to enhance activity towards non-phosphorylated polyhydroxylated substrates.6 Moreover, docking of L-arabinose in the linear form showed a possible interaction between the residue R520 and one of the OH groups (see ESI†), which may explain the higher specific activity for some R520 mutants.
Preparative scale
L-gluco-Heptulose 3 is a rare naturally occurring ketoheptose. While synthesis of the corresponding enantiomer D-gluco-heptulose has been reported in eight-steps,10 the synthesis and characterization of L-gluco-heptulose has not previously been reported. Variant H461Y was selected initially for the preparative-scale reaction due to its high conversion yields, and good enzyme stability. The reaction was stopped after 24 h and analysis of the crude product by 1H NMR spectroscopy showed approximately a 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 ratio between the remaining L-arabinose 2 and the L-gluco-heptulose 3 formed. The crude mixture was then filtered through Amberlite® IR120 (hydrogen form) to remove both Tris buffer and some remaining 2 to give L-gluco-heptulose in 45% isolated yield after column chromatography, which was comparable to the conversion yield noted (Table 2). In a similar way the reaction was then carried out using R520Y mutant with a final 40% isolated yield.
:50 ratio between the remaining L-arabinose 2 and the L-gluco-heptulose 3 formed. The crude mixture was then filtered through Amberlite® IR120 (hydrogen form) to remove both Tris buffer and some remaining 2 to give L-gluco-heptulose in 45% isolated yield after column chromatography, which was comparable to the conversion yield noted (Table 2). In a similar way the reaction was then carried out using R520Y mutant with a final 40% isolated yield.
Stereoselectivity and product isolation
L-gluco-Heptulose 3 can exist as an equilibrium mixture of α- and β-anomers. In the TK reaction one new stereogenic centre is formed, and spontaneous cyclisation can result in the generation of four isomers. However, GC-MS and NMR spectroscopy indicated that only one isomer was formed. Notably, NMR spectroscopic data of 3 recorded in D2O showed the presence of a single pyranose isomer for L-gluco-heptulose: the 1H and 13C NMR data were the same as that previously reported for the enantiomer of 3, D-gluco-heptulose.10 The large trans-coupling constant between the axial protons at 3-H and 4-H also confirmed that the hydroxyl group at C-3 was in an equatorial orientation giving an S-configurated centre at this position.10 The characterization data confirmed that the TK catalysed reaction showed exquisite diastereoselectivity leading to a single ketose with the D-threo configuration, and was consistent with the stereoselectivity observed in TK reactions with other aldose acceptors.12 Only the more stable α-anomer was generated and additionally, as previously reported, L-gluco-heptulose did not exhibit mutarotation behaviour: the optical rotation in aqueous solution remained constant over several days.10 Furthermore, the absolute configuration of 3 was confirmed by correlation of the optical rotation to its enantiomer10 and previous reports on the optical rotation of 3 in the literature.23Conclusions
An efficient TK-mediated asymmetric C–C bond forming reaction has been successfully applied to convert L-arabinose 1 into a ketoheptose with potential therapeutic applications in hypoglycaemia and cancer. The ready availability of L-arabinose, a major component of pectin in SBP and renewable starting material, and one step synthesis of L-gluco-heptulose avoiding the use of toxic or hazardous solvents and reagents, could serve as a stimulus for the development of other green chemistry applications using SBP waste. In this study, several E. coli TK mutants with enhanced activity toward the non-phosphorylated sugar 2 have been identified for the first time, leading to the target product 3 in good yields with total stereocontrol. A rapid high-throughput colorimetric screening assay to determine TK activity towards sugar-like substrates was also developed, which allowed the rapid identification of the higher performing mutants. The TK protocol developed could be performed on a preparative scale without further optimization, highlighting the potential for further synthetic applications in an industrial setting.Experimental
General experimental and analytical methods
All chemicals were obtained from commercial source and used as supplied, except Li-HPA 1 which was prepared by modification of a previously described procedure.24 TLC analysis was performed on Kieselgel 60 F254 precoated plates and compounds visualised by exposure to UV light, potassium permanganate or p-anisaldehyde stain. Flash column chromatography was carried out using silica gel (particle size 40–63 μm). NMR: 1H and 13C NMR spectra were recorded at 298 K at the field indicated using Bruker Avance 500 and Avance 600 machines. Coupling constants were measured in Hertz (Hz) and peaks referenced to the deuterated solvent used. Infrared spectra were recorded on a Perkin Elmer Spectrum 100 FTIR spectrometer.Transketolase preparation
Transketolase preliminary library investigations
Transketolases mutants (Table 1) were investigated for activity with L-arabinose 2 and Li-HPA 1via the formation of L-gluco-heptulose 3. Reaction conditions: Li-HPA 1 (33 mM), 2 (33 mM), MgCl2 (6 mM) and ThDP (1.7 mM) in Tris-HCl buffer (50 mM, pH 7.0) and using 33% v/v TK lysate. The reactions (300 μL) were incubated at 25 °C with 450 rpm orbital rotation.Docking experiments
ThDP-enamine and L-arabinose 2 (cyclic α-pyranose and linear form) structures were drawn in Chem3D Ultra v.13.0 and energy minimised by MM2 calculations. The energy-minimised ThDP-enamine was docked into the active site.6d,27 A rigid docking for the TK structure (PDB: 1QGD, E. coli) against 2 was performed using ligand docking and binding site analysis with PyMOL and Autodock Vina28,29 with the grid centered at −11.25, 25.858, 40.198 and a grid size of 28 Å × 28 Å × 28 Å. Conformations were selected on the basis of the lowest energy (standard deviation for the 9 conformations of the cyclic and linear forms was 0.2) and for the cyclic form from visual analysis and proximity to the ThDP-enamine. The binding affinity for cyclic L-arabinose (in Fig. 1) was −4.7 kcal mol−1 and for the linear form (in ESI Fig. 3†) −4.8 kcal mol−1.Colorimetric assay: assaying for TK-mediated synthesis of L-gluco-heptulose 3 (preliminary assay)
A 20 mL reaction mixture containing ThDP (1.7 mM), MgCl2 (6 mmol) and cell-free lysate (33% total reaction volume) in Tris-HCl (50 mM, pH 7.0) was incubated at 25 °C under slow magnetic stirring. After 20 min, 10 mL of the substrate solution (1 and 2, 100 mM at pH 7.0) was added and the reaction mixture incubated at 25 °C for 24 h. At time 0 and 24 h, 100 μL of the reaction mixture were quenched with 1 mL of Seliwanoff's reagent in a closed test tube and heated at 100 °C. After 5 minutes a cherry red coloration appeared in the solution quenched after 24 h, which became more intense after 10 min at 100 °C. No coloration was evident for the reaction quenched at 0 h, indicating there was no formation of 3 and no coloration interference from any of the reaction components. Moreover, three control experiments without either TK, 1 or 2, were performed in parallel and no coloration after treatment with Seliwanoff's reagent and heating was observed.Library selection for activity
Specific activities of selected mutants
Glycerol stocks of selected mutants were streaked from the library master plate onto LB agar containing 150 μg mL−1 ampicillin. Individual colonies were selected and grown in 10 mL LB with 150 μg mL−1 ampicillin for 18 h at 200 rpm, 37 °C. The cells were then transferred to a larger volume of fermentation and the cell lysate was prepared as previously described. The specific activities of selected TK mutants were obtained for reactions of 1 (33 mM) with 2 (33 mM), ThDP (1.7 mM), MgCl2 (6 mM), in Tris-HCl (50 mM) at pH 7.0. Lysate (33% (v/v)) was used in duplicate or triplicate reactions with a final volume of 900 μL. At a time point during the first 24 h, 50 μL of the solution was sampled and quenched by adding 50 μL of H2SO4 (0.5% v/v aqueous solution). These samples were then analysed by HPAC-PAD as described above. TK concentrations were determined by SDS-PAGE densitometry as previously described for soluble TK after clarification.6d Densitometry of samples electrophoresed on a 12% SDS-Page gel (Bio-Rad) was used. The Coomassie stained gel was imaged using Imagesoft software to calculate the TK band density. Total protein in the samples was determined using a standard Bradford assay. A range of concentrations of commercial BSA was run in each SDS-Page gel, and used to calculate a standard curve based on integrated optical density for calibration of the enzyme concentration.Preparative scale biocatalytic reaction
A 20 mL reaction mixture containing ThDP (1.7 mM), MgCl2 (6 mM) and cell-free lysate (33% total reaction volume, 0.39 and 1 mg mL−1 TK final concentration for R520Y and H461Y respectively) in Tris-HCl (50 mM, pH 7.0) was incubated at 25 °C under slow magnetic stirring. After 20 min, 10 mL of the substrate solution (1 and 2, 100 mM at pH 7.0 in Tris-HCl buffer (50 mM)) was added and the reaction mixture incubated at 25 °C. After 24 h, 60 mL of MeOH was added and the white precipitate formed removed by centrifugation (10 min, 4000 rpm). The sample was then filtered twice through a strong cationic resin (Amberlite® IR-120 hydrogen form, 40 mL) to remove both Tris buffer and some 2. The filtrate, containing L-gluco-heptulose 323 was dry-loaded on a silica gel column chromatography (EtOAc/2-PrOH/H2O) to give L-gluco-heptulose 3 as a colourless oil (95 mg, 45% for H461Y; 80 mg, 40% for R520Y). Rf 0.33 (EtOAc/2-PrOH/H2O, 7:3:1); [α]20D −67.0 (c 1, H2O), lit., [α]20D −67.8 (c 2.5, H2O);23 IR (film/cm−1) 3314, 2925, 2469, 1419; 1H NMR (D2O; 600 MHz) 3.37 (1H, t, J 9.6 Hz, 5-H), 3.46 (1H, d, J 9.6 Hz, 3-H), 3.49 (1H, d, J 12.0 Hz, 1-HH), 3.65 (1H, d, J 12.0 Hz, 1-HH), 3.68 (1H, t, J 9.6 Hz, 4-H), 3.70–3.75 (2H, m, 6-H, 7-HH), 3.79 (1H, m, 7-HH); 13C NMR (D2O; 151 MHz) 61.3 (C-7), 64.3 (C-1), 70.2 (C-5), 70.9 (C-3), 72.9 (C-6), 74.3 (C-4), 98.2 (C-2); m/z HRMS (ES+) found [MNa]+ 233.0645, C7H14O7Na requires 233.0637.
Acknowledgements
The authors would like to thank the UK Engineering and Physical Sciences Research Council (EPSRC) for financial support of this work (EP/K014897/1), for F. S. and M. C., as part of their Sustainable Chemical Feedstocks programme. Input and advice from the project Industrial Advisory Board is also acknowledged. Furthermore, we gratefully acknowledge K. Karu (UCL Mass Spectrometry Facility) and A. E. Aliev (UCL NMR Facility) in the Department of Chemistry and Maria F. Villegas-Torres in the Department of Biochemical Engineering.Notes and references
- (a) J. H. Clark, Green Chem., 1999, 1, 1–8 RSC; (b) G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS PubMed; (c) A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 4211–2502 CrossRef PubMed; (d) S. A. Sanchez-Vazquez, H. C. Hailes and J. R. G. Evans, Polym. Rev., 2013, 53, 627–694 CrossRef CAS.
- B. M. Nestl, S. C. Hammer, B. A. Nebel and B. Hauer, Angew. Chem., Int. Ed., 2014, 53, 3070–3095 CrossRef CAS PubMed.
- (a) U. Schörken and G. A. Sprenger, Biochim. Biophys. Acta, 1998, 1385, 229–243 CrossRef; (b) N. J. Turner, Curr. Opin. Biotechnol., 2000, 11, 527–531 CrossRef CAS PubMed; (c) R. Wohlgemuth, J. Mol. Catal. B: Enzym., 2009, 61, 23–29 CrossRef CAS; (d) H. C. Hailes, D. Rother, M. Müller, R. Westphal, J. M. Ward, J. Pleiss, C. Vogel and M. Pohl, FEBS J., 2013, 280, 6374–6394 CrossRef CAS PubMed; (e) A. Ranoux and U. Hanefeld, Top. Catal., 2013, 56, 750–764 CrossRef CAS.
- (a) J. Shaeri, R. Wohlgemuth and J. M. Woodley, Org. Process. Res. Dev., 2006, 10, 605–610 CrossRef CAS; (b) J. Shaeri, I. Wright, E. B. Rathbone, R. Wohlgemuth and J. M. Woodley, Biotechnol. Bioeng., 2008, 101, 761–767 CrossRef CAS PubMed.
- P. Srere, J. R. Cooper, M. Tabachnick and E. Racker, Arch. Biochem. Biophys., 1958, 74, 295–305 CrossRef CAS PubMed.
- (a) E. G. Hibbert, T. Senussi, W. Leia, M. E. B. Smith, J. M. Ward, H. C. Hailes and P. A. Dalby, J. Biotechnol., 2007, 131, 425–432 CrossRef CAS PubMed; (b) E. G. Hibbert, T. Senussi, M. E. B. Smith, S. J. Costello, J. M. Ward, H. C. Hailes and P. A. Dalby, J. Biotechnol., 2008, 134, 240–245 CrossRef CAS PubMed; (c) M. E. B. Smith, E. G. Hibbert, A. B. Jones, P. A. Dalby and H. C. Hailes, Adv. Synth. Catal., 2008, 350, 2631–2638 CrossRef CAS; (d) P. Payongsri, D. Steadman, J. Strafford, A. MacMurray, H. C. Hailes and P. A. Dalby, Org. Biomol. Chem., 2012, 10, 9021–9029 RSC; (e) D. Yi, T. Saravanan, T. Devamani, F. Charmantray, L. Hecquet and W. D. Fessner, Chem. Commun., 2015, 51, 480–483 RSC; (f) P. Payongsri, D. Steadman, H. C. Hailes and P. A. Dalby, Enzyme Microb. Technol., 2015, 71, 45–52 CrossRef CAS PubMed.
- (a) J. H. Roe and C. S. Hudson, J. Biol. Chem., 1936, 112, 443–449 CAS; (b) E. Simon and P. F. Kraicer, Arch. Biochem. Biophys., 1957, 69, 592–601 CrossRef CAS PubMed; (c) H. G. Coore and P. J. Randle, Biochem. J., 1964, 91, 56–59 CrossRef CAS PubMed; (d) E. P. Paulsen, L. Richenderfer and P. Winick, Nature, 1967, 214, 276–277 CrossRef CAS PubMed.
- M. Board, A. Colquhoun and E. A. Newsholme, Cancer Res., 1995, 55, 3278–3285 CAS.
- (a) J. C. Sowden, J. Am. Chem. Soc., 1950, 72, 3325–3325 CrossRef CAS; (b) R. Schaffner and H. S. Isbell, J. Org. Chem., 1962, 27, 3268–3270 CrossRef; (c) W. Nelkin, Acta Physiol. Hung., 1970, 38, 401–404 CAS; (d) A. Kampf and E. Dimant, Carbohydr. Res., 1974, 32, 380–382 CrossRef CAS; (e) B. Bessieres and C. Morin, J. Org. Chem., 2003, 68, 4100–4103 CrossRef CAS PubMed; (f) J. Cheng, Z. Fang, S. Li, B. Zheng and Y. Jiang, Carbohydr. Res., 2009, 344, 2093–2095 CrossRef CAS PubMed.
- D. Waschke, J. Thimm and J. Thiem, Org. Lett., 2011, 14, 3628–3631 CrossRef PubMed.
- (a) M. Bayashi, K. Funane, H. Ueyama, S. Ohya, M. Tanaka and Y. Kato, Biosci., Biotechnol., Biochem., 1993, 57, 998–1000 CrossRef; (b) M. Spagnuolo, C. Crecchio, M. D. R. Pizzigallo and P. Ruggiero, Biotechnol. Bioeng., 1999, 64, 685–691 CrossRef CAS PubMed; (c) S. Kühnel, H. A. Schols and H. Gruppen, Biotechnol. Biofuels, 2011, 4, 14 CrossRef PubMed; (d) R. Wu, H. Zhang, X. Zeng, J. Zhang and H. Xiong, Afr. J. Biotechnol., 2011, 10, 1907–1912 CAS.
- (a) Y. Kobori, D. C. Myles and G. M. Whitesides, J. Org. Chem., 1992, 57, 5899–5907 CrossRef CAS; (b) L. Hecquet, J. Bolte and C. Demuynck, Tetrahedron, 1994, 50, 8677–8684 CrossRef CAS; (c) G. A. Sprenger, U. Schorken, G. Sprenger and H. Sahm, Eur. J. Biochem., 1995, 230, 525–532 CrossRef CAS PubMed; (d) G. A. Sprenger and M. Pohl, J. Mol. Catal. B: Enzym., 1999, 6, 145–159 CrossRef CAS; (e) F. T. Zimmermann, A. Schneider, U. Schörken, G. A. Sprenger and W.-D. Fessner, Tetrahedron: Asymmetry, 1999, 10, 1643–1646 CrossRef CAS; (f) L. Hecquet, C. Demuynck, G. Schneider and J. Bolte, J. Mol. Catal. B: Enzym., 2001, 11, 771–776 CrossRef CAS; (g) F. Charmantray, V. Hélaine, B. Legeret and L. Hecquet, J. Mol. Catal. B: Enzym., 2009, 57, 6–9 CrossRef CAS.
- J. J. Villafranca and B. Axelrod, J. Biol. Chem., 1971, 10, 3126–3131 Search PubMed.
- D. Yi, T. Devamani, J. Abdoul-Zabar, F. Charmantray, V. Helaine, L. Hecquet and W. D. Fessner, ChemBioChem, 2012, 13, 2290–2300 CrossRef CAS PubMed.
- A. Ranoux, S. K. Karmee, J. Jin, A. Bhaduri, A. Caiazzo, I. W. C. E. Arends and U. Hanefeld, ChemBioChem, 2012, 13, 1921–1931 CrossRef CAS PubMed.
- A. Ranoux, I. W. C. E. Arends and U. Hanefeld, Tetrahedron Lett., 2012, 53, 790–793 CrossRef CAS.
- M. Rudrum and D. F. Shaw, J. Chem. Soc., 1965, 52–57 RSC.
- (a) J. A. Littlechild, N. J. Turner, G. R. Hobbs, M. D. Lilly, A. Rawas and H. Watson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1995, 51, 1074–1076 CrossRef CAS PubMed; (b) U. Nilsson, L. Meshalkina, Y. Lindqvist and G. Schneider, J. Biol. Chem., 1997, 272, 1864–1869 CrossRef CAS PubMed; (c) L. Hecquet, C. Demuynck, G. Schneider and J. Bolte, J. Mol. Catal. B: Enzym., 2001, 11, 771–776 CrossRef CAS.
- P. Asztalos, C. Parthier, R. Golbik, M. Kleinschmidt, G. Hubner, M. S. Weiss, R. Friedemann, G. Wille and K. Tittmann, Biochemistry, 2007, 46, 12037–12052 CrossRef CAS PubMed.
- A. Sevestre, V. Hélaine, G. Guyot, C. Martin and L. Hecquet, Tetrahedron Lett., 2003, 44, 827–830 CrossRef CAS.
- M. E. B. Smith, U. Kaulmann, J. M. Ward and H. C. Hailes, Bioorg. Med. Chem., 2006, 14, 7062–7065 CrossRef CAS PubMed.
- T. Seliwanoff, Chem. Ber., 1887, 20, 181–182 CrossRef.
- W. D. Maclay, R. M. Hann and C. S. Hudson, J. Am. Chem. Soc., 1942, 64, 1606–1609 CrossRef CAS.
- K. G. Morris, M. E. B. Smith, N. J. Turner, M. D. Lilly, R. K. Mitra and J. M. Woodley, Tetrahedron: Asymmetry, 1996, 7, 2185–2188 CrossRef CAS.
- T. Okuda, S. Saito and M. Hayashi, Carbohydr. Res., 1979, 68, 1–13 CrossRef CAS.
- P. M. Medeirosa and B. R. T. Simoneit, J. Chromatogr., A, 2007, 1141, 271–278 CrossRef PubMed.
- E. Fiedler, S. Thorell, T. Sandalova, R. Golbik, S. Konig and G. Schneider, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 591–595 CrossRef CAS PubMed.
- D. Seeliger and B. de Groot, J. Comput. Aided Mol. Des., 2010, 24, 417–422 CrossRef CAS PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: GC-MS chromatograms, screening plates and NMR spectra. See DOI: 10.1039/c5gc02660a |
| This journal is © The Royal Society of Chemistry 2016 |