
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Using imidazolium-based ionic liquids as dual solvent-catalysts for sustainable synthesis of vitamin esters: inspiration from bio- and organo-catalysis†
Yifeng
Tao‡
a,
Ruijuan
Dong‡
ab,
Ioannis V.
Pavlidis
c,
Biqiang
Chen
*a and
Tianwei
Tan
*a
aNational Energy R&D Center for Biorefinery, Beijing Key Lab of Bioprocess, Beijing University of Chemical Technology, No. 15 North 3rd Ring Rd East, 100029 Beijing, PR China. E-mail: chenbq@mail.buct.edu.cn; twtan@mail.buct.edu.cn; Tel: +86-10-64416691
bSchool of Preclinical Medicine, Beijing University of Chinese Medicine, No. 11 North 3rd Ring Rd East, 100029 Beijing, PR China
cDept. of Biochemistry, University of Kassel, Heinrich-Plett-Str. 40, 34132 Kassel, Germany
First published on 9th December 2015
Abstract
Vitamin E (VE) has significant biological activities and thus its acylation to increase its stability is of extreme interest. We developed an efficient and sustainable approach using imidazolium-based ionic liquids as dual solvent-catalysts for the esterification between α-tocopherol (the most active form of VE) and succinic anhydride. Although in literature it is reported that lipase can catalyze this reaction, hereby we demonstrate that the reaction observed in DMSO and DMF is catalyzed by the histidyl residues of the protein. Histidine and its analogue containing an imidazole ring were tested as organocatalysts for the production of α-tocopherol succinate. In light of the imidazole organocatalysis, commercially-available 3-alkyl-1-methyl imidazolium ILs [CnC1Im][X−] were investigated as dual solvent-catalysts for the esterification of α-tocopherol with succinic anhydride, and provided satisfactory yields and reaction rates. [C5C1Im][NO3−] can be recycled by water extraction, instead of organic solvent extraction to separate α-tocopherol succinate from [C5C1Im][NO3−], with an average yield of 94.1% for 4 subsequent batches, while the catalytic activity of the recycled ILs showed almost no loss after 4 batches. The developed protocol for the synthesis of α-tocopherol esters and IL recycling bears industrial potential due to the ease of use and the efficient recycling.
Introduction
The development of sustainable synthesis of chemicals is strongly required, owing to the continuously rising environmental concerns of conventional chemical approaches. The use of either biocatalysts or ionic liquids (ILs) as catalysts is widely deemed to be a sustainable alternative way to achieve the concept of green chemistry.1 For the case of ester synthesis via direct esterification, several green approaches are available both in terms of catalysts and green solvents, such as lipase/esterase-mediated catalysis,2 and using functionalized ionic liquids as dual solvent-catalysts.1b,c Lipase/esterase-mediated esterification in non-aqueous systems possess many advantages compared to acid/base catalysis,2 such as the mild reaction conditions, high selectivity, creation of less waste, possibility of solvent-free system and reuse of biocatalyst via immobilization. On other hand, the obstacles of biocatalytic approaches, especially the unsatisfactory stability of enzymes for industrial applications, should be overcome.2aRoom-temperature ionic liquids become alternative solvents and catalysts since from an environmental perspective they offer many advantages including negligible vapor pressure, designable properties, possibly simplified separation of products and potential reuse. The majority of ILs reported for esterification are imidazole or pyridine based derivatives. One typical and widely used family of this kind of IL is Brønsted acidic ionic liquids (BAILs); SO3H-fuctionalized imidazolium-based ILs with acidic counter anion and protonated N-alkylimidazolium cation have been highlighted as dual solvent-catalysts with satisfactory conversion rates and selectivity for esterification,3 although the complicated preparation of BAILs may limit their industrial applications. The produced hydrophobic esters were immiscible with the hydrophilic ILs so that esters could be easily separated from ILs by decantation. However, considering the solubility of substrates in ILs, so far the synthesized esters were mainly the products of short/medium-chain alcohols with saturated aliphatic acid: ethyl acetate,3a butyl acetate,4 glycerol triacetate5 and methyl oleate.6 Compared with ILs, most of these substrates investigated for esterification were of much lower viscosity.7
Elegant works on esterification in ILs have been carried out because it was found that most ILs do not inactivate enzymes like polar organic solvents do.8 In such system, ILs mostly served as the solvents while the enzyme acted as the catalysts so that structural-complex esters could be synthesized. For instance, a clean lipase-catalyzed process for producing flavor esters by direct esterification in switchable ILs/solid phases was described with almost 100% yield and the enzyme activity was practically unchanged over seven consecutive operation cycles.9 However, to our knowledge, a comparative study between biocatalysis and procedures using ILs as catalysts has not been carried out so far.
Vitamin E (VE) is a major natural antioxidant and an essential component of biological membranes. The term VE covers a group of 8 isoforms: α-, β-, γ-, δ-tocopherol and α-, β-, γ-, δ-tocotrienol. Among all these isoforms, α-tocopherol shows the highest VE activity.10 However, VE is unstable and its antioxidant value is reduced by light, air or oxidizing chemicals. Aiming to improve the stability of VE, acetylation of VE was suggested.11 The stable derivatives of VE, such as VE succinate, have the same biological activity as VE and have been highlighted as high-selective anticancer agents,10,12 efficiently inducing the apoptosis of cancer cells via the mitochondrial route.13 The hydrophobicity of VE due to the long alkyl chain leads to the immiscibility of VE in most hydrophilic BAILs, resulting in low or even no conversion of VE esterification in such ILs. The esterification of vitamins is a topic that draws significant attention in the hydrolase community, due to the ability of lipases to catalyze the acylation in non-conventional media. Lipase-catalyzed synthesis of VE succinate in molecular solvents is significantly affected by the solvent due to the opposite polarity between VE and succinic anhydride: high yields were obtained in aprotic polar solvents like dimethyl sulfoxide (DMSO) and N,N-dimethyl formamide (DMF), but almost no reaction occurred in conventional organic solvents.11,14 Until now, the catalytic activity in DMSO and DMF was believed to be due to the contribution of the lipase, while any possible chemical acylation reaction was neglected and never reported in these studies, although DMSO and DMF are believed to possess high denaturation capacity to the enzyme in most literature.15
Herein, we firstly demonstrated whether the reaction observed in DMSO and DMF is contributed by the catalytic activity of lipase or not. We found that the undergoing cause for such reactions in DMSO is the chemical catalysis of the histidyl residue in the protein, so that histidine and its analogue containing an imidazole ring were tested as organocatalysts for the production of α-tocopherol succinate. Bearing in mind green chemistry, an efficient and sustainable approach using commercially-available 3-alkyl-1-methyl-imidazolium ionic liquids as dual solvent-catalysts was investigated. To enhance the green impact of the IL catalyzed reaction, we developed a water extraction protocol, instead of an organic solvent extraction method, so that the ILs could be reused. The reaction efficiency, final yield and product separation of these strategies (Scheme 1) were compared and the distinct mechanisms of histidine and imidazolium-based ILs catalysis were proposed.
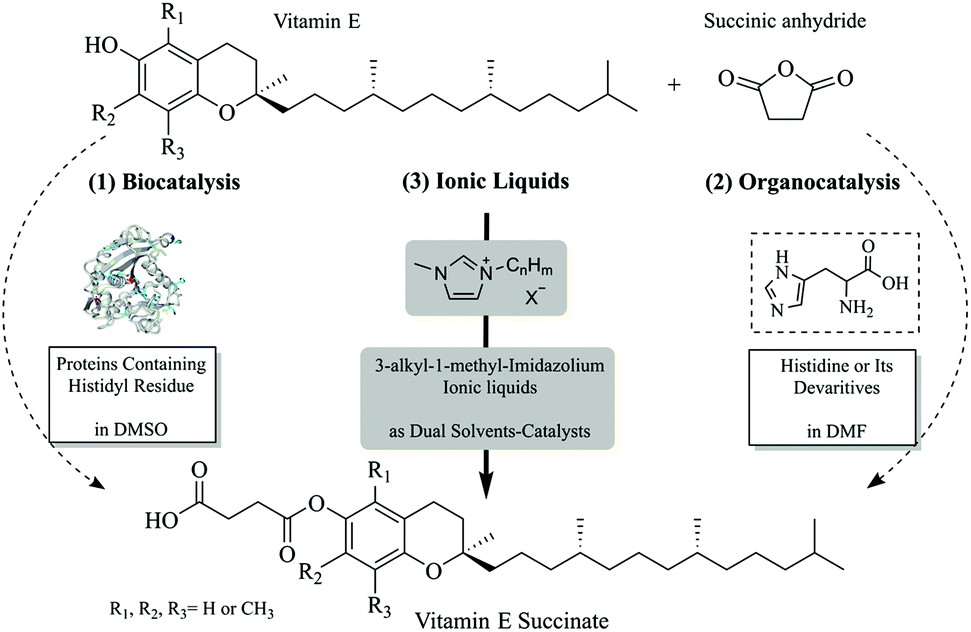 | ||
| Scheme 1 Three strategies for the esterification of vitamin E with succinic anhydride: (1) biocatalysis, (2) organocatalysis and (3) using ionic liquids as dual solvent-catalysts. | ||
Results and discussion
Plausible lipase-mediated synthesis of vitamin E succinate in DMSO
Over the past three decades, a remarkable number of works and publications concern lipase/esterase-mediated catalysis of transesterification or direct esterification have been reported since the essential paper16 of Zaks and Klibanov on enzymatic processes in organic solvents. We initially studied the lipase-mediated esterification of α-tocopherol with fatty acids, aiming to improve α-tocopherol stability. For the esterification of α-tocopherol with succinic anhydride, solvents have profound effects on the reaction possibility and rates,17 due to the different polarity of these two substrates. When screening organic solvents, the reactions were highly solvent-dependent (ESI, Table S1†), as described in literature.14c Commonly applied organic solvents such as hexane, tert-amyl alcohol, tert-butanol and acetonitrile are not good media for these reactions whereas the reactions were efficiently performed in DMSO and DMF.Over the last few years growing efforts on the improvement of the enzymatic processes in anhydrous DMSO and DMF via protein engineering, immobilization and computational chemistry were documented. It was reported that the lipases’ activities in neat DMSO could be maintained and even promoted through lipase modification14a,18 or immobilization.14b,19 However, DMSO and DMF, which are known as universal solvents with high denaturation capacity,15a not only strip essential water molecules from the enzyme, but also dissociate the enzyme tertiary structure leading to enzyme unfolding and subsequent deactivation.15b,c This conventional wisdom still holds owing to our unexpected results: several heat-inactivated (120 °C, 20 min) lipases from different sources can perform the esterification of α-tocopherol with succinic anhydride in neat DMSO, as shown in Fig. 1. More interestingly, heat inactivated proteins and enzymes of other classes, like glucose oxidase and catalase can perform the reaction (Table 1). Surprisingly, some of the heat-denatured samples exhibited higher activity than the native enzymes. It is expected that the unfolding, as a result of the heating process, renders the histidine residues of the protein core accessible, while in the folded protein only the histidine residues on the surface of enzyme were available for the catalysis.
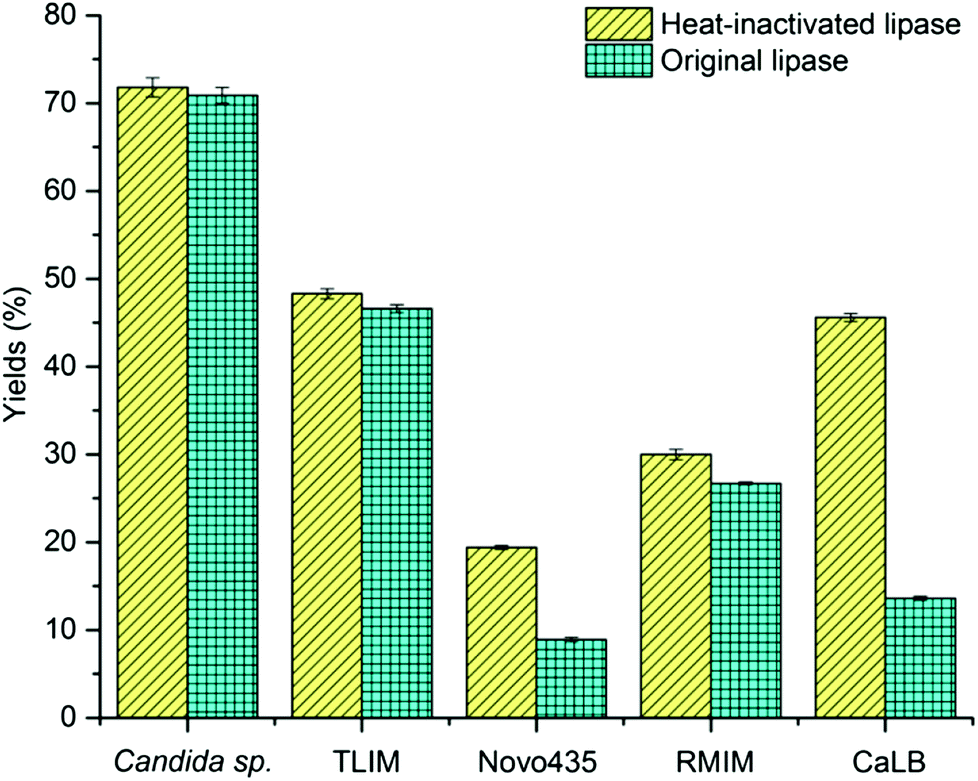 | ||
| Fig. 1 Comparison of heat-inactivated (120 °C, 20 min) and original lipase catalyzing the esterification of α-tocopherol succinate in DMSO. TLIM: lipozyme TLIM; RMIM: lipozyme RMIM; CaLB: Candida antarctica lipase B. | ||
Subsequently, we investigated the amino acid sequences and found that the activity of the heat inactivated protein was in line with the histidine content (Table 1); no reaction was observed for protamine, a protein without histidine residues. At the same time, enzymes that are not reported to catalyze esterification reactions, such as glucose oxidase, where quite efficient on this specific reaction, due to their histidine content. In summary, these results indicated that the underlying cause for esterification of α-tocopherol esters in DMSO is not a lipase-catalyzed reaction, but organocatalysis.
Organosynthesis of vitamin E succinate
Enzymatic transformations generally have a chemical catalysis counterpart, but amino acids as the building blocks composing the protein rarely fulfill their chemical counterpart roles. Typically, amino acids exhibit their catalytic potency after proper folding of the polypeptide chain, mostly due to the specific environment that the neighboring residues are producing. For instance, hydrolases perform their activities via a catalytic triad usually formed by serine, histidine and aspartate/glutamate residues.20 The results of the previous paragraph gave an insight that the histidine can be an effective esterification catalyst for the synthesis of vitamin esters.Reacting L-histidine (30%, mol towards α-tocopherol) in 1 mL DMF containing 0.2 M α-tocopherol and 0.8 M succinic anhydride at 50 °C for 24 h led to 40% yield of α-tocopherol succinate (Table 2, entry 8). This result was quite remarkable since histidine is slightly soluble in neat DMF (up to about 0.06 M histidine). The efficiency of histidine-catalyzed esterification in aprotic polar solvents followed the order: DMSO > DMF > dimethylacetamide > tetrahydro-1,3-dimethyl-2(1H)pyrimidine > 1,3-dimethyl-2-imidazolidinone. It needs to be stated that the medium of histidine-mediated esterification gradually became homogeneous in anhydrous DMSO, indicating a ring-open reaction of succinic anhydride with histidine. This was not observed in DMF, thus this solvent is expected to be more suitable regarding the efficiency while minimizing the ring opening of the anhydride.
| Entry | Compound | Initial ratea (10−2 min−1) | Yield after 24 hb (%) |
|---|---|---|---|
| a Initial rate refers to moles of product formed per moles of catalyst per minute. b Conditions: histidine derivatives (30%, mol of α-tocopherol) in 1 mL DMF containing 0.2 M α-tocopherol and 0.8 M succinic anhydride were incubated at 800 rpm and 50 °C in the dark and under nitrogen atmosphere for 24 h and the yield was determined through HPLC analysis, using an external standard. c Yield at 9 h. d These catalysts are slightly soluble in DMF. e Not detectable. | |||
| 1 |
|
10.8 ± 0.2 | 98.1 ± 1.3c |
| 2 |
|
4.4 ± 0.1 | 97.2 ± 1.5c |
| 3 |
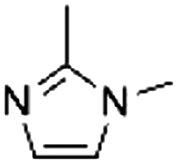
|
3.1 ± 0.2 | 91.7 ± 1.9c |
| 4 |
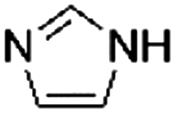
|
1.1 ± 0.1 | 89.1 ± 0.6 |
| 5 |
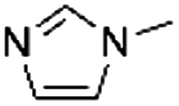
|
0.73 ± 0.06 | 86.9 ± 0.8 |
6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) d d |
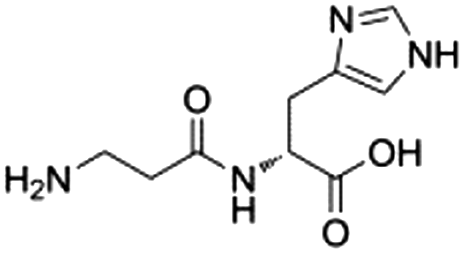
|
0.20 ± 0.02 | 45.7 ± 0.6 |
| 7 |
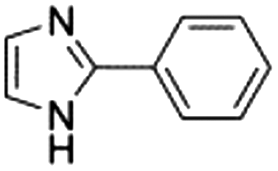
|
0.25 ± 0.03 | 45.3 ± 0.4 |
|
8d |
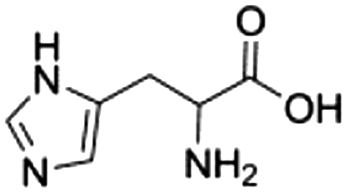
|
0.17 ± 0.01 | 39.8 ± 0.4 |
|
9d |
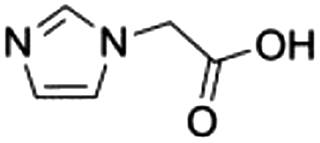
|
0.11 ± 0.01 | 28.5 ± 0.3 |
| 10 |
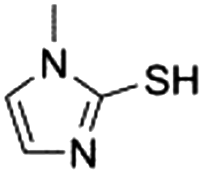
|
n.d.e | n.d.e |
| 11 | Xanthine; adenine, thymine, uracil, guanine | n.d.e | n.d.e |
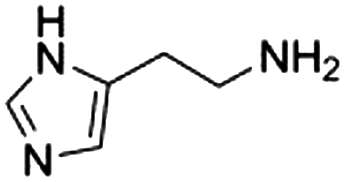
We also compared commercially available histidine analogues and derivatives under standard conditions with 30% mol (towards α-tocopherol) of catalyst (Table 2). Histamine (entry 1) exhibited high initial rate and yield; this could be due to the better solubility in DMF, owing to the absence of the carboxylic group. Although the final product (α-tocopherol succinate) was obtained and confirmed by NMR, we believed that exposure of histamine to succinic anhydride in DMF will form the terminal N-succinoyl derivative as an intermediate. It was reported that succinoylhistamine was formed with a yield of 90% at room temperature within 1.5 h in DMF and a white precipitate formed.21 However, in our cases, no white precipitate was observed at 50 °C, probably due to the further usage of the succinic moiety for the acylation. The possibly formed N-succinoyl derivative posed the question: what is the actual acylation catalyst? We assumed that both the imidazole ring and the amine group are essential for efficient catalysis to occur.
Modification of side chains of the imidazole ring (entries 1–5 and 7–10) resulted in the change of catalyst solubility in the organic solvents, but also in its reactivity: the presence of a carboxylic group led to the decrease in solubility (entries 6, 8 and 9), while the incorporation of a thiol group diminished any activity. Interestingly, neither purines nor pyrimidines (entry 11) exhibited any catalysis, although they possess a similar structure to the imidazole ring. Different kinds of anhydrides and fatty acids were tested as the acyl donor: only anhydrides could be activated in DMSO or DMF and thus act as the acyl donor, whereas none of fatty acids could (data not shown). Reactions of vitamin A and vitamin C with succinic anhydride (Scheme S1†) were further carried out to extend the applications of histidine-catalyzed esterification. Although that histidine was able to perform the esterification of both vitamins, no regioselectivity was observed in the case of vitamin C (Fig. S1†).
Although the use of histidine derivatives/analogues as catalysts for these acylation reactions brings several benefits related to their simplicity, low price and availability, the purification of the produced ester for organocatlysis is really difficult via an organic solvent extraction with DMSO or DMF. Without immobilization of the catalysts, the recycling of the chemical catalysts is not easy.
Imidazolium-based ionic liquids as dual solvent-catalysts for the synthesis of vitamin E succinate
The application of ILs as both solvent and catalysts for the esterification of α-tocopherol with succinic anhydride is an alternative sustainable approach since enzymatic process are not available for these substrates as discussed before. Considering the important IL properties, e.g. dissolving ability, viscosity, etc. for the potential for efficient α-tocopherol succinate synthesis, an appropriate choice of the anion and the cation of the ILs is necessary. BAILs containing an alkane-sulfonic acid group attached to an imidazole or pyridinium cation and bearing acid counteranions are of special importance as the most used ILs for the esterification process because they possess simultaneously the proton acidity and the characteristic properties of an ionic liquid.3a,22However, in light of organocatalysis using imidazole as catalyst, imidazolium based ILs without any modifications as shown in Table 3 were selected in our work to avoid the complicated preparation of functionalized BAILs. This simple IL family was previously reported mostly as the solvents for enzymatic catalysis and already commercially available.1b,23 The high viscosity of ILs limiting the mass transfer rate would be the first challenge to the use of the ILs during the synthesis of α-tocopherol succinate; the resulting ester with a free carboxylic group due to the use of anhydride may become miscible with ILs, which would increase the difficulty of the product separation and ILs recycling. Herein, we screened several imidazolium based ILs and developed an easy way to reuse the ILs via extraction using water.
| Entry | ILs | Name | Initial ratea (10−2 min−1) | Yield after 1.5 hb (%) |
|---|---|---|---|---|
| a Initial rate refers to moles of product formatted per moles of catalyst per minute. b Conditions: 4 mM ILs, 0.5 mM α-tocopherol and 1 mM succinic anhydride were incubated at 800 rpm and 50 °C in the dark and under nitrogen atmosphere for 3 h and the yield was determined through HPLC analysis, using an external standard. c Not detectable. | ||||
| 12 |
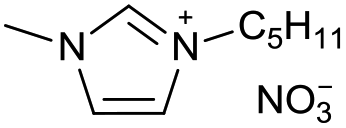
|
[C5C1Im][NO3−] | 0.69 ± 0.12 | 96.2 ± 1.6 |
| 13 |
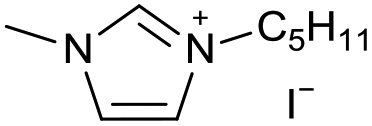
|
[C5C1Im][I−] | 0.56 ± 0.15 | 89.8 ± 2.1 |
| 14 |
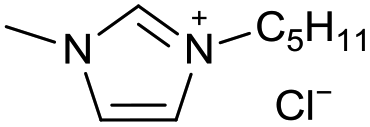
|
[C5C1Im][Cl−] | 0.17 ± 0.03 | 44.7 ± 1.6 |
| 15 |
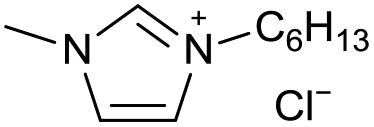
|
[C6C1Im][Cl−] | 0.33 ± 0.06 | 50.8 ± 1.5 |
| 16 |
|
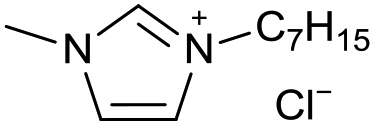
[C7C1Im][Cl−] | 0.62 ± 0.05 | 57.7 ± 2.1 |
| 17 |
|
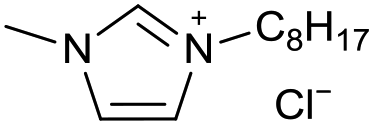
[C8C1Im][Cl−] | 0.05 ± 0.01 | 33.8 ± 0.9 |
| 18 |
|
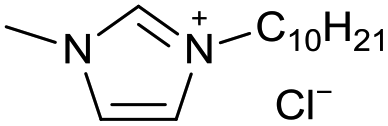
[C10C1Im][Cl−] | 0.20 ± 0.01 | 54.6 ± 1.2 |
| 19 |
|
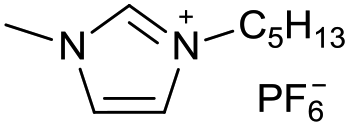
[C5C1Im][PF6−] | 0.10 ± 0.01 | 28.9 ± 0.9 |
| 20 |
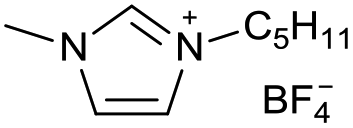
|
[C5C1Im][BF4−] | n.d.c | n.d.c |
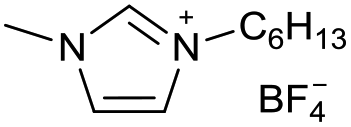
|
[C5C1Im][BF4−] | |||
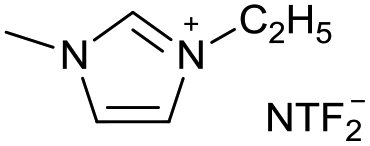
|
[C2C1Im][NTF2−] | |||
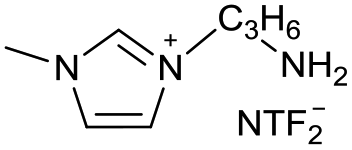
|
[C3NC1Im][NTF2−] | |||
Initial rates and yields of the selected imidazolium based ILs for the synthesis of α-tocopherol succinate is shown in Table 3. Investigation of ILs anions (entries 12–14, 19 and 20) on the efficiency of α-tocopherol esterification showed that the most effective nucleophilic anion was NO3− among all the studied anions although these anions may slightly change the ILs viscosity. α-Tocopherol was found to be immiscible with the hydrophilic ILs containing BF4− and NTF2−, resulting in no reactivity. It is worth pointing out that the initial rate depends on the substrates’ concentration: when the millimole ratio of α-tocopherol:succinic anhydride:ILs was 4:8:4, the initial rate when using [C5C1Im][NO3−] reached 2.86 × 10−2 min−1 (see ESI Table S1, entry S5†), which is comparable with the values of organocatalysts. However, with respect to the productivity, the approach using ILs as dual solvent-catalysts showed a higher efficiency than both biocatalysis and organocatalysis.
The change of substituted alkyl group on the imidazole ring of the cation mainly resulted in the change in IL viscosity: a previous study reported that an increase in the van der Waals forces primarily contributed to the increase in viscosity of the ILs.24 In agreement with this statement, in the 3-alkyl-1-methylimidazolium [PF6−] series, viscosity increases as the number of carbon atoms in the linear alkyl group is increased.25 However, in our work, the 3-alkyl-1-methylimidazolium [Cl−] series showed a more complicated behavior, resulting in the disordered reactivity: there was no linear relationship between reactivity and the substituted number of carbon atoms, while [C7C1Im][Cl−] showed the best reactivity for α-tocopherol esterification.
The reuse of ILs is necessary due to its relatively high cost and so far several ILs have been demonstrated with feasible recyclability merely via decantation of esters3c,5,26 because the hydrophobic esters were mostly immiscible with the used ILs. When using [C5C1Im][NO3−] as a dual solvent-catalyst for α-tocopherol esterification in the first batch, the reaction mixture tended to spontaneously separate into two layers, and the recycled yield of ILs was around 72.5 ± 1.8%; unfortunately and surprisingly, in the second batch, a homogenous mixture was observed even after centrifugation under 10000g for 30 min. 1-Alkoxymethylimidazolium lactates were synthesized and lactate could serve as the anion.27 Herein, we assumed that the resulted free carboxylic group of α-tocopherol succinate owing to the use of succinic anhydride may act as an anion to coordinate with the imidazolium cations. Consequently, this ionic liquid became miscible with the reactants.
Hence, extraction of products by solvents became an alternative choice. Firstly, we used organic solvents, like 10 volumes (versus total volume of reactant and ILs) ethyl acetate or diethyl ether to extract VE and α-tocopherol succinate from the IL, while succinic anhydride was firstly removed by centrifugation because it became insoluble in ILs with the decrease of temperature. Although a high isolated yield with 92.1 ± 1.2% of α-tocopherol succinate could be obtained, the extraction by organic solvent is not compatible to the concept of green chemistry. Water as a solvent is strongly environmentally favorable with respect to safety, cost and sustainability. To our delight, [C5C1Im][NO3−] is totally miscible with water while the other reactants are not. After the removal of succinic anhydride by centrifugation, the addition of a volume of water 2–3 times greater into the reaction mixture recycled almost 95.2 ± 2.1% of the ionic liquid after removing the water via evaporation under 80 °C and reduced pressure (around 100 Pa).
After the removal of water, the reusability of the [C5C1Im][NO3−] ionic liquid was investigated. As summarized in Table 4, about 94 ± 2% is the recovery of the IL after all batches were tested. The initial rate of α-tocopherol conversion into α-tocopherol succinate for 4 batches was similar, but the yield was slightly decreased due to the loss of ionic liquid in each batch. Meanwhile, the recovered [C5C1Im][NO3−] ionic liquid showed no structural difference to the original one by comparison of the 1H and 13C NMR spectra in DMSO-d6 (ESI, Fig. S4†). Thus, the developed protocol for the synthesis of α-tocopherol esters and IL recycling is practicable and applicable.
| Runs | Initial rate (10−2 min−1) | Yield after 1.5 h (%) | Recycled yield of ionic liquid (%) |
|---|---|---|---|
| 1 | 0.69 ± 0.12 | 96.2 ± 1.6 | 95.2 ± 2.1 |
| 2 | 0.67 ± 0.08 | 95.8 ± 1.2 | 96.3 ± 1.8 |
| 3 | 0.66 ± 0.09 | 93.8 ± 2.3 | 92.8 ± 1.5 |
| 4 | 0.66 ± 0.05 | 91.5 ± 1.0 | 93.5 ± 0.8 |
In order to highlight the applicability of the developed process, we investigated the synthesis of several vitamin esters with different acyl donors in [C5C1Im][NO3−]. Vitamin A, C and E could be easily modified by esterification with several acyl donors; however, it is clear that the formation as an anhydride is a pre-requirement for the success of the transformation (Table S3†). However, it needs to be mentioned that in the case of vitamin C, where multiple hydroxyl groups are available, no regioselectivity was observed.
Proposed catalytic mechanism
Lipase-mediated esterification occurs via the formation of a tetrahedral acyl-enzyme intermediate, which undergoes a nucleophilic attack from the second substrate, leading to the product formation and release of the product, regenerating the free enzyme.28 In the catalytic triad of lipases, the basic nitrogen of histidine abstracts a proton from serine to activate it as a nucleophile. As discussed before, the approach using lipase as catalyst for the esterification of vitamin E with succinic anhydride was not contributed to by the lipase but the imidazole ring of the histidyl residue in proteins. Thus, here we discuss the mechanism based on imidazole catalysis.Imidazole is known as an ester hydrolysis catalyst.29 Imidazole carbamates and ureas acting as catalysts and substrates mediated the chemoselective esterification and amidation of carboxylic acids in acetonitrile.30 Peptides or polymers containing histidine residues were reported to exhibit hydrolytic activity on p-nitrophenyl esters31 and in Michael additions,32 aldol reactions,33 oligomerisation,34etc. However, the proposed mechanisms for these catalysts were remarkably different from the one for the lipase-catalyzed reaction. Imidazole carbamate catalyzed esterification was attributed to a mechanism involving an activated ester intermediate.30b
Being cognizant of the earlier reported works,30b,31a,35 we currently propose this histidine-mediated esterification via acyl-imidazole intermediate (Scheme 2). The imidazole moiety is expected to be partly deprotonated in DMSO or DMF since the pKa of imidazole in DMSO is 18.6.36 As a result of the participation of the electron pair of the amide nitrogen in the π-electron system of the ring, tautomeric forms in five-membered N-heterocycles occur,37 leading to the possibility that nucleophilic attack can be alternatively accomplished by one of the electronegative nitrogen atoms in the imidazole ring. The first step of the substitution pathway, involving nucleophilic attack by the imidazole ring on the anhydride carbonyl and the leaving of the other carbonyl group, forms the acyl-imidazole intermediate. The subsequent transition begins with an SN2 nucleophilic attack on the carbonyl by the hydroxyl group of α-tocopherol and ends with the formation of a final product through expelling the catalyst. The amine group of histamine may be properly coordinating the acyl-imidazole intermediate, resulting in a significant increase in reaction rate.
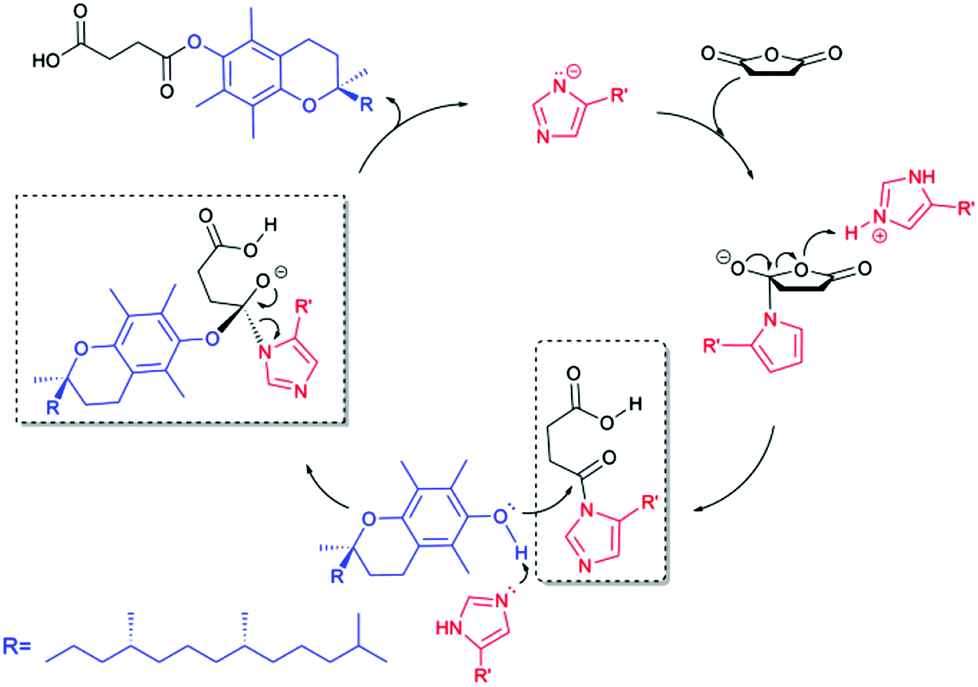 | ||
| Scheme 2 Proposed acyl-imidazole mechanism of the histidine-catalyzed esterification of vitamin E with succinic anhydride. | ||
Understanding the mechanism of catalysis by ILs at the molecular level is crucial for the rational design of ILs due to the impossibly experimental study of all ILs even a small fraction of the potential cations–anions combinations,38 but the mechanism of nucleophilic substitution by ILs seems more complicated. It was recognized that the C2 of the 1,3-alkyl-imidazolium anions is positively charged due to the electron deficit in the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond whereas the other carbons are practically neutral.38,39 This resulting acidity of the C2 hydrogen atom is the key to understanding the mechanism of IL catalysis.
N bond whereas the other carbons are practically neutral.38,39 This resulting acidity of the C2 hydrogen atom is the key to understanding the mechanism of IL catalysis.
As shown in Scheme 3, the imidazolium ILs initiate the esterification by donating the C2 proton to the oxygen atom of the anhydride, inducing the electrophilic activation of anhydride carbonyl. The subsequent nucleophilic attack on the carbonyl following a SN1 mechanism by the alcohol was proposed for the IL catalyzed synthesis of biodiesel40 or lipophilic esters41 in previous literatures, in which the nucleophilic role of anions was neglected. Chakraborti et al.42 proposed an “electrophile nucleophile dual activation” role of the [C4C1Im][CH3COO−] in catalyzing O-tert-butoxycarbonylation of 2-naphthol. They concluded that counteranions were also involved in the cooperative hydrogen bonds and charge–charge interactions with both substrates. Welton et al.43 proposed that the hydrogen bond basicity of the ILs, controlled by the anions, was the dominant factor in determining the esterification rate. In view of these crucial issues and considering the weak nucleophile attack ability of phenol hydroxyl group of α-tocopherol due to the shielding effect of two neighbor methyl groups, we promote that the anions in ILs perform the nucleophilic attack on the anhydride carbonyl in the first step, forming an anion-anhydride intermediate after the leaving of the other carbonyl group, and then the second step following an SN2 mechanism starts with a nucleophilic attack on the carbonyl by the hydroxyl group of α-tocopherol and ends with the formation of the final product through expelling the anion.
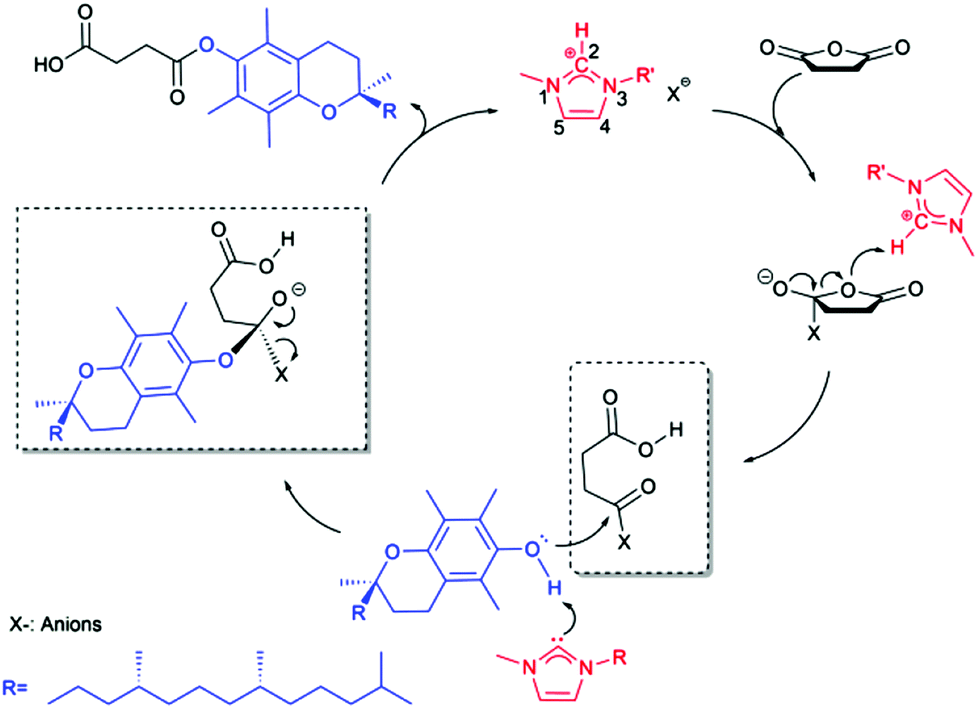 | ||
| Scheme 3 Proposed mechanism for esterification of α-tocopherol with succinic anhydride using 3-alkyl-1-methylimidazolium ionic liquids as dual solvent-catalysts. | ||
Experimental
General aspects
Vitamin E containing 96% α-tocopherol and succinic anhydride with a purity of 99% were obtained from Aladdin Industrial Inc. (Shanghai, China). Lipase formulation from Candida sp. was produced by Beijing CTA New Century Biotechnology Co., Ltd. Lipases including Lipozyme TLIM, Lipozyme RMIM, Candida antarctica lipase B were purchased from Novozymes®. The other proteins were commercially available. All 3-alkyl-1-methyl-imidazolium ionic liquids with a purity of 99% were purchased from the Centre for Green Chemistry and Catalysis, Lanzhou Institute of Chemical Physics, Chinese Academy of Sciences (Lanzhou, China). Methanol of chromatographic grade was purchased from Sigma-Aldrich. The other solvents and salts of analytical grade were obtained from Beijing Chemical Factory.Esterification reactions
For biocatalysis, 40 mg protein was added to 1 mL DMSO containing 0.2 M α-tocopherol and 0.8 M succinic anhydride at 50 °C for 24 h; for organocatalysis, reacting L-histidine or its analogues (30%, mol towards α-tocopherol) in 1 mL DMF containing 0.2 M α-tocopherol and 0.8 M succinic anhydride was carried out at 50 °C for 24 h; for experiments using ILs as dual solvent-catalysts, a standard mixture containing 0.5 mmol α-tocopherol, 1 mmol succinic anhydride and 4 mM ionic liquid was used and the reactions were carried out at 50 °C for 3 h. All reactions were investigated in the dark and under nitrogen atmosphere. Samples were obtained at scheduled times and yields were determined through HPLC analysis (Thermo Scientific Dionex Ultimate 3000 equipped with an AcclaimTM C18 column), using an external standard. The initial rate refers to moles of product formatted per moles of catalyst per minute when the yield was under 20%. α-Tocopherol succinate yield was calculated from the ratio between the observed and theoretical production of α-tocopherol succinate.Recyclability of ionic liquids
The usability of biocatalysts and organocatalysts were not investigated due to the difficulty of catalyst recycling. Considering the relative high cost of the ILs, the recyclability study of the ILs is important and necessary; however, decantation of products for recycling ILs was not available for our selected ILs. To our delight, [C5C1Im][NO3−] was totally miscible with water while the reactants were not. After the removal of succinic anhydride by centrifugation at room temperature, the reaction mixture was strongly mixed after the addition of 2–3 times volume deionized water. After removing water via evaporation under 80 °C and reduced pressure (around 100 Pa), the reusability of the [C5C1Im][NO3−] ionic liquid was investigated under the standard procedure.Purification
Purification of vitamin E succinate for biocatalysis and organocatalysis approaches proceeded as follows: (a) 1 mL of the reaction medium was added to 2 mL diethyl ether and incubated overnight under −18 °C; (b) after centrifugation at 8000g for 2 min, the organic phase was removed and added to 1.5 mL deionized water, and then mixed by vortex for 1 min; (c) after centrifugation at 8000g for 2 min, the organic phase was carefully transferred into a new tube and then dried with Na2SO4; (d) after the removal of solvent in a reduced-pressure rotary evaporator, 400 μL n-hexane was added to dissolve the unreacted α-tocopherol before further purification of the final isolated ester because α-tocopherol succinate is not soluble in n-hexane; (e) centrifuging at 8000g for 2 min again, the liquid was discarded and after being dried by nitrogen, a white powder was obtained for further characterization.For the approach using ILs as dual solvent-catalysts, a white solid was obtained after the removal of anhydride and the recycling of the ILs. 500 μL n-hexane was added to dissolve the unreacted α-tocopherol, further improving the purity of the final ester.
Conclusions
A comparative study between biocatalysis, organocatalysis and using ILs as dual solvent-catalysts for the direct esterification of vitamin E with succinic anhydride was carried out. Each approach possesses its own advantages and disadvantages. It is demonstrated that lipase-catalyzed esterification in DMSO or DMF was not attributed to the catalytic activity of lipase, but the chemical catalysis of the histidyl residue in protein. Although the use of histidine derivatives/analogues as catalysts for these acylation reactions brings several benefits in terms of their simplicity, low price and availability, the purification of the produced ester by organocatalysis is really difficult via an extraction with organic solvent from DMSO or DMF. The recycling of chemical catalysts is not easy but it can become even more appealing by immobilizing the catalyst to be able to reuse it, or even use cheap sources of histidine, such as commercial soymeal.Bearing green chemistry in mind and in light of the imidazole organocatalysis, we developed an efficient and sustainable strategy using 3-alkyl-1-methylimidazolium ILs as dual solvent-catalysts for the synthesis of α-tocopherol succinate, with satisfactory yields and reaction rates. The [C5C1Im][NO3−] ionic liquid can be recycled by water extraction with an average regeneration yield of 94.1%. The initial rate of α-tocopherol conversion into α-tocopherol succinate for 4 batches was almost similar but the yield was slightly decreased due to the small amount of loss of ionic liquid in each batch, indicating that the catalytic activity of the recycled ILs was unaffected. The developed protocol for synthesis of VE esters and IL recycling is practicable and applicable.
Although the imidazole ring is one of the main structural motifs in histidine and cations of imidazolium ILs, the catalytic mechanism significantly differs from histidine-catalyzed to imidazolium IL-catalyzed esterification. We proposed an acyl-imidazole intermediate for histidine catalyzed esterification, whereas for ILs catalyzed esterification, the role of anions of imidazolium ILs was promoted as performing the nucleophilic attack while the esterification was initiated by donating the proton of C2 on imidazole ring to the oxygen atom of anhydride.
Moreover, we have highlighted that the lipase-catalyzed esterification reactions taking place in aprotic polar solvents should be treated with caution, as the histidines of the protein (not necessarily the one of the active site) could perform the reaction as chemical catalysts. Thus, we would suggest that in such works, the imidazole concentration after purification of lipases via His-Tag should be titrated, in order to be able to exclude the potential of chemical catalysis via the imidazole used for the elution of the protein.
Acknowledgements
The authors acknowledge financial support of the National Basic Research Program of China (973 program: 2013CB733600), and of the National Natural Science Foundation of China (21436002, 21576019). The authors especially thank Prof. Dr Uwe T. Bornscheuer (Dept. of Biotechnology & Catalysis, Institute of Biochemistry, Greifswald, Germany) for valuable comments and fruitful discussions on this work.References
- (a) S. Wenda, S. Illner, A. Mell and U. Kragl, Green Chem., 2011, 13, 3007–3047 RSC; (b) H. Olivier-Bourbigou, L. Magna and D. Morvan, Appl. Catal., A, 2010, 373, 1–56 CrossRef CAS; (c) Q. Zhang, S. Zhang and Y. Deng, Green Chem., 2011, 13, 2619–2637 RSC.
- (a) M. T. Reetz, J. Am. Chem. Soc., 2013, 135, 12480–12496 CrossRef CAS PubMed; (b) A. I. Brígida, P. F. Amaral, M. A. Coelho and L. R. Gonçalves, J. Mol. Catal. B: Enzym., 2014, 101, 148–158 CrossRef.
- (a) A. C. Cole, J. L. Jensen, I. Ntai, K. L. T. Tran, K. J. Weaver, D. C. Forbes and J. H. Davis, J. Am. Chem. Soc., 2002, 124, 5962–5963 CrossRef CAS PubMed; (b) T. Joseph, S. Sahoo and S. Halligudi, J. Mol. Catal. A: Chem., 2005, 234, 107–110 CrossRef CAS; (c) X. Han, H. Du, C. Hung, L. Liu, P. Wu, D. Ren, S. Huang and S. Liu, Green Chem., 2014, 17, 499–508 RSC.
- H. Zhu, F. Yang, J. Tang and M. He, Green Chem., 2003, 1, 38–39 RSC.
- X. Liu, H. Ma, Y. Wu, C. Wang, M. Yang, P. Yan and U. Welz-Biermann, Green Chem., 2011, 13, 697–701 RSC.
- A. H. M. Fauzi and N. A. S. Amin, Energy Convers. Manage., 2013, 76, 818–827 CrossRef.
- (a) S. Zhang, N. Sun, X. He, X. Lu and X. Zhang, J. Phys. Chem. Ref. Data, 2006, 35, 1475–1517 CrossRef CAS; (b) G. Yu, D. Zhao, L. Wen, S. Yang and X. Chen, AIChE J., 2012, 58, 2885–2899 CrossRef CAS.
- (a) S. Park and R. J. Kazlauskas, Curr. Opin. Biotechnol., 2003, 14, 432–437 CrossRef CAS PubMed; (b) C. Aouf, E. Durand, J. Lecomte, M. C. Figueroa-Espinoza, E. Dubreucq, H. Fulcrand and P. Villeneuve, Green Chem., 2014, 16, 1740–1754 RSC.
- (a) P. Lozano, J. M. Bernal and A. Navarro, Green Chem., 2012, 11, 3026–3033 RSC; (b) P. Lozano, J. M. Bernal, E. Garcia-Verdugo, G. Sanchez-Gomez, M. Vaultier, M. I. Burguete and S. V. Luis, Green Chem., 2015, 17, 3706–3717 RSC.
- K. N. Prasad, B. Kumar, X.-D. Yan, A. J. Hanson and W. C. Cole, J. Am. Coll. Nutr., 2003, 22, 108–117 CrossRef CAS PubMed.
- P. Torres and D. Reyes, Process Biochem., 2008, 43, 145–153 CrossRef CAS.
- (a) Y. Zhang, J. Ni, E. M. Messing, E. Chang, C.-R. Yang and S. Yeh, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 7408–7413 CrossRef CAS PubMed; (b) N. Duhem, F. Danhier and V. Préat, J. Controlled Release, 2014, 182, 33–44 CrossRef CAS PubMed.
- (a) J. Truksa, L. Dong, J. Rohlena, J. Stursa, M. Vondrusova, J. Goodwin, M. Nguyen, K. Kluckova, Z. Rychtarcikova and S. Lettlova, Antioxid. Redox Signaling, 2015, 22, 883–900 CrossRef CAS PubMed; (b) B. Yan, M. Stantic, R. Zobalova, A. Bezawork-Galeta, M. Stapelberg, J. Stursa, K. Prokopova, L. Dong and J. Neuzil, BMC Cancer, 2015, 15, 401–412 CrossRef PubMed.
- (a) C. Yin, Z. Cong and M. Gao, Chin. J. Chem. Eng., 2011, 19, 135–139 CrossRef CAS; (b) Y. Hu, X. Jiang, S. Wu, L. Jiang and H. Huang, Chin. J. Catal., 2013, 34, 1608–1616 CrossRef CAS; (c) X. Jiang, H. Yi, L. Jiang, J. Gong and H. Huang, Chem. Res. Chin. Univ., 2013, 29, 223–226 CrossRef CAS.
- (a) Y. L. Khmelnitsky, V. V. Mozhaev, A. B. Belova, M. V. Sergeeva and K. Martinek, Eur. J. Biochem., 1991, 198, 31–41 CrossRef CAS PubMed; (b) U. R. Desai and A. M. Klibanov, J. Am. Chem. Soc., 1995, 117, 3940–3945 CrossRef CAS; (c) P. P. Wangikar, P. C. Michels, D. S. Clark and J. S. Dordick, J. Am. Chem. Soc., 1997, 119, 70–76 CrossRef CAS.
- (a) A. Zaks and A. M. Klibanov, Science, 1984, 224, 1249–1251 CAS; (b) A. Zaks and A. M. Klibanov, Proc. Natl. Acad. Sci. U. S. A., 1985, 82, 3192–3196 CrossRef CAS PubMed.
- A. M. Klibanov, Nature, 2001, 409, 241–246 CrossRef CAS PubMed.
- (a) P. Yedavalli and N. M. Rao, Protein Eng., Des. Sel., 2013, 26, 317–324 CrossRef CAS PubMed; (b) M. T. Reetz, P. Soni, L. Fernandez, Y. Gumulya and J. D. Carballeira, Chem. Commun., 2010, 46, 8657–8658 RSC.
- J. Ge, D. Lu, J. Wang and Z. Liu, Biomacromolecules, 2009, 10, 1612–1618 CrossRef CAS PubMed.
- (a) L. Brady, A. M. Brzozowski, Z. S. Derewenda, E. Dodson, G. Dodson, S. Tolley, J. P. Turkenburg, L. Christiansen, B. Huge-Jensen and L. Norskov, Nature, 1990, 343, 667–770 CrossRef PubMed; (b) J. D. Schrag, Y. Li, S. Wu and M. Cygler, Nature, 1991, 351, 761–764 CrossRef CAS PubMed.
- H. Marcel, Org. Biomol. Chem., 2013, 11, 5162–5172 Search PubMed.
- A. R. Hajipour and F. Rafiee, Org. Prep. Proced. Int., 2010, 42, 285–362 CrossRef CAS.
- J. P. Hallett and T. Welton, Chem. Rev., 2011, 111, 3508–3576 CrossRef CAS PubMed.
- X. Wu, L. M. Wang, R. A. Nieman and C. A. Angell, J. Phys. Chem. B, 2003, 107, 11749–11756 CrossRef.
- S. V. Dzyuba and R. A. Bartsch, ChemPhysChem, 2002, 3, 161–166 CrossRef CAS PubMed.
- H. Zhang, F. Xu, X. Zhou, G. Zhang and C. Wang, Green Chem., 2007, 9, 1208–1211 RSC.
- J. Pernak, I. Goc and I. Mirska, Green Chem., 2004, 6, 323–329 RSC.
- H. Beer, G. Wohlfahrt, J. Mccarthy, D. Schomburg and R. Schmid, Protein Eng., 1996, 9, 507–517 CrossRef CAS PubMed.
- W. P. Jencks and J. Carriuolo, J. Biol. Chem., 1959, 234, 1272–1279 CAS.
- (a) S. T. Heller and R. Sarpong, Org. Lett., 2010, 12, 4572–4575 CrossRef CAS PubMed; (b) S. T. Heller and R. Sarpong, Tetrahedron, 2011, 67, 8851–8859 CrossRef CAS.
- (a) K. G. Byler, Y. Li, R. A. Houghten and K. Martinez-Mayorga, Org. Biomol. Chem., 2013, 11, 2979–2987 RSC; (b) G. Chadha and Y. Zhao, Org. Biomol. Chem., 2013, 11, 6849–6855 RSC; (c) Y. Liu, X. Meng, J. Li and X. Li, Colloids Surf., A, 2013, 436, 839–845 CrossRef CAS; (d) H. Yang and J. C. Sherman, Bioorg. Med. Chem. Lett., 2013, 23, 1752–1753 CrossRef CAS PubMed.
- K. Akagawa, N. Sakai and K. Kudo, Angew. Chem., Int. Ed., 2015, 54, 1822–1826 CrossRef CAS PubMed.
- S. Bayat, B. A. Tejo, A. B. Salleh, E. Abdmalek, Y. M. Normi and M. B. A. Rahman, Chirality, 2013, 25, 726–734 CrossRef CAS PubMed.
- R. Wieczorek, M. Dörr, A. Chotera, P. L. Luisi and P.-A. Monnard, ChemBioChem, 2013, 14, 217–223 CrossRef CAS PubMed.
- L. Mandell, J. Moncrief and J. Goldstein, Tetrahedron, 1963, 19, 2025–2030 CrossRef CAS PubMed.
- F. G. Bordwell, Acc. Chem. Res., 1988, 21, 456–463 CrossRef CAS.
- (a) W. F. Reynolds, I. R. Peat, M. H. Freedman and J. R. Lyerla, J. Am. Chem. Soc., 1973, 95, 328–331 CrossRef CAS PubMed; (b) H. Saito, Y. Tanaka and S. Nagata, J. Am. Chem. Soc., 1973, 95, 324–328 CrossRef CAS.
- W. Hermann, Angew. Chem., Int. Ed., 2008, 47, 654–670 CrossRef PubMed.
- R. S. Mohan, S. Chowdhury and J. L. Scott, Tetrahedron, 2007, 63, 2363–2389 CrossRef.
- K. Li, Z. Yang, J. Zhao, J. Lei, X. Jia, S. H. Mushrif and Y. Yang, Green Chem., 2015, 17, 4271–4280 RSC.
- A. Arfan and J. P. Bazureau, Org. Process Res. Dev., 2005, 9, 743–748 CrossRef CAS.
- A. K. Chakraborti and R. Sudipta Raha, J. Am. Chem. Soc., 2009, 131, 6902–6903 CrossRef CAS PubMed.
- (a) T. P. Wells, J. P. Hallett, C. K. Williams and W. Tom, J. Org. Chem., 2008, 73, 5585–5588 CrossRef CAS PubMed; (b) C. Lorna, F. Ruben, L. N. Llewellyn, L. M. Veronica and W. Tom, J. Org. Chem., 2006, 71, 8847–8853 CrossRef PubMed; (c) G. Ranieri, J. P. Hallett and T. Welton, Ind. Eng. Chem. Res., 2007, 47, 638–644 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Supplementary figures and tables, and characterization of products. See DOI: 10.1039/c5gc02557e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2016 |