
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBio-renewable enantioselective aldol reaction in natural deep eutectic solvents†
Regina
Martínez
,
Lucía
Berbegal
,
Gabriela
Guillena
* and
Diego J.
Ramón
*
Instituto de Síntesis Orgánica, and Dpto de Química Orgánica. Universidad de Alicante. Apdo. 99, E-03080-Alicante, Spain. E-mail: gabriela.guillena@ua.es; djramon@ua.es
First published on 5th November 2015
Abstract
Among the deep eutectic solvents (DES), natural deep eutectic solvents (NADES) formed by D-glucose and racemic malic acid are suitable media to perform the enantioselective L-proline catalyzed intermolecular aldol reaction, creating simultaneously and selectively a C–C bond and a new stereocenter. The scope of the reaction was found to be broad, with products being obtained with good levels of diastereo- and enantioselectivities. Furthermore, when the reaction was performed at a large scale, the catalyst together with the reaction media can be recovered by simple water extraction and reused at least three times affording similar results. Therefore, the use of NADES as reaction media to carry out a VOC-free selective process has been demonstrated for the first time. The process is clean, cheap, simple and scalable and meets most of the criteria to be considered as a sustainable and bio-renewable process, with the reaction media and catalyst arising directly from Nature.
Introduction
Organocatalyzed enantioselective reactions1 have been proposed as ideal green processes,2 as these procedures use only small organic molecules as catalysts in the absence of any type of metal. The epitome, among these processes, is the enantioselective cross-aldol reaction.3 However, a close look at the reaction, as well as the synthetic protocols, forecasts some aspects that must not be obviated, concerning the twelve principles of Green Chemistry.4The first reported organocatalyzed enantioselective cross-aldol reaction5 showed that natural proline is an excellent catalyst for the reaction between acetone (1a) and 4-nitrobenzaldehyde (2a), rendering the expected product 3a in good yield (Scheme 1). The chosen solvent, to improve the catalyst solubility, was DMSO; although the large excess of acetone used might also play a role as a solvent. The presence of large amounts of volatile organic compounds (VOCs) as the reaction medium makes the whole process not so “green” from an environmental point of view. This is due to the intrinsic toxicity of DMSO,6 and the minimum sustainability, since the organic solvent comes from a finite resource such as petroleum.
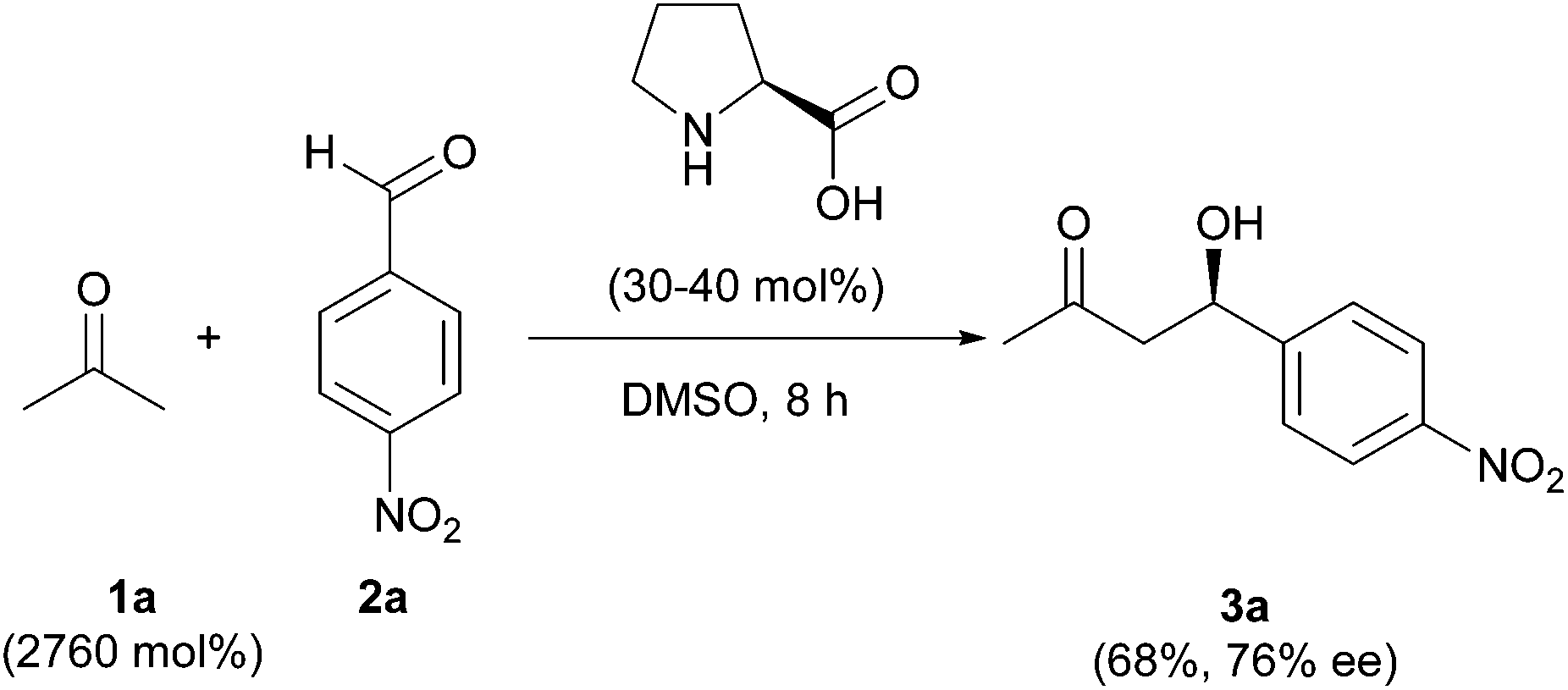 | ||
| Scheme 1 Enantioselective cross-Aldol reaction. | ||
Organic chemists solved quickly the problem of pollutants by performing the reaction under solvent-free conditions.7 A similar reaction performed under ball mill conditions gave the same aldol product 3a in 73% yield and 56% ee.8 However, in this new protocol, the final work-up using a large amount of diethyl ether (about 80 mL mmol−1) returns us the initial problems associated with the use of VOCs. Other organocatalysts different from proline used under solvent-free conditions did not overcome this work-up problem, since the final extraction using VOCs is needed to separate the products from reagents and catalysts.9
Water10 is another ideal medium, from the environmental point of view, with a similar aldol reaction being performed in a minimum amount of this solvent.11 In this case, using 10 mol% of proline in the reaction between cyclohexanone (1c) and aldehyde 2a gave the expected compound 3j in 73% yield and 99% ee, after 96 h. But, as in previous cases, the use of a large amount of solvent for work-up (ethyl acetate) was inexorable, with the use of other organocatalysts, under similar reaction conditions, not solving this problem.12
On one hand, organic solvents seem to be the major environmental issue for the aldol reaction, since proline is a biorenewable catalyst. On the other hand, very recently, a new type of solvent, generically called deep eutectic solvents (DES), has emerged as an alternative in organic synthesis.13 The initial attempts to perform an organocatalysed enantioselective Diels–Alder reaction using L-proline as the catalyst in the low-melting mixture carnitine![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :urea media failed.14 The final product was obtained with 93% yield as a racemate. Even the use of the ionic liquid choline (2S)-2-pyrrolidinecarboxylate15 gave the aldol product in a racemic way. Only very recently, the tandem enzyme-organocatalyst aldol reaction using acetaldehyde, as the unique nucleophilic partner, and (S)-α,α-bis[3,5-bis(trifluoromethyl)phenyl]-2-pyrrolidine-methanol, as the organocatalyst, in glycerol:choline chloride and isopropanol as the medium gave the expected aldol products with good yields and enantioselectivities.16 However, the use of a non-biorenewable catalyst, isopropanol as the co-solvent and the final ethyl acetate workup highlight the need for improving the conditions to achieve a sustainable process.
:urea media failed.14 The final product was obtained with 93% yield as a racemate. Even the use of the ionic liquid choline (2S)-2-pyrrolidinecarboxylate15 gave the aldol product in a racemic way. Only very recently, the tandem enzyme-organocatalyst aldol reaction using acetaldehyde, as the unique nucleophilic partner, and (S)-α,α-bis[3,5-bis(trifluoromethyl)phenyl]-2-pyrrolidine-methanol, as the organocatalyst, in glycerol:choline chloride and isopropanol as the medium gave the expected aldol products with good yields and enantioselectivities.16 However, the use of a non-biorenewable catalyst, isopropanol as the co-solvent and the final ethyl acetate workup highlight the need for improving the conditions to achieve a sustainable process.
In this study, following our studies on DES catalysed reactions, we introduce an L-proline aldol process in a deep eutectic medium,17 which can be carried out on a gram scale, thus avoiding the use of volatile organic solvents, and fulfilling all the requirements of a bio-sustainable reaction.
Results and discussion
The L-proline (30 mol%) catalysed reaction between acetone (1a, 5 equiv.) and p-nitrobenzaldehyde (2a) at room temperature was chosen as a model to evaluate several deep eutectic mixtures as the solvent for this process (Table 1). All the tested solvents were based in choline chloride (ChCl) as a hydrogen bond acceptor (HBA) component, changing the hydrogen bond donor (HBD) source in each case. The best results in terms of conversion and enantioselectivity were obtained using the mixture ChCl:glycerol as the solvent (Table 1, entry 3), being superior to those achieved in ChCl:urea (Table 1, entry 2). In fact, the performance of L-proline in ChCl:glycerol was comparable to those achieved under solvent-free conditions (Table 1, entry 1). The use of resorcinol as the HBD gave a solvent where the aldol product 3a was formed with good enantioselectivity (59% ee), together with the condensation product coming from the dehydration of product 3a (Table 1, entry 4). When the HBD component was changed to an acid molecule, such as malic acid, only the dehydration product was achieved (Table 1, entry 5). Other HBD acids such as D/L tartaric acid or oxalic acid gave a DES where the catalyst was inactive, probably due to the protonation of the nitrogen of the L-proline. The need for 30 mol% L-proline as the catalyst loading using ChCl:glycerol as reaction media was checked by reducing the amount to 15 mol%. Although the level of enantioselectivity was maintained, the conversion sharply decreased under these reaction conditions (Table 1, compare entries 3 and 8). Also, the possible catalytic activity of the solvent was evaluated by carrying out the reaction in the absence of proline, with no product being formed under these conditions (Table 1, entry 9). As the addition of a small amount of water in organocatalyzed reaction has sometimes a beneficial effect on the results18 and decreases the viscosity of the DES,19 its effect was tested in the process using ChCl:glycerol as the reaction medium (Table 1, entry 10). However, in this new reaction medium, the addition of only 2 equivalents of water caused a drop in the achieved enantioselectivity. Finally, the use of glycerol as the solvent medium, for the same process, was evaluated (entry 11, Table 1). However, the results in terms of conversion and enantioselectivity were clearly lower than those achieved in the ChCl:glycerol mixture.
| Entry | Solvent | Conversionb (%) | eec (%) |
|---|---|---|---|
| a Reaction between p-nitrobenzaldehyde (1 mmol) and acetone (5 equiv.), catalyzed by L-proline (30 mol%) at 25 °C for 24 h, unless otherwise stated. b Conversion calculated by 1H-NMR using N,N-diphenylformamide as the internal standard. c Determined by chiral HPLC. d Reaction carried out under solvent free conditions. e 41% of the condensation product was obtained. f Only the condensation product (4-(4-nitrophenyl)but-3-en-2-one) was obtained. g Only 15 mol% was used as the catalyst. h Reaction carried out in the absence of L-proline. i 2 equiv. of water was added in the reaction media. | |||
| 1d | — | 82 | 56 |
| 2 | ChCl:urea (1:2) |
32 | 38 |
| 3 | ChCl:glycerol (1:2) |
80 | 54 |
| 4 | ChCl:resorcinol (1:1) |
40e | 59 |
| 5 | ChCl:L-malic acid (1:1) |
80f | — |
| 6 | ChCl:D/L tartaric acid (1:1) |
— | — |
| 7 | ChCl:oxalic acid (1:1) |
— | — |
| 8g | ChCl:glycerol (1:2) |
40 | 52 |
| 9h | ChCl:glycerol (1:2) |
— | — |
| 10i | ChCl:glycerol (1:2) |
82 | 12 |
| 11 | Glycerol | 40 | 12 |
Once the best reaction conditions were found using ChCl:glycerol, several organocatalysts were tested in the reaction model (Fig. 1).
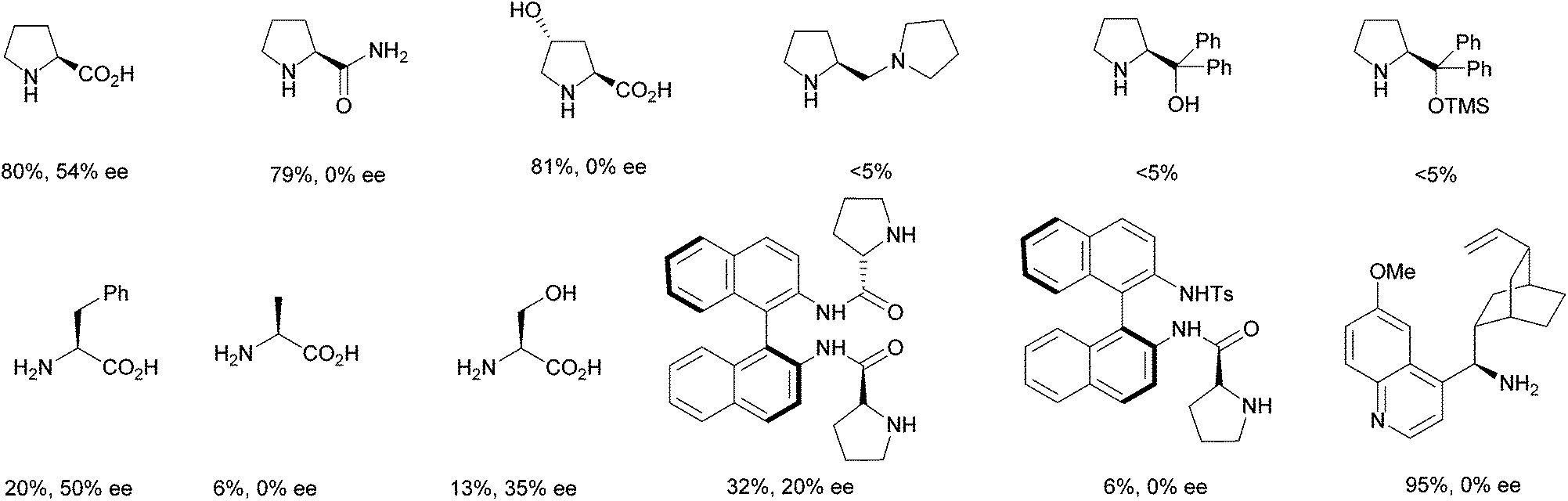 | ||
| Fig. 1 Catalyst tested in the cross-Aldol reaction in ChCl:glycerol. | ||
L-Prolinamide and (S)-4-trans-hydroxyprolinol were active rendering product 3a but as a racemic mixture, while proline amine bearing a pyrrolidine ring or a diphenylmethanol moiety showed to be inactive. Primary α-amino acids such as (S)-phenylalanine and (S)-serine gave product 3a with moderate enantioselectivities albeit in low conversion, whereas (S)-alanine leads to a very low conversion. Finally, two different 1,1′-binaphthyl-2,2′-diamine (BINAM) prolinamide derivatives were used as the catalyst, but lower yields compared to those achieved by using L-proline were encountered. While this study was performed, the use of chiral primary amines 9-amino-9-deoxy-epi-cinchone derivatives as organocatalysts to perform several conjugate addition processes in DES was successfully reported.20 Therefore, (9R)-9-amino-9-deoxyquinidine trihydrochloride was used as the catalyst in this reaction. This afforded only 33% of the dehydrated product. Also, free primary amines, obtained by treatment with a base, were tested in the aldol process, giving the expected aldol but as a racemic mixture.
As the results in ChCl based DES were not very good, we explored the use of a natural deep eutectic solvent21 (NADES) as the reaction medium22 to perform this transformation. Thus, a mixture of D-glucose and racemic malic acid in 1:1 proportion forms a highly viscous natural eutectic solvent. Regarding the sustainability of the process, this mixture was found to be ideal since both components come from biorenewable sources and are completely biodegradable and nontoxic. Thus, the model reaction between acetone (1a, 5 equiv.) and p-nitrobenzaldehyde (2a) catalyzed by L-proline (30 mol%) at room temperature was carried out in this medium (Table 2, and Scheme 2).
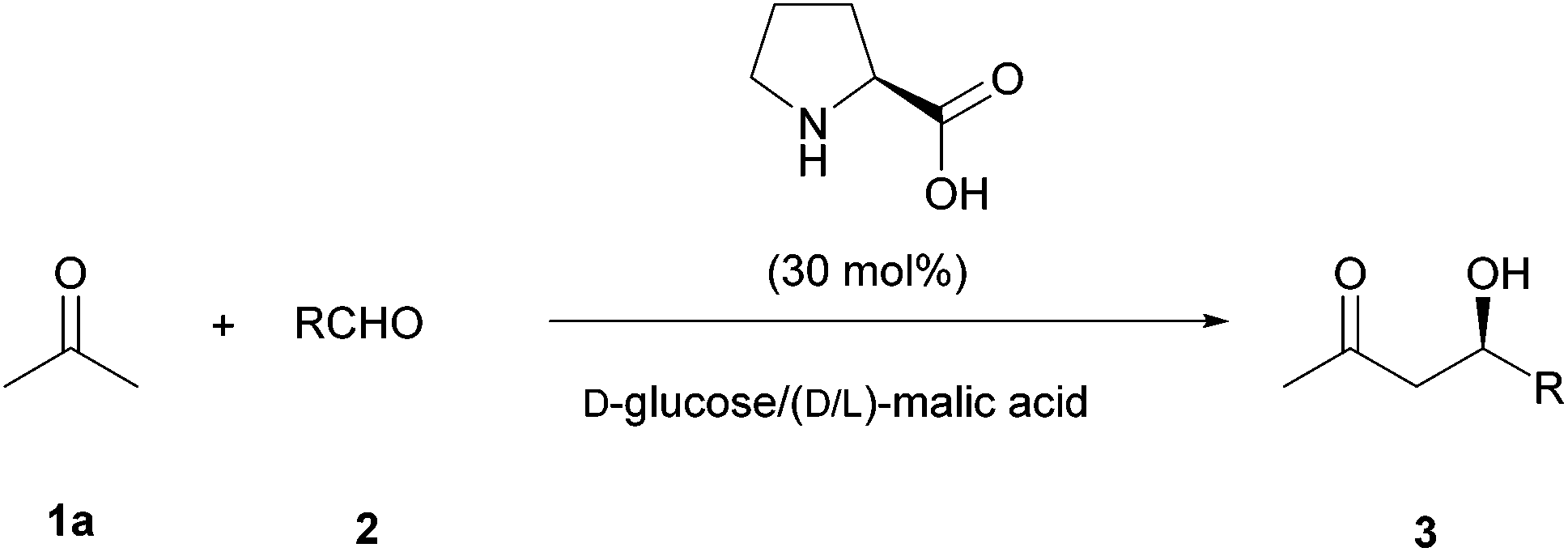 | ||
| Scheme 2 Enantioselective cross-Aldol reaction between acetone and aldehydes in NADES. | ||
| Entry | Solvent | R | Product | Conversionb (%) | eec (%) |
|---|---|---|---|---|---|
| a Reaction between aldehyde (1 mmol) and acetone (5 equiv.), catalyzed by L-proline (30 mol%) at 25 °C for 24 h, unless otherwise stated. b Conversion calculated by 1H-NMR using N,N-diphenylformamide as the internal standard. c Determined by chiral HPLC. d Reaction carried out in the absence of proline. e Only 15 mol% of the catalyst was used. f 48 h were required for completion. g 7 days were required for completion. | |||||
| 1 |
D-Glucose/(D/L)-malic acid (1:1) |
4-O2NC6H4 | 3a | 85 | 70 |
| 2d |
D-Glucose/(D/L)-malic acid (1:1) |
4-O2NC6H4 | 3a | <5 | — |
| 3e |
D-Glucose/(D/L)-malic acid (1:1) |
4-O2NC6H4 | 3a | 45 | 70 |
| 4 |
D-Glucose/D-malic acid (1:1) |
4-O2NC6H4 | 3a | 50 | 68 |
| 5 |
D-Glucose/L-malic acid (1:1) |
4-O2NC6H4 | 3a | 40 | 66 |
| 6 |
D-Glucose/(D/L)-malic acid (1:2) |
4-O2NC6H4 | 3a | 22 | 70 |
| 7 |
D-Glucose/(D/L)-malic acid (2:1) |
4-O2NC6H4 | 3a | 78 | 60 |
| 8f |
D-Glucose/(D/L)-malic acid (1:1) |
2-O2NC6H4 | 3b | 91 | 78 |
| 9f |
D-Glucose/(D/L)-malic acid (1:1) |
3-O2NC6H4 | 3c | 86 | 60 |
| 10 |
D-Glucose/(D/L)-malic acid (1:1) |
4-NCC6H4 | 3d | 79 | 68 |
| 11 |
D-Glucose/(D/L)-malic acid (1:1) |
4-F3CC6H4 | 3e | 85 | 75 |
| 12 |
D-Glucose/(D/L)-malic acid (1:1) |
2-ClC6H4 | 3f | 90 | 60 |
| 13 |
D-Glucose/(D/L)-malic acid (1:1) |
C6H5 | 3g | 40 | 65 |
| 14 |
D-Glucose/(D/L)-malic acid (1:1) |
4-MeC6H4 | 3h | 20 | 72 |
| 15g |
D-Glucose/(D/L)-malic acid (1:1) |
C6H11 | 3i | 58 | 60 |
To our delight the standard aldol product 3a was obtained after 24 h in 85% conversion and 70% ee (Table 2, entry 1). The inactivity of the reaction media as a catalyst for this reaction was proved by performing the process in the absence of L-proline (Table 2, entry 2). A decrease in the catalyst loading caused a decrease in the conversion but not in the enantioselectivity (Table 2, entry 3).
The influence of the stereochemistry of malic acid on the stereochemical outcome of the reaction was studied by carrying out the reaction using D-glucose/D-malic acid or D-glucose/L-malic acid (1:1) as a solvent, with similar results in terms of enantiomeric excess being found to the results using D-glucose/(D/L)-malic acid, albeit lower conversions were obtained for product 3a (Table 2, compare entries 1 with 4 and 5). Also the optimal proportion between D-glucose and racemic malic acid was stabilised to be 1:1, since a 1:2 or 2:1 proportion between these two components of the eutectic mixture led to worse results in terms of conversion and enantioselectivities (Table 2, compare entries 1 with 6 and 7). Once the best reaction conditions were found using D-glucose/(D/L)-malic acid as the solvent, the scope of the reaction between acetone and several aldehydes was studied (Table 2, entries 8–13). From the results, it can be concluded that the same levels of enantioselectivities are achieved with aromatic aldehydes bearing electron-withdrawing or electron-donating substituents, with only the conversions being affected by their electronic nature. Also, aliphatic aldehydes such as cyclohexanecarbaldehyde can be used as an electrophile, but lower conversion and enantioselectivity were obtained, with a longer reaction time being required (Table 2, entry 15).
Although the mechanism for the aldol reaction seems to be well established, the role of self aggregation of the catalyst as well as the possible autocatalytic effect depending on the reaction conditions has been stressed. In order to clarify the possible pathway of the reaction, the effect of the enantiomeric excess of the proline catalyst on the enantioselectivity of the reaction leading to product 3a was examined (Fig. 2). A clear negative non-linear effect was detected. This effect might be explained as a consequence of a kinetic conglomerate phase effect in which the proline dissolution occurs simultaneously with turnover an asymmetric reaction,23 and discarding the possible kinetic resolution of catalyst by the chiral hydroxyl aldehyde present as solvent (D-glucose).24
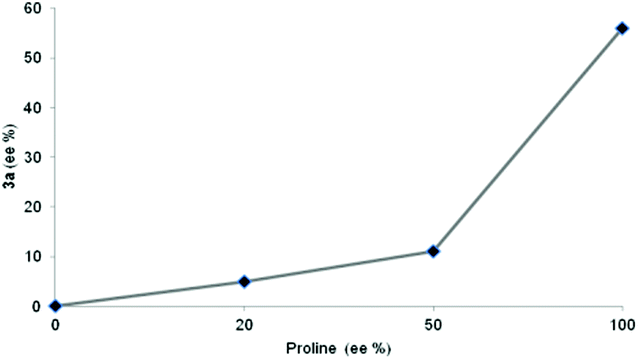 | ||
| Fig. 2 Non-linear effect. | ||
Once the best reaction conditions for this transformation were determined, the possible recovery and reuse of the NADES together with the catalyst was explored. First, this recyclability study was carried out by washing out the formed product 3a with a small amount (3 × 1 mL) of several volatile organic solvents such as ethyl acetate, acetone, diethyl ether, hexane and 2-butanol. In all cases, a sharp decrease of the conversion was detected after the second cycle (third cycle in the case of 2-butanol, Fig. 3) but the enantioselectivity remained constant with the reaction cycles. In such cases, once the excess of ketone was evaporated, the remaining crude mixture was percolated through a small pad of silica in order to remove the unreacted aldehyde, affording the pure product 3a. Alternatively, the product can be purified by recrystallization from hexane/ethyl acetate. Therefore, only a little amount of volatile organic solvents was required in order to obtain the pure product. When the work-up of the reaction was done using an organic solvent, the organic layer was analysed by CG-MS in order to investigate the composition of the extract. In the chromatogram, only the excess of the starting ketone, the unreacted aldehyde and the final product were detected, with no traces of the components of the DES mixture being identified.
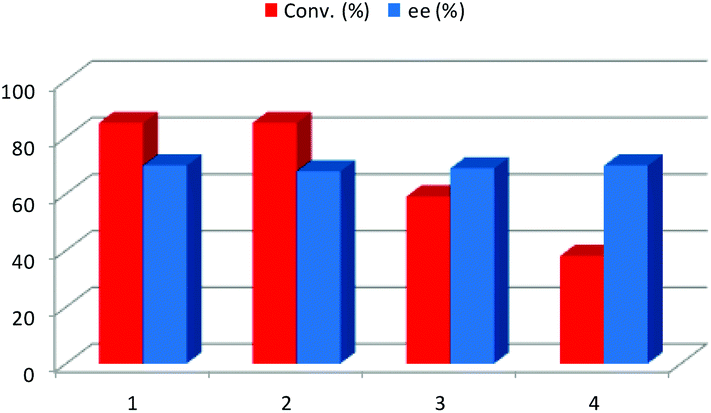 | ||
| Fig. 3 Recycling studies using volatile organic solvents (2-butanol). | ||
But our final goal was to be able to recycle the NADES and the catalyst without using volatile organic solvents. For this purpose, the reaction was scaled up in order to obtain approximately 5 g of the final product 3a.
After reaction completion, water was added to the reaction media, dissolving the D-glucose, racemic malic acid and L-proline, and the organic layer formed only by the product 3a was separated. Once the excess of acetone was removed by evaporation, the final product was purified by recrystallization. The water of the aqueous layer was evaporated in order to recover the solvent and the catalyst and a new batch of reagents was added. This procedure was repeated three times having the same conversion and enantioselectivity (Fig. 4). Therefore, the possibility of carrying out an aldol reaction in a biorenewable and natural solvent mixture and catalyst was demonstrated, this being a good example of the potential application of NADES as solvents to perform sustainable and complex chemical processes. In order to further study the reusability, avoiding possible catalyst saturation, the amount of L-proline and the amount of DES were reduced to a fourth. Under these new reaction conditions, after 24 h, only 55% of conversion was achieved with the enantioselectivity being 68% ee. After three recycling experiments, following the above described procedure, both conversion and enantioselectivities remained almost the same.
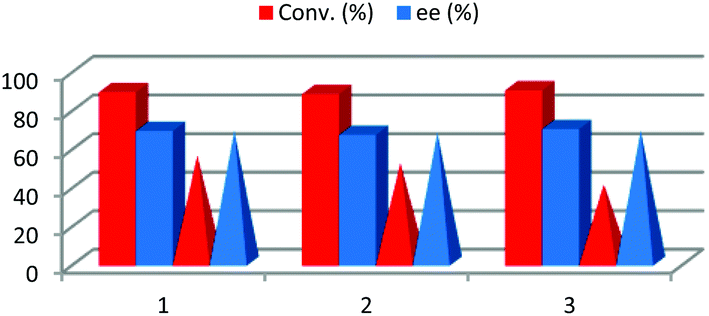 | ||
| Fig. 4 Recycling (bar chart) and recyclability (pyramid based chart) of NADES/L-proline by water extraction. | ||
Furthermore, the versatility of this transformation in NADES was evaluated by using different ketones (Scheme 3 and Fig. 5). When cyclic ketones were used as nucleophiles, two different diastereoisomers can be obtained, so selective control of the diastereo- and enantioselectivity of the reaction would be desirable. In fact, for cyclohexanone and tetrahydro-4H-thiopyran-4-one derivatives 3j–3n, the main achieved diastereoisomer has an anti-configuration, with moderate to good levels of diastereoselectivity being accomplished (50–80%). Also, moderate to excellent enantioselectivity was obtained for these products depending on the used electrophile. Meanwhile, the main diastereoisomer obtained with cyclopentanone depended on the character of the electrophile. While with p-nitrobenzaldehyde the main isomer was syn-3o, with decanal the major isomer was anti-3p, the latter being the diastereoisomer of the required intermediate for the synthesis of the oviposition attractant pheromone of the female Culex Mosquito.25 Also, α-alkoxyketones can be used as nucleophiles. For these ketones, three possible isomers (anti/syn and iso) could be achieved. However, under the applied reaction conditions, only the anti and syn-diastereoisomers were achieved for products 3q and 3r, with the anti-isomer being the major one. For both cases, high conversion and moderate enantioselectivity were accomplished.
 | ||
| Scheme 3 Enantioselective cross-Aldol reaction between cyclic ketones and aldehydes in NADES. | ||
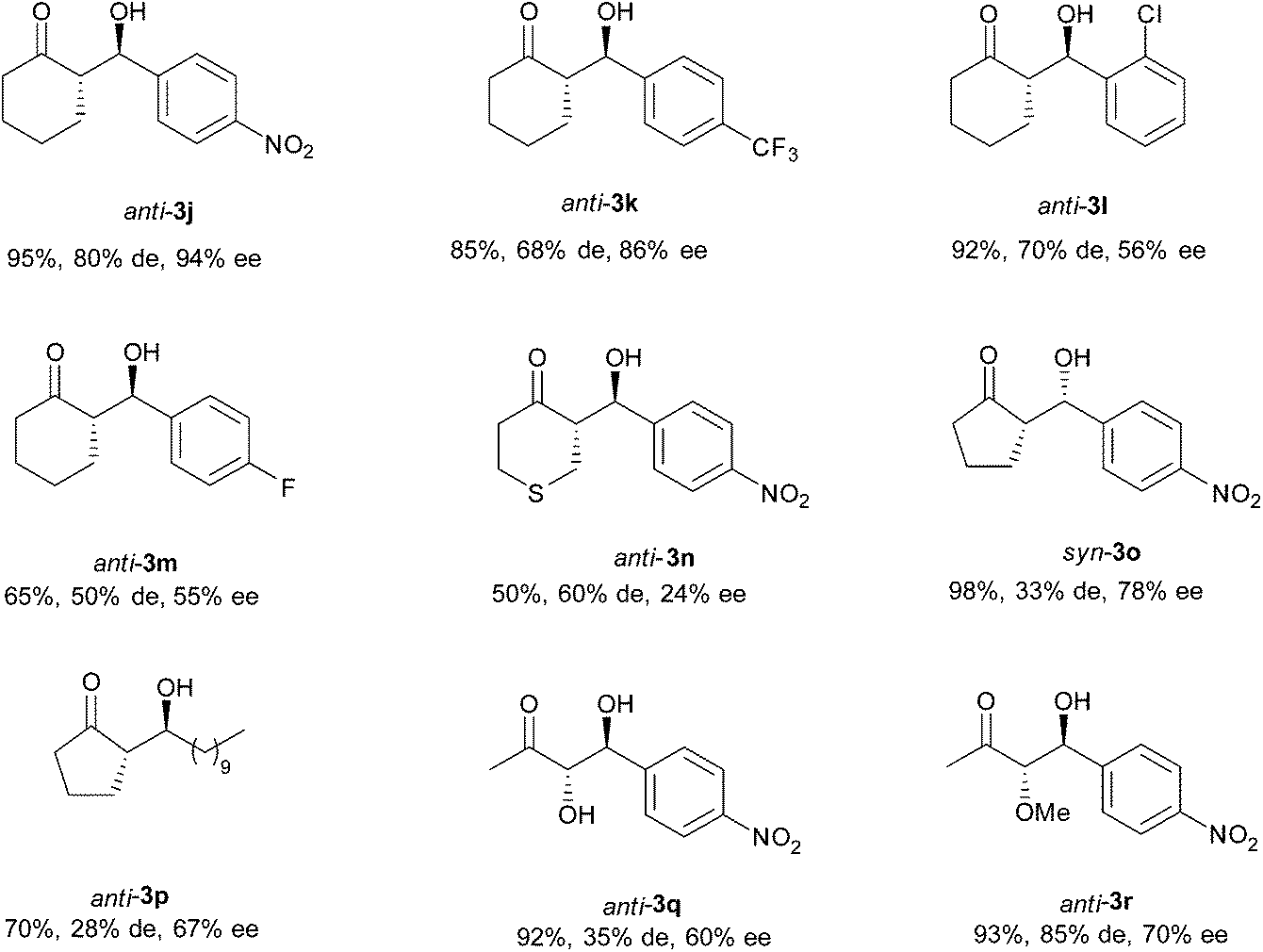 | ||
| Fig. 5 Products of the cross-Aldol reaction between cyclic ketones and aldehydes. | ||
Finally, this reaction could be extended to the aldol reaction between a non-enolizable aldehyde and an aliphatic aldehyde such as propanal (source of nucleophile). In these cases, longer reaction times were required (5 days) to achieve the full conversion. After reduction of the aldol product to the corresponding chiral 1,3-diol, products 5 (Scheme 4) were obtained with moderate to good yields and diastereoselectivities but with excellent enantioselectivities.
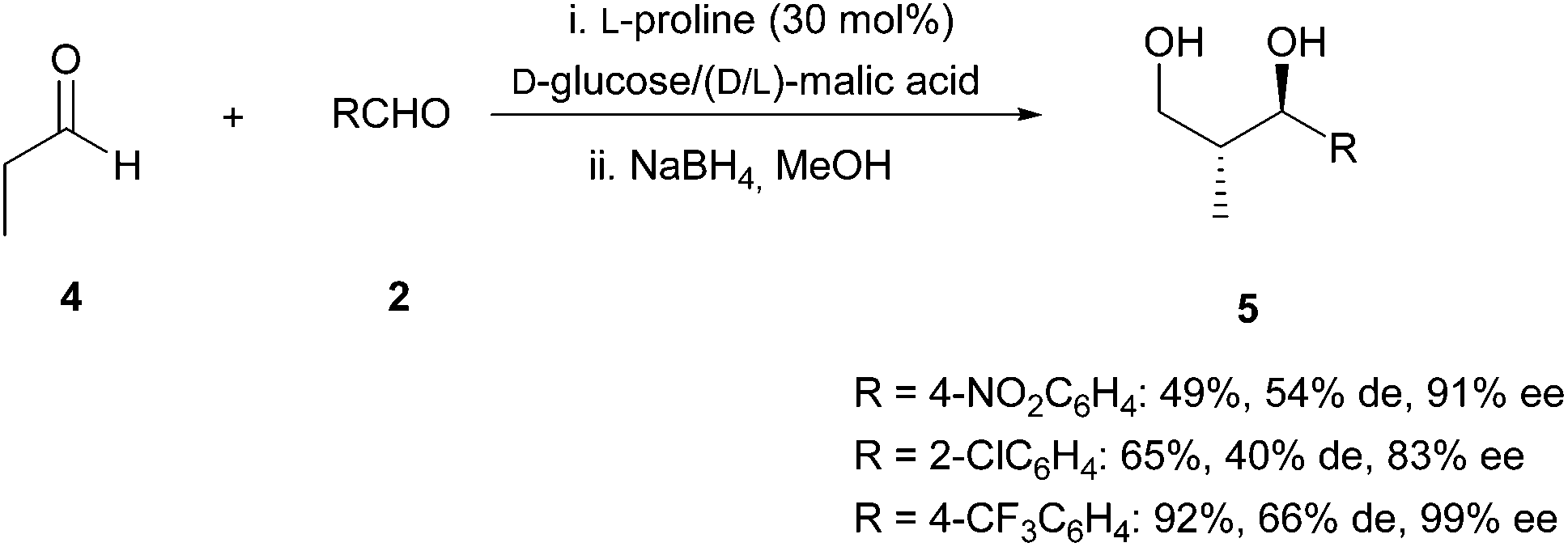 | ||
| Scheme 4 Products of the cross-Aldol reaction between propanal and aldehydes. | ||
Experimental
General
All reactions were carried out under argon. All reagents were commercially available and used without further purification. 1H NMR (300 MHz, 400 MHz) and 13C NMR (75 MHz) spectra were obtained on a Bruker AC-300 using CDCl3 as the solvent and TMS as the internal standard, unless otherwise stated. HPLC analyses were performed on an Agilent 1100 series equipped with a chiral column (details of each compound are given in the ESI†), using mixtures of n-hexane/isopropyl alcohol (IPA) as the mobile phase, at 25 °C. Analytical TLC was performed on Schleicher & Schuell F1400/LS silica gel plates and the spots were visualised under UV light (λ = 254 nm). For chromatography we employed Merck silica gel 60 (0.063–0.2 mm). For recycling experiments, an Edwards T-station equipped with a diaphragm pump 75 was used for water evaporation.General procedure for the preparation of DES
The corresponding solid components of the desired DES in the correct proportion were placed in a 50 mL round-bottom flask. The resulting mixture was heated to 80 °C (from 1 to 3 h) under an argon atmosphere with stirring until a clear colourless liquid was obtained.General procedure for the aldol reaction in a deep eutectic solvent
To around 1 mL of the corresponding solvent in a vessel under an argon atmosphere, L-proline (0.035 g, 30 mol%) and the corresponding aldehyde (1 mmol) were added. Then the source of nucleophile was charged (5 mmol in the case of acetone and propanal, 1 mmol for cyclohexanone and 2 mmol for the other ketones). The reaction mixture was stirred under an argon atmosphere for 24 h to 5 days (see Tables 1 and 2, Scheme 3 and text) at room temperature. Then, 2 mL of water were added and the mixture was extracted with ethyl acetate (3 × 1 mL). The resulting organic phase was dried over anhydrous magnesium sulphate, and the solvent was evaporated under reduced pressure. The resulting crude material was purified by percolation through a small pad of silica gel with 1:1 ethyl acetate/hexane mixtures. In the case of using propanal, after extraction, the resulting organic phase was dried over anhydrous magnesium sulphate, and the solvent was evaporated under reduced pressure. The resulting crude was treated with sodium borohydride (5 mmol, 190 mg) in methanol (3 mL). The reaction mixture was stirred for 2 h at 0 °C. After the reaction, phosphate buffer (2 mL) was added, and the mixture was extracted with ethyl acetate (3 × 1 mL). The resulting organic phase was dried over anhydrous magnesium sulphate, and the solvent was evaporated under reduced pressure. The resulting crude material was purified by percolation through a small pad of silica gel with 1:1 ethyl acetate/hexane mixtures.
All compounds 3 and 5, which were previously described in the literature, were fully characterized by spectroscopy means (1H and 13C NMR) and the data are given in the ESI.†
Recover and reuse of the catalyst and DES
The corresponding solvents [approx. 3 mL: D-glucose (2.7 g) and D/L-malic acid (2.1 g)] were placed in a vessel under an argon atmosphere. L-Proline (0.175 g) and the corresponding aldehyde (5 mmol) were added. Then the source of nucleophile was charged (25 mmol). The reaction mixture was stirred under an argon atmosphere for 24 h. Then, 10 mL of water was added and the resulting organic upper layer was collected through a pipette for the gram scale procedure. The resulting organic phase was dried over anhydrous magnesium sulphate, and the solvent was evaporated under reduced pressure. The resulting crude material was purified by recrystallization from ethyl acetate/hexane mixtures. The aqueous layer was evaporated under reduced pressure. Water traces were eliminated for the residue using a high-vacuum membrane pump system over 24 hours. Then, the flask containing the DES and L-proline was charged with a new batch of aldehyde and acetone.Conclusions
The enantioselective aldol reaction between several ketones and aldehydes catalysed by simple L-proline has been performed in a natural deep eutectic solvent formed by the combination of D-glucose and racemic malic acid. The levels of selectivity achieved in this medium are good and higher than those obtained under other green conditions such as under solvent-free conditions. The procedures are clean, simple, cheap, scalable and safe. Furthermore, the catalyst and reaction media can be recovered and reused at least three times, achieving similar results. Organic volatile compounds (VOC) were only used in order to obtain a pure analytical sample of the product. All these facts pointed out the possibility of carrying out enantioselective organocatalyzed organic reactions using natural deep eutectic solvents as reaction media, this being a clear example of a green, bio-renewable and sustainable process.Acknowledgements
This work was supported by the University of Alicante (VIGROB-173 and UAUSTI13-09).Notes and references
- See for instance: (a) A. Berkessel and H. Gröger, Asymmetric Organocatalysis: From Biomimetic Concepts to Applications in Asymmetric Synthesis, Wiley-VCH, Weinheim, 2005 Search PubMed; (b) Enantioselective Organocatalysis: Reactions and Experimental Procedures, ed. P. I. Dalko, Wiley-VCH, Weinheim, 2007 Search PubMed; (c) I. R. Shaikh, J. Catal., 2014, 402860 Search PubMed (35 pages).
- (a) Green Chemistry: Designing Chemistry for the Environment, ed. P. T. Anastas and T. C. Williamson, ACS, Washington DC, 1996 Search PubMed; (b) R. A. Sheldon, I. Arends and U. Hanefeld, Green Chemistry and Catalysis, Wiley-VCH, Weinheim, 2007 Search PubMed; (c) R. A. Sheldon, Chem. Soc. Rev., 2012, 41, 1437–1451 RSC.
- G. Guillena, in Modern Methods in Stereoselective Aldol Reactions, ed. R. Mahrwald, Wiley-VCH, Weinheim, 2013, pp. 155–268 Search PubMed.
- P. T. Anastas and J. B. Zimmerman, Environ. Sci. Technol., 2003, 37, 94A–101A CrossRef PubMed.
- B. List, R. A. Lerner and C. F. Barbas III, J. Am. Chem. Soc., 2000, 122, 2395–2396 CrossRef CAS.
- J. Galvao, B. Davis, M. Tilley, E. Normando, M. R. Duchen and M. F. Cordeiro, FASEB J., 2014, 28, 1317–1330 CrossRef CAS PubMed.
- For a review, see: A. Bañón-Caballero, G. Guillena and C. Nájera, Mini-Rev. Org. Chem., 2014, 11, 118–128 CrossRef.
- (a) B. Rodríguez, T. Rantanen and C. Bolm, Angew. Chem., 2006, 118, 7078–7080 ( Angew. Chem., Int. Ed. , 2006 , 45 , 6924–6926 ) CrossRef; (b) B. Rodríguez, A. Bruckmann and C. Bolm, Chem. – Eur. J., 2007, 13, 4710–4722 CrossRef PubMed.
- See for instance: (a) G. Guillena, M. C. Hita, C. Nájera and S. F. Viózquez, Tetrahedron: Asymmetry, 2007, 18, 2300–2304 CrossRef CAS; (b) G. Guillena, M. C. Hita, C. Nájera and S. F. Viózquez, J. Org. Chem., 2008, 73, 5933–5943 CrossRef CAS PubMed; (c) G. Guillena, C. Nájera and S. F. Viózquez, Synlett, 2008, 3031–3035 CrossRef CAS; (d) J. G. Hernández and E. Juaristi, J. Org. Chem., 2011, 76, 1464–1467 CrossRef PubMed; (e) T. P. Kumar, N. C. Vavle, V. Patro and K. Haribabu, Tetrahedron: Asymmetry, 2014, 25, 457–461 CrossRef CAS; (f) E. Machuca and E. Juaristi, Tetrahedron Lett., 2015, 56, 1144–1148 CrossRef CAS.
- For a review, see: M. Raj and V. K. Singh, Chem. Commun., 2009, 6687–6703 RSC.
- Y. Hayashi, S. Aratake, T. Itoh, T. Okano, T. Sumiya and M. Shoji, Chem. Commun., 2007, 957–959 RSC.
- See for instance: (a) Y. Hayashi, T. Sumiya, J. Takahashi, H. Gotoh, T. Urushima and M. Shoji, Angew. Chem., 2006, 118, 972–975 ( Angew. Chem., Int. Ed. , 2006 , 45 , 958–961 ) CrossRef; (b) N. Mase, Y. Nakai, N. Ohara, H. Yoda, K. Takabe, F. Tanaka and C. F. Barbas III, J. Am. Chem. Soc., 2006, 128, 734–735 CrossRef CAS PubMed; (c) S. S. V. Ramasastry, K. Albertshofer, N. Utsumi and C. F. Barbas III, Org. Lett., 2008, 10, 1621–1624 CrossRef CAS PubMed; (d) R. Pedrosa, J. M. Andrés, R. Manzano, D. Román and S. Téllez, Org. Biomol. Chem., 2011, 9, 935–940 RSC; (e) J. G. Hernández and E. Juaristi, Tetrahedron, 2011, 67, 6953–6959 CrossRef; (f) C. Shen, F. Shen, G. Zhou, H. Xia, X. Chen, X. Liu and P. Zhang, Catal. Commun., 2012, 26, 6–10 CrossRef CAS; (g) N. Khiar, R. Navas, E. Elhalem, V. Valdivia and I. Fernández, RSC Adv., 2013, 3, 3861–3864 RSC; (h) S. Paladhi, J. Das, P. K. Mishra and J. Dash, Adv. Synth. Catal., 2013, 355, 274–280 CAS; (i) L. Qin, L. Zhang, Q. Jin, J. Zhang, B. Han and M. Liu, Angew. Chem., 2013, 125, 7915–7919 ( Angew. Chem., Int. Ed. , 2013 , 52 , 7761–7765 ) CrossRef; (j) R. Pedrosa, J. M. Andrés, A. Gamarra, R. Manzano and C. Pérez-López, Tetrahedron, 2013, 69, 10811–10819 CrossRef CAS; (k) S. Eymur, E. Akceylan, O. Sahin, A. Uyanik and M. Yilmaz, Tetrahedron, 2014, 70, 4471–4477 CrossRef CAS; (l) A. Bisticha, I. Triandafillidi and C. G. Kokotos, Tetrahedron: Asymmetry, 2015, 26, 102–108 CrossRef CAS.
- For reviews about different aspects of DES, see: (a) C. Ruβ and B. König, Green Chem., 2012, 14, 2969–2982 RSC; (b) Q. Zhing, K. De Oliveira Vigier, S. Royer and F. Jérôme, Chem. Soc. Rev., 2012, 41, 7108–7146 RSC; (c) M. Francisco, A. van den Bruinhorts and M. C. Kroon, Angew. Chem., 2013, 125, 3152–3163 ( Angew. Chem., Int. Ed. , 2013 , 52 , 3074–3085 ) CrossRef; (d) B. Tang and K. H. Row, Monatsh. Chem., 2013, 144, 1427–1454 CrossRef CAS; (e) J. García-Álvarez, Deep Eutectic Solvents and their Applications as New Green and Biorenewable Reaction Media, in Handbook of Solvents, ed. G. Wypych, ChemTec Publishig, Toronto, 2014, vol. 2, pp. 813–844 Search PubMed; (f) E. L. Smith, A. P. Abbott and K. S. Ryder, Chem. Rev., 2014, 114, 1160–11082 Search PubMed.
- F. Ilgen and B. König, Green Chem., 2009, 11, 848–854 RSC.
- S. Hu, T. Jiang, Z. Zhang, A. Zhu, B. Han, J. Song, Y. Xie and W. Li, Tetrahedron Lett., 2007, 48, 5613–5617 CrossRef CAS.
- C. R. Müller, I. Meiners and P. Domínguez de María, RSC Adv., 2014, 4, 46097–46101 RSC.
- (a) J. M. Pérez and D. J. Ramón, ACS Sustainable Chem. Eng., 2015, 3, 2343–2343 CrossRef; (b) X. Marset, J. M. Pérez and D. J. Ramón, Green Chem., 2015 10.1039/C5GC01745A.
- (a) A. I. Nyberg, A. Usano and P. M. Pinko, Synlett, 2004, 1891–1896 CAS; (b) M. Gruttadauria, F. Giacalone and R. Noto, Adv. Synth. Catal., 2009, 351, 33–57 CrossRef CAS.
- (a) A. Yadav and S. Pandey, J. Chem. Eng. Data, 2014, 59, 2221–2229 CrossRef CAS; (b) Y. Daia, G.-J. Witkamp, R. Verpoortea and Y. H. Choi, Food Chem., 2015, 187, 14–19 CrossRef PubMed.
- E. Massollo, S. Palmieri, M. Benaglia, V. Capriati and F. M. Perna, Green Chem., 2015 10.1039/c5gc01855b.
- For reviews on NADES, see: (a) Y. Dai, J. van Spronsen, G.-J. Witkamp, R. Verpoorte and Y. H. Choi, Anal. Chim. Acta, 2013, 766, 61–68 CrossRef CAS PubMed; (b) A. Pavia, R. Craveiro, I. Aroso, M. Martins, R. L. Reis and A. R. C. Duarte, ACS Sustainable Chem. Eng., 2014, 2, 1063–1071 CrossRef.
- For reviews on the use of other bio-based solvents, see: Y. Gu and F. Jérôme, Chem. Soc. Rev., 2013, 42, 9550–9570 RSC.
- M. Klussmann, S. P. Mathew, H. Iwamura, D. H. Wells, A. Armstrong and D. G. Blackmond, Angew. Chem., 2006, 118, 8157–8160 ( Angew. Chem., Int. Ed. , 2006 , 45 , 7989–7992 ) CrossRef.
- R. Ríos, P. Schyman, H. Sundén, G.-L. Zhao, F. Ullah, L.-J. Chen, A. Laaksonen and A. Córdova, Chem. – Eur. J., 2010, 16, 13935–13940 CrossRef PubMed.
- H. Ikishima, Y. Sekiguchi, Y. Ichikawa and H. Kotsuki, Tetrahedron, 2006, 62, 311–316 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H, 13C-NMR and HPLC data for all compounds 3 and 5. See DOI: 10.1039/c5gc02526e |
| This journal is © The Royal Society of Chemistry 2016 |