
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Eco-friendly production of high quality low cost graphene and its application in lithium ion batteries
Ali Reza
Kamali
Department of Materials Science and Metallurgy, University of Cambridge, 27 Charles Babbage RD, Cambridge CB3 0FS, UK. E-mail: ark42@cam.ac.uk; alirezakam@yahoo.com
First published on 17th December 2015
Abstract
Large scale production of low cost and high quality graphene from abundant raw materials using eco-friendly methods is a critical step towards the widespread and sustainable use of this so-called “wonder material”. This paper for the first time reports a single step molten salt electrochemical method for the high yield preparation of graphene nanosheets having all the characteristics mentioned above. This process uses readily available commercial graphite electrodes as the carbon source which is both abundant and cheap. Surprisingly, apart from graphite, the other consumables are H2 and electricity, and no by-product is produced. This method is not only eco-friendly but also very efficient. It offers a production rate of 450 g graphene per litre of molten salt per day. A molten salt volume of 10 L should be able to produce 4.5 kg graphene in a day. The graphene product showed a high conductivity of 5.8 × 105 S m−1. The bench-scale production of high quality graphene, on a scale of tens of grams, was achieved using a novel two working electrode electrolysis cell, operating at a current density of about 1 A cm−2 which is at least an order of magnitude higher than any other electrochemical exfoliation method which has been used so far for the preparation of graphene. The mechanism involved in the process is discussed. The graphene nanosheets showed a high oxidation temperature of 663 °C when heated in air at 40 °C min−1. A simple and green strategy was developed to anchor SnO2 nanocrystals on the graphene nanosheets, and the lithium storage performance of the composite obtained was investigated. The composite displayed a high and stable lithium capacity of 1016 mA h g−1 after 100 cycles of lithiation and de-lithiation.
Introduction
Graphite is one of the most versatile non-metallic minerals in the world which has been known and used for at least 2500 years. However, the hexagonal structure of graphite, which consists of sp2 hybridised carbon atoms in a hexagonal arrangement, was identified no earlier than 1924. In 2004, a single layer of carbon atoms (so-called graphene) was separated from graphite using adhesive tape, demonstrating that graphene can be stable in an isolated state. Subsequent studies showed that graphene possesses superior mechanical, electronic, thermal and tribological properties.1–3 The combination of these properties as well as low bulk density, high surface area and good chemical stability make graphene extremely attractive for many applications including electron conductive additives for Li-ion battery anode4 and cathode5 materials, corrosion prevention,6 conducting inks,7 lubricants,8 more efficient solar cells,9 novel antibiotics,10 new catalyst material for fuel cells,11 supercapacitor electrode material,12 and oxygen reduction reactions,13 fillers in new ultra-high performance polymer-,14 ceramic-15 and metal-based composites,16 and electronic contacts.17 In addition to these, graphene/semiconductor nanocomposites are a promising new class of catalysts for the photodegradation of dye pollutants.18 Graphene also provides new opportunities to advance water desalination technologies,19 and challenges the current existing adsorbents employed for the removal of low concentrated contaminants from aqueous solutions.20In fact, this amazing wide range of diverse applications has been the driving force for governments to generously fund graphene research and innovation worldwide, including the European funding of €1 billion. Despite its importance, however, there is no process available for sustainable large-scale production of bulk graphene and without this, most of the potential applications cannot be fulfilled. High quality graphene can be obtained by rubbing graphite on a surface21 which is not technologically scalable. Other methods including the chemical oxidation of graphite,22–25 solution-phase exfoliation of graphite in solvents,26–30 epitaxial growth,31 chemical vapour deposition,32–34 ball milling35 and the arc discharge method36 suffer from one or more drawbacks such as a low rate of production, the low quality of the graphene product and the use of hazardous oxidants, reductants or solvents.
Chemical oxidation of graphite followed by exfoliation and reduction treatments is the most widely used approach for the preparation of chemically converted graphene. This approach uses graphite as the starting material, but is time consuming and involves the excessive use of strong oxidising and reducing agents such as KMnO4, KClO3, NaBH4 and hydrazine hydrate. As a consequence, the graphene product (so called reduced graphene oxide) is heavily damaged resulting in a poor conductivity.22–25
Graphene of higher quality can be produced by liquid phase exfoliation of graphite, using solvents such as N,N-dimethylformamide (DMF),26 2,2,6,6-tetramethylpiperidine-1-oxyl,37N-methyl-2-pyrrolidone38 and cetyltrimethylammonium bromide.39 It should be mentioned that most of these solvents are considered as hazardous due to their impacts on the environment. For example, a short-term exposure to DMF has been observed to damage the liver in animals and in humans.40
Savaram et al.41 reported an eco-friendly method for the preparation of graphene avoiding the use of chemical oxidants and reductants. Although some success was achieved, the method proposed, however, required multi-steps and used the SO42−-graphite intercalation compound as the carbon source. Moreover, the whole process is time-consuming and may take several days to be completed.41
Exfoliation of graphite may also be achieved by applying a potential to graphite feed materials immersed in an electrolyte. In most of these methods, the graphite feed material immersed in a room temperature electrolyte is connected to the positive pole of a power source. It leads to the oxidation of the graphite, allowing the intercalation of anions from the electrolyte, followed by the exfoliation of the graphite.42–47 However, the anodic oxidation of graphite leads to the formation of a significant amount of oxygen-containing groups which cannot be avoided due to the over-oxidation of the graphite.
Hence, the cathodic reduction of graphite electrodes has the advantage of the absence of oxidising conditions thereby preventing the generation of defects in the product.48 However, there have not been many investigations on the cathodic reduction of graphite to produce graphene because the high yield exfoliation of graphite without any oxidation process is difficult.48 The cathodic exfoliation of graphite was investigated at low temperatures in electrolytes such as propylene carbonate,49 tetrabutylammonium tetrafluoroborate,50 and the solution of lithium chloride and/or triethylamine hydrochloride in dimethyl sulfoxide.51 These electrochemical methods for the preparation of graphene suffer from some limitations. First, the reported methods employ specific grades/sizes of graphite as the carbon feed material including very small pieces of natural graphite flakes,44,45 highly oriented pyrolytic graphite (HOPG),50 porous pellets of graphite and poly(vinylidene fluoride),51 high purity iso-molded graphite47 and graphite foil.44 These types of graphite materials are difficult and/or expensive to be employed in large scale electrochemical operations due to the technical complications and/or costs associated with the manufacturing of the electrodes required. The second limitation of the electrochemical methods explained above is that these processes will typically result in the production of multi-layered graphene or even graphite chunks. Hence, there is a fundamental need to re-engineer the electrochemical setups so as to allow effectively application of the electrochemical driving force to graphite materials.53 It should also be noted that the electrochemical exfoliation of graphite at low temperatures (<100 °C) usually is carried out by applying 4–10 V between a graphite working electrode and a counter electrode.44–52 However, the high resistance of the electrolyte/electrode systems used results in a very low current density on the graphite electrode with values such as 1 mA,50 100 mA (ref. 47) or 50 mA cm−2.51 The low electrode current density causes a low rate of the electrochemical reactions at the electrodes creating insufficient exfoliation of graphite. Therefore, the products obtained contained a high quantity of multi-layered graphite chunks.53
Xu et al.52 found that lithium intercalated graphite reacts with HCl solution, resulting in the release of H2 in the interlayer space of graphite which subsequently leads to exfoliation of graphite. Although success was achieved in producing high quality few layer graphene, Li-intercalated graphite was first required to be produced.
In a completely different method, hydrogen can be directly formed in the graphite lattice by an electrochemical method. In contrast to the low temperature electrochemical methods, we have recently presented evidence which suggests that hydrogen cations dissolved in molten LiCl can be discharged on graphite cathodes, and then intercalate into the graphite structure, leading to the exfoliation of the graphite material into graphene nanosheets.54 In this process, the graphite electrodes are employed as the cathode, avoiding the drawbacks associated with the excessive oxidation of the graphite.
This method has the potential of producing high quality graphene on a large scale. However, there are still some open questions which need to be answered before the technology can be implemented on the industrial scale.
First, the current method by which hydrogen cations are formed in molten salt is based on the hydrolysis of molten LiCl brought about by the presence of water in the atmosphere of the electrolysis cell. However, this process involves the oxidation of oxygen anions (formed by the hydrolysis reaction) on the graphite anode of the cell. It subsequently leads to the consumption of the graphite anode to form CO2 which then reacts with Li2O dissolved in molten LiCl to form lithium carbonate. As a result, the as-produced carbonaceous material contains lithium carbonate and therefore needs further purification steps to produce pure graphene.54 Therefore, whilst the presence of hydrogen cations in the molten salt is essential for the promotion of the exfoliation process at the graphite cathode, the formation of oxygen anions in the molten salt is not desirable. It should be mentioned that the possibility of preventing the formation of oxygen anions significantly enhances the overall performance of the process, by avoiding the electrochemical oxidation and thus consumption of graphite anodes, eliminating purification steps and also avoiding the formation of unnecessary by-products. In this paper, a novel mechanism is presented for the formation of hydrogen cations without introducing oxygen anions to the molten salt, leading to an eco-friendly method for sustainable production of graphene. The scalability of the molten salt approach towards large scale preparation of graphene is also demonstrated. Furthermore, a novel green one-step method was developed to anchor SnO2 nanocrystals on the graphene nanosheets produced. The resultant composite material showed an excellent lithium-storage performance.
Experimental
Preparation of graphene
A modified electrochemical method was used for the bench scale preparation of graphene nanosheets. A schematic representation of the set-up used for the electrochemical process is shown in Fig. 1. The apparatus is comprised of a vertical tubular Inconel reactor, which is positioned inside a resistance furnace. The upper end of the reactor is closed with a stainless steel lid sealed with an O-ring and compression fittings. The lid is equipped with apertures for electrode leads and the thermocouple as well as with alumina tubes for the gas inlet and outlet. For electrolysis purposes, 1.2 kg of anhydrous lithium chloride, LiCl (Sigma-Aldrich), was placed in an alumina crucible with an internal diameter of 10 cm and a height of 20 cm. Two industrial-grade graphite rods (Goodfellow 809-013-12, diameter 1.3 cm, length 30 cm, purity 99.997%) were used as the cathode, and a graphite rod with the diameter of 2 cm was employed as the anode. The graphite electrodes were connected to a power supply (QPX600DP Dual 600 watt) with alumina shielded copper rods of 6 mm in diameter. At first, the temperature was raised to about 800 °C, where the LiCl is in the molten state, by a ramp of 5 °C min−1, under a flow of 200 cm3 min−1 of a gas mixture Ar–4%H2. Then the electrochemical process was carried out. First, the DC current diverter shown in Fig. 1 was adjusted so that only one of the 1.3 cm diameter graphite rods served as the working electrode, whilst the 2 cm diameter graphite rod served as the counter electrode. Under these conditions, a constant direct current of 40 A, corresponding to a cathode current density of about 1 A cm−2, was applied between two electrodes. Then in the intervals of about 60 min and for a total of 240 min, the power supply was turned off, and the other 1.3 cm diameter graphite rod was connected to the negative pole of the power supply by the application of a manual current diverter (see Fig. 1). Thereafter, the cell was cooled to room temperature, and the product obtained was retrieved from the solidified salt by washing with copious amounts of distilled water and vacuum filtering. The black powder obtained was dried at 100 °C. The final product was 70 g graphene nanosheets in the form of black fluffy powder.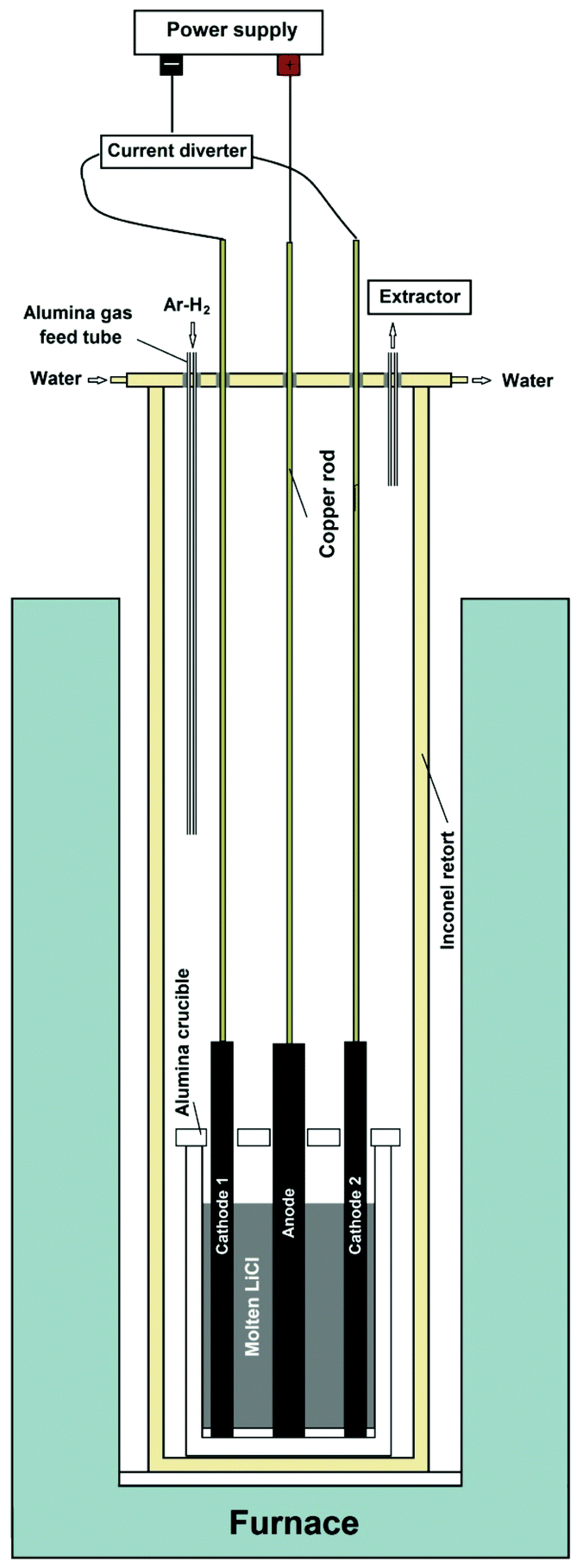 | ||
| Fig. 1 Schematic of the modified experimental setup used for the preparation of graphene. | ||
Preparation of SnO2 nanocrystals-loaded graphene nanosheets
Ten grams of anhydrous SnCl2 (Sigma Aldrich, 452335) was pressed into a pellet of 20 mm in diameter. The pellet was placed in an alumina crucible of 22 mm in diameter and 50 mm in height. 0.2 g graphene in the form of fluffy powder was placed on the top of the SnCl2 pellet in the crucible. A tube furnace equipped with an alumina tube was used for the reaction. The tube had a diameter of 80 mm and a length of 1.2 m, from which 0.9 m was inside the furnace. First, the furnace was heated up until the temperature at the centre of the tube rose to 580 °C, at which the temperature at either ends of the tube was about 50 °C. Then an air flow of 20 L min−1 was applied through the tube, and the crucible containing SnCl2 and graphene was pulled from one end of the alumina tube to the other end in about 25 min, corresponding to a heating/cooling rate of about 40 °C min−1. The product obtained was washed with distilled water in order to remove unreacted SnCl2, and then vacuum filtered and dried.Characterization methods
The morphology of the carbon materials was examined by using an FEI Nova Nano-SEM, a 200 kV JEOL 2000FX analytical transmission electron microscope (TEM), and a 200 kV FEI Tecnai F20 field emission gun high resolution TEM (HRTEM). A Philips 1710 X-ray diffractometer (XRD) with Cu-Kα radiation (wavelength = 1.54 Å) was used to record the diffraction patterns. XRD data were analysed using the X'Pert High Score Plus program. Raman data were collected using a Renishaw 1000 Ramanscope with a He–Ne ion laser of a wavelength of 633 nm (red, 1.96 eV). The electrical conductivity of graphene nanosheets produced was measured at room temperature using a four-probe conductivity measuring device (Guangzhou Kunde Technology Co. Ltd, China).In order to investigate the lithium storage performance of the SnO2-loaded graphene, an electrode was prepared using the SnO2-loaded graphene as the active material, and tested as anode for lithium ion batteries. For this, a slurry was prepared by mixing 85 wt% active materials, 7 wt% carbon black, and 8 wt% polyvinylidene fluoride in N-methyl pyrrolidinone, and pasted on a copper foil. After drying, the coated foil was calendared, and subsequently punched into a disk electrode with a diameter of 13 mm. The electrode was assembled into a 2025 coin-type cell in an Ar-filled glove box using Li-foil as the counter electrode and Celgard 2400 as the separator. The electrolyte was composed of 1 M LiPF6 dissolved in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) mixture of ethylene carbonate and diethyl carbonate. The cell was galvanostatically cycled between 0.01 and 3 V vs. Li/Li+ at a 1 C rate using a Neware multichannel battery tester.
:1 (v/v) mixture of ethylene carbonate and diethyl carbonate. The cell was galvanostatically cycled between 0.01 and 3 V vs. Li/Li+ at a 1 C rate using a Neware multichannel battery tester.
The graphene product and SnCl2 material were investigated by means of non-isothermal differential scanning calorimetry (DSC) using an SDT Q600 analyser equipped with alumina crucibles.
Results and discussion
Characterization of the graphite electrode material
The electrochemical conversion of graphite electrodes into graphene under an atmosphere of Ar–H2 is reported in this paper. A small piece of a graphite electrode used in the process was ground into powder with an agate mortar and pestle, and the powder obtained was investigated by means of SEM, XRD and Raman spectroscopy. The SEM micrographs, shown in Fig. 2, indicate the presence of planar grains of graphite flakes with diameters between 1 and several micrometres as well as more irregularly shaped carbon particles with diameters in the sub-micrometre range. | ||
| Fig. 2 SEM micrographs of the powdered graphite electrode material. | ||
The XRD spectrum recorded on the sample is shown in Fig. 3b. The reflection peaks were observed at 26.58°, 42.41°, 44.59°, 54.65°, 77.73° and 83.61° which could be attributed to the (002), (100), (101), (004), (110) and (112) crystalline reflections of hexagonal graphite, respectively. Based on the data obtained for the most intense (002) peak of the powdered graphite electrode material, the average crystalline domain size in the direction perpendicular to the (002) planes could be calculated to be 36.4 nm using the Scherrer's equation.55 The XRD pattern of highly crystalline natural graphite flakes (Alfa Aesar) is also shown in Fig. 3c for comparison. As can be seen, the intensity of the (002) reflection of natural graphite flakes is about 20 times more than that of the graphite electrode material, with an average crystalline domain size d002 of 41.5 nm, calculated from the XRD data. Considering the fact that the same instrument and the same sample holder were used for the XRD measurement, the much weaker (002) diffraction peak intensity in the powdered graphite electrode material indicates the presence of lower dimensional graphite crystals in the electrode material in comparison with natural graphite flakes which consist of highly oriented graphite crystallites. As suggested by the SEM and XRD results, the graphite electrode can be characterised by the presence of graphitic domains which are randomly orientated in the bulk graphite.
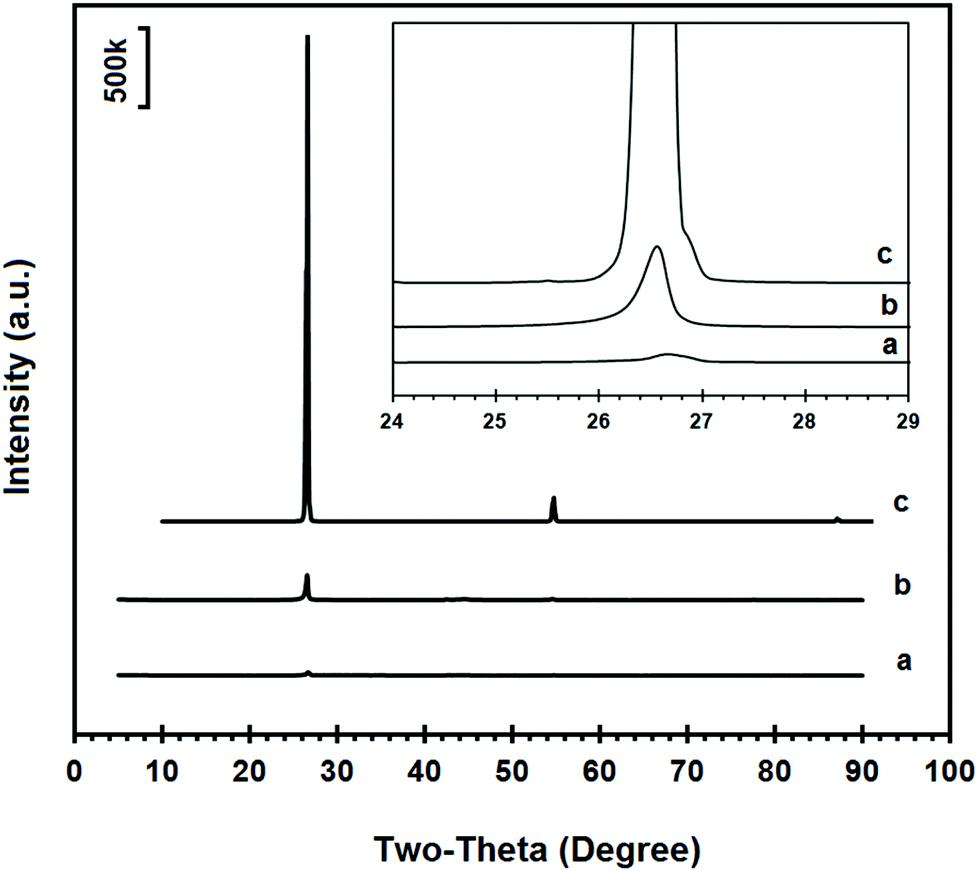 | ||
| Fig. 3 XRD diffraction spectra of (a) the graphene product, (b) powdered graphite electrode material and (c) natural graphite flakes. The inset reveals the higher magnification of XRD patterns around the most intense (002) peak. | ||
Raman spectroscopy provides useful information about the structural properties of carbon materials.56 The raw Raman spectra of the powdered graphite material and natural graphite flakes in the wavenumber range 100–3200 cm−1 are presented in Fig. 4b and c, respectively. The spectrum of the powdered graphite electrode is characterized by the presence of the so-called G, D and 2D bands. The Raman G band is related to the in-plane vibrational mode of the graphitic lattice. The D band is related to crystal defects and the 2D band is the second order of the D band. The Raman data obtained for the carbon materials are presented in Table 1. In graphitic materials, disorder is caused by the presence of lattice defects such as dislocations, crystallite boundaries, impurities and edges. Also the relative intensity ratio of the G band to the D band (IG/ID) indicates the in-plane structural order of carbon materials.56 The IG/ID value for the powdered graphite material is shown in Table 1. As can be seen, the D band is almost absent in the Raman spectrum of the natural graphite flakes, indicating that graphite flakes are graphite crystals with a very low density of structural defects. On the other hand, from the XRD and the Raman results, the graphite electrode material possesses hexagonal graphitic stacks of rather random orientation with crystalline defects.
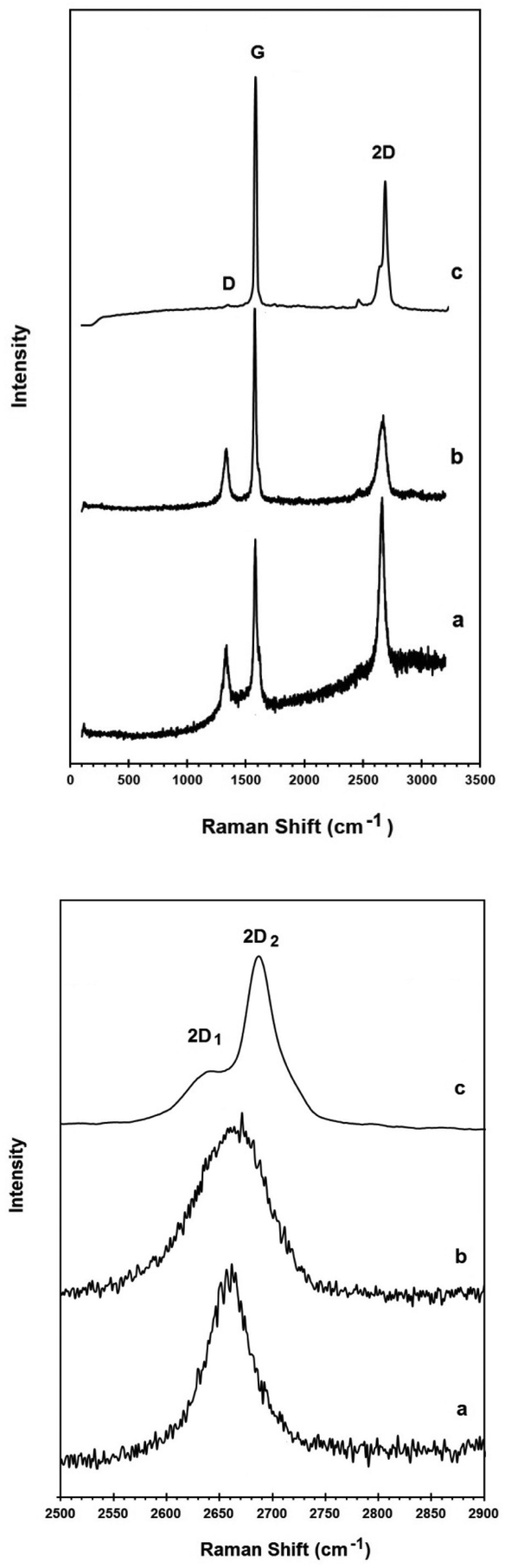 | ||
| Fig. 4 Raman spectrum of (a) the graphene product, (b) the powdered graphite electrode material and (c) natural graphite flakes. The upper panel shows the spectra in the wavenumber range 100–3200 cm−1. The down panel shows the 2D bands at a higher magnification presenting the peak shape in more detail. | ||
| Graphite electrode material | Natural graphite flakes | Graphene product | |
|---|---|---|---|
| D line frequency (cm−1) | 1330 | — | 1332 |
| G line frequency (cm−1) | 1576 | 1580 | 1580 |
| 2D line frequency (cm−1) | 2665 | 2687 | 2660 |
| I G/ID | 3.6 | — | 2.5 |
Fabrication of graphene nanosheets
Two graphite rods of 13 mm in diameter were used as the carbon source in the molten salt preparation of graphene. The apparatus used in this study is shown in Fig. 1, and explained in the Experimental section of the paper. As is shown, the electrochemical cell includes two working electrodes and one counter electrode. Fig. 5a shows the arrangement of graphite electrodes in the electrolysis cell. The alumina crucible containing the electrodes and LiCl was loaded into the molten salt reactor, shown in Fig. 1, and heated to 800 °C where LiCl is in the molten state. Then, the two graphite electrodes were alternatively connected to the negative pole of a DC power source. A third graphite rod (20 mm in diameter) was used as the counter electrode. The electrochemical process was carried out under a flow of argon containing 4% hydrogen. The process was begun by applying a constant electric current of 40 A between one of the working electrodes and the counter electrode. The current applied corresponded to a cathode current density of about 1 A cm−2. During the process, the two working electrodes were alternately connected to the negative pole of the power source in intervals. Fig. 6 shows the potential difference between the graphite electrodes and a Mo pseudo reference electrode immersed in the molten salt. | ||
| Fig. 5 (a) Photograph of the electrolysis cell. Two graphite rods served as alternative cathodes during the molten salt process. The arrangement of the electrodes in an alumina crucible containing LiCl, before loading into the molten salt reactor can be seen. (b) Photograph of the graphite electrodes after being used as the cathode during the molten salt process. Part of the graphite cathodes exposed to the molten salt (about 11.5 cm) was completely exfoliated into graphene. Distilled water was added to the alumina crucible in order to dissolve the solidified LiCl, resulting in the retrieval of 70 g graphene material, which was stored in a jar after vacuum filtration and drying. | ||
 | ||
| Fig. 6 The potential difference between two graphite cathodes (red and green lines) and a Mo pseudo-reference electrode. The blue line is the potential difference between the graphite counter electrode and the reference electrode. The electrodes were polarized at a constant current of 40 A. | ||
After the process, the molten salt was allowed to cool. It was observed that the solidified salt in the crucible was completely black and that the part of graphite cathodes exposed to the molten salt had disappeared as shown in Fig. 5b. These observations provided evidence that the cathode electrodes exposed to the molten LiCl were completely exfoliated, and the graphene product was thoroughly mixed with the salt. A sufficient amount of distilled water was added to the alumina crucible in order to dissolve solidified LiCl. The graphene product was retrieved from the back dispersion by vacuum filtering, and then allowed to dry.
Characterization of the graphene product
Fig. 7 and 8 show SEM and bright-field TEM images of the graphene nanosheets produced, respectively. The micrographs indicate the preparation of high yield randomly oriented graphene nanosheets with a lateral dimension up to several micrometres and an extremely high quality in appearance. | ||
| Fig. 7 SEM micrographs of the graphene product obtained by the molten salt process under a flow of Ar–H2. | ||
 | ||
| Fig. 8 Typical bright field TEM micrographs of graphene nanosheets produced in the molten salt under a flow of Ar–H2. A selected area diffraction pattern recorded on the edge of a nanosheet is shown as the inset exhibiting the typical six-fold symmetry expected for graphene. | ||
From high-resolution TEM observations, it was noted that the carbon material produced comprised few-layer graphene, typically between 1–10 layers. Fig. 9 shows a high resolution TEM image of the graphene nanosheets from which the presence of a number of single and double layer graphene sheets can be observed. A ten-layer graphene with a large number of lattice dislocations can also be seen in this micrograph. The presence of structural defects in both the graphite feed material and the graphene product was confirmed by the presence of the D band in the corresponding Raman spectra (Fig. 4).
 | ||
| Fig. 9 A high resolution TEM image of graphene nanosheets produced in molten salt under a flow of Ar–H2. | ||
The exfoliation of graphite in the molten salt will be discussed to be due to the diffusion of hydrogen into the interlayer space of graphite. It may be assumed that lattice dislocations impose a significant barrier for the diffusion of hydrogen into the structure of graphite. Thus further exfoliation of graphene flakes is difficult in regions with higher density of dislocations. It is, therefore, anticipated that a higher yield of single and double layer graphene can be produced by using a graphite feed material containing a lower density of lattice defects. It is currently being explored in our laboratory.
Fig. 3a compares the XRD pattern of the graphene nanosheets produced with the powdered graphite electrode material, as shown in Fig. 3b. The magnified (002) peaks are presented in the inset of Fig. 3. As can be seen, the intensity of the (002) peak in the graphene product is about one tenth of that of the powdered graphite material, indicating much less abundance of the close-packed hexagonal structure of carbon. This result reveals that the graphite electrode material was highly exfoliated to individual graphene sheets, although a small fraction of less-exfoliated graphene flakes might still exist in the sample.
Fig. 4 compares the Raman spectra of the powdered graphite electrode material, the graphene product and natural graphite flakes. The Raman data obtained are presented in Table 1.
As already mentioned, the D peak is absent in the Raman spectrum of the natural graphite, which is a characteristic of perfect hexagonal graphite crystals. This mode only becomes active in the presence of disorder and defects.60
In contrast, the D band is present in the Raman spectra of both the graphite electrode and the graphene product. The IG/ID ratio of the graphene product was calculated to be 2.5. Considering the fact that the edge of the graphene sheets contributes to the recorded Raman intensity of the D peak, the slightly smaller value of the IG/ID ratio in the graphene product in comparison with that of the powdered graphite material (3.6), therefore, is attributed to the higher density of graphene edges in the graphene product. However, the IG/ID ratio in the graphene product is still high and suggests that the nanosheets produced are composed of carbon crystallites with a large degree of crystallinity. It should be noted that, to the best of the author's knowledge, most of the methods used for the preparation of graphene from graphite employ specific grades of high quality graphite crystals.44,45,47,50,51,57–59 In the current paper, however, electrode grade graphite was employed as the carbon source. Despite this, the crystallinity of the graphene product is still high which demonstrates the high capability of the proposed approach in producing high quality graphene.
It is anticipated that graphene of various qualities can be produced by using graphite electrode materials with different morphological and structural qualities.
It is known that the 2D peak of graphitic materials is extremely sensitive to the number of layers. The 2D peak of bulk graphite materials is asymmetric consisting of two components, whilst the 2D peak of single-layer graphene is composed of a red shifted single peak.62
The down panel of Fig. 4 compares the 2D bands of the graphene product with the powdered graphite electrode material and natural graphite flakes. As can be seen, the 2D band in natural graphite flakes consists of the well-known 2D1 and 2D2 components, which is a characteristic feature of crystalline graphite. However, the 2D peak of the powdered graphite electrode material is more symmetric and also shifted to lower frequencies compared to that of the natural graphite, confirming the low dimensionality of its structure. This 2D Raman feature can lead to the conclusion that the powdered graphite electrode is basically made of stacks of a limited number of graphene layers, which is in excellent agreement with the X-ray diffraction results (Fig. 3). For single layer graphene, the 2D peak is expected to be a single symmetric peak.61 The Raman spectrum of the graphene product presented in Fig. 4, therefore, provides evidence that the product is mostly single or few layer graphene.
Possible mechanism involved in the formation of graphene
The formation of graphene nanosheets in molten salt, presented in this paper, can be explained by the effective exfoliation of graphite electrodes. At a constant current of about 1 A cm−2 and an applied voltage of average 5 V (Fig. 6), the electrolysis process is initiated by the decomposition of molten LiCl,| Li+ + e = Li (at the cathode) | (1) |
| Cl− = e + 1/2Cl2 (at the anode) | (2) |
The chlorine gas evolved from the anode reacts with H2 in the atmosphere above the molten salt to produce HCl, which can be subsequently dissolved in molten LiCl, according to
| 1/2H2 (g) + 1/2Cl2 (g) = [HCl]in LiCl | (3) |
It is known that HCl is highly soluble in LiCl-based molten salts and the dissolved HCl is ionised to produce protons and chloride ions.63–65 Hydrogen ions formed can then be reduced on the graphite cathode under the cathodic potential to form hydrogen atoms which can subsequently intercalate into the interlayer space between graphene layers of the graphite electrode. The combination of hydrogen atoms between the graphene layers of graphitic carbon forms hydrogen molecules which can lead to the peeling-off the graphene sheets due to their high kinetic energy. The mechanism proposed is illustrated in Fig. 10.
 | ||
| Fig. 10 Illustration of the mechanism involved in the preparation of graphene from graphite in molten LiCl. (a) Chlorine gas evolved from the anode during the initial electrolysis of LiCl reacts with H2 present in the atmosphere to form HCl, which can be subsequently dissolved in molten LiCl. Hydrogen ions formed can then be reduced on the cathode to form hydrogen atoms which can subsequently intercalate into the interlayer space between graphene layers of the graphite electrode. Combination of hydrogen atoms to form H2 in the interlayer space of graphite leads to the peeling-off the graphene sheets. The size of atoms and molecules of hydrogen permits their intercalation into the van der Waals gaps between graphene layers, as depicted in (b). | ||
It should be pointed out that the consumption of LiCl during the process is very small supporting the idea that the process mainly proceeds by the cathodic discharge of H+ and not Li+. To further confirm this, the molten salt process was conducted under the same conditions as in Fig. 1 and 5 with the only difference that pure dry argon was used instead of Ar–4%H2. Under these conditions, the final product contained carbon nanoparticles of mainly less than 100 nm and carbon nanotubes with a wide diameter range of 2–200 nm, as can be depicted from Fig. 11. This result is in agreement with the literature66 confirming that the interaction of lithium from molten LiCl with graphite cathodes immersed in the molten salt can lead to the formation of carbon nanotubes and nanoparticles. However, under dry argon conditions no graphene nanosheets, as observed in Fig. 7 and 8, could be produced. Thus, it is straightforward to attribute the formation of graphene nanosheets to the presence of hydrogen in the atmosphere.
 | ||
| Fig. 11 SEM (upper panel) and TEM (down panel) micrographs from carbon nanomaterials produced by the electrochemical erosion of graphite electrodes immersed in molten LiCl in dry pure argon. The product contains carbon nanoparticles and nanotubes with a wide diameter range of 2–200 nm, with no indication of the formation of graphene nanosheets as observed in Fig. 7 and 8. | ||
It is worth mentioning that the formation of chlorine bubbles on the graphite anode surface (reaction (2)) may increase the electrode resistance causing peaks in the anode potential-time curve, as observed in Fig. 6. This effect was reduced by selecting an appropriate graphite anode with a higher surface area than that of the graphite cathodes, as explained in the Experimental section.
It is important to note that the rate of the exfoliation process at the graphite cathode, leading to the formation of graphene, is very high. Graphite electrodes with an exposed surface area of 1 m2 are expected to produce about 2 kg graphene per hour. This high rate of exfoliation is caused by a high cathodic current density of 1 A cm−2, achievable at a low cell potential of about 5 V. It must be noted that the high current density reported here is at least an order of magnitude higher than that of the room temperature electrochemical exfoliation processes.44–52 This unique feature of the method proposed in this paper is due to the high diffusion coefficients of the species involved in the electrochemical reaction occurred. The diffusion coefficient of the protons arising from the dissolved HCl in molten LiCl is an order of magnitude higher than most other solutes in molten salts.63–65,67 Therefore, the hydrogen ions can easily travel to the graphite cathode. On the other hand, the diffusion coefficients of hydrogen atoms and molecules in graphite at 800 °C are very high having values of 3.3 × 10−5 cm2 s−1 and 3.5 × 10−4 cm2 s−1 respectively.68
It is well known that the room temperature electrolysis of water with graphite electrodes leads to the evolution of hydrogen at the graphite cathode. The question here is why the room temperature electrolysis doesn't lead to the intercalation of hydrogen in graphite and thus the exfoliation of graphite? To answer this question it should be pointed out that at room temperature, it takes 15 days for hydrogen atoms to diffuse several angstroms in the interlayer space of graphite.69 Therefore, hydrogen atoms formed at the graphite surface are very likely to combine to form hydrogen gas which then escapes from the surface. At 800 °C, however, hydrogen atoms need much less than a millisecond for the same length of diffusion.69 Therefore, hydrogen atoms reduced on the cathode surface are very likely to diffuse into the graphite cathode before getting combined to form H2. Formation of hydrogen molecules in the interlayer space of graphite then causes the exfoliation of graphite to form graphene. It is, in fact, the main reason why the efficient electrochemical exfoliation of graphite by hydrogen can be achieved only at high temperatures by the assistance of molten salts, as presented in this paper.
The electrical conductivity of graphene nanosheets produced was measured to be 5.8 × 105 S m−1, which is much greater than that of graphene oxide (0.5 S m−1),70 and also graphene powders prepared by the reduction of graphene oxide (2.0 × 104 S m−1),70 mechanical exfoliation of graphite (1.0 × 103 S m−1)71 and room temperature electrochemical exfoliation of expanded graphite (2.4 × 104).72 The excellent conductivity of graphene nanosheets produced makes the material a promising candidate as a key component of anode materials in high capacity lithium ion batteries, as explained in the next section.
Cycle performance of SnO2-loaded graphene as anode material for Li-ion batteries
Lithium-ion batteries are the first choice for personal electronics and most electric cars because of their high energy density and excellent cycling performance. The latter is mainly attributed to the excellent cycle stability of graphite which is traditionally used as anode in commercial lithium-ion batteries. However, the lithium storage capacity of graphite which is limited to 372 mA h g−1 cannot fulfil new requirements needing a high energy density. A number of materials with a Li storage capacity higher than graphite have been investigated as possible anode materials. Among them, SnO2 is one of the most promising candidates due to its high theoretical specific capacity of 789 mA h g−1. It, however, suffers from large volume changes as much as 300% associated with full lithium insertion and extraction processes leading to the loss of electrical contact and therefore failure of the electrode. The other limitation of SnO2 in this application associates with its poor electronic conductivity which negatively affects the electrochemical performance of the electrode. An effective strategy to tackle these restrictions is the incorporation of graphene with SnO2 nanoparticles.73–75 The cycling performance of SnO2 nanocrystals anchored on graphene nanosheets produced in molten salt is presented here.A green and simple strategy was used to prepare SnO2 loaded graphene nanosheets. This strategy is based on the recent findings that the oxidation of SnCl2 may lead to the formation of highly crystalline SnO2 nanostructures, which is explained by a gas–solid phase transition at the heating rates equal to or greater than 20 °C min−1 (ref. 76, 77) as follows:
| SnCl2 (g) + O2 (g) → SnO2 (s) + Cl2 (g) |
| ΔG° = −182 kJ (at 580 °C) | (4) |
Moreover, it is known that graphite particles with a surface area of 5 m2 g−1 can provide an appropriate surface for reaction (4) to occur.78 It was straightforward to assume that graphene nanosheets produced in this paper with a surface area of more than 500 m2 g−1 could efficiently catalyse reaction (4) in order to produce a high yield of SnO2-loaded graphene. The mechanism by which SnO2 nanocrystals are loaded on graphene sheets is schematically represented in Fig. 12a, and the experimental setup used to implement this mechanism is discussed in the Experimental section. The process is based on the evaporation and subsequent oxidation of SnCl2 on graphene sheets to form SnO2 anchored graphene. The thermal stability in air of the graphene nanosheets as well as the phase transformation temperatures of SnCl2 can be seen from their DSC thermograms shown in Fig. 12b. The air oxidation of graphene nanosheets can be measured from the corresponding DSC curve to be 663 °C. A weight loss of about 96% was observed at 1000 °C, and Fig. 13 shows the SEM micrograph of the residue. This sample is characterised by micrometre-sized particles forming aggregated structures, and bears no morphological resemblance to graphene nanosheets. Energy-dispersive X-ray analysis of the residue demonstrated the presence of C, O, Cl, K, Ca and S, which are likely to originate from graphene, the oxygen from the environment, salt and also impurities in the graphite raw material.
 | ||
| Fig. 12 (a) The mechanism of the formation of SnO2 nanocrystals anchored on graphene nanosheets. SnCl2 thermally evaporates and rises through the graphene nanosheets. Graphene surfaces act as the catalyst for the oxidation of SnCl2 to form SnO2 nanocrystals attached to the graphene layers. (b) DSC curves of graphene and SnCl2. The endothermic peaks are upward. The curves were obtained in 100 ml min−1 air flow at 40 °C min−1. | ||
 | ||
| Fig. 13 SEM micrograph of the residue remaining after heating the graphene nanosheets to 1000 °C in 100 ml min−1 air flow at 40 °C min−1. | ||
The DSC thermogram of SnCl2 shows two endothermic peaks at 269 and 568 °C which are due to the melting and evaporation of SnCl2, respectively. These indicate that graphene nanosheets are stable at temperatures below 600 °C and therefore can play a catalytic role in enhancing reaction (4). This method is not only simple, inexpensive and green, but also produces a high quality composite material in which highly crystalline SnO2 nanocrystals of 5–20 nm in size are anchored on graphene nanosheets, as can be depicted from Fig. 14a–d. The X-ray diffraction pattern of the prepared hybrid material is shown in Fig. 14e showing the diffraction peaks of tetragonal SnO2.
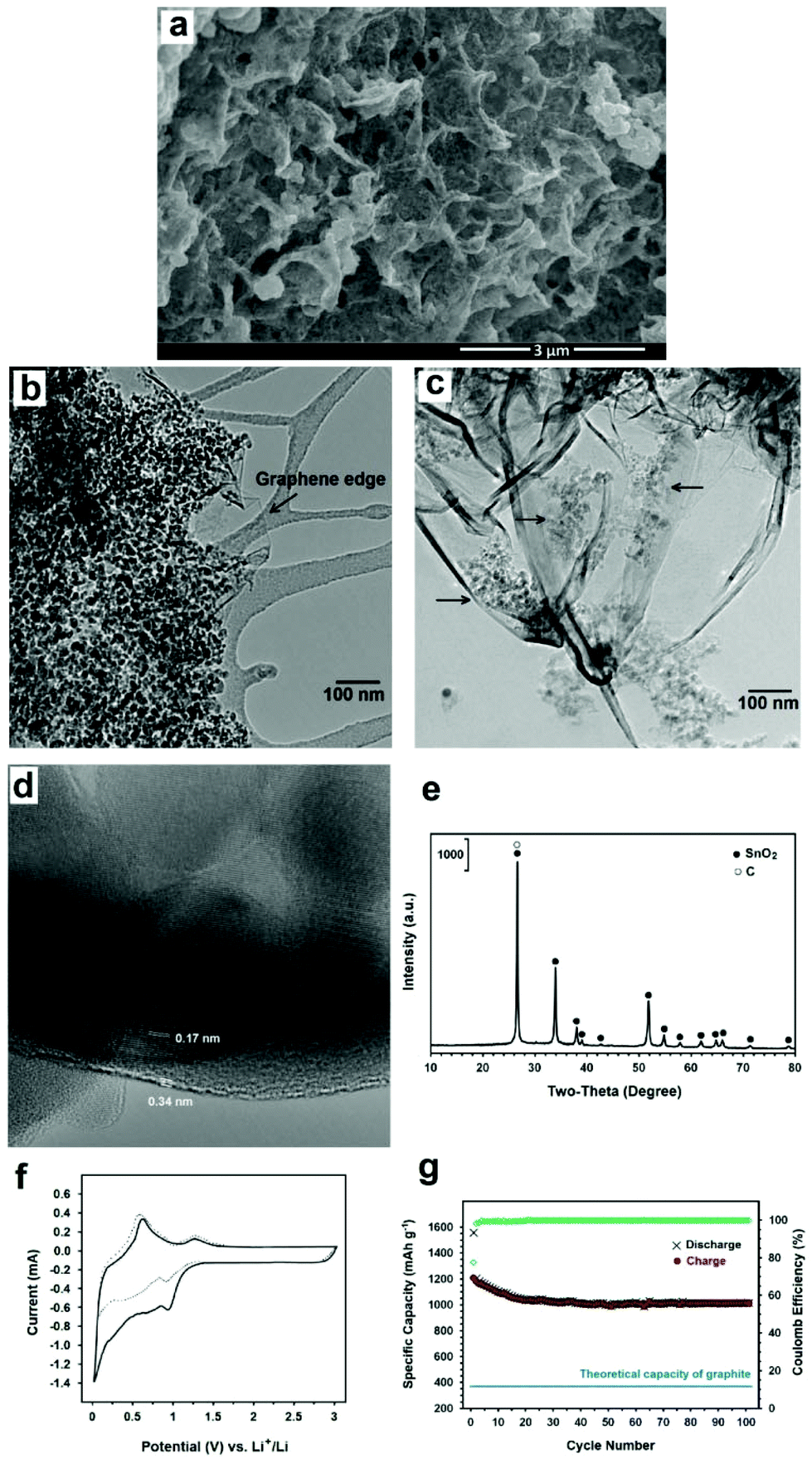 | ||
| Fig. 14 Morphology and electrochemical performance of the SnO2 anchored graphene. (a) SEM and (b and c) TEM micrographs. An unloaded edge of graphene is indicated in (b). A less loaded section of the sample is exhibited in (c) showing separated clusters of SnO2 nanocrystals. The clusters are still in electronic contact with each other through the graphene sheets. (d) A high resolution TEM micrograph exhibiting the presence of a SnO2 nanocrystal on a few layer graphene sheet. It demonstrates close contact between the SnO2 nanoparticle and the graphene layer. The interplanar distances of 0.17 and 0.34 nm are attributed to the (211) and (002) planes of tetragonal SnO2 and hexagonal graphite, respectively. (e) XRD pattern. (f) CV curves of the first (solid line) and the second (dotted line) cycles of the electrode. (g) Lithium charge–discharge performance and coulomb efficiency in 100 cycles at 1 C. | ||
The electrochemical performance of the composite material produced as the anode active material for Li ion batteries was characterised using a coin-cell with lithium metal as the counter-electrode.
Fig. 14f shows the cyclic voltammograms of the electrode containing SnO2-loaded graphene during the first and second sweep at a scanning rate of 500 mA g−1 in the potential range of 0.003–3 V versus Li+/Li. In the first cycle, two cathodic waves are observed. The first cathodic peak at about 0.90 V can be attributed to the reduction of SnO2 to Sn and the synchronous formation of Li2O as well as the formation of solid electrolyte interface (SEI) layers at the surface of active materials. The second cathodic peak at about 0.02 V is ascribed to the formation of LixSn intermetallics up to Li4.4Sn stoichiometry. Moreover, two anodic peaks can be seen during the first scanning process. The first anodic peak at about 0.62 V corresponds to the lithium extraction from graphene layers and the decomposition of LixSn intermetallics. The second anodic peak at about 1.27 is attributed to the reaction between Li2O and Sn to form SnO2..73–75 The coulomb efficiency of the first cycle is 77% which increases to above 99% after 4 cycles.
Fig. 14g exhibits the cycling performance of the composite. The discharge specific capacity at a current density of 1 C is 1016 mA h g−1 after 100 cycles, which is much higher than the theoretical capacity of graphite, the commercial anode material in Li-ion batteries.
It should be mentioned that although SnO2 nanocrystals are known to have a high theoretical specific capacity, their electrochemical performance is poor, reaching less than 200 mA h g−1 after 100 cycles79 due to the poor conductivity and also the large volume change of SnO2 during charge–discharge processes, leading to fatigue failure and disintegration of the electrode.
The high performance of the SnO2-graphene composite prepared in this paper is attributed to the presence of graphene nanosheets which provide excellent electronic contact between individual SnO2 particles and clusters, overcoming the loss of the mechanical and electronic integrity of the active material over charge–discharge cycling.
The method presented in this paper can provide a green, effective, and economical strategy for the preparation of high quality graphene, with high electronic conductivity and thermal stability. It is also worth mentioning that consumables used for the production of graphene by this method comprise graphite, hydrogen and electrical energy, bearing in mind that LiCl is not considerably consumed during the process and therefore can be recovered and reused. The specific energy consumption can be estimated to be approximately 25 kWh kg−1. If we consider the current average world price of electricity and the graphite electrode to be about 20 US cents per kWh and US $4000 per metric tons, respectively, it might be possible to estimate the cost of producing high quality graphene to be about US $10–20 kg−1. These characteristics can make the graphene product attractive for many applications.
The graphene product exhibited impressive performance in other applications such as graphene–Si composite anode materials for advanced Li-ion batteries, supercapacitor electrode materials, graphene–polymer and graphene–ceramic composites. These applications will be discussed separately in detail in subsequent publications.
Conclusions
Industrial-grade graphite electrodes can be peeled off into single or few layer graphene when cathodically polarized in molten LiCl under an atmosphere of Ar–H2. At first, LiCl is decomposed under the influence of the potential difference to form Cl2, which evolves from the anode and subsequently reacts with H2 present in the atmosphere to form HCl. The dissolution of HCl in molten LiCl leads to the formation of H+ which is subsequently reduced on the graphite cathode to form atomic and then molecular hydrogen, leading to the exfoliation of graphite to high quality graphene nanosheets. Catalytic oxidation of SnCl2 vapour on the graphene nanosheets led to the formation of SnO2 nanocrystals anchored on the graphene nanosheets. The resultant composite exhibited an impressive lithium storage performance.References
- P. Avouris and C. Dimitrakopoulos, Mater. Today, 2012, 15, 86 CrossRef CAS
.
- A. H. Castro Neto, Mater. Today, 2010, 13, 12 CrossRef
- D. Berman, A. Erdemir and A. V. Sumant, Mater. Today, 2014, 17, 32 CrossRef
- B. Zhang, Y. Yu, Y. Liu, Z. D. Huang, Y. He and J. K. Kim, Nanoscale, 2013, 5, 2100 RSC
- G. Kucinskis, G. Bajars and J. Kleperis, J. Power Sources, 2013, 240, 66 CrossRef CAS
- D. Prasai, J. C. Tuberquia, R. R. Harl, G. K. Jennings and K. I. Bolotin, ACS Nano, 2012, 6, 1102 CrossRef CAS PubMed
- L. Huang, Y. Huang, J. Liang, X. Wan and Y. Chen, Nano Res., 2011, 4, 675 CrossRef CAS
- D. Berman, A. Erdemir and A. V. Sumant, Mater. Today, 2014, 17, 32 CrossRef
- Z. Yin, J. Zhu, Q. He, X. Cao, C. Tan, H. Chen, Q. Yan and H. Zhang, Adv. Energy Mater., 2014, 4, 1300574 Search PubMed
- Y. Tu, M. Lv, P. Xiu, T. Huynh, M. Zhang, M. Castelli, Z. Liu, Q. Huang, C. Fan, H. Fang and R. Zhou, Nat. Nanotechnol., 2013, 8, 594 CrossRef CAS PubMed
- H. J. Choi, S. M. J. Min Seo, D. W. Chang, L. Dai and J. B. Baek, Nano Energy, 2012, 1, 534 CrossRef CAS
- L. Weinstein and R. Dash, Mater. Today, 2013, 16, 356 CrossRef
- Y. Zhou, C. H. Yen, S. Fu, G. Yang, C. Zhu, D. Du, P. C. Wo, X. Cheng, J. Yang, C. M. Wai and Y. Lin, Green Chem., 2015, 17, 3552 RSC
- T. Kuilla, S. Bhadra, D. Yao, N. H. Kim, S. Bose and J. H. Lee, Prog. Polym. Sci., 2010, 35, 1350 CrossRef CAS
- H. Porwal, S. Grasso and M. J. Reece, Adv. Appl. Ceram., 2013, 112, 443 CrossRef CAS
- H. Wu, H. Li, G. Sun, S. Ma and X. Yang, J. Mater. Chem. C, 2015, 3, 5457 RSC
- S. Yang, P. Zhou, L. Chen, Q. Sun, P. Wang, S. Ding, A. Jiang and D. Wei Zhang, J. Mater. Chem. C, 2014, 2, 8042 RSC
- K. M. Cho, K. H. Kim, H. O. Choi and H. T. Jung, Green Chem., 2015, 17, 3972 RSC
- K. A. Mahmoud, B. Mansoor, A. Mansour and M. Khraisheh, Desalination, 2014, 356, 209 Search PubMed
- J. G. Yu, L. Y. Yu, H. Yang, Q. Liu, X. H. Chen, X. Y. Jiang, X. Q. Chen and F. P. Jiao, Sci. Total Environ., 2015, 502, 70 CrossRef CAS PubMed
- A. K. Geim, Science, 2009, 324, 1530 CrossRef CAS PubMed
- V. Nicolosi, M. Chhowalla, M. G. Kanatzidis, M. S. Strano and J. N. Coleman, Science, 2013, 340, 1226419 CrossRef
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228 RSC
- Y. Hong, Z. Wang and X. Jin, Sci. Rep., 2013, 3, 3439 Search PubMed
- J. Chen, B. Yao, C. Li and G. Shi, Carbon, 2013, 64, 225 CrossRef CAS
- M. Yi and Z. Shen, Carbon, 2014, 78, 622 CrossRef CAS
- Y. Hernandez, V. Nicolosi, M. Lotya, F. M. Blighe, Z. Sun, S. De, I. T. McGovern, B. Holland, M. Byrne, Y. K. Gun-Ko, J. J. Boland, P. Niraj, G. Duesberg, S. Krishnamurthy, R. Goodhue, J. Hutchison, V. Scardaci, A. C. Ferrari and J. N. Coleman, Nat. Nanotechnol., 2008, 3, 563 CrossRef CAS PubMed
- J. S. Bunch, Y. Yaish, M. Brink, K. Bolotin and P. L. McEuen, Nano Lett., 2005, 5, 287 CrossRef CAS PubMed
- P. Blake, P. D. Brimicombe, R. R. Nair, T. J. Booth, D. Jiang, F. Schedin, L. A. Ponomarenko, S. V. Morozov, H. F. Gleeson, E. W. Hill, A. K. Geim and K. S. Novoselov, Nano Lett., 2008, 8, 1704 CrossRef PubMed
- J. N. Coleman, Acc. Chem. Res., 2013, 46, 14 CrossRef CAS PubMed
- W. A. de Heer, C. Berger, X. Wu, P. N. First, E. H. Conrad, X. Li, T. Li, M. Sprinkle, J. Hass, M. L. Sadowski, M. Potemski and G. Martinez, Solid State Commun., 2007, 143, 92 CrossRef CAS
- K. S. Kim, Y. Zhao, H. Jang, S. Y. Lee, J. M. Kim, K. S. Kim, J. H. Ahn, P. Kim, I. Y. Choi and B. H. Hong, Nature, 2009, 457, 706 CrossRef CAS PubMed
- P. W. Sutter, J. I. Flege and E. A. Sutter, Nat. Mater., 2008, 7, 406 CrossRef CAS PubMed
- J. Li, X. Y. Wang, X. R. Liu, Z. Jin, D. Wang and L.-J. Wan, J. Mater. Chem. C, 2015, 3, 3530–3535 RSC
- I. Y. Jeon, H. J. Choi, S. M. Jung, J. M. Seo, M. J. Kim, L. Dai and J. B. Baek, J. Am. Chem. Soc., 2013, 135, 1386–1393 CrossRef CAS PubMed
- Y. Chen, H. Zhao, L. Sheng, L. Yu, K. An, J. Xu, Y. Ando and X. Zhao, Chem. Phys. Lett., 2012, 538, 72 CrossRef CAS
- B. Subramanya and D. K. Bhat, J. Power Sources, 2015, 75, 90 CrossRef
- K. R. Paton,
et al.
, Nat. Mater., 2014, 13, 624 CrossRef CAS PubMed
- Y. Lin, J. Jin, O. Kusmartsevab and M. Song, J. Phys. Chem. C, 2013, 117, 17237 CAS
- EPA (U.S. Environmental Protection Agency), N,N-Dimrthylformide, Revised in 2000, Integrated Risk Information System[online].
- K. Savaram, M. Kalyanikar, M. Patel, R. Brukh, C. R. Flach, R. Huang, M. R. Khoshi, R. Mendelsohn, A. Wang, E. Garfunkel and H. He, Green Chem., 2015, 17, 869 RSC
- M. K. P. Kumar, M. Nidhi and C. Srivastava, RSC Adv., 2015, 5, 24846 RSC
- A. Wang, E. Garfunkel and H. He, Green Chem., 2015, 17, 869 RSC
- K. Parvez, Z. S. Wu, R. Li, X. Liu, R. Graf, X. Feng and K. S. Mullen, J. Am. Chem. Soc., 2014, 136, 6083 CrossRef CAS PubMed
- K. Chen and D. Xue, J. Colloid Interface Sci., 2014, 436, 41 CrossRef CAS PubMed
- K. Parvez, R. Li, S. R. Puniredd, Y. Hernandez, F. Hinkel, S. Wang, X. Feng and K. Müllen, ACS Nano, 2013, 7, 3598 CrossRef CAS PubMed
- A. Taheri Najafabadi and E. Gyenge, Carbon, 2014, 71, 58 CrossRef
- C. T. J. Low, F. C. Walsh, M. H. Chakrabarti, M. A. Hashim and M. A. Hussain, Carbon, 2013, 54, 1 CrossRef CAS
- J. Wang, K. K. Manga, Q. Bao and K. P. Loh, J. Am. Chem. Soc., 2011, 113, 8888 CrossRef PubMed
- A. J. Cooper, N. R. Wilson, I. A. Kinloch and R. A. W. Dryfe, Carbon, 2014, 66, 340 CrossRef CAS
- A. M. Abdelkader, I. A. Kinloch and R. A. W. Dryfe, ACS Appl. Mater. Interfaces, 2014, 6, 1632 CAS
- M. Xu, H. Sun, C. Shen, S. Yang, W. Que, Y. Zhang and X. Song, Nano Res., 2015, 8, 801 CrossRef CAS
- Y. L. Zhong, Z. Tian, G. P. Simon and D. Li, Mater. Today, 2015, 18, 73 CrossRef CAS
- A. R. Kamali and D. J. Fray, Nanoscale, 2015, 7, 11310 RSC
- J. I. Langford and A. J. C. Wilson, J. Appl. Crystallogr., 1978, 11, 102 CrossRef CAS
- M. S. Dresselhaus, A. Jorio and R. Saito, Annu. Rev. Condens. Matter Phys., 2010, 1, 89 CrossRef CAS
- N. I. Kovtyukhova, Y. Wang, A. Berkdemir, R. Cruz-Silva, M. Terrones, V. H. Crespi and T. E. Mallouk, Non-oxidative intercalation and exfoliation of graphite by Brønsted acids, Nat. Chem., 2014, 6, 957 CrossRef CAS PubMed
- Y. Sugiyama, O. Kubo, R. Omura, M. Shigehara, H. Tabata, N. Mori and M. Katayama, Appl. Phys. Lett., 2014, 105, 123116 CrossRef
- C. Shih, A. Vijayaraghavan, R. Krishnan, R. Sharma, J. Han, M. Ham, Z. Jin, S. Lin, G. L. C. Paulus, N. F. Reuel, Q. H. Q. H. Wang, D. Blankschtein and M. S. Strano, Nat. Nanotechnol., 2011, 6, 439 CrossRef CAS PubMed
- J. C. Meyer, A. K. Geim, M. I. Katsnelson, K. S. Novoselov, T. J. Booth and S. Roth, Nature, 2007, 446, 60 CrossRef CAS PubMed
- A. C. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter, 2001, 61, 14095 CrossRef
- A. C. Ferrari, Solid State Commun., 2007, 143, 47 CrossRef CAS
- N. Q. Minh and B. J. Welch, Aust. J. Chem., 1975, 28, 965 CrossRef
- N. Q. Minh and B. J. Welch, Aust. J. Chem., 1975, 28, 2579 CrossRef
- N. Q. Minh and B. J. Welch, J. Electroanal. Chem., 1978, 92, 179 CrossRef
- A. R. Kamali and D. J. Fray, Carbon, 2014, 77, 835 CrossRef CAS
- J. D. Van Norman and R. J. Tivers, J. Electrochem. Soc., 1971, 118, 258 CrossRef CAS
- C. P. Herrero and R. Ramirez, J. Phys. D: Appl. Phys., 2010, 43, 255402 CrossRef
- M. Warrier, R. Schneider, E. Salonen and K. Nordlund, Nucl. Fusion, 2007, 47, 1656 CrossRef CAS
- Z. Ji, J. Chen, L. Huang and G. Shi, Chem. Commun., 2015, 51, 2806 RSC
- W. Gao, L. B. Alemany, L. Ci and P. M. Ajayan, Nat. Chem., 2009, 1, 403 CrossRef CAS PubMed
- L. Wu, W. Li, P. Li, S. Liao, S. Qiu, M. Chen, Y. Guo, Q. Li, C. Zhu and L. Liu, Small, 2014, 10, 1421 CrossRef CAS PubMed
- P. Roy and S. K. Srivastava, J. Mater. Chem. A, 2015, 3, 2454 CAS
- A. R. Kamali and D. J. Fray, J. New Mater. Electrochem. Syst., 2010, 13, 147 CAS
- J. S. Chen and X. W. Lou, Small, 2013, 9, 1877 CrossRef CAS PubMed
- A. R. Kamali, J. Therm. Anal. Calorim., 2014, 118, 99 CrossRef CAS
- A. R. Kamali, G. Divitini, C. Ducati and D. J. Fray, Ceram. Int., 2014, 40, 8533 CrossRef CAS
- A. R. Kamali and D. J. Fray, Mater. Sci. Eng., B, 2012, 177, 819 CrossRef CAS
- W. Zhou, J. Wang, F. Zhang, S. Liu, J. Wang, D. Yin and L. Wang, Chem. Commun., 2015, 51, 36603662 Search PubMed
| This journal is © The Royal Society of Chemistry 2016 |