
Efficient conversion of carbon dioxide at atmospheric pressure to 2-oxazolidinones promoted by bifunctional Cu(II)-substituted polyoxometalate-based ionic liquids†
Mei-Yan
Wang
,
Qing-Wen
Song
,
Ran
Ma
,
Jia-Ning
Xie
and
Liang-Nian
He
*
State Key Laboratory and Institute of Elemento-Organic Chemistry, Collaborative Innovation Center of Chemical Science and Engineering, Nankai University, Tianjin 300071, P. R. China. E-mail: heln@nankai.edu.cn; Fax: (+86) 22-23503878; Tel: (+86) 22-23503878
First published on 23rd October 2015
Abstract
Copper(II) substituted polyoxometalate-based ionic liquids e.g. [(nC7H15)4N]6[α-SiW11O39Cu] were successfully developed as halogen-free bifunctional catalysts for the carboxylative cyclization of propargylic amines with CO2. Such a CO2 fixation protocol proceeded smoothly at atmospheric pressure under solvent-free conditions, in an environmentally benign and low energy-input manner. Notably, various propargylic amines could react smoothly to afford 2-oxazolidinones as the target products in high to quantitative yields. Furthermore, the dual activation of both propargylic amine and CO2 by [(nC7H15)4N]6[α-SiW11O39Cu] was studied using NMR techniques and control experiments.
Introduction
Carbon dioxide chemistry has drawn much attention in the last two decades as CO2 is an abundant, nontoxic, non-flammable, easily available, and renewable C1 resource. Various useful chemicals, such as dimethyl carbonate,1 urethanes,2 formic acid,3 methanol,4 cyclic carbonates,5 polycarbonates,6 and others, have been prepared by using CO2 as a feedstock. However, compared with traditional C1 sources (e.g., carbon monoxide and phosgene), CO2 is thermodynamically stable and kinetically inert, which is still the main obstacle in developing practical processes to convert CO2 into useful chemicals. Therefore, the exploration of efficient catalysts would be crucial to realize CO2 conversion.2-Oxazolidinones are important heterocyclic chemicals playing a significant role as chemical intermediates7 and chiral auxiliaries8 in organic synthesis, and as antibacterial drugs9 in pharmaceutical chemistry. Therefore, many efforts have been made to synthesize these useful heterocycles, such as catalytic asymmetric intermolecular aminopalladation10 and a formal [3 + 2] cycloaddition reaction.11 Recently, the employment of CO2 as a C1 building block to construct oxazolidinone motifs has attracted considerable attention.12 One of the most promising examples of CO2 fixation to access 2-oxazolidinones is the carboxylative cyclization of propargylic amines with CO2 (Scheme 1), which represents an important clean and atom economical reaction. As early as in 1964, Dimroth and Pasedach developed a practicable copper-promoted carboxylative cyclization protocol with benzene or tetrahydrofuran as a solvent.13 Subsequently, numerous catalytic systems including super bases,14 silver,15t-BuOI,16 and N-heterocyclic carbene–Au complexes17 have been developed. Very recently, Han and co-workers reported an effective protic ionic liquid as both a non-metal catalyst and solvent for the carboxylative cyclization of propargylic amines with CO2.12a They demonstrated that the activation of the amino group is crucial and the intramolecular cyclization is the rate-determining step. Despite the significant advances in the past few decades, the example of CO2 utilization at atmospheric pressure is limited. Therefore, development of alternative methodologies for the synthesis of 2-oxazolidinones using CO2 under mild conditions is still highly desirable.
 | ||
| Scheme 1 Carboxylative cyclization of propargylic amines with CO2. | ||
Polyoxometalate (POM), as a class of large clusters of metal–oxide units, has been widely used in many fields due to their excellent properties.18 For example, behaving as an oxygen-rich structure, heteropoly acid anions have rich negative charge on the O atom and strong nucleophilicity, leading to being able to activate CO2 molecules via the formation of the CO2-adduct.19,20 Recently, polyoxometalate-based ionic liquids have received considerable attention in epoxidation,21 deep desulfurization of fuels,22 esterification,23 and so on.24 Notably, excellent POM-based catalysis has also been applied in the cycloaddition of epoxides and CO2,25 direct conversion of styrene into styrene carbonate,26 and reduction of CO2.19
On the other hand, copper salts are widely employed as activators of the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bond in organic synthesis.27 We envisioned that the POM anion could activate both the N–H bond of propargylic amines and CO2, leading to the generation of the carbamate intermediate, the CC bond of which is activated by the Cu(II) species, and thus promotes the intramolecular nucleophilic cyclization to afford 2-oxazolidinones. As expected, [(nC7H15)4N]6[α-SiW11O39Cu] could serve as a well-defined single-component bifunctional catalyst for simultaneously activating propargylic amines as well as CO2 molecules (Scheme 2), and thus helped the reaction to efficiently proceed at low CO2 pressure. Furthermore, the cations and anions of [(nC7H15)4N]6[α-SiW11O39Cu] have synergistic effects in catalyzing the reaction.
C bond in organic synthesis.27 We envisioned that the POM anion could activate both the N–H bond of propargylic amines and CO2, leading to the generation of the carbamate intermediate, the CC bond of which is activated by the Cu(II) species, and thus promotes the intramolecular nucleophilic cyclization to afford 2-oxazolidinones. As expected, [(nC7H15)4N]6[α-SiW11O39Cu] could serve as a well-defined single-component bifunctional catalyst for simultaneously activating propargylic amines as well as CO2 molecules (Scheme 2), and thus helped the reaction to efficiently proceed at low CO2 pressure. Furthermore, the cations and anions of [(nC7H15)4N]6[α-SiW11O39Cu] have synergistic effects in catalyzing the reaction.
 | ||
| Scheme 2 Interaction of [(nC7H15)4N]6[α-SiW11O39Cu] with CO2 and propargylic amines. | ||
Results and discussion
A series of transition-metal-substituted polyoxometalate-based ionic liquids [(nC7H15)4N]6[α-SiW11O39M] (M = Cu(II), Co(II), Fe(II), Ni(II), Zn(II), Mn(II)) were synthesized according to the published procedures (see the ESI†).20a,28 The carboxylative cyclization of N-benzylprop-2-yn-1-amine (1a) was initially chosen as the model reaction under a CO2 balloon in the absence of any solvent at 60 °C as summarized in Table 1. The reaction without a catalyst was carried out only to recover the starting material quantitatively (entry 1). (nC7H15)4NBr and K8[α-SiW11O39] were found to be ineffective (entries 2 and 3). Interestingly, K6[α-SiW11O39Cu], CuCl2, and a mixture of CuCl2 and (nC7H15)4NBr showed moderate activity (entries 4–6), thus indicating the promotion effect of Cu(II) as expected. It was also worth mentioning that K8[α-SiW11O39] allowed the reaction to produce the carbamate rather than 2-oxazolidinone 2a (entry 3, see ESI, Scheme S1†), also supporting the activation of both propargylic amines and CO2 by POM anions. These inspired us to investigate the catalysis of [(nC7H15)4N]6[α-SiW11O39Cu]. Gratifyingly, 96% yield of 3-benzyl-5-methyleneoxazolidin-2-one (2a) was obtained under the given conditions (entry 7). In this context, several transition metal-substituted polyoxometalate-based ionic liquids [(nC7H15)4N]6[α-SiW11O39M] [M = Co(II), Fe(II), Ni(II), Zn(II), Mn(II)] were also screened. Among the tested [(nC7H15)4N]6[α-SiW11O39M], [(nC7H15)4N]6[α-SiW11O39Cu] showed much higher catalytic activity than Co(II), Fe(II), Ni(II), Zn(II), Mn(II) counterparts under identical reaction conditions (entry 7 vs. 8–12), indicating efficient activation ability of Cu(II) in the [α-SiW11O39Cu]6− on the CC bond.
|
|
|||
|---|---|---|---|
| Entry | Cat./% | Conv./% | Yieldb/% |
| a Unless otherwise specified, the reactions were performed with 1a (145.2 mg, 1.0 mmol), catalyst (molar percentage to 1a, 2.5 mol%), a CO2 balloon, 60 °C, 20 h. b Determined by the 1H NMR technique, using 1,1,2,2-tetrachloroethane as an internal standard. c A mixture of CuCl2 (2.5 mol%) + (nC7H15)4NBr (15 mol%). d 25 °C. | |||
| 1 | — | 0 | 0 |
| 2 | (nC7H15)4NBr | 0 | 0 |
| 3 | K8[α-SiW11O39] | 0 | 0 |
| 4 | K6[SiW11O39Cu] | 45 | 43 |
| 5 | CuCl2 | 78 | 67 |
| 6c | CuCl2 + (nC7H15)4NBr | 91 | 68 |
| 7 | [(nC7H15)4N]6[α-SiW11O39Cu] | >99 | 96 |
| 8 | [(nC7H15)4N]6[α-SiW11O39Co] | 70 | 66 |
| 9 | [(nC7H15)4N]6[α-SiW11O39Fe] | 18 | 18 |
| 10 | [(nC7H15)4N]6[α-SiW11O39Ni] | 18 | 16 |
| 11 | [(nC7H15)4N]6[α-SiW11O39Zn] | 45 | 45 |
| 12 | [(nC7H15)4N]6[α-SiW11O39Mn] | 47 | 43 |
| 13 | [(nC4H9)4N]6[α-SiW11O39Cu] | >99 | 95 |
| 14 | [(nC16H33)(CH3)3N]6[α-SiW11O39Cu] | 94 | 86 |
| 15d | [(nC7H15)4N]6[α-SiW11O39Cu] | 79 | 73 |
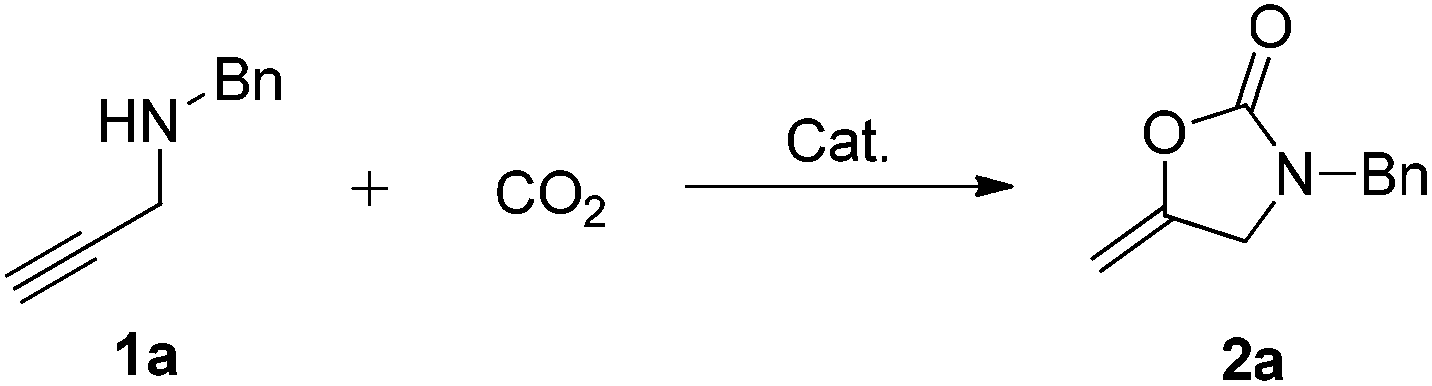
On the other hand, the (nC7H15)4N+ was found to be favorable for this reaction in comparison with potassium cations (entry 7 vs. 4). In addition, other bulky tetraalkylammonium cations such as (nC4H9)4N+ and (nC16H33)(CH3)3N+ also gave good results (entries 13 and 14). This is understandable because the bulky tetraalkylammonium cation can not only stabilize the carbamate intermediate, but enhance its solubility and O-nucleophilicity, and thus promote subsequent intramolecular nucleophilic cyclization resulting in the formation of 2-oxazolidinones.29 In addition, the bulky tetraalkylammonium cation increases the solubility of the polyoxometalate compared with the potassium cation, leading to higher catalytic activity. Moreover, owing to efficient catalytic activity of [(nC7H15)4N]6[α-SiW11O39Cu], the reaction could also proceed well even at ambient temperature (entry 15).
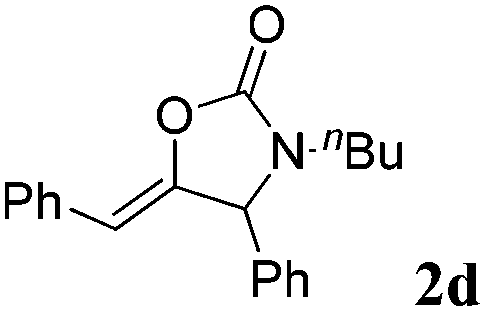
The carboxylative cyclization for a variety of propargylic amines was then undertaken to explore the scope of this well-developed [(nC7H15)4N]6[α-SiW11O39Cu] catalytic system. As shown in Table 2, both terminal and internal propargylic amines performed smoothly to give the corresponding 2-oxazolidinones in excellent yields (entries 1–7). In general, substituents at the propargylic position of propargylic amines have a remarkable effect on the reaction outcome, where the geminal dialkyl inhibited the reaction, probably due to steric hindrance (entry 8). Notably, para-substituents at the benzene ring of the terminal position increased the reactivity for the carboxylative cyclization reaction for an obviously shortened reaction time (entries 6, 7 vs. 3).
|
|
|||
|---|---|---|---|
| Entry | Substrate | Product | Yielde/% |
| a Unless otherwise stated, all the reactions were carried out with 1 mmol of propargylic amine, [(nC7H15)4N]6[α-SiW11O39Cu] (130.0 mg, 0.025 mmol), CO2 (99.999%, balloon), 60 °C, 20 h. b 0.3 mL EtOH as the solvent. c 2 h. d 10 h. e Determined by the 1H NMR technique. | |||
| 1 |

|

|
96 |
| 2 |
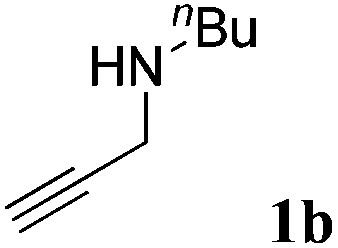
|

|
92 |
| 3 |
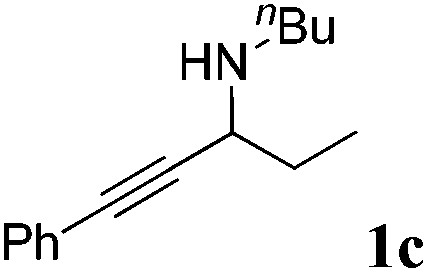
|
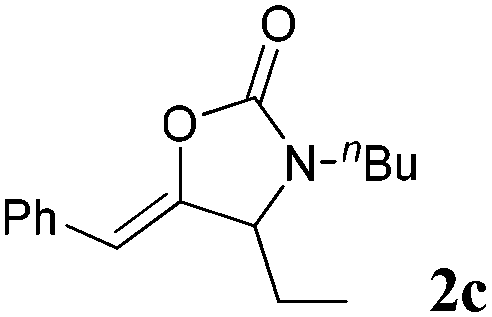
|
95 |
| 4b |
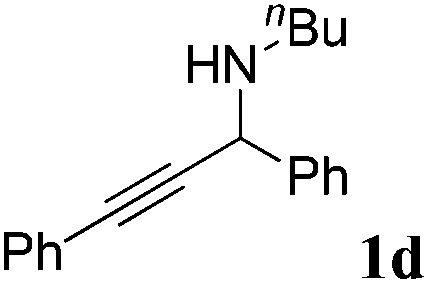
|
|
94 |
| 5 |
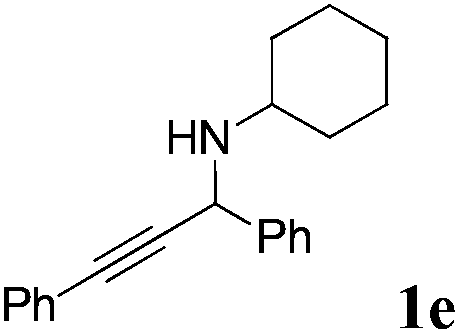
|

|
93 |
| 6b,c |

|
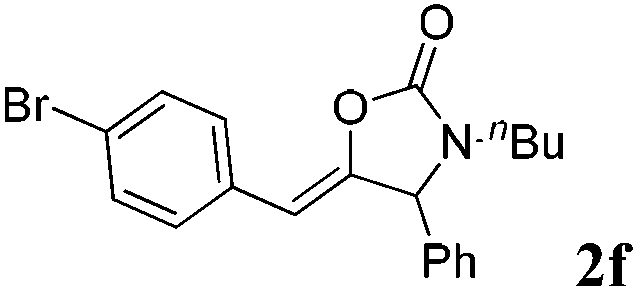
|
92 |
| 7b,d |

|
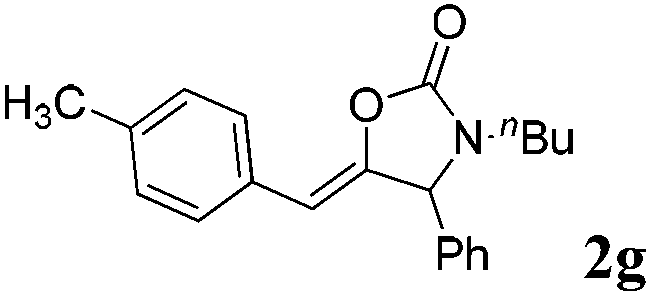
|
99 |
| 8b |

|

|
0 |
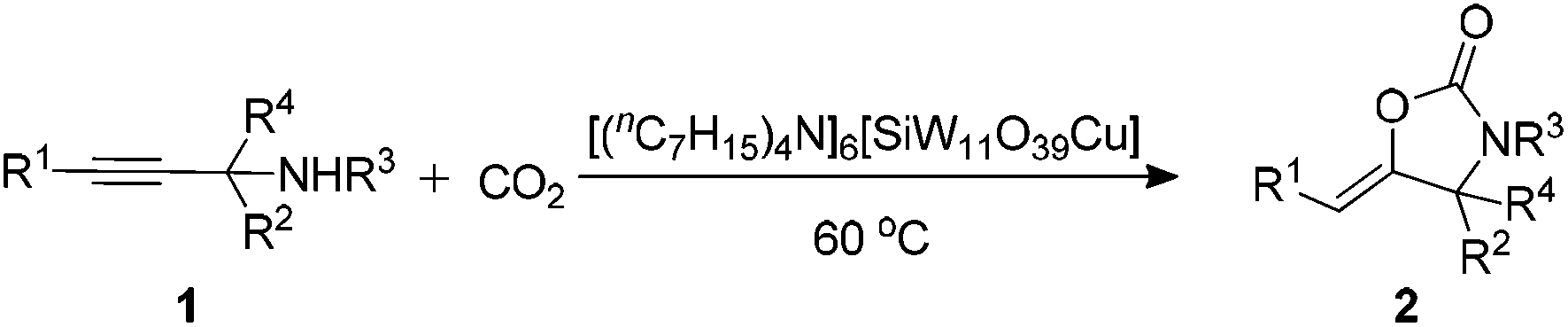
To further verify our initial assumption that both the propargylic amine and CO2 could be activated by [(nC7H15)4N]6[α-SiW11O39Cu], the interaction of [(nC7H15)4N]6[α-SiW11O39Cu] with CO2 and 1a was monitored respectively by 1H and 13C NMR techniques as depicted in Fig. 1 and 2. In the 1H NMR spectrum, the N–H signal of 1a appeared at δ = 1.68 ppm (Fig. 1b). However, this signal disappeared when [(nC7H15)4N]6[α-SiW11O39Cu] was added to 1a (Fig. 1c), thus illustrating that the propargylic amine is activated by [(nC7H15)4N]6[α-SiW11O39Cu] through the formation of N–H⋯O hydrogen bonds.30 Two new signals appeared at δ = 157.9 and 124.7 ppm when CO2 was bubbled into a dry solution of [(nC7H15)4N]6[α-SiW11O39Cu] and CDCl3 (Fig. 2). The newly-appeared peak at 157.9 ppm assigned to the carbonyl carbon of the monodentate carbonate presumably supports the CO2 activation by [α-SiW11O39Cu]6−,31 whilst the signal at 124.7 ppm is due to the physical absorption of CO2.
 | ||
| Fig. 1 1H NMR investigation. (a) [(nC7H15)4N]6[α-SiW11O39Cu] (50 mg, 0.01 mmol) in 0.3 mL of CDCl3. (b) 1a (55 mg, 0.4 mmol) in 0.3 mL of CDCl3; (c) 1a (55 mg, 0.4 mmol), [(nC7H15)4N]6[α-SiW11O39Cu] (50 mg, 0.01 mmol) in 0.3 mL of CDCl3. | ||
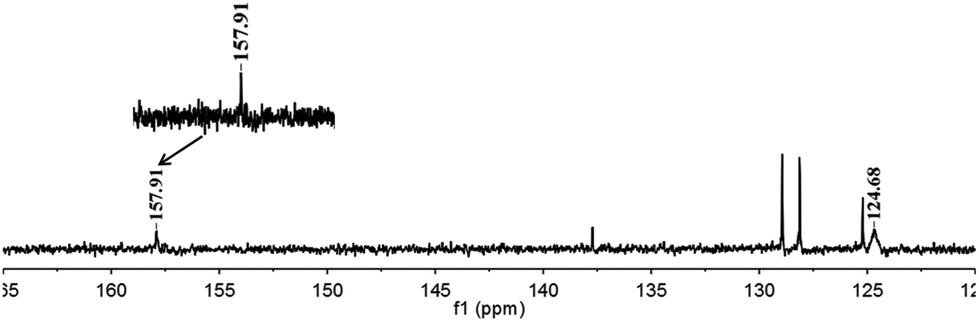 | ||
| Fig. 2 13C NMR study. [(nC7H15)4N]6[α-SiW11O39Cu] (containing a small amount of toluene) in CDCl3 at atmospheric CO2 (25 °C). | ||
On the basis of experimental and NMR studies, a plausible mechanism for the [(nC7H15)4N]6[α-SiW11O39Cu] promoted fixation of CO2 with propargylic amines is proposed as illustrated in Scheme 3, taking the carboxylative cyclization of 1a as an example. Propargylic amines and CO2 are activated by [(nC7H15)4N]6[α-SiW11O39Cu] in the form of species I. Then, the activated propargylic amine and CO2 are further converted into the carbamate II under the participation of another molecular propargylic amine which obtains a proton to be converted into the ammonium cation [1a + H]+. Compared with [1a + H]+, the bulky tetraheptylammonium cation (Q) can not only stabilize the carbamate anion, but also enhance the O-nucleophilicity, so it could exchange with [1a + H]+ to form a favorable carbamate III. Subsequently, with the aid of activation of CC bonds by Cu(II), intramolecular nucleophilic cyclization of III gives the intermediate IV. Finally, proto-demetallation with the help of a proton carrier [1a + H]+ affords the product 2a along with the regeneration of [(nC7H15)4N]6[α-SiW11O39Cu].
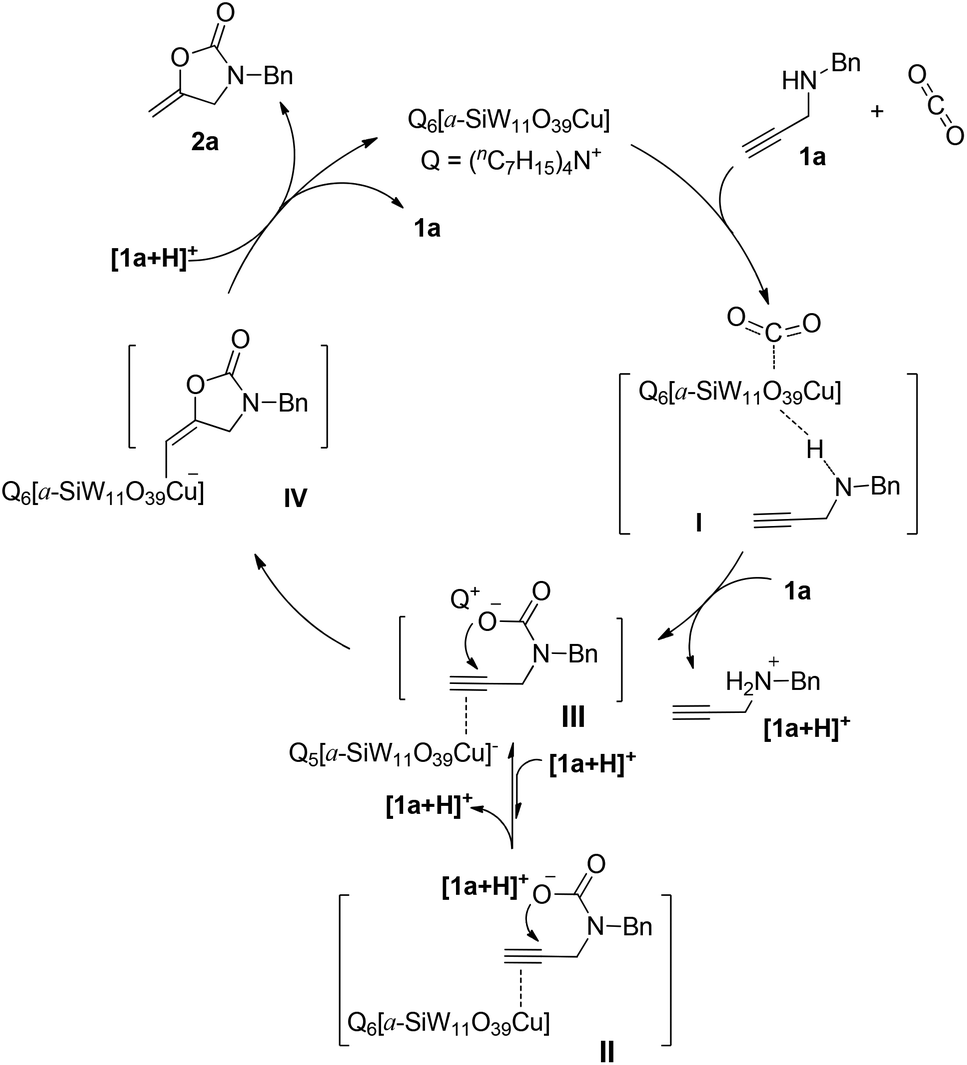 | ||
| Scheme 3 Plausible mechanism for the [(nC7H15)4N]6[α-SiW11O39Cu]-catalyzed carboxylative cyclization of propargylic amines and CO2. | ||
Conclusions
In summary, we have described a cost-competitive catalytic process promoted by robust bifunctional [(nC7H15)4N]6[α-SiW11O39Cu] which can simultaneously activate both CO2 and propargylic amines for efficient chemical fixation of CO2 at atmospheric pressure to produce 5-alkylideneoxazolidin-2-ones at room temperature in excellent yields. This protocol also features a simple process with low energy consumption and avoidance of complicated preparation of the catalyst. Such findings may be of great interest to develop cost-effective, halogen-free and efficient bifunctional catalysts for the preparation of the CO2-based products under mild reaction conditions.Experimental section
Transition-metal-substituted polyoxometalate-based ionic liquids were synthesized according to previous reports.20a,28 Taking the synthesis of [(nC7H15)4N]6[α-SiW11O39Cu] as an example: 25 mL of 20 mM aqueous K8[α-SiW11O39] solution and 50 mL of a 10 mM aqueous CuCl2 solution were mixed and stirred for 15 min. Then the solution was treated with tetraheptylammonium bromide (2.47 g, 3 mmol) dissolved in 60 mL toluene. After the two phases were allowed to settle for 10 minutes, the organic layer was decanted and washed seven times with 20 mL water portions. The wet toluene was removed by heating to 40 °C under vacuum. Then 30 mL dry toluene was added and evaporated under reduced pressure to remove the traces of water. [(nC7H15)4N]6[α-SiW11O39Cu] was obtained as a blue viscous liquid. IR (neat, KBr): 2955, 2927, 2856, 1638, 1618, 1465, 1383, 1265, 949, 898, 798, 739 cm−1. Anal. Calcd for [(nC7H15)4N]6[α-SiW11O39Cu] (MW = 5202): C 38.78, H 6.98, N 1.62, Found: C 38.19, H 6.80, N 1.66.General procedure for the synthesis of 5-alkylideneoxazolidin-2-ones
[(nC7H15)4N]6[α-SiW11O39Cu] (130.0 mg, 0.025 mmol) and propargylic amine (1 mmol) were added to a 10 mL Schlenk tube equipped with a magnetic stir bar. The flask was capped and sealed. Then a gas-exchanging process was conducted using the “freeze–pump–thaw” method. The reaction mixture was stirred at 60 °C for 20 h under a CO2 balloon (99.999%). Upon completion, product yield was determined by the 1H NMR technique using 1,1,2,2-tetrachloroethane as an internal standard. Then the reaction mixture was diluted with dichloromethane (2 mL). The organic phase was collected and then purified by column chromatography on silica gel using petroleum ether/ethyl acetate as an eluent to afford the desired product. All the products were characterized by NMR and MS and compared with the data available in the literature.12e,32Acknowledgements
We are grateful to the National Natural Science Foundation of China, the Specialized Research Fund for the Doctoral Program of Higher Education (20130031110013), the MOE Innovation Team (IRT13022) of China, and the “111” Project of the Ministry of Education of China (project no. B06005) for financial support.References
- (a) M. He, Y. Sun and B. Han, Angew. Chem., Int. Ed., 2013, 52, 9620–9633 CrossRef CAS PubMed; (b) P. Tundo and M. Selva, Acc. Chem. Res., 2002, 35, 706–716 CrossRef CAS PubMed; (c) M. Honda, M. Tamura, Y. Nakagawa and K. Tomishige, Catal. Sci. Technol., 2014, 4, 2830–2845 RSC; (d) A.-H. Liu, Y.-N. Li and L.-N. He, Pure Appl. Chem., 2012, 84, 581–602 CrossRef CAS.
- M. Yoshida, N. Hara and S. Okuyama, Chem. Commun., 2000, 151–152 RSC.
- R. Tanaka, M. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2009, 131, 14168–14169 CrossRef CAS PubMed.
- S. Wesselbaum, T. vomStein, J. Klankermayer and W. Leitner, Angew. Chem., Int. Ed., 2012, 51, 7499–7502 Search PubMed.
- (a) M. Honda, M. Tamura, K. Nakao, K. Suzuki, Y. Nakagawa and K. Tomishige, ACS Catal., 2014, 4, 1893–1896 Search PubMed; (b) Q.-W. Song, W.-Q. Chen, R. Ma, A. Yu, Q.-Y. Li, Y. Chang and L.-N. He, ChemSusChem, 2015, 8, 821–827 CrossRef CAS PubMed; (c) B. A. Vara, T. J. Struble, W. Wang, M. C. Dobish and J. N. Johnston, J. Am. Chem. Soc., 2015, 137, 7302–7305 Search PubMed.
- D. J. Darensbourg, A. I. Moncada, W. Choi and J. H. Reibenspies, J. Am. Chem. Soc., 2008, 130, 6523–6533 CrossRef CAS PubMed.
- L. Aurelio, R. T. C. Brownlee and A. B. Hughes, Chem. Rev., 2004, 104, 5823–5846 CrossRef CAS PubMed.
- C. W. Y. Chung and P. H. Toy, Tetrahedron: Asymmetry, 2004, 15, 387–399 CrossRef CAS.
- T. A. Mukhtar and G. D. Wright, Chem. Rev., 2005, 105, 529–542 CrossRef CAS PubMed.
- L. E. Overman and T. P. Remarchuk, J. Am. Chem. Soc., 2002, 124, 12–13 CrossRef CAS PubMed.
- Y. Fukata, K. Asano and S. Matsubara, J. Am. Chem. Soc., 2013, 135, 12160–12163 CrossRef CAS PubMed.
- (a) J.-Y. Hu, J. Ma, Q.-G. Zhu, Z.-F. Zhang, C.-Y. Wu and B.-X. Han, Angew. Chem., Int. Ed., 2015, 54, 5399–5403 CrossRef CAS PubMed; (b) A. W. Miller and S. T. Nguyen, Org. Lett., 2004, 6, 2301–2304 CrossRef CAS PubMed; (c) Z.-Z. Yang, Y.-N. Li, Y.-Y. Wei and L.-N. He, Green Chem., 2011, 13, 2351–2353 RSC; (d) R. Juarez, P. Concepcion, A. Corma and H. Garcia, Chem. Commun., 2010, 46, 4181–4183 RSC; (e) Q.-W. Song, B. Yu, X.-D. Li, R. Ma, Z.-F. Diao, R.-G. Li, W. Li and L.-N. He, Green Chem., 2014, 16, 1633–1638 RSC; (f) B.-S. Wang, E. H. M. Elageed, D.-W. Zhang, S.-J. Yang, S. Wu, G.-R. Zhang and G.-H. Gao, ChemCatChem, 2014, 6, 278–283 CrossRef CAS; (g) D. Adhikari, A. W. Miller, M.-H. Baik and S. T. Nguyen, Chem. Sci., 2015, 6, 1293–1300 RSC.
- P. Dimroth and H. Pasedach, DE Pat, 1164411, 1964 Search PubMed.
- (a) M. Costa, G. Paolo Chiusoli, D. Taffurelli and G. Dalmonego, J. Chem. Soc., Perkin Trans. 1, 1998, 1541–1546 RSC; (b) M. Costa, G. P. Chiusoli and M. Rizzardi, Chem. Commun., 1996, 1699–1700 RSC; (c) R. Nicholls, S. Kaufhold and B. N. Nguyen, Catal. Sci. Technol., 2014, 4, 3458–3462 RSC.
- (a) S. Yoshida, K. Fukui, S. Kikuchi and T. Yamada, Chem. Lett., 2009, 38, 786–787 CrossRef CAS; (b) S. Kikuchi, S. Yoshida, Y. Sugawara, W. Yamada, H.-M. Cheng, K. Fukui, K. Sekine, I. Iwakura, T. Ikeno and T. Yamada, Bull. Chem. Soc. Jpn., 2011, 84, 698–717 CrossRef CAS; (c) M. Yoshida, T. Mizuguchi and K. Shishido, Chem. – Eur. J., 2012, 18, 15578–15581 CrossRef CAS PubMed.
- Y. Takeda, S. Okumura, S. Tone, I. Sasaki and S. Minakata, Org. Lett., 2012, 14, 4874–4877 CrossRef CAS PubMed.
- S. Hase, Y. Kayaki and T. Ikariya, Organometallics, 2013, 32, 5285–5288 CrossRef CAS.
- (a) A. B. Bourlinos, K. Raman, R. Herrera, Q. Zhang, L. A. Archer and E. P. Giannelis, J. Am. Chem. Soc., 2004, 126, 15358–15359 CrossRef CAS PubMed; (b) S.-S. Wang and G.-Y. Yang, Chem. Rev., 2015, 115, 4893–4962 CrossRef CAS PubMed.
- M. Girardi, S. Blanchard, S. Griveau, P. Simon, M. Fontecave, F. Bedioui and A. Proust, Eur. J. Inorg. Chem., 2015, 2015, 3642–3648 CrossRef CAS.
- (a) S. H. Szczepankiewicz, C. M. Ippolito, B. P. Santora, T. J. Vande Ven, G. A. Ippolito, L. Fronckowiak, F. Wiatrowski, T. Power and M. Kozik, Inorg. Chem., 1998, 37, 4344–4352 CrossRef CAS PubMed; (b) G.-G. Gao, F.-Y. Li, L. Xu, X.-Z. Liu and Y.-Y. Yang, J. Am. Chem. Soc., 2008, 130, 10838–10839 CrossRef CAS PubMed.
- (a) N. S. Antonova, J. J. Carbó, U. Kortz, O. A. Kholdeeva and J. M. Poblet, J. Am. Chem. Soc., 2010, 132, 7488–7497 CrossRef CAS PubMed; (b) Y. Leng, J. Zhao, P. Jiang and J. Wang, ACS Appl. Mater. Interfaces, 2014, 6, 5947–5954 CrossRef CAS PubMed.
- W.-L. Huang, W.-S. Zhu, H.-M. Li, H. Shi, G. Zhu, H. Liu and G.-Y. Chen, Ind. Eng. Chem. Res., 2010, 49, 8998–9003 CrossRef CAS.
- Y. Leng, J. Wang, D.-R. Zhu, X.-Q. Ren, H.-Q. Ge and L. Shen, Angew. Chem., Int. Ed., 2009, 48, 168–171 CrossRef CAS PubMed.
- (a) T. Yamase, Chem. Rev., 1998, 98, 307–326 CrossRef CAS PubMed; (b) S. Herrmann, M. Kostrzewa, A. Wierschem and C. Streb, Angew. Chem., Int. Ed., 2014, 53, 13596–13599 CrossRef CAS PubMed.
- (a) M. Sankar, N. H. Tarte and P. Manikandan, Appl. Catal., A, 2004, 276, 217–222 CrossRef CAS; (b) H. Yasuda, L.-N. He, T. Sakakura and C. Hu, J. Catal., 2005, 233, 119–122 CrossRef CAS; (c) J. Langanke, L. Greiner and W. Leitner, Green Chem., 2013, 15, 1173–1182 RSC.
- J.-L. Wang, J.-Q. Wang, L.-N. He, X.-Y. Dou and F. Wu, Green Chem., 2008, 10, 1218–1223 RSC.
- M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS PubMed.
- (a) A. Tézé and G. Hervé, J. Inorg. Nucl. Chem., 1977, 39, 999–1002 CrossRef; (b) D. E. Katsoulis and M. T. Pope, J. Chem. Soc., Dalton Trans., 1989, 1483–1489 RSC.
- (a) T. Ema, Y. Miyazaki, S. Koyama, Y. Yano and T. Sakai, Chem. Commun., 2012, 48, 4489–4491 RSC; (b) T. Ema, Y. Miyazaki, J. Shimonishi, C. Maeda and J.-y. Hasegawa, J. Am. Chem. Soc., 2014, 136, 15270–15279 CrossRef CAS PubMed.
- (a) C. P. Pradeep, D.-L. Long, G. N. Newton, Y.-F. Song and L. Cronin, Angew. Chem., Int. Ed., 2008, 47, 4388–4391 CrossRef CAS PubMed; (b) T. Akutagawa, F. Kudo, R. Tsunashima, S.-i. Noro, L. Cronin and T. Nakamura, Inorg. Chem., 2011, 50, 6711–6718 CrossRef CAS PubMed.
- (a) C.-X. Guo, B. Yu, J.-N. Xie and L.-N. He, Green Chem., 2015, 17, 474–479 RSC; (b) T. Kimura, K. Kamata and N. Mizuno, Angew. Chem., Int. Ed., 2012, 51, 6700–6703 CrossRef CAS PubMed.
- W.-J. Yoo and C.-J. Li, Adv. Synth. Catal., 2008, 350, 1503–1506 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: General procedures, spectral data or other electronic format. See DOI: 10.1039/c5gc02311d |
| This journal is © The Royal Society of Chemistry 2016 |