
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHomogeneous and heterogenised masked N-heterocyclic carbenes for bio-based cyclic carbonate synthesis†
Joseph A.
Stewart
a,
Roland
Drexel
a,
Bjørnar
Arstad
b,
Erik
Reubsaet
a,
Bert M.
Weckhuysen
a and
Pieter C. A.
Bruijnincx
*a
aInorganic Chemistry and Catalysis, Debye Institute for Nanomaterials Science, Utrecht University, Universiteitsweg 99, 3584 CG, Utrecht, The Netherlands. E-mail: p.c.a.bruijnincx@uu.nl; Fax: +31 302511027; Tel: +31 302537400
bDepartment of Sustainable Energy Technology, SINTEF, Forskningsveien 1, 0314, Oslo, Norway
First published on 9th November 2015
Abstract
(Multifunctional) cyclic carbonates are generating much interest, with bio-based bis-cyclic compounds attracting attention from the polymer sector as potential renewable monomers for systems such as non-isocyanate polyurethanes. Here, the efficient synthesis of one such substrate, diglycerol dicarbonate, utilising CO2-masked N-heterocyclic carbene (NHC) organocatalysts is demonstrated. The 1,3-dialkylimidazole-2-carboxylate pre-catalyst, which can be produced both in and ex situ, yields the desired cyclic product, expressing full conversion within 3 h when using the ex situ synthesised pre-catalyst with 5 mol% loading, but can also operate with 1 mol% loading efficiently. Substituted derivatives of the imidazole-based organocatalyst have also been investigated to gauge the sensitivity of the system. A number of bio-based diols are also investigated, with 1,2-, 2,3- and 1,3-diols yielding five- and six-membered cyclic products, respectively; 1,3-diols are significantly more reluctant to cyclisation, yielding both 1- and 3-monocarbonates, dicarbonates and the cyclic products. A more in depth study was also carried out on glycerol as a substrate, both in its pure a crude form, providing insight into how impurities impact on the activity of the carbene catalyst. Through 13C-labelled reagent experiments, a mechanism is proposed for the conversion of diols to their cyclic carbonate analogues. Finally, the organocatalyst was immobilized on siliceous mesostructured cellular foam (MCF). Using an alternative activation procedure, a supported, masked NHC catalyst is achieved and characterised with DRIFTS, TGA and 13C solid-state NMR. This heterogenised catalyst can be easily recovered and reused up to three times expressing its original activity if properly regenerated by a simple ion exchange procedure. Of important note, this system can also successfully convert crude glycerol with high selectivity observed for the cyclic product.
Introduction
The multitude of applications of cyclic carbonates include their use as aprotic solvents, precursors to polymeric materials, electrolytes and as intermediates for a number of fine chemicals to name but a few. Cyclic compounds are typically synthesised from either the diol or epoxide precursor, with both pathways having received ample interest over the past decade and most studies concentrating on short-chain, terminally functionalised substrates.1–5 One substrate that has received considerable attention in this respect is glycerol. Glycerol is a renewable raw material derived from triglycerides and is industrially produced via saponification, hydrolysis and transesterification. Glycerol itself can be converted into a number of useful chemicals, with routes explored including glyceric acid by oxidation, propylene glycol and 1,3-propanediol by hydrogenolysis and acrolein by acid-catalysed dehydration.6–8 In addition, extensive research efforts have focussed on glycerol carbonate (GC) production from glycerol.9 Indeed, GC has highly desirable physical and chemical properties. It is biodegradable, water-soluble, non-toxic and non-flammable and can be utilised in a number of ways, from solvents and beauty product component to building eco-composites and as a carrier in lithium batteries.10,11 Phosgene has previously been used for the synthesis of GC from glycerol,12 but its highly toxic nature has led to research into more environmentally friendly processes. A number of different catalytic systems have been developed for this process, making use of a range of “CO” sources including CO2, CO and O2, urea, ethylene carbonate and dimethyl or diethyl carbonate (DMC, DEC), of which each can be considered renewable.9,13The different CO sources have been tested in combination with numerous homogeneous, heterogeneous and biological catalysts for the synthesis of GC.9 Focussing on those examples that make use of the alkyl carbonates, it can be seen that di(m)ethyl carbonate typically gives higher conversions and selectivities (95+%) than ethylene carbonate. Systems explored include Mg–Al hydrotalcites,14 Mg–Al mixed oxides,15 Mg–Al–Zr mixed oxides,16 calcium oxide17,18 and calcium hydroxide19 for DMC/DEC, compared with zeolites and basic resins,20 Mg-mixed oxides,3 immobilised ionic liquids21 and Li-hydrotalcites3 that use ethylene carbonate. Lipases have also yielded good catalytic results, with up to 100% yields being reported, albeit after long reaction times.22,23
Organocatalysts have recently also been reported for GC formation in particular and cyclic carbonate formation in general. Sakai et al., for instance, employed organocatalysts with an ammonium betaine framework for the conversion of various epoxides to cyclic carbonates, including the conversion of glycidol to GC.24 Lu et al. showed that CO2 adducts of N-heterocyclic olefins and alkoxide-functionalised imidazolium betaines are highly active for the carboxylative cyclisation of propargylic alcohols under mild CO2 pressures.25,26
Naik et al. in turn used DMC as the CO source with 1,3-dialkyl-substituted imidazolium-2-carboxylate catalysts for the conversion of glycerol to glycerol carbonate.27 The catalyst could also be obtained in situ by methylation and carboxylation of the appropriate 1-alkylimidazole precursor with DMC. The authors tentatively suggest that in solution, this (pre-)catalyst undergoes decarboxylation to yield the active N-heterocyclic carbene (NHC), although do not evidence this. Such imidazole-based betaines, which have also been used as (pre-)catalysts for other reactions26,28,29 and as precursors for N-heterocyclic carbene transfer agents, can be synthesised via a number of routes as demonstrated by Crabtree et al.30 It has been shown that such imidazole-2-carboxylates perform as “masked” NHCs, in which the carboxylate actually acts as a protecting group for the active carbene, with decarboxylation in solution preceding the catalytic reaction.31–34 Relatedly, Palencia et al. recently demonstrated the NHC-catalysed synthesis of glycerol carbonate from glycerol and DMC. In this study, the NHC is generated in situ from the imidazolium halide by the addition of potassium tert-butoxide.35
Naik et al. furthermore found the alkyl chain length on the 1-position of the imidazole to influence catalyst activity, with a butyl chain being optimal.27 The organocatalyst system was significantly faster than the benchmark catalyst K2CO3,36 and, in contrast to K2CO3, proved effective for the conversion of a crude glycerol. The substrate scope of the imidazolium-2-carboxylates studies was limited to glycerol, however. It is clearly of interest to see how such readily accessible organocatalysts would perform with other diol substrates, such as ethylene diol, 1,2- and 1,3-propanediol and 1,2-, 2,3- and 1,3-butanediol as well as diglycerol.2,37–39
Dicyclics synthesis is also of considerable interest, for instance to the polymer sector. In particular, ring-opening of cyclic carbonates by amines provides an interesting, non-isocyanate-based alternative to the current production routes to polyurethanes.9,11,40 Indeed, the current use of isocyanates for polyurethane production is associated with environmental concerns, not only because the isocyanates are highly toxic themselves, but also because they are produced from the highly toxic compound, phosgene. To mitigate these toxicity concerns, a number of dicyclic monomers have been highlighted to replace isocyanates for the production of non-isocyanate polyurethanes (NIPUs). Examples include limonene dicarbonate,41 fatty acid-based bis-cyclic carbonates,42,43 di(glycerol (di)carbonate)s11,44 and others.40 Diglycerol thus is an attractive substrate for cyclocarbonation, given its potential as precursor to NIPU monomers. To date, there is only one reported catalytic synthesis of diglycerol dicarbonate (DGDC) from diglycerol, utilising Mg–Al hydrotalcites.39
In this paper, the synthesis of DGDC is explored further utilising analogues of the imidazole-derived organocatalysts reported by Naik et al.27 A number of other renewable diol substrates (Fig. 1(b)) are also used to explore the general applicability and efficiency of this system as well as its use for the conversion of an industrial crude glycerol. Reactions performed with 13C-labelled dimethyl carbonate furthermore provided insight into the mechanism of cyclic carbonate formation. Based on this system, a heterogeneous, silica-supported imidazole-based catalyst is prepared and tested and its reuse demonstrated.
 | ||
| Fig. 1 (a) Reaction overview, (b) renewable diols used as substrates in this research. DG – diglycerol, G – (crude) glycerol, ED – ethylene diol, PD – propanediol, BD – butanediol. | ||
Results and discussion
Homogeneous NHCs
Building on the example reported by Naik et al. of glycerol carbonate synthesis using 1-butyl-3-methylimidazolium-2-carboxylates, we investigated a range of 1-n-alkyl-3-methylimidazolium-2-carboxylates and their precursors for cyclic carbonate synthesis utilising DMC, concentrating first on the synthesis of DGDC from DG under mild, solvent-free conditions. The three stages of the imidazole-based catalyst are defined in Fig. 2, and will be referred to throughout this study as stated. | ||
| Fig. 2 The three stages of the imidazole-based catalyst (R = alkyl chain). | ||
The results show that DGDC can also be synthesised efficiently with these organocatalysts (Fig. 3). For example, 1-butylimidazole gave a DGDC yield of 65%, with 80% selectivity after 18 h at 74 °C. A dimethyl carbonate to glycerol ratio of 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 is used to both aid with the higher viscosity of DG and to take into account the fact that it contains two hydroxyl pairs per molecule. K2CO3 was also tested as benchmark catalyst, yielding 10% diglycerol dicarbonate and 32% diglycerol monocarbonate (DGMC) at 44% conversion, under the same conditions. The pre-catalyst is formed in situ by reaction of the relevant 1-alkylsubstituted imidazole precursor with the excess of dimethyl carbonate. This activation process has previously been studied computationally and was suggested to first involve a rate-determining methyl transfer from DMC to the alkylimidazole, which results in an ion pair. Proton transfer then induces C–O bond cleavage to give an N-heterocyclic carbene (NHC), CO2 and MeOH. The third and final step is nucleophilic attack of CO2 by the NHC forming the new C–C bond.30
:1 is used to both aid with the higher viscosity of DG and to take into account the fact that it contains two hydroxyl pairs per molecule. K2CO3 was also tested as benchmark catalyst, yielding 10% diglycerol dicarbonate and 32% diglycerol monocarbonate (DGMC) at 44% conversion, under the same conditions. The pre-catalyst is formed in situ by reaction of the relevant 1-alkylsubstituted imidazole precursor with the excess of dimethyl carbonate. This activation process has previously been studied computationally and was suggested to first involve a rate-determining methyl transfer from DMC to the alkylimidazole, which results in an ion pair. Proton transfer then induces C–O bond cleavage to give an N-heterocyclic carbene (NHC), CO2 and MeOH. The third and final step is nucleophilic attack of CO2 by the NHC forming the new C–C bond.30
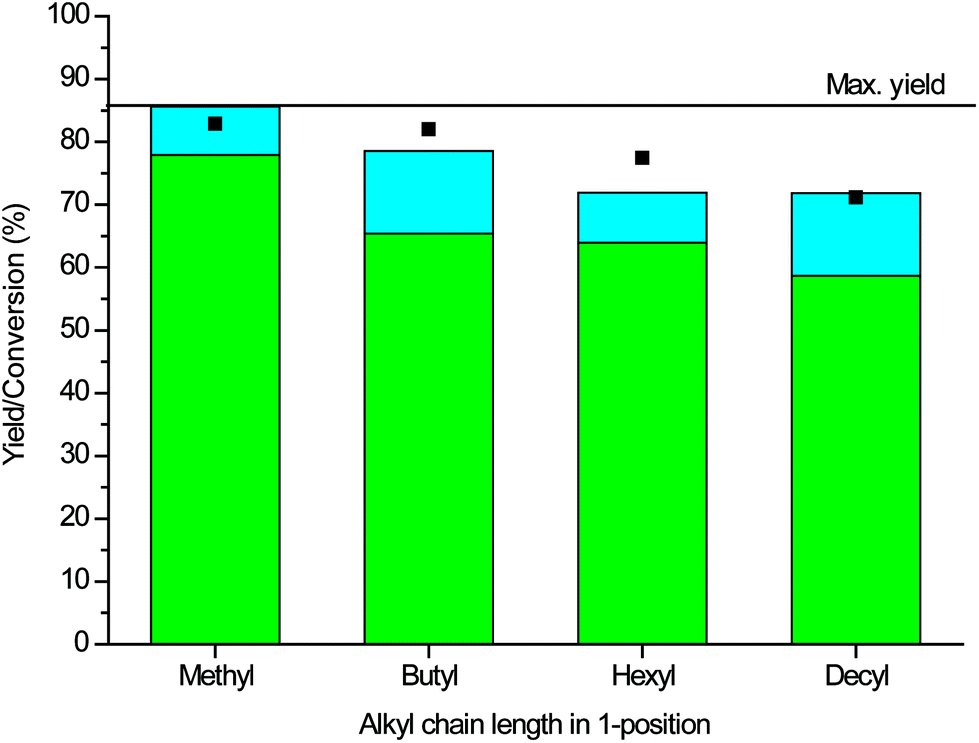 | ||
| Fig. 3 Influence of alkyl chain length on conversion of DG (squares) and yield of DGDC (green) and DGMC (blue), utilising 5 mol% 1-alkyl-3-methylimidazolium-2-carboxylate synthesised in situ from its 1-alkylsubstituted imidazole precursor. Conditions: DG:DMC 6:1, 74 °C, 18 h. Max. yield is the theoretical maximum yield of DGDC due to the purity of feedstock, see ESI.† | ||
As previously observed by Naik et al., variation of the 1-alkyl chain length influenced the catalytic activity of the active carbene form, with activity decreasing with an increase in alkyl chain length, Fig. 3. This could be the result of (a combination of) two factors; namely the longer induction period required to generate the carboxylate form and the stability of the formed carboxylate w.r.t. to the formation of the active carbene. As generation of the carboxylate actually proceeds via the active carbene, the relative stability of the carbene and the carboxylate thus plays a pivotal role in the expressed activity. Each of the 1-alkylimidazoles produced DGDC within 18 h with a 5 mol% loading, with the 1-methyl-substituted catalysts expressing the highest yield (78% with 94% selectivity). This yield is close to the maximum that can be obtained as the purity of the starting diglycerol is only 86% (see ESI† for definitions), with no by-products being observed, except for small amounts of the intermediate DGMC. The largest impurity in the starting material is glycerol, and glycerol carbonate is indeed observed in the reaction mixture. With the inclusion of glycerol carbonate, the carbon balance amounts to 95% for the reaction with 1-methylimidazole. The longer hexyl and decyl alkyl chains gave yields of 64 and 59% of the dicarbonate, respectively (both with 83% selectivity). The optimal chain length for DGDC is thus different from the butyl one found by Naik et al. for GC. They attributed the influence of the chain length to a balance between the stability of the pre-catalyst, the methyl-substituted carboxylate being too stable, and the rate at which the carboxylate form is made, i.e. longer alkyl chains decreasing the rate of formation.27 The stability of the carboxylate can be gauged from the torsional angle the carboxylate makes with the imidazolium ring; for 1,3-dimethylimidazolium-2-carboxylate, this angle is 29.03°, which still allows for a certain degree of π-orbital overlap and hence stabilisation. As alkyl chain length increases, the torsional angle increases, decreasing the π-orbital overlap and allowing for more facile decarboxylation.45 Thus, the decrease in activity that we observed with increasing chain length may be a combination of lower activity of the carbene catalyst and lower amounts of the pre-catalyst being formed from the precursor due to slower kinetics. The fact that Naik et al. observed that butyl was optimal for glycerol conversion can be associated with steric hindrance of the larger chain approaching the substrate being more apparent for the diglycerol substrate.27 It should also be noted that imidazole as catalyst precursor gave a DGDC yield of only 4%. Variation of the molar ratio of DMC:substrate for the best catalyst to either of 3 and 9:1 resulted in a drop in activity to 64 and 63%, respectively (Table S1†). This observation is attributed to the necessity to overcome the viscosity of the substrate and a balance between the system becoming diluted when too large an excess of reagent is added. Equilibrium considerations will also play a role.
1-Methylimidazoles with various electron-donating and withdrawing substituents in the 2- or 5-position, depicted in Fig. S1,† were also tested. These substitutions proved to have a rather dramatic effect on activity, as each of the 5-substituted precursors tested gave no diglycerol dicarbonate, with only low amounts of diglycerol monocarbonate being detected (Table S2†). This reduction in activity can be attributed to two factors; electron-withdrawing groups are thought to influence the formation of the carboxylate pre-catalyst detrimentally, affecting the rate-determining methylation of the imidazole.46 The introduction of electron-donating groups on the other hand stabilise the pre-catalyst carboxylate, and hence reduce the extent to which the active carbene is formed.45 As discussed in more detail below, the 4-carboxylate isomer can also form when reactions are run at higher temperatures. 1,2-Dimethylimidazole, i.e. a precursor for which the 2-carboxylate cannot be formed and any reaction has to go through the 4-carboxylate and the resulting mesoionic carbene,47 was also tested. Although significantly less active, it did show the highest conversion (29%) of all the substituted 1-methylimidazoles. It was also the only derivative tested that was able to produce the desired dicyclic product, although in very low yields (3%).
The results discussed thus far were achieved using the 1-alkylimidazole precursor rather than the (substituted) 1,3-dialkylimidazolium carboxylate pre-catalyst, which is generated in situ. The pre-catalyst, however, can also be synthesised and isolated in its crystalline form and used as such.30 In fact, it has already been noted that the pre-catalyst synthesis conditions are similar to those of the reaction; indeed, the choice of reaction temperature might have consequences if the pre-catalyst has to be formed in situ, as syntheses carried out below 95 °C predominantly yield the 2-carboxylate, while above this temperature the formation of the 4-carboxylate becomes increasingly favoured, and dominates above 120 °C.27,30 As already shown above and further detailed below, the mesoionic carbene obtained from the 4-carboxylate is less active than the one obtained from the 2-carboxylate.47 The use of the as-synthesised 1,3-dimethylimidazolium-2-carboxylate (1) pre-catalyst has a significant impact on the reaction rate, as can be clearly seen from Fig. 4; the induction period seen for the precursor is absent when the pre-catalyst is used. This induction period is in line with separate synthesis times of 2 h required to achieve quantitative conversion of the precursor to the active catalyst ex situ.30 Reaction times are now decreased significantly with the pre-catalyst, with a yield of 71% already being achieved after 3 h, with a maximum yield of 75% after 6 h; in contrast, the reaction with the precursor required 18 h to get to a similar yield of 78%. This finding is also significant, as our previously reported method for catalytic synthesis of DGDC requires 6 h to achieve high yields utilising 30 wt% of Mg–Al hydrotalcites under less mild conditions.39
 | ||
| Fig. 4 Yield of DGDC against time for reactions starting with the precursor (red) or pre-catalyst (black). Conditions: 5 mol% 1-methyl-substituted pre-catalyst or catalyst precursor, 74 °C, DMC:DG 6:1 molar ratio. | ||
Reactions with a 1 mol% loading of the pre-catalyst were also investigated, achieving a yield of 55% within 18 h, with 20% of the monocarbonate being observed as an intermediate product. Three other carboxylate derivatives were also synthesised, isolated and tested (2–4, Fig. 5). 2 expressed higher activity than 1 when compared using 1 mol% loading reaching full conversion and expressing 91% selectivity towards the dicyclic product in 18 h, Table 1. These results indicate that 1 is the most active form for this reaction when starting with the precursor, as formation of the carboxylate is more facile with small substituents in the 1- and 3-positions. However, larger substituents destabilise the pre-catalyst, and thus, the 1-butyl form shows higher activity when the as-synthesised pre-catalysts are compared.35
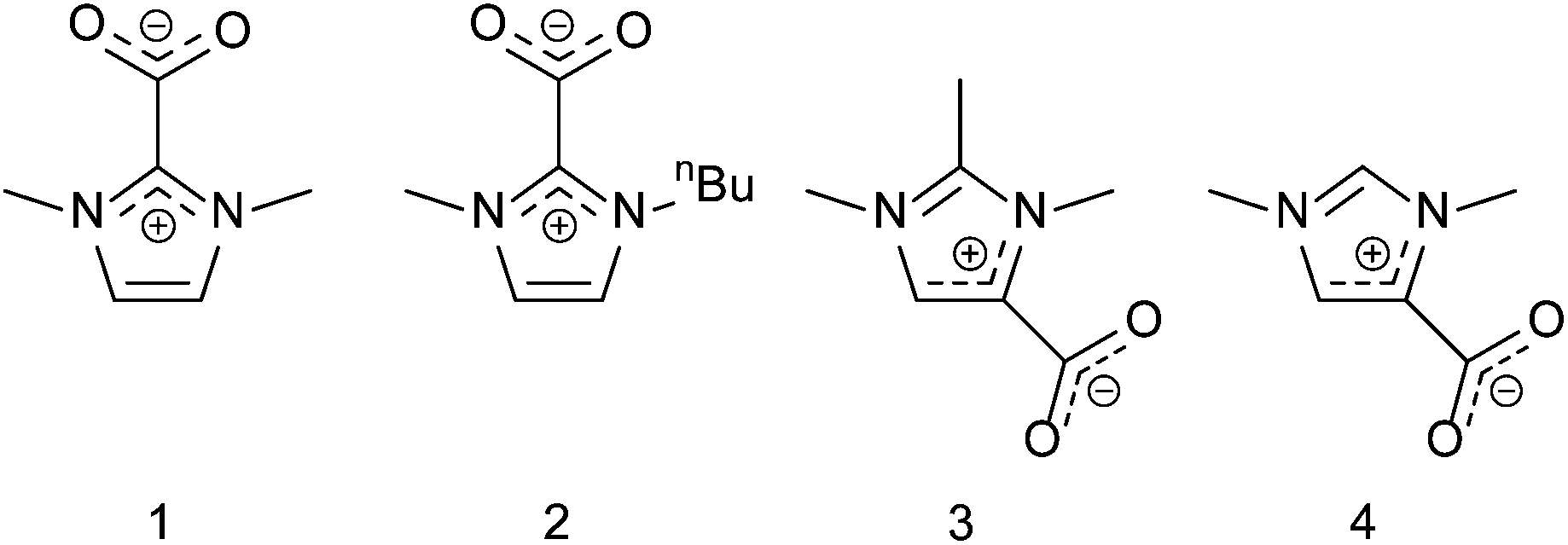 | ||
| Fig. 5 Synthesised pre-catalysts for cyclocarbonation reactions. | ||
| Pre-catalyst | Catalyst loading (mol%) | Conversion (%) | Dicyclic yield (%) | Dicyclic selectivity (%) | Mono-cyclic carbonate yield (%) | Mono-cyclic carbonate selectivity (%) | C-balance (%) |
|---|---|---|---|---|---|---|---|
| Conditions: 0.5 g DG, 18 h, 6:1 DMC:DG, 74 °C, mol% pre-catalyst loading with respect to DG. |
|||||||
| 1 | 5 | 100 | 72 | 83 | 16 | 18 | 102 |
| 1 | 1 | 93 | 55 | 69 | 20 | 25 | 94 |
| 2 | 1 | 100 | 79 | 91 | 9 | 11 | 102 |
| 3 | 5 | 76 | 50 | 77 | 15 | 23 | 100 |
| 4 | 5 | 15 | 0 | — | 8 | 60 | 94 |
The 4-carboxylate pre-catalysts 3 and 4 were considerably less active than 1 and 2. The lower activity can be associated with 4-carboxylates being thermodynamically more stable than the 2-carboxylates. As exemplified by the torsion angles of the carboxylates with the imidazolium ring,47 the carboxylate group is more weakly bound in the 2-carboxylate, thus leading to the active carbene species more easily. The addition of an extra methyl group in the 2-position, (3), did have a drastic effect on activity, compared to 4. The latter exhibited only 15% conversion (no dicarbonate, 8% monocarbonate), whereas the trimethyl derivative was able to express 76% conversion, with 50% yield of DGDC compared to just 8% monocarbonate after 18 h for the 4. This can be attributed to the electron-donating effect of the extra methyl group in the 2-position, decreasing the stability of the carboxylate and increasing the nucleophilicity (σ-donating ability) of the resulting carbene species.48 Under our conditions, 4 may indeed be too stable and not yield the active carbene.47
Cyclocarbonation of various 1,2-diols, 1,3-diols and 2,3-diols was tested with 1 (Table 2), allowing for a comparison of the activity of terminal versus internal, primary versus secondary alcohols and five- or six-membered ring formation. Cyclic carbonate products were achieved for all the substrates tested, with the highest yield of 83% achieved for glycerol when using a DMC:substrate ratio of 3:1 after 3 h with 1 mol% 1. Yields of the cyclic product were found to increase with increasing alkyl chain length of the substrate. Ethylene diol was converted to give the cyclic carbonate in 55% yield, while 1,2-propanediol and 1,2-butanediol showed improved yields of 74 and 79% and high selectivities of 96 and 99%, respectively.
| Substrate | DMC molar ratio | Conversion (%) | Cyclic product yield (%) | Cyclic product selectivity (%) | Mono-cyclic carbonate yield (%) | 13/16 yield (%) | 14/17 + 18 yield (%) | C-balance (%) |
|---|---|---|---|---|---|---|---|---|
| Conditions: 0.5 g substrate, 1 mol% 1 was utilised as the pre-catalyst for 3 h at 74 °C with DMC as “CO” source. Products are denoted as: 12 – propane 1,3-carbonate, 13 – 3-methoxycarbonyloxypropan-1-ol, 14 – propane-1,3-diyl dimethyl dicarbonate, 15 – butane 1,3-carbonate, 16 – 3-methoxycarbonyloxybutan-1-ol, 17 – 3-methoxycarbonyloxybutan-3-ol, 18 – butane-1,3-diyl dimethyl dicarbonate. | ||||||||
| Glycerol | 6 | 79 | 72 | 93 | — | — | — | 95 |
| Glycerol | 3 | 88 | 83 | 94 | — | — | — | 95 |
| Diglycerol | 6 | 58 | 21 | 37 | 32 | — | — | 95 |
| ED | 3 | 61 | 55 | 90 | — | — | — | 94 |
| 1,2-PD | 3 | 76 | 74 | 96 | — | — | — | 97 |
| 1,3-PD | 3 | 49 | 2 | 3 | — | 36 (13) | 7 (14) | 96 |
| 1,2-BD | 3 | 80 | 79 | 99 | — | — | — | 100 |
| 1,3-BD | 3 | 40 | 2 | 4 | — | 26 (16) | 13 (17 + 18) | 101 |
| 2,3-BD | 3 | 51 | 41 | 80 | — | — | — | 90 |
The products from the 1,3-diol reactions could not be identified unambiguously from the crowded standard 1H NMR spectra (Fig. S3 and S5†), but could from their 2D NMR spectra (Fig. S4, S6 (both 1H–1H TOCSY) and S7 (1H–1H COSY)†). In Table 2, products 17 and 18 are presented together due to the fact that their spectra are indistinguishable in the NMR spectra. As expected, much less cyclic carbonate formation is observed for 1,3-butanediol and 1,3-propanediol (2% in both cases), with six-membered rings being less favourable than the five-membered ones formed from their 1,2-counterparts. The conversion of these 1,3-diols was still relatively high, however, and in agreement with Selva et al.,38 open carbonates were detected instead (Fig. S2†).
For 1,3-propanediol yields, 36% and 7%, of the open mono- (13) and dicarbonate (14) were formed. For 1,3-butanediol, the yield of the open carbonate of the primary alcohol (16, 26%) was significantly higher than the combination of the 17 and 18, i.e. those carbonates involving the secondary alcohol (13%). Comparing the results of the three butanediols tested, both 1,2- and 2,3-butanediol can be converted to their respective five-membered cyclic carbonates, however the yield of the internal cyclic carbonate is lower than of the terminal counterpart. For 1,3-butanediol the yield of the six-membered cyclic carbonate is very small, with the open carbonates being favoured.
The conversion of ethylene diol, 1,3-propanediol and 2,3-butanediol was also studied over time, Table 3. The reaction appears to be equilibrium-limited, with its position dependent on the substrate. For example, ethylene diol yielded 32% (100% selectivity) of the cyclic carbonate after 20 min, however only produced a yield of 58% after 6 h, with similar trends observed with different substrates. Indeed, addition of the equivalent amount of methanol that would be produced upon full conversion, from the start to a reaction of glycerol and 1 mol% 1 at 74 °C for 3 h led to a 13% reduction in both conversion and yield, but with the very high selectivity (99%) maintained. A reaction run in methanol as solvent yielded only 11% after 3 h compared with 83% without added methanol. 1,3-Propanediol was initially converted to the open carbonate, with yields of the cyclic product being only 2% after 1 h. However, as the reaction progressed, the yield of the open dicarbonate (14) increased from 4% after 1 h to 11% after 6 h, whilst still maintaining high selectivity (70–75%) towards the 1-mono open product (13), Table 3. This again highlights the higher reactivity of the diol substrate compared to the open monocarbonate product and that the open carbonates are favoured over cyclic carbonates for 1,3-diols. Without catalyst, only starting material is recovered for both 1,3-butanediol and 1,3-propanediol under standard reaction conditions. The fact that no open products are observed for substrates that yield the five-membered cyclic products indicated that under our conditions, the ring closing reaction is fast and driven by the stability of the product.38
| Substrate | Time (h) | Temp. (°C) | Conversion (%) | 1-Open mono-carbonate yield (%) | DGMC yield (%) | 14/18 and 17 yield (%) | Cyclic carbonate yield (%) | Cyclic carbonate selectivity (%) | C-balance (%) |
|---|---|---|---|---|---|---|---|---|---|
| Conditions: 0.5 g substrate, stirred under argon for the indicated reaction time at indicated temperature. 1 mol% 1 was utilised as the pre-catalyst with 3:1 DMC:substrate ratio. Products are denoted as: 12 – propane 1,3-carbonate, 13 – 3-methoxycarbonyloxypropan-1-ol, 14 – propane-1,3-diyl dimethyl dicarbonate, 15 – butane 1,3-carbonate, 16 – 3-methoxycarbonyloxybutan-1-ol, 17 – 3-methoxycarbonyloxybutan-3-ol, 18 – butane-1,3-diyl dimethyl dicarbonate.a DG:DMC ratio was 6:1. |
|||||||||
| ED | 0.33 | 74 | 32 | — | — | — | 32 | 100 | 100 |
| ED | 1 | 74 | 59 | — | — | — | 54 | 91 | 95 |
| ED | 3 | 74 | 61 | — | — | — | 55 | 90 | 94 |
| ED | 6 | 74 | 63 | — | — | — | 58 | 92 | 95 |
| 1,3-PD | 1 | 74 | 35 | 25 | — | 4 | 2 | 6 | 96 |
| 1,3-PD | 3 | 74 | 49 | 36 | — | 7 | 1 | 3 | 96 |
| 1,3-PD | 6 | 74 | 58 | 43 | — | 11 | 10 | 2 | 97 |
| 2,3-BD | 1 | 74 | 33 | — | — | — | 28 | 83 | 95 |
| 2,3-BD | 3 | 74 | 51 | — | — | — | 41 | 80 | 90 |
| 2,3-BD | 6 | 74 | 56 | — | — | — | 50 | 88 | 93 |
| G | 3 | 90 | 95 | 0 | — | — | 98 | 103 | 103 |
| DGa | 3 | 90 | 74 | — | 21 | — | 45 | 60 | 92 |
| 1,3-BD | 3 | 90 | 69 | 41 | — | 26 | 3 | 4 | 101 |
| 2,3-BD | 3 | 90 | 67 | — | — | — | 69 | 103 | 102 |
Results reported thus far have been for reactions carried out at 74 °C. Increasing the temperature to 90 °C for reactions with a few selected substrates, did lead to higher activity, Table 3. Glycerol reached 95% conversion within 3 h, which is a slight increase of 7% compared with the reaction run at 74 °C. Diglycerol saw its yield double to 45%, with an increase in selectivity towards the dicyclic product, 60% compared with 37% at 74 °C, with less mono-cyclic product yielded. 1,3-Butanediol did not show a significant change in the amount of cyclic product yielded at the higher reaction temperature, however an increase in the products involving the secondary alcohol (17 and 18) was noted. For the internal diol of 2,3-butanediol, yield of cyclic product increased by 28% at the increased temperature. Care must be taken with further increasing the temperature, as above 95 °C the formation of the less active 4-carboxylate becomes favoured over the 2-carboxylate.
 | ||
| Fig. 6 Glycerol conversion (black symbols) and glycerol carbonate yield (red symbols) against time for the conversion of pure (circles) and crude (squares) glycerol with 1. Conditions: 1 mol% loading of pre-catalyst, 3:1 DMC:(crude) glycerol, 74 °C. | ||
:1 molar ratio run in DMSO-d6 provided more insight into the mechanism of reaction. 1H, 13C and HMBC 2D NMR experiments allowed the identification of a 13C-labelled methyl ester group attached to the imidazolium compound and a signal attributed to 13C-labelled methanol (Fig. 7(a), S8 and S9†). The HMBC NMR spectra confirmed that the 13C-labelled ester group was indeed attached to the imidazole ring, through multiple bond correlation. The detection of the labelled carbonyl carbon and methyl ester carbon attached to the imidazole framework indicates that decarboxylation of the unlabelled CO2 had occurred, followed by attack of the carbonyl carbon of the 13C3-DMC, with subsequent loss of one 13C-labelled methoxy group. The observation of such a labelled ester species allowed other mechanisms, such as one in which the carboxylate itself acts as a base to deprotonate the alcohol group of the substrate, to be ruled out and suggests that an NHC species is indeed involved in the catalytic cycle. The formation of 13C-labelled methanol is thought to be the result of the methoxy anion reacting with trace amounts of water in the NMR solvent, and hence the counter ion is a hydroxyl group in this experiment.
 | ||
| Fig. 7 (a) 13C-NMR of reaction between 13C3-DMC and 1,3-dimethylimidazolium-2-carboxylate (1) at 74 °C for 18 h, in DMSO-d6, X representing OH. * represents 13C-labelled carbon atoms. (b) Proposed mechanism for the conversion of vicinal diol substrates to cyclic carbonates. | ||
As the NHC attacks the carbonate and is not protonated by the water present, this suggests that the NHC itself does not deprotonate the alcohol either, but that is the role of the methoxy group generated following the nucleophilic attack of the NHC on the carbonyl group of DMC. Taking into account previous literature and the reaction run with 13C3-DMC, we propose the mechanism shown in Fig. 7(b), in which the in situ generated carbene attacks the carbon of the carbonyl group of DMC. In doing so, one methoxy anion is cleaved, and in turn deprotonates the primary alcohol group of the substrate. The substrate alkoxide can then attack the carbonyl group attached to the imidazole complex and the other methoxy anion is lost. This free anion can then deprotonate the secondary alcohol group, which can then attack the carbonyl carbon of the imidazole complex regenerating the active carbene species. Based solely on literature evidence,47,48 Palencia et al. have recently proposed a similar mechanism starting from 1-(2,6-dimethylphenyl)-3-hexadecylimidazolium bromide, which requires the addition of a base to yield the carbene in situ.35
Catalyst immobilisation
To allow for facile separation and potential reuse the organocatalyst was immobilised on a support. Carbene-based catalysts and their masked forms, including the 2-carboxylates, have been integrated into a number of polymer systems for a variety of reactions using different approaches.52–54 Notably, it has been shown that a differently masked NHC, i.e. the hydrogen carbonate protected form ((H)HCO3−), allows facile handling and this system can be easily recovered and reused, maintaining high activity.31,55,56 A silica-supported hydrogen carbonate masked NHC catalyst was, for example, used in the epoxidation of olefins with H2O2.57 For these reasons we have attempted to synthesise such protected NHCs on silica for the purpose of cyclic carbonate formation. Based on the results detailed above and previous studies in which the active carbene catalyst is protected by either CO2 or (H)HCO3− to allow facile handling of the catalyst,52,55 1-methyl-3-(3-triethoxysilylpropyl) imidazolium chloride was immobilised on self-synthesised mesostructured cellular foam (MCF) silica (IL1). IL1 was subsequently converted to generate two differently masked supported-NHCs; either the supported CO2-masked NHC (IL2) via deprotonation and treatment with CO2 or (H)HCO3-masked NHC (IL3), via ion exchange with KHCO3. 1-Methyl-3-(3-triethoxysilylpropyl) imidazolium chloride was chosen for grafting as it can allow a route to both IL2 and IL3 and due to the fact that initial attempts to activate supported 1-propylimidazole with DMC, as with the organocatalyst system, proved unsuccessful. Siliceous MCF was chosen due to its large surface area and pore volume as well as the fact that imidazolium halide compounds had previously been successfully grafted to it.58Degree of functionalisation and textual properties. The MCF used was characterised by a high surface area (738 m2 g−1) and average pore size of 12.1 nm. IL1 was prepared by grafting 0.93 mmol g−1 of 1-methyl-3-(3-triethoxysilylpropyl) imidazolium chloride (as determined by TGA) on the MCF support, which was accompanied by a reduction in surface area and pore volume, in accordance with literature.59–62 Conversion of IL1 to IL2 and IL3 resulted in functionalisations of 0.59 and 0.34 mmol g−1, respectively. The results presented in Table 4, show that pore volume of IL2 is similar to that of IL1, but increases upon converting IL1 to IL3, which can be attributed to the partial loss of functionalisation that occurs. The same trend is observed when comparing surface area, which is similar for IL1 and IL2, but increases for IL3. This could be associated with the lower degree of functionalisation, with higher degrees of functionalisation known to decrease surface area.62 This trend is similar to the one observed by Park et al.59
The degrees of functionalisation of IL1, IL2 and IL3 were analysed both with TGA and elemental analysis (EA), Fig. S11† and Table 4. For IL1, TGA measurements showed a loss of 17.5% above 180 °C, with distinct weight losses at 247 °C, and 494 °C, which are associated with the loss of ethoxy groups left on the silica tether and the imidazole group, respectively.63 For IL2 and IL3 the weight losses above 180 °C were 11.6% and 8.3% respectively, which equates to loadings of 0.59 mmol g−1 and 0.34 mmol g−1. The weight loss at higher temperatures for IL2 is less sharp, and can be associated with partial hydrolysis of the ethoxy group of the tether during functionalisation. The loss in this region for IL3 comes at a higher temperature than the other functionalised samples, which is attributed to full hydrolysis of the ethoxy groups of the tether due to the exposure to water during the synthesis route.63 The degree of functionalisation obtained by elemental analysis and TGA differed considerably, with EA giving higher values of 1.40, 0.96, and 0.53 mmol g−1 for IL1, IL2 and IL3, respectively. For parity, catalyst loading was calculated using the TGA data. Elemental analysis also indicated that around 80% of the inactive chloride was exchanged for hydrogen carbonate in the ion exchange step for the synthesis of IL3. The significantly lower degree of functionalisation seen for IL3 can be attributed to the use of methanol and water in each of the procedures respectively. It has been observed that the Si–O bond is labile when in the presence of water and alcohols.64 When IL1 is stirred in water to yield IL3 the loss of functionalisation is more extreme than when IL2 is washed with methanol.
DRIFTS and solid-state NMR analysis. IL1, IL2, IL3 and pristine MCF were analysed by means of DRIFTS, as shown in Fig. S12.†IL1 exhibited the characteristic bands of aromatic C–H stretching (2930, 2960, 3100 and 3150 cm−1), ring stretching of the imidazolium group (1575, 1525 and 1460 cm−1) and the C–Si stretching (620 cm−1) vibrations, indicating, in accordance with literature, successful grafting had taken place.59 As well as the additional signals attributed to C–H, C–C and C–N bonds, functionalisation was also indicated by a reduction in the free silanol signal at 3740 cm−1.65IL2 and IL3 also contain the characteristic signals of the organic stretching vibrations as in IL1, indicating that functionalisation remains after their respective treatments. The band at 1312 cm−1 in the spectra of IL3 can be tentatively assigned to the symmetrical stretching vibration of the hydrogen carbonate counter ion,66 however due to the strong absorption bands of the silica support, the carboxylate and (H)HCO3 forms of the supported system could not be confirmed using DRIFTS analysis. 13C MAS-NMR measurements of the IL1, IL2 with and without methanol washing and IL3 (Fig. 8) confirmed the nature of the groups tethered to the MCF silica. In the NMR spectra, all the signals of the molecule are accounted for and in accordance with literature, with the addition of some residual signals attributed to unreacted ethoxy groups at 59 and 16 ppm.59,63,67,68 To enhance the signal of the quaternary carboxylate carbon, IL2 was prepared using 13C-labelled CO2.
 | ||
| Fig. 8 13C MAS-NMR of (a) IL1, (b) IL2 without washing, (c) IL2 washed with methanol (d) IL3. IL2 was prepared with 13C-labelled CO2 to enhance the signal of the quaternary carbon of the carboxylate. * represents 13C-labelled carbon atoms. | ||
The initial measurement of a sample of IL2 (Fig. 8(c)) prepared as described in the ESI† with a final methanol washing step, to remove the potassium chloride salt, did not show the anticipated carboxylate signal at around 160 ppm. This could either be due to unsuccessful carboxylation or be the result of carboxylate loss due to reaction with moisture in the methanol, hence forming the hydrogen carbonate derivative.45,69 The latter is in accordance with literature that demonstrated the interchange of the carboxylate and hydrogen carbonate forms in wet solvents.56,69 For the IL2 sample that was not washed with methanol (Fig. 8(b)), a signal at 162 ppm was indeed seen, corresponding to the carbon of the carboxylate.51 Further 1H–13C cross-polarisation experiments varying the Hartmann–Hahn contact time, Fig. S13,† show this peak to indeed belong to a quaternary carbon, and thus that the carboxylation was successful. In fact, the NMR spectrum of the methanol-washed IL2 sample is very similar to the one of IL3, which contains a hydrogen carbonate counter ion, further evidencing that the carboxylate had formed and reacted with trace amounts of water. The signal of hydrogen carbonate expected around 160 ppm is not observed in the spectra, however this signal is often weak compared to the other carbons of the imidazolium compound in solution NMR and given the relatively low loading, the HCO3 carbon was not resolved.31,55,56 Signals attributed to the ethoxy group of the silicon tether are reduced in those samples exposed to wet conditions (Fig. 8(c) and (d)). The peak seen around −1 ppm in each of the IL2 samples was identified as being quaternary, but its origin is not clear. These NMR findings thus indicate that for catalytic testing IL2 and IL3 can in fact be considered to be the same (H)HCO3 protected carbenes, which differ in the degree of functionalisation.
Synthesis of glycerol carbonate. The supported hydrogen carbonate-masked NHCs were studied for the conversion of glycerol to glycerol carbonate. Reaction temperature and DMC
:substrate ratio were assessed and results are presented in Table 5. A reaction temperature of 90 °C gave the highest yield (37%) and selectivity (104%) when run with a DMC:substrate ratio of 5 for 6 h. This temperature is just below the point at which the 4-carboxylate begins to be favoured over the 2-carboxylate, as shown for the homogeneous system earlier. The 5:1 ratio yielded the highest amount of product, 37%, compared to ratios of 2.5:1, 7.5:1 and 10:1, which yielded 29%, 30% and 22%, respectively, under the same conditions. The DMC:substrate ratio thus had a significant effect on the activity of the catalyst, as was also observed for the homogeneous system. The lower activity observed for the reaction run with IL2 at 110 °C may be associated with the formation of the inactive 4-carboxylate form. Reactions without any catalyst, with only pristine MCF or with the catalyst precursor IL1 showed no conversion under these conditions (Table 5).
:glycerol ratio and reaction time on the synthesis of glycerol carbonate
| Catalyst | DMC molar ratio | Temp. (°C) | Time (h) | Conversion (%) | Cyclic yield (%) | Cyclic selectivity (%) | C-balance (%) |
|---|---|---|---|---|---|---|---|
| Conditions: 0.25 g glycerol with 1 mol% loading of IL2. | |||||||
| — | 5 | 90 | 6 | 0 | 0 | 0 | 101 |
| MCF | 5 | 90 | 6 | 0 | 0 | 0 | 99 |
| IL1 | 5 | 90 | 6 | 0 | 0 | 0 | 103 |
| IL2 | 5 | 90 | 6 | 35 | 37 | 104 | 101 |
| IL2 | 5 | 74 | 6 | 19 | 17 | 90 | 98 |
| IL2 | 5 | 110 | 6 | 27 | 21 | 79 | 94 |
| IL2 | 2.5 | 90 | 6 | 32 | 29 | 90 | 97 |
| IL2 | 7.5 | 90 | 6 | 37 | 30 | 81 | 93 |
| IL2 | 10 | 90 | 6 | 22 | 22 | 98 | 100 |
| IL2 | 5 | 90 | 24 | 83 | 75 | 91 | 92 |
Monitoring conversion and selectivity against time for both IL2 and IL3 at the same (tethered) catalyst loading (Fig. 9), showed both systems to efficiently convert glycerol to glycerol carbonate at the same rate, once the active carbene is formed, with TOFs of 4.2 for both IL2 and IL3. This further confirms the NMR results, which indicated IL2 and IL3 essentially to be the same, only differing in the degree of functionalisation. The activity of the supported carbene studied here is significantly lower than the homogeneous imidazolium-carboxylates described above and by Naik et al.27 The homogeneous organocatalyst does not allow for facile recovery and reuse of the catalyst, however. This supported imidazolium system under study can be easily recovered, and as shown below, recycled. Having established that IL2 and IL3 are the same species, further experiments were run using IL3 only.
 | ||
| Fig. 9 Comparison of IL2 (black) and IL3 (red) for the conversion of glycerol (squares) to glycerol carbonate (circles) over time. | ||
 | ||
| Fig. 10 Recycling experiments of IL3. Conditions: 1 mol% loading of catalyst, 90 °C, 8 h, DMC:glycerol ratio of 5. Spent IL3 was washed with KHCO3 before reuse for Regen. result. For all other results, the spent catalyst was used without further treatment. | ||
Spent IL3 for run 1 was analysed using DRIFTS and TGA. TGA of IL3, Fig. S14(b),† indicated an increase in weight loss of just 2% compared to the fresh catalyst and signals attributed to glycerol carbonate were detected in DRIFTS (Fig. S14(a)†). However, the high mass balance together with the results of both the DRIFTS and TGA results show that little organics are deposited on the solid.
:diglycerol ratio of 10.39 The low selectivity towards the dicyclic product observed at shorter reaction times is due to the fact that the DGMC is formed first, as was the case for the hydrotalcite system39 and the homogeneous NHC catalysts described above. The catalytic activity of IL3 towards diglycerol is considerably lower than that observed for the homogeneous 1,3-imidazolium-2-carboxylate (1) and expresses turnover numbers around one tenth of the only other known heterogeneous catalyst for diglycerol conversion, around 152 compared with 1887.39 Of particular note is that the catalytic system also efficiently converts crude glycerol, Table 6. Similar yields and selectivities were achieved for both the pure and crude glycerol, and similar yields as that of the homogeneous system, although obtained at a slightly more elevated temperature. This is in contrast with the results of the homogeneous system, which expressed reduced activity when converting the same crude glycerol.
| Substrate | DMC: substrate ratio | Time (h) | Conversion (%) | Cyclic yield (%) | Cyclic selectivity (%) | DGMC yield (%) | C-balance (%) |
|---|---|---|---|---|---|---|---|
| Conditions: 1 mol% IL3, at 90 °C with DMC as “CO” source. | |||||||
| DG | 10 | 6 | 51 | 8 | 15 | 38 | 95 |
| DG | 10 | 24 | 83 | 68 | 82 | 16 | 101 |
| 1,2-BD | 5 | 6 | 48 | 48 | 101 | — | 100 |
| 2,3-BD | 5 | 6 | 15 | 15 | 98 | — | 100 |
| Crude G | 5 | 6 | 52 | 44 | 85 | — | 92 |
Conclusions
Imidazolium carboxylates and hydrogen carbonates were studied as masked NHCs and found to successfully catalyse the synthesis of cyclic carbonates from their respective diols. The organocatalyst system expressed high activity for the conversion of diglycerol to diglycerol dicarbonate, with loadings as low as 1 mol%, operating under solvent-free and mild conditions. The substrate scope of this system was extended to different renewable diol-substrates, containing both terminal and internal diols. Cyclic carbonate products were yielded in high amounts with good selectivity for systems that contain vicinal diols, however the 1,3-diol systems we investigated favoured open carbonates over the six-membered cyclic product. Notably, the system is able to efficiently convert crude glycerol, expressing high selectivity towards glycerol carbonate, albeit at a reduced rate compared to reactions with pure glycerol. Structural variation of the organocatalyst illustrated the sensitivity of the system to the introduction of either electron-withdrawing or donating groups, affecting either the initial methylation of the precursor or the relative stability of the carbene catalyst vs. the carboxylate pre-catalyst. A mechanism for this system was furthermore postulated based on studies with 13C-labelled dimethyl carbonate, demonstrating that the system indeed involves a carbene as active catalyst.The masked NHC system has also been heterogenised on siliceous MCF, protecting the NHC with hydrogen carbonate. This heterogeneous catalyst could efficiently convert both pure and crude glycerol feedstocks, as well as a number of other vicinal diols under mild conditions and without the use of an additional solvent. Catalyst recycling studies with glycerol as substrate showed that the catalyst can be easily recovered and reused if properly reactivated by a straightforward ion-exchange procedure between runs.
Experimental
Details of materials, instrumentation, substituted imidazole synthesis, MCF synthesis and product analysis can be found in the ESI.†Typical homogeneous NHC testing
:1 molar ratio of dimethyl carbonate to diol. The vials were placed under argon and stirred under heating for the assigned time.
Typical heterogenised NHC testing
:1 molar ratio of dimethyl carbonate. The vials were placed under argon, sealed and stirred under heating for the assigned time.
Acknowledgements
The authors would like to thank the Biobased Performance Materials (BPM) research programme for funding, project no. BPM-013.References
- M. North, R. Pasquale and C. Young, Green Chem., 2010, 12, 1514–1539 RSC.
- S.-Y. Huang, S.-G. Liu, J.-P. Li, N. Zhao, W. Wei and Y.-H. Sun, J. Fuel Chem. Technol., 2007, 35, 701–705 CrossRef CAS.
- M. J. Climent, A. Corma, P. De Frutos, S. Iborra, M. Noy, A. Velty and P. Concepción, J. Catal., 2010, 269, 140–149 CrossRef CAS.
- C. Martín, G. Fiorani and A. W. Kleij, ACS Catal., 2015, 5, 1353–1370 CrossRef.
- M. Cokoja, M. E. Wilhelm, M. H. Anthofer, W. A. Herrmann and F. E. Kühn, ChemSusChem, 2015, 8, 2436–2454 CrossRef CAS PubMed.
- H. W. Tan, A. R. Abdul Aziz and M. K. Aroua, Renewable Sustainable Energy Rev., 2013, 27, 118–127 CrossRef CAS.
- P. Cintas, S. Tagliapietra, E. Calcio Gaudino, G. Palmisano and G. Cravotto, Green Chem., 2014, 16, 1056–1065 RSC.
- M. Pagliaro, R. Ciriminna, H. Kimura, M. Rossi and C. Della Pina, Angew. Chem., Int. Ed., 2007, 46, 4434–4440 CrossRef CAS PubMed.
- M. O. Sonnati, S. Amigoni, E. P. Taffin de Givenchy, T. Darmanin, O. Choulet and F. Guittard, Green Chem., 2013, 15, 283–306 RSC.
- A. Behr, J. Eilting, K. Irawadi, J. Leschinski and F. Lindner, Green Chem., 2008, 10, 13–30 RSC.
- J. R. Ochoa-Gómez, O. Gómez-Jiménez-Aberasturi, C. Ramírez-López and M. Belsué, Org. Process Res. Dev., 2012, 16, 389–399 CrossRef.
- F. Strain, US Pat, US2446145, 1948 Search PubMed.
- W. K. Teng, G. C. Ngoh, R. Yusoff and M. K. Aroua, Energy Convers. Manage., 2014, 88, 484–497 CrossRef CAS.
- A. Takagaki, K. Iwatani, S. Nishimura and K. Ebitani, Green Chem., 2010, 12, 578–581 RSC.
- P. Liu, M. Derchi and E. J. M. Hensen, Appl. Catal., A, 2013, 467, 124–131 CrossRef CAS.
- M. Malyaadri, K. Jagadeeswaraiah, P. S. Sai Prasad and N. Lingaiah, Appl. Catal., A, 2011, 401, 153–157 CrossRef CAS.
- J. Li and T. Wang, J. Chem. Thermodyn., 2011, 43, 731–736 CrossRef CAS.
- F. S. H. Simanjuntak, T. K. Kim, S. D. Lee, B. S. Ahn, H. S. Kim and H. Lee, Appl. Catal., A, 2011, 401, 220–225 CrossRef CAS.
- J. Li and T. Wang, React. Kinet., Mech. Catal., 2011, 102, 113–126 CrossRef CAS.
- Z. Mouloungui, J.-W. Yoo, C.-A. Gachen, A. Gaset and G. Vermeersch, EP0739888, 1996 Search PubMed.
- H.-J. Cho, H.-M. Kwon, J. Tharun and D.-W. Park, J. Ind. Eng. Chem., 2010, 16, 679–683 CrossRef CAS.
- K. Lee, C.-H. Park and E. Lee, Bioprocess Biosyst. Eng., 2010, 33, 1059–1065 CrossRef CAS PubMed.
- S. Kim, Y. Kim, H. Lee, D. Yoon and B. Song, J. Mol. Catal. B: Enzym., 2007, 49, 75–78 CrossRef CAS.
- Y. Tsutsumi, K. Yamakawa, M. Yoshida, T. Ema and T. Sakai, Org. Lett., 2010, 12, 5728–5731 CrossRef CAS PubMed.
- Y.-B. Wang, Y.-M. Wang, W.-Z. Zhang and X.-B. Lu, J. Am. Chem. Soc., 2013, 135, 11996–12003 CrossRef CAS PubMed.
- Y.-B. Wang, D.-S. Sun, H. Zhou, W.-Z. Zhang and X.-B. Lu, Green Chem., 2014, 16, 2266–2272 RSC.
- P. U. Naik, L. Petitjean, K. Refes, M. Picquet and L. Plasseraud, Adv. Synth. Catal., 2009, 351, 1753–1756 CrossRef CAS.
- H. Zhou, W. Z. Zhang, C. H. Liu, J. P. Qu and X. B. Lu, J. Org. Chem., 2008, 73, 8039 CrossRef CAS PubMed.
- I. Tommasi and F. Sorrentino, Tetrahedron Lett., 2005, 46, 2141–2145 CrossRef CAS.
- A. M. Voutchkova, M. Feliz, E. Clot, O. Eisenstein and R. H. Crabtree, J. Am. Chem. Soc., 2007, 129, 12834–12846 CrossRef CAS PubMed.
- M. Fèvre, P. Coupillaud, K. Miqueu, J.-M. Sotiropoulos, J. Vignolle and D. Taton, J. Org. Chem., 2012, 77, 10135–10144 CrossRef PubMed.
- S. Naumann and M. R. Buchmeiser, Catal. Sci. Technol., 2014, 4, 2466–2479 CAS.
- M. Hans, L. Delaude, J. Rodriguez and Y. Coquerel, J. Org. Chem., 2014, 79, 2758–2764 CrossRef CAS PubMed.
- D. M. Denning and D. E. Falvey, J. Org. Chem., 2014, 79, 4293–4299 CrossRef CAS PubMed.
- B. Hervert, P. D. McCarthy and H. Palencia, Tetrahedron Lett., 2014, 55, 133–136 CrossRef CAS.
- G. Rokicki, P. Rakoczy, P. Parzuchowski and M. Sobiecki, Green Chem., 2005, 7, 529–539 RSC.
- J. George, Y. Patel, S. M. Pillai and P. Munshi, J. Mol. Catal. A: Chem., 2009, 304, 1–7 CrossRef CAS.
- M. Selva, A. Caretto, M. Noe and A. Perosa, Org. Biomol. Chem., 2014, 12, 4143–4155 CAS.
- J. A. Stewart, B. M. Weckhuysen and P. C. A. Bruijnincx, Catal. Today, 2015, 257, 274–280 CrossRef CAS.
- M. S. Kathalewar, P. B. Joshi, A. S. Sabnis and V. C. Malshe, RSC Adv., 2013, 3, 4110–4129 RSC.
- M. Bahr, A. Bitto and R. Mulhaupt, Green Chem., 2012, 14, 1447–1454 RSC.
- A. Boyer, E. Cloutet, T. Tassaing, B. Gadenne, C. Alfos and H. Cramail, Green Chem., 2010, 12, 2205–2213 RSC.
- L. Maisonneuve, A. S. More, S. Foltran, C. Alfos, F. Robert, Y. Landais, T. Tassaing, E. Grau and H. Cramail, RSC Adv., 2014, 4, 25795–25803 RSC.
- J. L. J. van Velthoven, L. Gootjes, D. S. van Es, B. A. J. Noordover and J. Meuldijk, Eur. Polym. J., 2015, 70, 125–135 CrossRef CAS.
- B. R. Van Ausdall, J. L. Glass, K. M. Wiggins, A. M. Aarif and J. Louie, J. Org. Chem., 2009, 74, 7935–7942 CrossRef CAS PubMed.
- A. R. Katritzky, H. Yang, D. Zhang, K. Kirichenko, M. Smiglak, J. D. Holbrey, W. M. Reichert and R. D. Rogers, New J. Chem., 2006, 30, 349–358 RSC.
- A. Schmidt, A. Beutler, M. Albrecht, B. Snovydovych and F. J. Ramirez, Org. Biomol. Chem., 2008, 6, 287–295 CAS.
- S. Okamoto, H. Ishikawa, Y. Shibata and Y.-i. Suhara, Tetrahedron Lett., 2010, 51, 5704–5707 CrossRef CAS.
- B. Gabriele, R. Mancuso, G. Salerno, L. Veltri, M. Costa and A. Dibenedetto, ChemSusChem, 2011, 4, 1778–1786 CrossRef CAS PubMed.
- G. A. Grasa, R. M. Kissling and S. P. Nolan, Org. Lett., 2002, 4, 3583–3586 CrossRef CAS PubMed.
- A. Hoppe, F. Sadaka, C.-H. Brachais, G. Boni, J.-P. Couvercelle and L. Plasseraud, Beilstein J. Org. Chem., 2013, 9, 647–654 CrossRef CAS PubMed.
- J. Pinaud, J. Vignolle, Y. Gnanou and D. Taton, Macromolecules, 2011, 44, 1900–1908 CrossRef CAS.
- H. Zhou, W.-Z. Zhang, Y.-M. Wang, J.-P. Qu and X.-B. Lu, Macromolecules, 2009, 42, 5419–5421 CrossRef CAS.
- G. M. Pawar and M. R. Buchmeiser, Adv. Synth. Catal., 2010, 352, 917–928 CrossRef CAS.
- P. Coupillaud, J. Pinaud, N. Guidolin, J. Vignolle, M. Fèvre, E. Veaudecrenne, D. Mecerreyes and D. Taton, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 4530–4540 CAS.
- M. Fèvre, J. Pinaud, A. Leteneur, Y. Gnanou, J. Vignolle, D. Taton, K. Miqueu and J.-M. Sotiropoulos, J. Am. Chem. Soc., 2012, 134, 6776–6784 CrossRef PubMed.
- C. Yuan, Z. Huang and J. Chen, Catal. Commun., 2012, 24, 56–60 CrossRef CAS.
- K. H. Park, S. Kim and Y. K. Chung, Bull. Korean Chem. Soc., 2008, 29, 2057–2060 CrossRef CAS.
- L. Han, S.-W. Park and D.-W. Park, Energy Environ. Sci., 2009, 2, 1286–1292 CAS.
- Y. Han, S. S. Lee and J. Y. Ying, Chem. Mater., 2007, 19, 2292–2298 CrossRef CAS.
- P. Schmidt-Winkel, W. W. Lukens, P. Yang, D. I. Margolese, J. S. Lettow, J. Y. Ying and G. D. Stucky, Chem. Mater., 2000, 12, 686–696 CrossRef CAS.
- H. Kim, J. C. Jung, S. H. Yeom, K.-Y. Lee, J. Yi and I. K. Song, Mater. Res. Bull., 2007, 42, 2132–2142 CrossRef CAS.
- S. Udayakumar, V. Raman, H.-L. Shim and D.-W. Park, Appl. Catal., A, 2009, 368, 97–104 CrossRef CAS.
- A. Monge-Marcet, R. Pleixats, X. Cattoen and M. Wong Chi Man, Catal. Sci. Technol., 2011, 1, 1544–1563 CAS.
- E. Borodina, S. I. Karpov, V. F. Selemenev, W. Schwieger, S. Maracke, M. Fröba and F. Rößner, Microporous Mesoporous Mater., 2015, 203, 224–231 CrossRef CAS.
- E. Garand, T. Wende, D. J. Goebbert, R. Bergmann, G. Meijer, D. M. Neumark and K. R. Asmis, J. Am. Chem. Soc., 2010, 132, 849–856 CrossRef CAS PubMed.
- B. Karimi, D. Elhamifar, J. H. Clark and A. J. Hunt, Chem. – Eur. J., 2010, 16, 8047–8053 CrossRef CAS PubMed.
- S. Udayakumar, M.-K. Lee, H.-L. Shim, S.-W. Park and D.-W. Park, Catal. Commun., 2009, 10, 659–664 CrossRef CAS.
- N. J. Bridges, C. C. Hines, M. Smiglak and R. D. Rogers, Chem. – Eur. J., 2007, 13, 5207–5212 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5gc02046h |
| This journal is © The Royal Society of Chemistry 2016 |