
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Molecular structure, morphology and growth mechanisms and rates of 5-hydroxymethyl furfural (HMF) derived humins†
George
Tsilomelekis
,
Michael J.
Orella
 ,
Zhexi
Lin
,
Ziwei
Cheng
,
Weiqing
Zheng
,
Vladimiros
Nikolakis
and
Dionisios G.
Vlachos
*
,
Zhexi
Lin
,
Ziwei
Cheng
,
Weiqing
Zheng
,
Vladimiros
Nikolakis
and
Dionisios G.
Vlachos
*
Department of Chemical and Biomolecular Engineering, Catalysis Center for Energy Innovation, University of Delaware, Newark, DE 19716, USA. E-mail: vlachos@udel.edu
First published on 17th November 2015
Abstract
We apply ATR-FTIR spectroscopy, Scanning Electron Microscopy (SEM) and Dynamic Light Scattering (DLS) experiments to investigate the molecular structure, morphology and growth mechanism of 5-hydroxymethyl furfural (HMF) derived humins as a function of HMF conversion. Our FTIR data support a reaction pathway in which humins form either through a ring opening mechanism and/or through substitution at the α or β position via nucleophilic attack. The addition of DMSO as a co-solvent leads to significant changes in the FTIR spectra of humins. We find that the nucleophilic attack pathway is suppressed in the presence of DMSO co-solvent and rationalizes the very small humin particles (∼100 nm) observed in SEM images contrary to the large particles (with multimodal size distribution and largest particles of up to 3–4 μm) observed in neat water. DLS experiments under several reaction conditions further confirm the particle size distribution observed via SEM. A plausible reaction network for humin formation, which rationalizes qualitatively our experimental results as well as those reported in the literature, is also postulated.
1. Introduction
The overdependence of the developed world on finite energy resources in conjunction with the increasing CO2 emissions and the recurring uncertainty in the price of crude oil motivate research efforts for the development of processes that use alternative or renewable sources for the production of fuels and chemicals. There is a general consensus that the abundant biomass is an alternative carbon containing carrier to augment production of petroleum-based fuels and chemicals. 5-Hydroxymethylfurfural (HMF) is an important, highly functionalized, bio-based chemical building block, produced from the dehydration of hexoses (Scheme 1), which can play a key role in the production of biomass-derived intermediates, such as 2,5-furandicarboxylic acid,1 2,5-dimethylfuran,2,3 adipic acid and levulinic acid (LA),4etc. A number of studies have been performed on the kinetics of HMF formation from various feedstocks (glucose, fructose, oligo- and poly-saccharides) as summarized elsewhere.5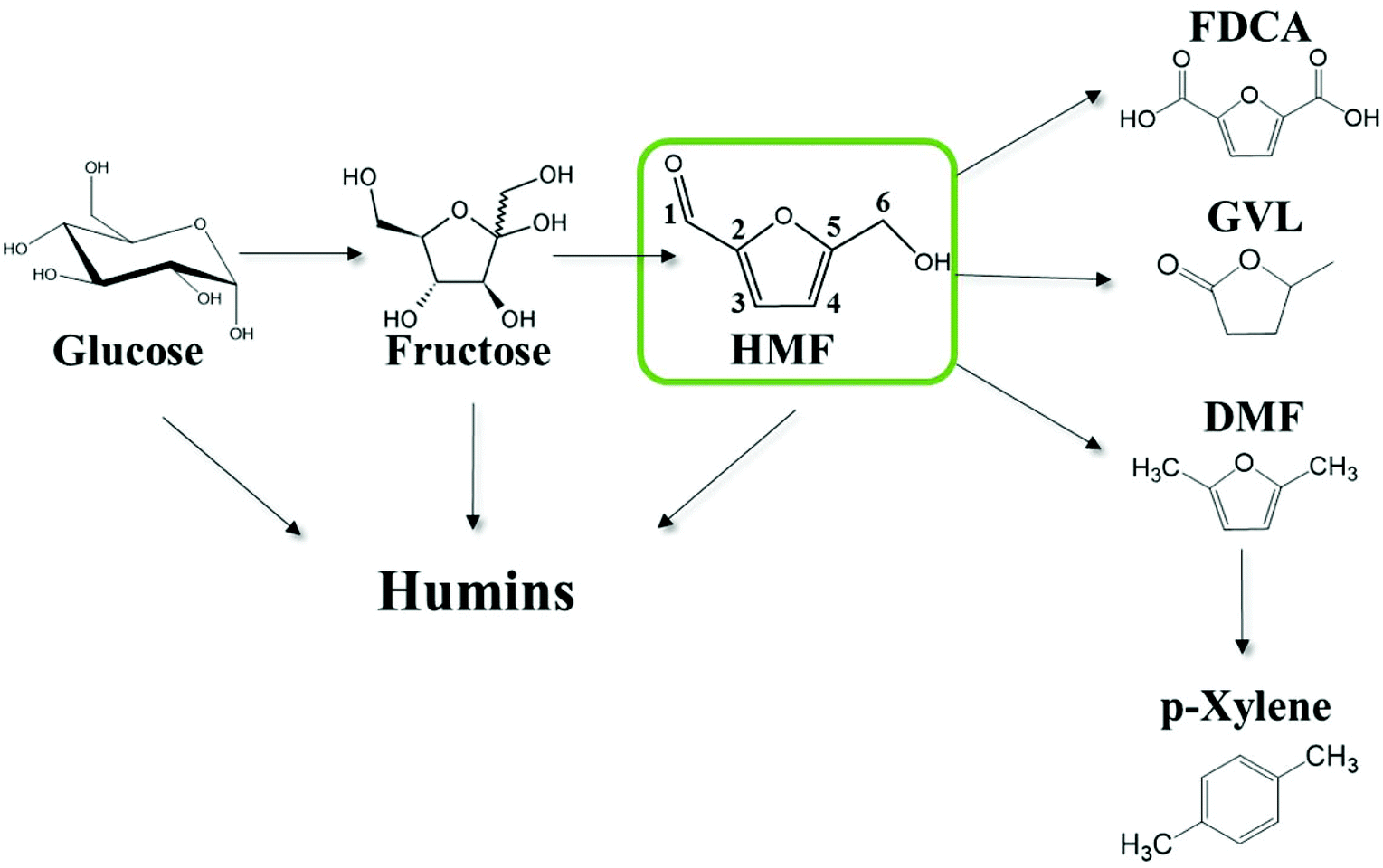 | ||
| Scheme 1 Thermocatalytic reaction network of the solution phase acid catalyzed cellulosic biomass-derivatives conversion to fuels, chemicals and humins. Carbons are numbered for convenience. | ||
The high functionality of biomass-derived chemicals is also responsible for side reactions leading to undesirable products. Techno-economic analysis of the production of renewable p-xylene from biomass suggests that suppressing the side reactions occurring in the production of HMF is key to its economic viability.6 In general, the acid catalyzed conversion of biomass is unavoidably accompanied by the formation of complex and recalcitrant carbonaceous materials, the so-called humins.7–12
Existing knowledge about the molecular structure of these carbonaceous materials is based primarily on studies of hydrothermal carbons (HTC), formed by reacting sugar solutions in a non-acidic environment, and to a smaller extent on studies of humins, formed during the acid catalyzed conversion of carbohydrates.13–16 These could possess similar structural features. Previous studies indicate that humins consist of a furan-rich polymer network containing different oxygen functional groups.15,17 The structure strongly depends on the reaction conditions, such as feedstock structure, reactant concentration, reaction temperature, and time.13,14,18,19 Humic materials adapt a spherical structure consisting of a condensed, hydrophobic core and a less dense, hydrophilic shell.13,14,20 This work focuses on humins made during the acid catalyzed biomass processing.
Sumerskii et al. have suggested that the humins consist of a network of 60% furan rings linked with 20% ether or acetal aliphatic linkers formed via polycondensation.21 Lund et al.22,23 proposed that humins originate from 2,5-dioxo-6-hydroxyhexanal (DHH), formed by the rehydration of HMF, which undergoes aldol condensation with the carbonyl group of HMF. An extensive conjugated network of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and furan rings with aldehyde or ketone functional groups was proposed based on IR spectroscopy. The proposed aldol addition/condensation pathway was further corroborated by reacting HMF with benzaldehyde as a model compound. Lund and co-workers also suggest that levulinic acid (LA) is not involved in the humin formation reaction. The extent of incorporation of HMF in the humin structure is dependent on the accumulation of HMF during the acid-catalyzed conversion of the sugars. They also suggested that humins could not be directly formed from sugars. van Zandvoort et al.18 suggested that the elemental composition of humins results from condensation reactions between sugars, HMF, and intermediates during the dehydration of carbohydrates. Condensation reactions between HMF molecules occur mainly through linkages at the α position or through substitutions at the β position of HMF via nucleophilic attack. In addition, the rehydration product, LA, is not significantly incorporated in humins even though aldol condensation of HMF with the ketone group is possible. Recent NMR data (1D cross polarization and direct excitation 13C solid-state NMR, 2D 13C-detected double-quantum, 2D 1H-detected heteronuclear correlation) revealed that the major linkages in humins, formed in acid catalyzed glucose dehydration, involve Cα–Caliphatic and Cα–Cα bonds while Cβ–Caliphatic and Cβ–Cβ were observed to a smaller extent.24 Ketone groups present were shown to originate from the incorporation of LA in the molecular structure through covalent bonds or from sugar dehydration and HMF rehydration intermediates.
C and furan rings with aldehyde or ketone functional groups was proposed based on IR spectroscopy. The proposed aldol addition/condensation pathway was further corroborated by reacting HMF with benzaldehyde as a model compound. Lund and co-workers also suggest that levulinic acid (LA) is not involved in the humin formation reaction. The extent of incorporation of HMF in the humin structure is dependent on the accumulation of HMF during the acid-catalyzed conversion of the sugars. They also suggested that humins could not be directly formed from sugars. van Zandvoort et al.18 suggested that the elemental composition of humins results from condensation reactions between sugars, HMF, and intermediates during the dehydration of carbohydrates. Condensation reactions between HMF molecules occur mainly through linkages at the α position or through substitutions at the β position of HMF via nucleophilic attack. In addition, the rehydration product, LA, is not significantly incorporated in humins even though aldol condensation of HMF with the ketone group is possible. Recent NMR data (1D cross polarization and direct excitation 13C solid-state NMR, 2D 13C-detected double-quantum, 2D 1H-detected heteronuclear correlation) revealed that the major linkages in humins, formed in acid catalyzed glucose dehydration, involve Cα–Caliphatic and Cα–Cα bonds while Cβ–Caliphatic and Cβ–Cβ were observed to a smaller extent.24 Ketone groups present were shown to originate from the incorporation of LA in the molecular structure through covalent bonds or from sugar dehydration and HMF rehydration intermediates.
Based on the existing literature, it is clear that HMF is a key species in the formation of humins during the acid catalyzed conversion of cellulosic biomass. The absence of extensive reported kinetics on HMF degradation, especially in different solvents has hampered progress in understanding the stability of HMF under acidic conditions. Several plausible mechanisms for the rehydration of HMF have been suggested based on NMR experiments25 and thermochemistry calculations.26 The mechanism of humin formation and growth is even less understood.18,21,25,27 It has been reported that protonating HMF at C6, could result in dehydration followed by further HMF addition through the formation of acetal and hemiacetal bonds.21 Consequently, the acid concentration is expected to play an important role in the formation of humins. In addition, the regioselective 2,3 addition of water to HMF results in DHH that may react with HMF to form humins25,27 but mechanistic details are lacking. Besides the acid and water concentration, temperature is also expected to play an important role on HMF degradation. Most of the previous studies focused mainly on the thermal decomposition of HMF in subcritical or supercritical water as well as on the acid catalyzed hydrolysis of HMF in aqueous systems.28–30 Previous studies, summarized elsewhere,5 showed that the activation energy for the HMF degradation to humins is around ∼100 kJ mol−1 while the corresponding activation energy for the rehydration to levulinic acid spans a range of 50–225 kJ mol−1 underlining the sensitivity of the levulinic acid production on temperature. However, kinetic experiments at milder conditions (similar to those reported herein) have shown that the selectivity to levulinic acid relative to humins is not a strong function of temperature.23
Herein, we focus on the understanding of HMF derived humins during Brønsted acid HMF degradation. Our work builds on our research endeavors in providing molecular level understanding about the stability of HMF towards degradation reactions.31–33 Specifically, the goal of the present study is to investigate the effect of reaction conditions, HMF conversion and acid concentration on the molecular structure, morphology and particle growth rates of humins. To this end, we utilize ATR-FTIR spectroscopy, Scanning Electron Microscopy, and in situ Dynamic Light Scattering in order to provide fundamental insights into humin formation at the 50–140 °C temperature range. The present study shows for the first time the FTIR spectra of humins at an early stage of their formation (low HMF conversion) and their evolution with reaction progress. We also unravel the effect of co-solvents on humin molecular structure and morphology. Emphasis is also placed on disentangling the effect of the aforementioned parameters on humin growth by suggesting a plausible reaction network for the HMF degradation.
2. Experimental
2.1. Materials and reaction conditions
5-(Hydroxymethyl)furfural (98% purity, Sigma Aldrich), levulinic acid (98% Sigma Aldrich liquid form), formic acid (88% Fisher Scientific), dimethyl sulfoxide (≥99.5%, Sigma Aldrich), hydrochloric acid (37 wt%, Fisher Scientific) were used without any further purification. Solutions were prepared by mixing with deionized water prepared using a Millipore water purification system (model: Direct-Q3 UV R).5 wt% HMF solutions were prepared by dissolving HMF in 50 ml of 0.1 M HCl aqueous solution. The solution was mixed well in order to ensure complete dissolution of HMF. HMF degradation experiments were carried out in 10 ml thick-walled glass vials heated in a temperature-controlled oil bath at 50–140 °C. The lower than typical temperature range is due to limitations of our in situ Dynamic Light Scattering cell (see below). Furthermore, to parallel the scattering experiments, reactions were conducted without stirring (see section 2.3). The volume of all reaction mixtures was constant at 3 ml. Vials with reaction mixture were removed periodically, quenched in an ice-bath and filtered using 0.2 μm filter before collecting a sample for analysis. To investigate the effect of DMSO as a co-solvent (50–50 mole fraction with water) on the degradation of HMF, a set of experiments was carried out at 120 °C by keeping constant the total HCl concentration at 0.1 M. The purification procedure of humins is shown in Scheme 2. The post reaction solution usually contains HMF, formic and levulinic acids as well as humins; therefore, after reaction, depending on the size of the humin particles, the product solution was filtered or centrifuged to separate the solid humins and the supernatant liquid. The remaining solid humins were thoroughly washed with ∼2 L of DI water. The pH of the water was measured to ensure that any acid residue was effectively removed (Fig. S1 of the ESI†). For large humin particles, Soxhlet extraction was also used with water as the solvent according to a humins-washing protocol reported previously.18 The synthesized humins were dried in an oven at 80 °C for 24 hours before further characterization.
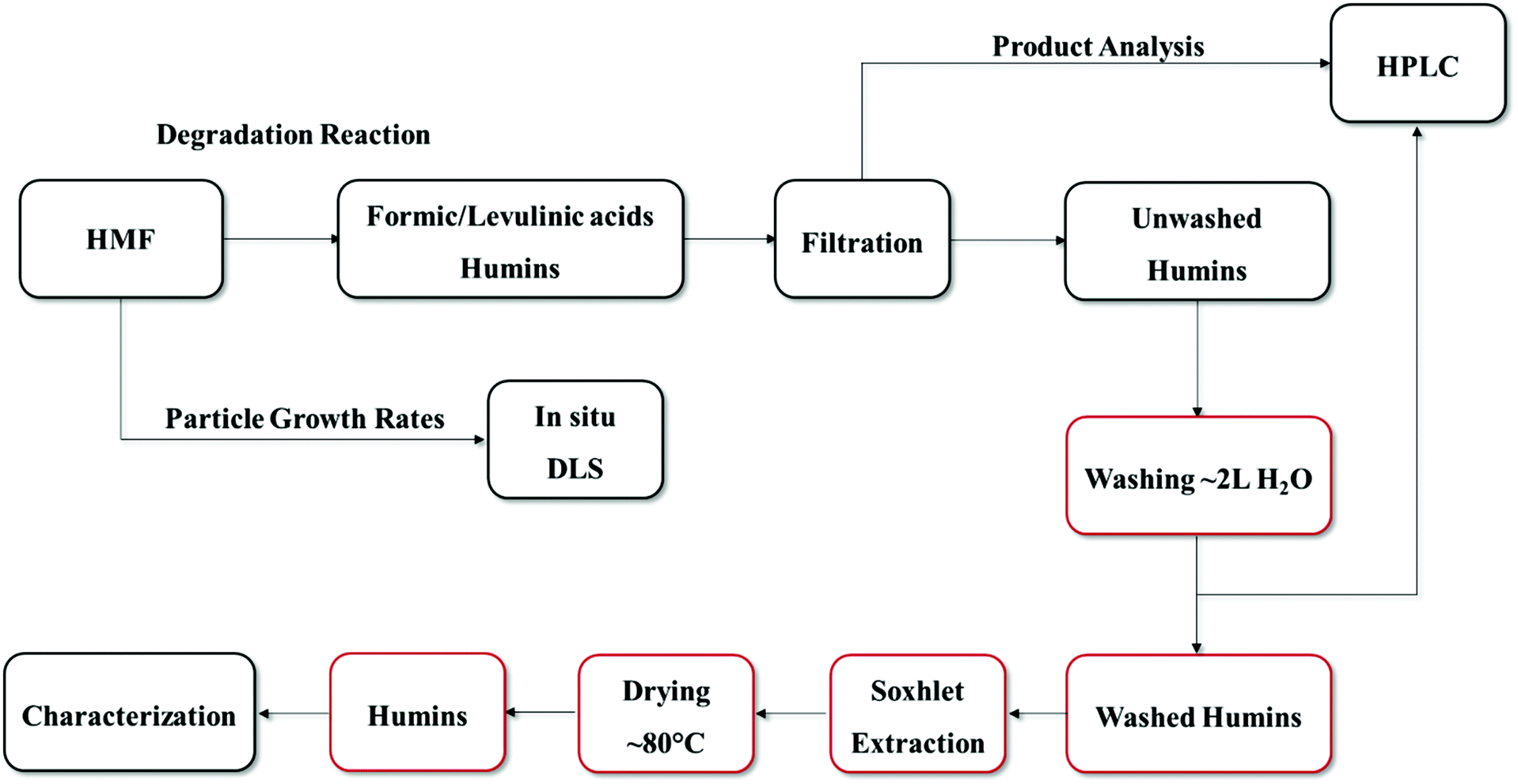 | ||
| Scheme 2 Preparation and purification procedure of HMF-derived humins. Boxes in red color indicate the washing procedure which was followed. | ||
Samples from the reactors were further diluted tenfold with distilled water and were analyzed with High Performance Liquid Chromatography (Waters e2695 with Aminex HPX-87H column and 2414 refractive index detector). A 5 mM solution of sulfuric acid was used as mobile phase with a flow rate of 0.5 mL min−1. The temperature of the column oven was 50 °C and the temperature of the refractive index (RI) detector was 35 °C. Calibration curves were built for all of the known compounds and used for the quantification of reactants and products.
2.2. ATR-FTIR spectroscopy and scanning electron microscopy (SEM)
A Nicolet 8700 FTIR spectrometer equipped with a DTG detector and a Golden Gate single-reflection diamond ATR was used for all spectroscopic studies. The instrument was constantly purged with de-humidified air and CO2 depleted air. The resolution of the instrument was set at 4 cm−1 for the whole set of measurements and the number of scans varied from 16 to 32 scans. The FTIR spectra of all derived humins have been normalized with respect to the ∼1520 cm−1 band that is ascribed to the stretching vibration of CC bonds in furanic rings.
The morphology of the samples was investigated using an AURIGATM 60 microscope (Carl Zeiss NTS GmbH, Germany) equipped with a Schottky Field Emission Gun (FEG). All samples were deposited on adhesive carbon tape and sputtered by a DESK IVTM sputter unit (Denton Vacuum Inc., NJ, U.S.A.) equipped with Au/Pd target.
2.3. Dynamic light scattering (DLS)
In situ dynamic DLS measurements at 90° were conducted using a BI-200SM goniometer and the BI-9000AT digital correlator, from Brookhaven Instruments, Inc. The samples were irradiated using a 389 mW, 532 nm solid state laser from CNI Optoelectronics Technology Co., Ltd. A heating jacket was used to control the temperature of the sample. Samples were prepared in 20 mL scintillation vials. Prior to each experiment, the solution was then filtered, using a 0.2 μm filter, into a 10 mL Wheaton glass reactor vial without a stir bar present. The vial was sealed with a crimped septum top before placing into the goniometer. Measurements were carried out to ensure no particles were present initially. Once it was verified that the initial solution had no particles, the reaction was started and data was collected in 10 minute intervals.The autocorrelation functions were fitted using the CONTIN method as implemented in the Brookhaven Dynamic Light Scattering Software ver. 5.84. The value of the viscosity of water at reaction temperature was used to calculate the particle size distributions.
3. Results
3.1. Effect of washing procedure on humins from various substrates
The procedure used for the preparation and purification of the HMF-derived humins is shown in Scheme 2.It has been reported that humins collected after the dehydration of sugars or HMF degradation have to be washed thoroughly before characterization.18,22,23 In order to ensure that any further characterization would not be affected by post reaction species possibly adsorbed on the humins (HMF, formic acid and LA), we have established a protocol for washing the humins collected after HMF degradation that is similar to that utilized previously.18 In Fig. S2,† the ATR-FTIR spectra of the “as prepared” humins (Fig. S2a†) are compared to those of the “washed humins” (Fig. S2b†). The spectrum of the washed humins shows differences at several specific vibrational bands encompassing the 1665, 1395, 1160, 1064 and 981 cm−1 bands. We ascribe the observed differences to the contribution of vibrational bands of unreacted HMF, formic and levulinic acids which are adsorbed on humins (see Fig. S2c–e†). Analysis of the filtrate of the washed humins using HPLC revealed the presence of a small amount of HMF, formic and levulinic acids, further corroborating the above point. The washing procedure allowed us to reproducibly prepare humin particles with consistent morphology and molecular structure.
Previously reported IR spectra of fructose-, glucose- and HMF-derived humins revealed more similarities than significant differences.18,19,22,23,34–36 However, the preparation conditions used in each study differed significantly. As a result, the origin of the differences in the spectra (namely, nature of the substrate or different conditions) and their impact on the aforementioned mechanisms are unclear. In an effort to resolve this point and further assess the effectiveness of our proposed washing procedure, humins formed from fructose, glucose and HMF were prepared at identical conversion of the corresponding substrate (∼85–90% conversion). The pertinent FTIR results are compared in Fig. 1. The FTIR spectra of the glucose and fructose derived humins are very similar to those reported in the literature. The appearance of two bands at 1625 and 1710 cm−1 as well as differences at the absorbance of the 1525 and 1030 cm−1 peaks were observed when carbohydrates were used as substrates. The latter was attributed to the different amount of HMF incorporated in the humins.22 Such differences were not observed in our spectra. On the other hand, we observed some differences in the 1100–1200 cm−1 region that could be ascribed to multiple C–OH stretching bonds.
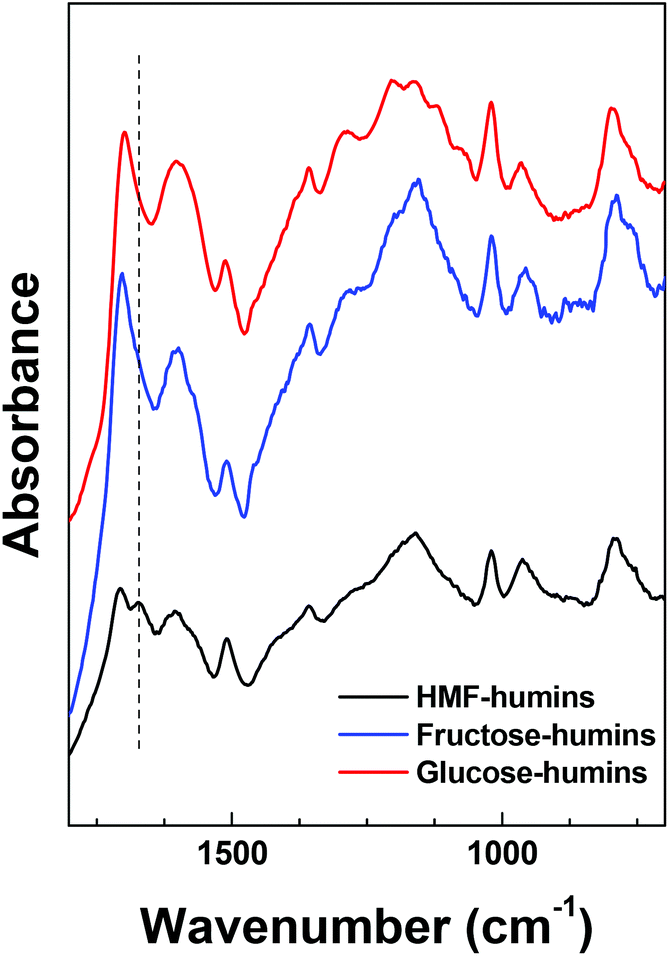 | ||
| Fig. 1 ATR-FTIR spectra of HMF-, fructose- and glucose-derived humins at high reactant conversion. | ||
The most significant difference between the spectra corresponds to the ∼1670 cm−1 band, which is clearly observed only in the HMF-derived humins. This band is ascribed to the CO stretching band of the aldehyde group of HMF-type units that are incorporated in the humin structure. The absence of this band in the fructose- and glucose-derived humins could be ascribed to the low fraction of HMF-type units in the humin structure probably due to the low HMF yields during reaction. This band was also not observed in the IR spectrum of glucose-derived humins doped with HMF, reported by van Zandvoort et al.18 We believe that this difference is due to the lower concentration of HMF in their mixture or to different reaction conditions. The HMF derived humins herein have been prepared with pure HMF as feedstock and thus, a strong contribution of the remaining aldehyde groups in their ATR-FTIR spectra is expected. It is also possible that sugars-derived humins form via a path that does not involve HMF formation. However, the broad spectral envelope of the ∼1700 cm−1 band could overlap with a possible contribution of the ∼1670 cm−1 (with low absorbance) band. Given that the stretching vibration of CC bonds of furanic rings is present in all samples, we propose that all humins form mainly through HMF (or another furanic component) as an intermediate. Along these lines, we may also infer that the CO bond attributed to the 1670 cm−1 band is consumed with the substrates or reaction intermediates. Further insights into this topic can be provided by examining the spectral evolution of humins throughout the reaction history discussed below. Since fructose- and glucose-derived humins appear to have similar molecular structure based on our FTIR results, next we discuss only the effect of HMF conversion on the infrared spectra as well as the morphology of HMF-derived humins in aqueous as well as mixed DMSO–H2O reaction media.
3.2. Effect of HMF conversion on the molecular structure of humins
It has been reported previously that the spectra of humins derived from glucose or fructose do not change appreciably as a function of conversion.22 Since HMF is an important intermediate in the formation of humins, the molecular structure of HMF-derived humins in aqueous as well as mixed DMSO–H2O reaction media was examined using ATR-FTIR spectroscopy as a function of HMF conversion. The corresponding results are shown in Fig. 2. In contrast to previous results using sugars as substrates,22 significant changes with increasing conversion (from 10% up to complete conversion) were observed when HMF was used as substrate in aqueous solutions. Upon increasing HMF conversion, the following observations can be made from the corresponding ATR-IR spectra:(i) The absorbance of the 1702 cm−1 band gradually increases relative to that of the CC stretching vibration of the furanic rings and finally obscures the contribution of the 1668 cm−1 band. Alternatively the absorbance of 1670 cm−1 might also decrease with increasing conversion.
(ii) The absorbance of the 758 cm−1 band decreases with increasing conversion and almost disappears at 100% conversion.
(iii) The absorbance in the spectral envelope between 1100 cm−1 and 1300 cm−1 wavenumber region increases with a new, intense and well defined band appearing at ∼1160 cm−1.
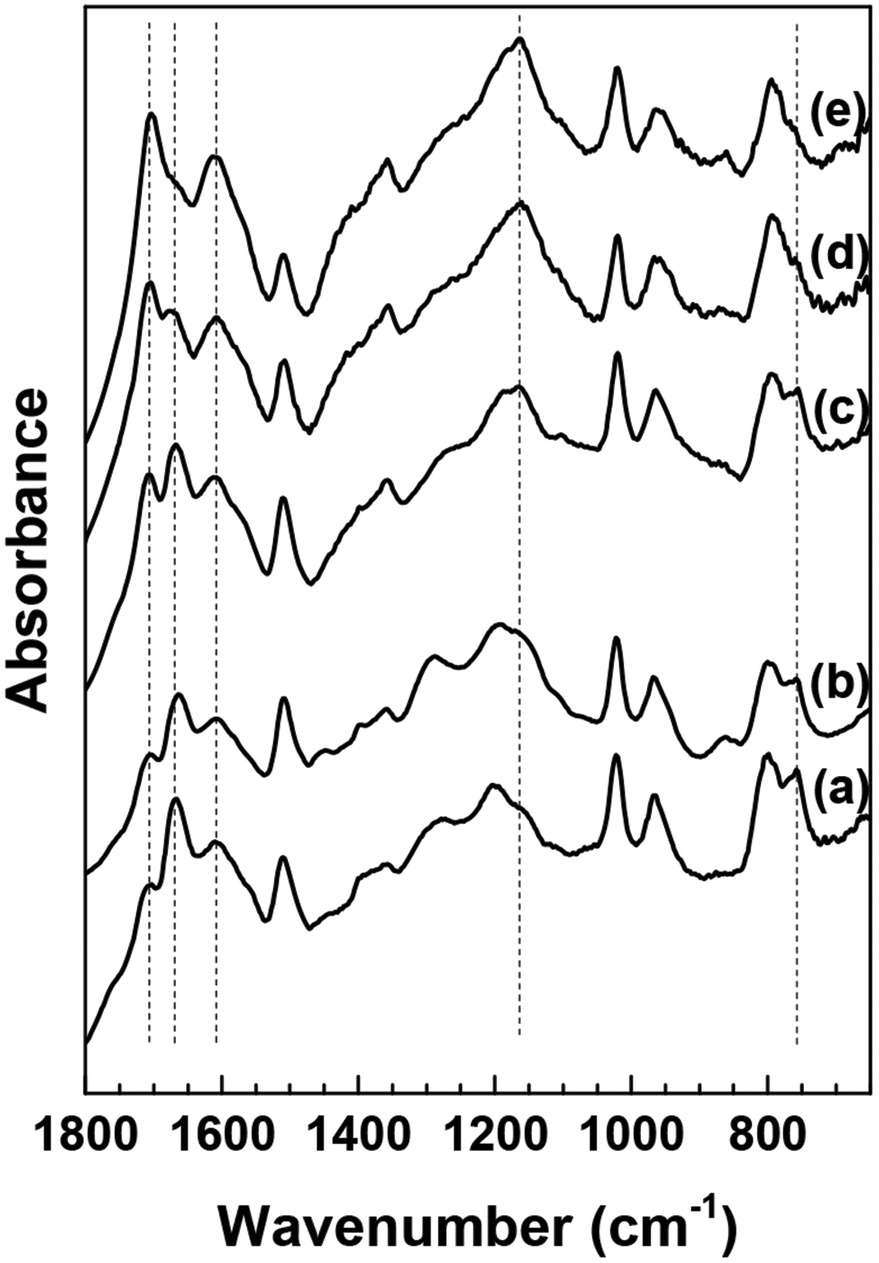 | ||
| Fig. 2 ATR-FTIR spectra of washed HMF derived humins at (a) 10%, (b) 40%, (c) 70%, (d) 80% and (e) 100% HMF conversion in water. | ||
Based on the postulated mechanisms, aldol condensation/addition reactions are implicated in the formation of humins in which the reactants, namely glucose, fructose or HMF, must first be converted to 2,5-dioxo-6-hydroxyhexanal (DHH)22,23 shown in Scheme 3. This hypothesis is based on the ring opening mechanism of HMF (acid catalyzed hydrolysis) leading to DHH proposed originally by Horvat.25 However, DHH was not spectroscopically identified suggesting that it is a highly reactive intermediate. The formation of the DHH could explain the broad spectral envelop in the 1665–1760 cm−1 region due to the different types of CO present in its structure. The stretching vibration of CO functional groups absorbs in the 1665–1760 cm−1 region. The 1702 cm−1 could possibly be attributed to carbonyls in saturated aliphatic ketones (e.g., DHH-like structure, or levulinic acid24), while the 1668 cm−1 corresponds to an aldehyde group similar to that in HMF. The decrease of the 1668 cm−1 band simultaneously with a corresponding increase of the 1702 cm−1 band with increasing HMF conversion is consistent with the proposed aldol condensation reactions between the enols from DHH and the carbonyl group of HMF leading to humins.22,23 Our data indicate also a monotonic decrease in the intensity of the 758 cm−1 band with increasing HMF conversion. This band is ascribed to C–H out of plane vibrations of furan rings. This observation together with a relative decrease of the peak at ∼1510 cm−1, ascribed to furan ring stretches, points to a decrease of the H atoms attached to furanic rings after long reaction time (high HMF conversion). The recent insights from advanced NMR techniques on the Cα–Caliphatic and Cα–Cα linkages in humins during the acid catalyzed glucose dehydration (Cβ–Caliphatic and Cβ–Cβ were minimal)24 underscore the complexity in the growth mechanism and that advanced and complementary spectroscopic techniques should be utilized. Our proposed mechanism is based solely on the evolution of the FTIR spectra of humins from low to complete HMF conversions. Our FTIR data are consistent with the model proposed by van Zandvoort et al.18 in which humins are formed also through substitution at the β or α position via nucleophilic attack.
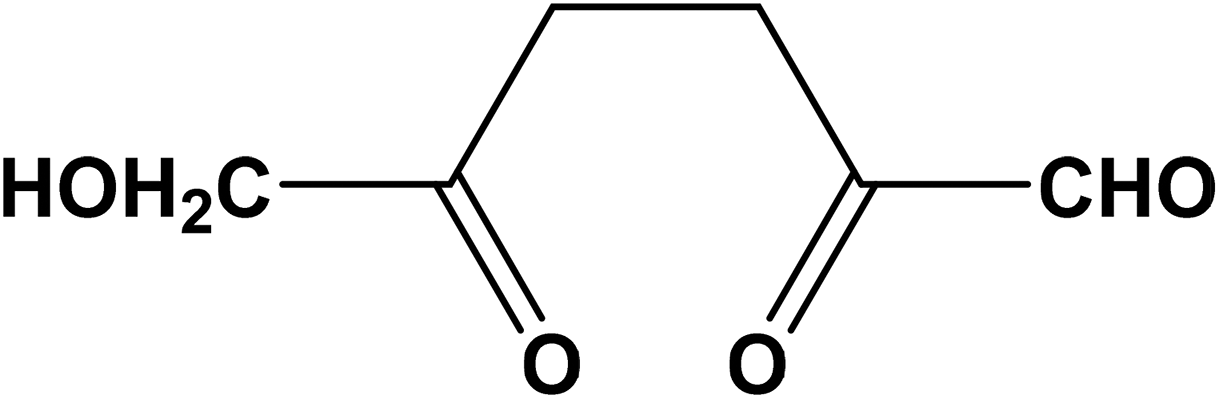 | ||
| Scheme 3 2,5-Dioxo-6-hydroxyhexanal (DHH) as the intermediate in the formation of humins proposed by Horvat.25 | ||
The increased absorbance in the spectral envelope, between 1100–1300 cm−1 wavenumber region with the new intense and well defined band appearing at ∼1160 cm−1, could arise from the presence of ether bonds. Although the possibility of etherification reactions was previously reported,21 complementary spectroscopic techniques are required to clarify this point. Recently, CP-DQSQ experiments24 for glucose derived humins revealed a correlation between aliphatic carbons and a C–O group, which might be due to alcohols, ether or acetal bonds. It was also underlined that the presence of ether or acetal bonds could partially explain the solubilization and reduction in molecular weight of the humins upon alkaline treatment.35 In this perspective, our data indicates that humin formation involves multiple parallel reaction pathways in addition to aldol condensations.
The aforementioned reactions involving nucleophilic attack could definitely be altered by co-solvents. We hypothesize that this fact relates to the beneficial effect of polar aprotic co-solvents as reaction media in maximizing HMF yields10,37–39 by minimizing its degradation reactions. The addition of co-solvent could modify solvent–reactant or solvent–solvent interactions and dynamics, which in turn affect the local solvation as well as the free energy landscape between reactants and products of each elementary step. In order to provide insights into the role of solvent in the molecular structure of humins and their formation mechanisms, humins were collected during the HMF degradation in a water/dimethyl sulfoxide (DMSO) mixture (an aqueous/polar aprotic co-solvent mixture). The color of the post reaction mixtures diluted using neat water was dark yellow with precipitated solid black particles (humins). When DMSO was used as co-solvent the mixture was black, even after filtration and tenfold dilution with a DMSO/water/HCl solution (Fig. 3a). On the other hand, dilution with water/DMSO or with water alone results in particle precipitation (Fig. 3b and c). These observations indicate that humins are partially soluble in DMSO or that their particle size is significantly smaller than that of the cut-off radius of the filter. Furthermore, the observed precipitation when the sample is diluted with a non-acidified DMSO/water mixture indicates that these small particles have sufficient surface charge that prohibits their aggregation and precipitation, leading to dependence on the presence of HCl. DLS measurements in solutions diluted with DMSO/water/HCl reveal the existence of particles with diameter ∼150 nm.
 | ||
| Fig. 3 Post reaction mixture of HMF degradation (∼40% conversion) in DMSO/water 10× diluted with (a) DMSO/water/HCl (initial reaction mixture), (b) DMSO/water (no HCl), and (c) with water. | ||
In an attempt to determine the influence of co-solvent on the humin molecular structure, we compare the HMF-derived humins prepared in DI water at 40% conversion (Fig. 4a) with those made in the DMSO/water mixtures at 40% and 75% conversion (Fig. 4b and c). The spectra shown in Fig. 4a and b have generally similar features. A noticeable difference is the absorbance of the 757 cm−1 band, ascribed to C–H out of plane vibrations of furan rings, which appears to be higher in the humins prepared in the water/DMSO mixture than those prepared in pure water. Based on the aforementioned hypothesis, we suggest now that the use of a polar aprotic co-solvent, such as DMSO, suppresses the path proposed by van Zandvoort et al.18 which involves nucleophilic attack of the furan ring on the carbonyl group of HMF. The lighter color of the HMF derived humins in the presence of DMSO (Fig. 4) could possibly indicate the existence of higher O/C ratio which could also explain the higher intensity of the CO band at 1668 cm−1.19
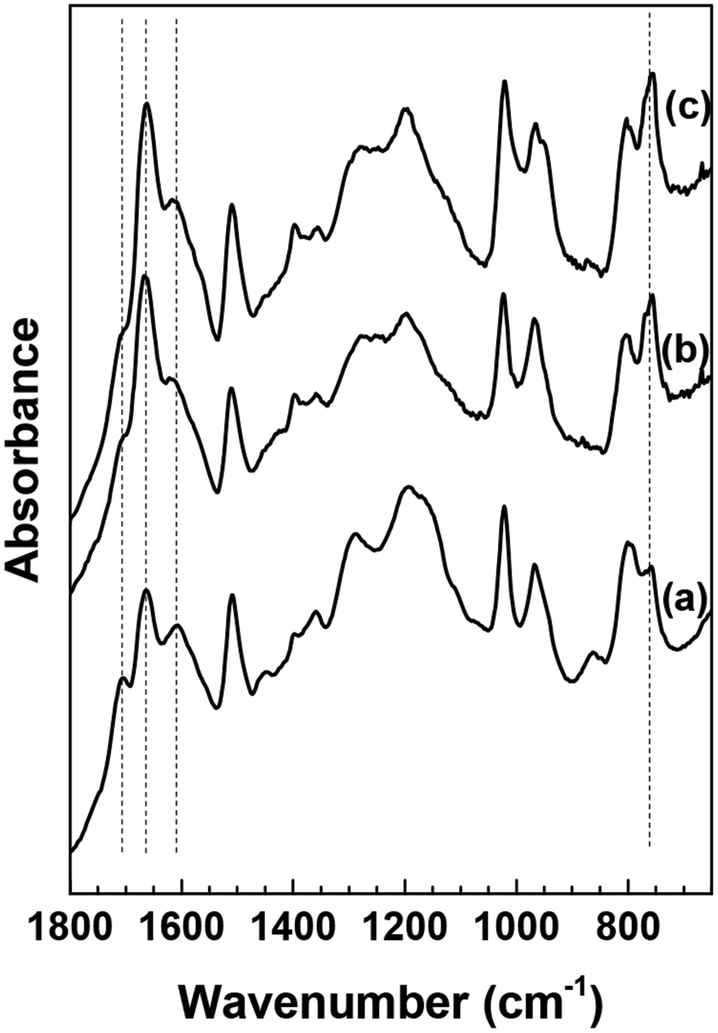 | ||
| Fig. 4 ATR-FTIR spectra of washed HMF derived humins in (a) water (40% HMF conversion) and a 50–50 DMSO/water solution at (b) 40% HMF conversion and (c) 75% HMF conversion. | ||
The above findings are further corroborated by our recent vibrational spectroscopy along with ab initio calculations that show that the carbonyl group of HMF, in a DMSO/water mixed solvent, preferentially interacts with DMSO even in the presence of ∼30–40% mole fraction of water.31 Frontier molecular orbital theory revealed that the interaction of DMSO with the carbonyl of HMF increases the LUMO energy of HMF which consequently can influence its susceptibility either to protonation or nucleophilic attack. Based on these findings, we postulate that the preferential solvation of HMF or of one of the reaction intermediates by polar aprotic co-solvents could be an important mechanism by which HMF is stabilized at its carbonyl group against nucleophilic attack. The infrared spectra at a higher HMF conversion of 75% indicate no significant changes (Fig. 4c), which underscores the importance of polar aprotic co-solvents in suppressing the evolution of the humin molecular structure and further supports our hypothesis.
3.3. Effect of HMF conversion on the morphology of humins
Fig. 5 shows representative scanning electron micrographs of humin samples collected under different reaction conditions. The humins formed from HMF are well defined spherical interconnected particles. The size of particles formed in an aqueous solution increases with increasing the conversion of HMF (up to 2–5 μm). High magnification images show a multimodal distribution of humin particles underscoring that new humin particles with particle size ∼200 nm are formed throughout HMF conversion. | ||
| Fig. 5 SEM micrographs of humins prepared at different conditions: (a) 10% HMF conversion in water, (b) 100% HMF conversion in water, (c) 40% HMF conversion in water/DMSO, and (d) 75% HMF conversion in water/DMSO. | ||
Interconnected spherical particles have also been reported for hydrothermal carbon from biomass derivatives.16,17 The reported hydrothermal carbon particle size appeared to depend on the nature of the feedstock (pentoses, hexoses, HMF and furfural) with the HMF derived carbon microstructure is composed of small particles (<200 nm).17 The aforementioned hydrothermal carbons were prepared in the absence of an acid catalyst as well as at higher temperature. Despite the differences in conditions, the common structural features of hydrothermal carbons and humins reported elsewhere18,24,35,36 stimulate some discussion. The hydrothermal carbon particle sizes appear, in general, to be smaller than the humin particles formed during acid catalyzed dehydration. Fig. 5c and d shows the SEM images of humins formed in a mixed solvent at 40 and 75% HMF conversion, respectively. Contrary to the well-defined multimodal particle distribution observed in humins prepared in water, the particles prepared in the presence of DMSO are spherical particles of 50–100 nm in size at all HMF conversions. These results are consistent with the aforementioned FTIR analysis that shows no significant change in the molecular structure of humins with increasing conversion. In the presence of DMSO, humin nanoparticles form but their further growth is retarded. This finding indicates distinct reaction paths for the formation of humin nanoparticles and of large particles. Based on the analysis of the FTIR data, we postulate that the growth of larger particles involves nucleophilic attack of HMF carbonyl at the α- or β-position of the furan rings. Finally, these results show that the morphology of humin byproducts strongly depends on the reaction conditions as well as on the nature of the solvent employed.
3.4. Dynamic lights scattering (DLS) experiments and growth rates
The SEM micrographs of the HMF derived humins revealed particles of size ∼100 nm–5 μm in water and of ∼100 nm in the presence of DMSO. This is in agreement with our DLS experiments showing a very narrow distribution of particle size of ∼150 nm in water/DMSO mixtures. Since the ATR-FTIR and SEM images do not show appreciable changes in the molecular structure and morphology of humins in water/DMSO mixtures as a function of HMF conversion, next we focus on understanding particle growth in water via DLS under in situ reaction conditions. Specifically, we investigate the effect of acid concentration and temperature due to being key parameters in humin formation.18,23 Due to instrument limitations, the reactions were carried out at a lower temperature range (50–70 °C). It was not possible to detect any nanoparticles via DLS until after several minutes following the initiation of the reaction when particles of 100–200 nm were observed, consistent with the smallest particles in our SEM images. This induction period strongly depends on the acid concentration as well as the reaction temperature. For example, at pH = 0, an induction of ∼10 min was observed at 50 °C while at higher pH = 1, the induction period was ∼60 min. At longer reaction times, several measurements have shown two exponential decay times indicating a bimodal distribution in the particle size, in qualitative agreement with the aforementioned SEM results.For each set of conditions examined, there is a time period during which the particle size increases almost rapidly with time after the induction period. At longer times, the particle growth rate decreases with time (Fig. 6). This can be either due to the fact that larger particles precipitate, and as a result are not detected by DLS, or to contributions from particles that nucleated at later times. The almost linear dependence of the particle size on the square root of time (Fig. S3 of the ESI†) provides support for diffusion limited growth, which is common to crystal growth when sizes become sufficiently large. To avoid these complications the discussion below focuses on the initial particle growth regime (short times after induction time). For instance, at pH = 1 and a temperature of 50 °C, the particle growth rate was estimated to be ∼2.1–2.9 nm min−1. Fig. 7a summarizes the humin short time growth rates as a function of acid concentration and temperature: the higher the temperature and/or the lower the pH, the higher the growth rate of the humin particles.
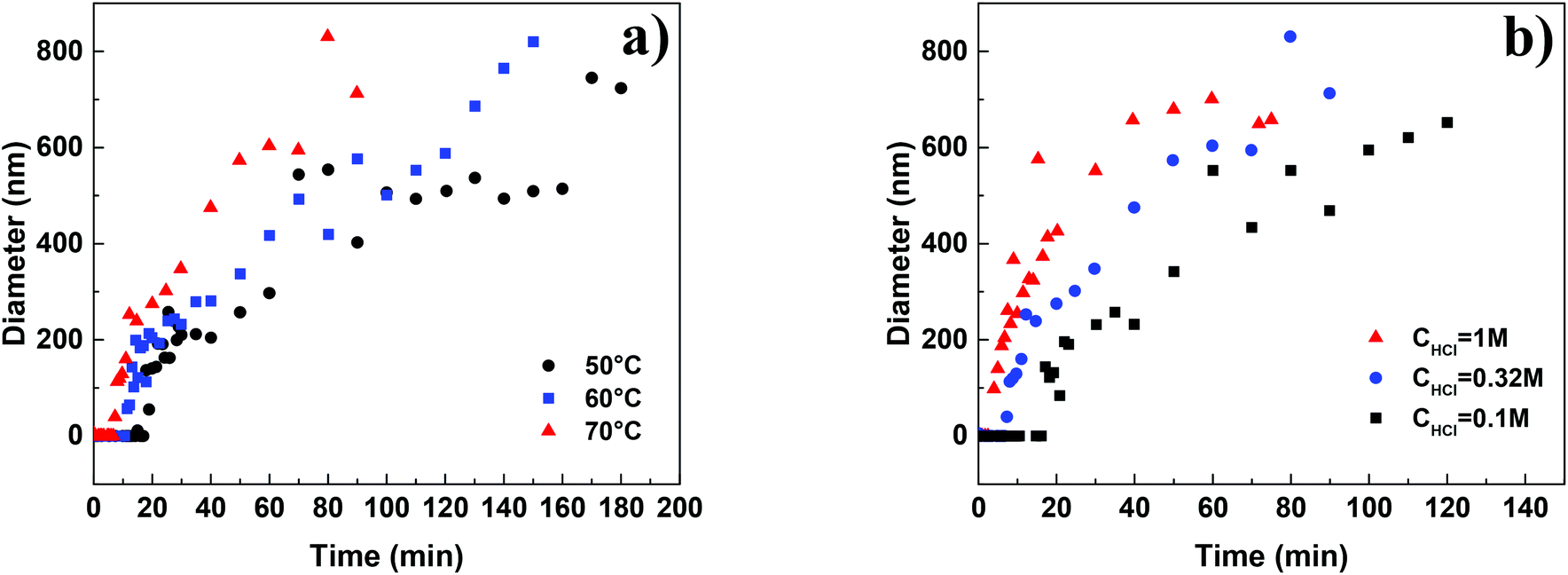 | ||
| Fig. 6 Particle growth of humins as a function of (a) temperature at pH = 0.5 and (b) HCl concentration at 70 °C. | ||
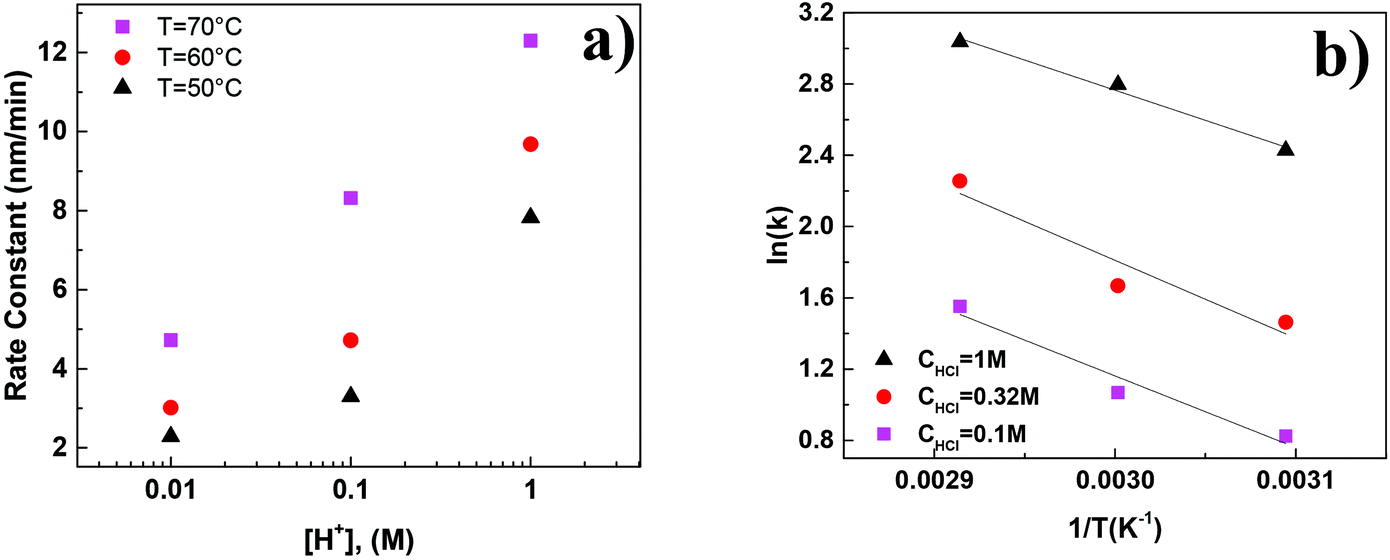 | ||
| Fig. 7 (a) Initial growth rate of humin formation as a function of acid concentration and temperature and (b) Arrhenius plot of the growth rates of humins. | ||
Fig. 7b shows an Arrhenius plot using the growth rates presented in Fig. 7a. The estimated apparent activation energy of ∼28–36 kJ mol−1 is lower than the literature reported values which lie in the 85–130 kJ mol−1 range.12,40,41 The lower values can potentially be attributed to transport effects (it was not possible to mix the reactants to avoid the disturbance of the in situ DLS experiments) as discussed above (see Fig. S3†). An additional source can include limited temperature data set, due to the restricted temperature range of the in situ DLS measurements, leading to large uncertainty in the estimation.
Another possible reason is the direct measurement of humin formation (particle growth), instead of indirectly approximating the humin formation rate through a carbon balance (common approach used in literature). The lack of mechanistic studies on the formation of humins has hampered progress in unravelling the reaction network and consequently, depending on the formation mechanism, the corresponding rate constants could be significantly different when approximated by closing the mass balance or by direct measurement. In this direction, we combine the results from our ATR-FTIR, SEM and in situ DLS measurements to introduce a plausible simplified reaction network (Scheme 4) for the formation and agglomeration of humins during the acid catalyzed degradation of HMF. Initially soluble oligomeric humins form via aldol condensation22,23,25 and/or etherification reactions21 in which HMF reacts with DHH-like intermediates or other HMF molecules. The particles grow in size through nucleophilic attack as proposed by van Zandvoort et al.;18 however this path is significantly suppressed in the presence of polar aprotic co-solvents, such as DMSO. At high HMF conversion, we suggest that direct HMF addition to the primary particles could take place as well, while aggregation of the primary particles leads to the precipitation of large, insoluble in water particles of several microns in size. Each of the proposed pathway finally contributes to the overall humin formation rate constant usually calculated based on a carbon balance approach. The postulated humin formation reaction network rationalizes qualitatively our experimental results as well as those reported in the literature. Further exploitation of advanced experimental techniques for disentangling the contribution of each path to the overall lumped observed rate constant would be important to further elucidate the humin formation mechanism.
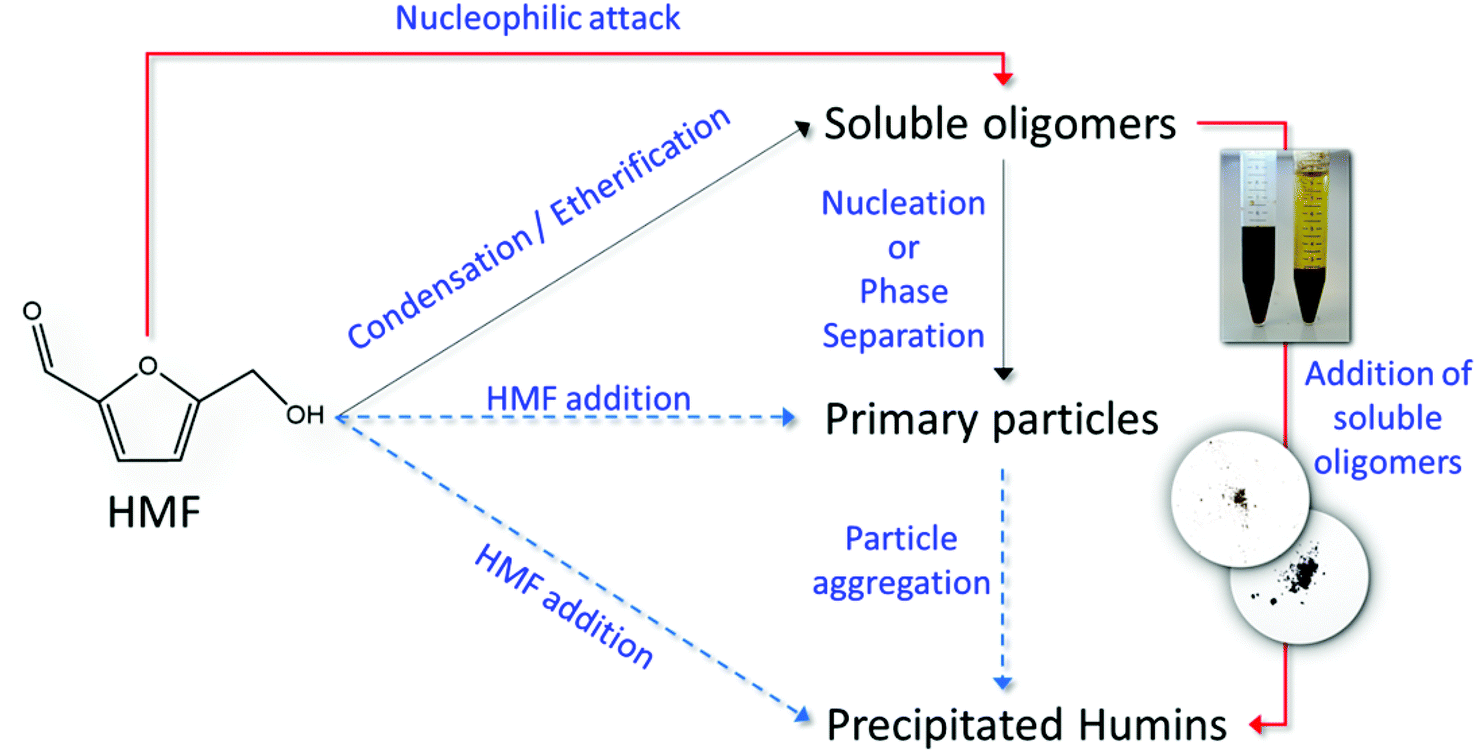 | ||
| Scheme 4 Reaction network of the formation of humins in the acid catalyzed degradation of HMF. | ||
4. Conclusions
The molecular structure, morphology and formation mechanisms and growth rates of humins in the Brønsted acid catalyzed degradation of HMF were investigated using ATR-FTIR spectroscopy, scanning electron microscopy and in situ dynamic light scattering. The evolution of the humin molecular structure as a function of HMF conversion was revealed from analysis of the FTIR spectra. A decrease of the absorbance of the C–H out of plane mode from H atoms attached to furanic ring at long reaction times is consistent with previous proposed reaction mechanism involving nucleophilic attack of an HMF carbonyl group to the α- or β-position of the furanic ring. A decrease of the 1668 cm−1 bands of CO stretches supports further this mechanism. However, the rich in information CO stretching region of all the humins prepared cannot exclude other reaction pathways such as aldol addition/condensation. Interestingly, we find that the nucleophilic attack pathway is suppressed in the presence of DMSO co-solvent and rationalizes the very small humin particles observed in SEM images. A multimodal distribution was observed in the SEM images for the humins made in water, where larger particles consist of smaller ones, in excellent agreement with DLS results. All the present data have been combined in a simplified reaction network which qualitative explains our experimental results as well as from prior work.
Given the importance of the stability of biomass derived intermediates, such as 5-HMF, to the development of bio-refineries, research efforts in eliminating or valorizing the humin by-products are crucial. This work has further elucidated the formation and structure of humins by providing, to the best of our knowledge, for the first time direct measurement of growth rates of the HMF derived humin particles and information on their molecular structure and morphology as a function of HMF conversion. Moreover, insights into the effect of DMSO on the molecular structure and morphology of humins might eventually be used to minimize humin formation.
Acknowledgements
The work was financially supported by the Catalysis Center for Energy Innovation, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Award Number DE-SC0001004.References
- J. J. Pacheco and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 8363–8367 CrossRef CAS PubMed.
- J. Jae, W. Q. Zheng, R. F. Lobo and D. G. Vlachos, ChemSusChem, 2013, 6, 1158–1162 CrossRef CAS PubMed.
- Y. Roman-Leshkov, C. J. Barrett, Z. Y. Liu and J. A. Dumesic, Nature, 2007, 447, 982–U985 CrossRef CAS PubMed.
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539–554 RSC.
- R. J. van Putten, J. C. van der Waal, E. de Jong, C. B. Rasrendra, H. J. Heeres and J. G. de Vries, Chem. Rev., 2013, 113, 1499–1597 CrossRef CAS PubMed.
- Z. Lin, M. Ierapetritou and V. Nikolakis, AIChE J., 2013, 59, 2079–2087 CrossRef CAS.
- B. Girisuta, B. Danon, R. Manurung, L. P. B. M. Janssen and H. J. Heeres, Bioresour. Technol., 2008, 99, 8367–8375 CrossRef CAS PubMed.
- B. Girisuta, L. P. B. M. Janssen and H. J. Heeres, Green Chem., 2006, 8, 701–709 RSC.
- B. Girisuta, L. P. B. M. Janssen and H. J. Heeres, Ind. Eng. Chem. Res., 2007, 46, 1696–1708 CrossRef CAS.
- B. F. M. Kuster, Carbohydr. Res., 1977, 54, 177–183 CrossRef CAS.
- B. F. M. Kuster and L. M. Tebbens, Carbohydr. Res., 1977, 54, 159–164 CAS.
- B. F. M. Kuster and H. M. G. Temmink, Carbohydr. Res., 1977, 54, 185–191 CrossRef CAS.
- M. Sevilla and A. B. Fuertes, Carbon, 2009, 47, 2281–2289 CrossRef CAS.
- M. Sevilla and A. B. Fuertes, Chem. – Eur. J., 2009, 15, 4195–4203 CrossRef CAS PubMed.
- N. Baccile, G. Laurent, F. Babonneau, F. Fayon, M. M. Titirici and M. Antonietti, J. Phys. Chem. C, 2009, 113, 9644–9654 CAS.
- Y. Z. Mi, W. B. Hu, Y. M. Dan and Y. L. Liu, Mater. Lett., 2008, 62, 1194–1196 CrossRef CAS.
- M. M. Titirici, M. Antonietti and N. Baccile, Green Chem., 2008, 10, 1204–1212 RSC.
- I. van Zandvoort, Y. H. Wang, C. B. Rasrendra, E. R. H. van Eck, P. C. A. Bruijnincx, H. J. Heeres and B. M. Weckhuysen, ChemSusChem, 2013, 6, 1745–1758 CrossRef CAS PubMed.
- S. Reiche, N. Kowalew and R. Schlogl, ChemPhysChem, 2015, 16, 579–587 CrossRef CAS PubMed.
- C. Yao, Y. Shin, L. Q. Wang, C. F. Windisch, W. D. Samuels, B. W. Arey, C. Wang, W. M. Risen and G. J. Exarhos, J. Phys. Chem. C, 2007, 111, 15141–15145 CAS.
- I. V. Sumerskii, S. M. Krutov and M. Y. Zarubin, Russ. J. Appl. Chem., 2010, 83, 320–327 CrossRef CAS.
- S. K. R. Patil, J. Heltzel and C. R. F. Lund, Energy Fuels, 2012, 26, 5281–5293 CrossRef CAS.
- S. K. R. Patil and C. R. F. Lund, Energy Fuels, 2011, 25, 4745–4755 CrossRef CAS.
- I. van Zandvoort, E. J. Koers, M. Weingarth, P. C. A. Bruijnincx, M. Baldus and B. M. Weckhuysen, Green Chem., 2015, 17, 4383–4392 RSC.
- J. Horvat, B. Klaic, B. Metelko and V. Sunjic, Tetrahedron Lett., 1985, 26, 2111–2114 CrossRef CAS.
- G. Yang, E. A. Pidko and E. J. M. Hensen, J. Catal., 2012, 295, 122–132 CrossRef CAS.
- S. K. R. Patil and C. R. F. Lund, Abstr. Pap., Jt. Conf. – Chem. Inst. Can. Am. Chem. Soc., 2012, 243 Search PubMed.
- Q. Jing and X. Y. Lu, Chin. J. Chem. Eng., 2008, 16, 890–894 CrossRef CAS.
- G. C. A. Luijkx, F. Vanrantwijk and H. Vanbekkum, Carbohydr. Res., 1993, 242, 131–139 CrossRef CAS.
- A. Chuntanapum, T. L. K. Yong, S. Miyake and Y. Matsumura, Ind. Eng. Chem. Res., 2008, 47, 2956–2962 CrossRef CAS.
- G. Tsilomelekis, T. R. Josephson, V. Nikolakis and S. Caratzoulas, ChemSusChem, 2014, 7, 117–126 CrossRef CAS PubMed.
- T. R. Josephson, G. Tsilomelekis, C. Bagia, V. Nikolakis, D. G. Vlachos and S. Caratzoulas, J. Phys. Chem. A, 2014, 118, 12149–12160 CrossRef CAS PubMed.
- S. H. Mushrif, S. Caratzoulas and D. G. Vlachos, Phys. Chem. Chem. Phys., 2012, 14, 2637–2644 RSC.
- A. Chuntanapum and Y. Matsumura, Ind. Eng. Chem. Res., 2009, 48, 9837–9846 CrossRef CAS.
- I. van Zandvoort, E. R. H. van Eck, P. de Peinder, H. J. Heeres, P. C. A. Bruijnincx and B. M. Weckhuysen, ACS Sustainable Chem. Eng., 2015, 3, 533–543 CrossRef CAS.
- T. M. C. Hoang, E. R. H. van Eck, W. P. Bula, J. G. E. Gardeniers, L. Lefferts and K. Seshan, Green Chem., 2015, 17, 959–972 RSC.
- D. W. Brown, A. J. Floyd, R. G. Kinsman and Y. Roshanali, J. Chem. Technol. Biotechnol., 1982, 32, 920–924 CrossRef CAS.
- M. A. Mellmer, C. Sener, J. M. Gallo, J. S. Luterbacher, D. M. Alonso and J. A. Dumesic, Angew. Chem., Int. Ed., 2014, 53, 11872–11875 CrossRef CAS PubMed.
- S. Jia, Z. Xu and Z. C. Zhang, Chem. Eng. J., 2014, 254, 333–339 CrossRef CAS.
- R. J. van Putten, J. C. van der Waal, E. de Jong, C. B. Rasrendra, H. J. Heeres and J. G. de Vries, Chem. Rev., 2013, 113, 1499–1597 CrossRef CAS PubMed.
- T. D. Swift, C. Bagia, V. Choudhary, G. Peklaris, V. Nikolakis and D. G. Vlachos, ACS Catal., 2014, 4, 259–267 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5gc01938a |
| This journal is © The Royal Society of Chemistry 2016 |