
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mesoporous sulfonic acid silicas for pyrolysis bio-oil upgrading via acetic acid esterification†
Jinesh C.
Manayil
,
Carlos V. M.
Inocencio
,
Adam F.
Lee
 * and
Karen
Wilson
*
* and
Karen
Wilson
*
European Bioenergy Research Institute, Aston University, Birmingham B4 7ET, UK. E-mail: a.f.lee@aston.ac.uk; k.wilson@aston.ac.uk; Tel: +44 (0)121 2044036
First published on 9th October 2015
Abstract
Propylsulfonic acid derivatised SBA-15 catalysts have been prepared by post modification of SBA-15 with mercaptopropyltrimethoxysilane (MPTMS) for the upgrading of a model pyrolysis bio-oil via acetic acid esterification with benzyl alcohol in toluene. Acetic acid conversion and the rate of benzyl acetate production was proportional to the PrSO3H surface coverage, reaching a maximum for a saturation adlayer. Turnover frequencies for esterification increase with sulfonic acid surface density, suggesting a cooperative effect of adjacent PrSO3H groups. Maximal acetic acid conversion was attained under acid-rich conditions with aromatic alcohols, outperforming Amberlyst or USY zeolites, with additional excellent water tolerance.
1. Introduction
Technologies to utilise renewable resources for the sustainable production of transportation fuels and chemicals is of great current interest due to growing concerns over the depletion of fossil fuel reserves and associated climate change.1 Promising solutions are offered through the thermochemical processing of lignocellulosic biomass through pyrolysis or gasification, and transesterification of non-edible and waste plant/algal oils and fats.1–3 Fast pyrolysis of waste agricultural/forestry biomass for the production and subsequent upgrading of bio-oils to liquid transportation fuels has received considerable attention in this regard.4,5 However, the direct use of fast pyrolysis bio-oils is limited by its low heating value due to the high oxygen content, thermally instability, and strong acidity with associated high water content.6–8 Typical bio-oils are a mixture of acids, alcohols, furans, aldehydes, esters, ketones, sugars and multifunctional compounds such as hydroxyacetic acid, hydroxyacetaldehyde and hydroxyacetone (derived from cellulose and hemicellulose), together with 3-hydroxy-3-methoxy benzaldehyde, phenols, guaiacols and syringols derived from the lignin component.9,10 Biomass pyrolysis routes to transportation fuels are therefore only economically viable if the bio-oil is subjected to upgrading treatments to improve their physicochemical properties.5,11–14There are several promising catalytic processes for bio-oil upgrading/pre-treatment, including ketonisation, aldol condensation and hydro-deoxygenation. Acetic acid is one of the major impurities present in bio-oils (around 1–10%),7 and its removal via esterification affords a simple and effective method to raise pH and improve bio-oil stability.4 Strong mineral acids such as H2SO4 are effective at catalysing low temperature esterification, but their corrosive and hazardous nature hinders handling and storage, while as soluble catalysts, resulting upgraded bio-oils require subsequent neutralisation generating large quantities of aqueous waste for clean-up. Solid acid catalysts can circumvent these disadvantages, however new tailored solid acids are desirable with superior acid site accessibility and stability in aqueous environments, and compatible with high molecular weight hydrocarbons present in bio-oil that may lead to pore-blockage of microporous solid acids such as zeolites.15,16
Catalytic esterification of acetic acid has been previously investigated with a range of alcohols, notably methanol and ethanol which do not occur at any significant concentration in pyrolysis bio-oils, and must therefore derive from an additional external carbon source, impacting upon the overall process sustainability.14,17–19 If a continuous process were to operate via reactive distillation, it would be desirable for both water and the ester is removed continuously to drive the equilibria to completion. For this purpose alcohols with boiling points higher than water are required to avoid alcohol loss by evaporation.20 Aromatic esters find application in a wide range of areas spanning fine, pharmaceutical and agrochemicals sectors,21 thus in the context of a biorefinery, in addition to improving bio-oil stability, the production of benzyl acetate from esterification with benzyl alcohol could add value to the overall process.22,23 There has been previous interest in catalysing acetic acid esterification with benzyl alcohol and cresol over zeolites, zirconia, alumina, and silica,22,24 however such microporous catalysts are ill-suited to the liquid phase transformation of viscous bio-oils containing sterically challenging components. Sulfonated mesoporous carbons25 and silicotungstic/zirconia functionalised mesoporous SBA-15 silica18 have also shown promise for acetic acid esterification with benzyl alcohol, but of idealised mixtures unrepresentative of pyrolysis bio-oils and under inert atmospheres. Sulfonic acid silicas are an important class of solid acid catalysts26–30 well known for their applications in biodiesel production, but have rarely been exploited for bio-oil upgrading and even then only for acetic acid esterification with pure ethanol,17,31 or in the presence of light aldehyde components present in pyrolysis bio-oils which hinder ester formation at low reaction temperatures.32 Real pyrolysis oils, whether from fast, intermediate or catalytic routes, contain significant (∼25 wt%) non-polar lignin derivatives,8 for which toluene may be considered a representative solvent. Here we report the first investigation of acetic acid esterification with benzyl alcohol in toluene as a simulated bio-oil over propylsulfonic acid functionalised SBA-15 (PrSO3H/SBA-15), which exhibits outstanding water tolerance. Catalyst formulation and the impact of reaction conditions are optimised for maximal activity and ester yield and significantly outperform commercial solid acid catalysts.
2. Experimental
2.1 Catalyst synthesis
SBA-15 was synthesised adopting the protocol of Zhao and co-workers.33 Typically 10 g of Pluronic P123 triblock copolymer was dissolved in 75 ml of water and 250 ml of 2 M HCl solution. The mixtures were stirred at 35 °C for dissolution and then 23 ml of TEOS were added, maintained at 35 °C for 20 h under stirring. The resulting gel was then aged at 80 °C for 24 h. Finally, the solid product was filtered, washed with water and calcined statically in air at 550 °C for 5 h.A series of sulfonic acid functionalised SBA-15 were prepared following the method reported elsewhere.34 The thiol coverage was varied from low to saturation (based on hydroxyl density). A stock solution of MPTMS in toluene was initially prepared as precursor for grafting on SBA-15. Specific amount of MPTMS in toluene solution (0.01 < MPTMS/SBA-15 < 1) was added per gram of material. The overall volume of toluene was kept constant as 30 ml with further addition of toluene and the mixture refluxed for 24 hours. The resulting thiol functionalised samples were then filtered washed with methanol and dried at 80 °C. Oxidation of thiol groups was carried out with H2O2 at room temperature for 24 h (30 mL of 33 wt% H2O2 per gram of material). The samples are denoted as PrSO3H(xML)/SBA-15, x gives the volume of MPTMS per g of SBA-15, ML for saturation (monolayer coverage).
2.2 Characterisation
Physicochemical properties of the as-synthesised catalysts were fully characterised. Low angle XRD patterns were recorded on a Bruker D8 Advance diffractometer fitted with an X'celerator detector and Cu Kα (1.54 Å) source over the range 2θ = 0.3–10°. Nitrogen porosimetry was measured on a Quantachrome Nova 4000 porosimeter and analysed with NovaWin software. Samples were degassed at 120 °C for 4 h prior to analysis at −196 °C. Thermogravimetric analysis (TGA) was performed on a Mettler Toledo, TGA/DSC2 Stare system under flowing nitrogen during heating at 10 °C min−1 from 40–800 °C. Total sulfur loadings were calculated from the TGA weight loss between 200–600 °C, and verified by XRF analysis on a Bruker S8 Tiger. DRIFTS measurements were conducted using a Thermo Scientific Nicolet environmental cell and smart collector accessory on a Thermo Scientific Nicolet iS50 FT-IR Spectrometer with MCT detector. The catalysts diluted in KBr (10 wt%) were loaded in the environmental cell and subjected to evacuation at 200 °C for 2 h to remove physisorbed water/moisture. Analyses were performed at 200 °C. Ex situ pyridine adsorption studies were made by wetting the samples with pyridine. Excess pyridine was removed overnight in vacuo at 50 °C, with subsequent in vacuo analysis by DRIFTS at 50 °C. Acid sites concentrations were measured by NH3 pulse chemisorption using a Quantachrome ChemBET 3000 instrument interfaced to an MKS Minilab mass spectrometer (MS). Samples were degassed at 120 °C overnight under helium prior to NH3 pulse titration at 100 °C. Temperature-programmed desorption (TPD) was subsequently performed on ammonia saturated samples between 100–500 °C.2.3 Esterification
Esterifications were performed in batch at atmospheric pressure using a Radley carousel reaction station.35 Reactions were performed at 100 °C employing 0.05 g of catalyst, 10 mmol of acetic acid, 5 mmol of benzyl alcohol (acid![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :alcohol mole ratio 2:1), and 1.28 mmol of dihexyl ether as an internal standard in 10 ml toluene. Aliquots were withdrawn periodically from the reaction mixture and analysed by off-line GC after dilution with dichloromethane using a Varian 450-GC equipped with a CP-Sil 5 CB 15 m × 0.25 mm × 0.25 μm capillary column. The impact of acid:alcohol molar ratio was investigated for a fixed 10 mmol of acetic acid, adjusting the toluene accordingly to maintain a constant reaction volume. Turnover Frequencies (TOFs) were calculated by normalising initial rates derived from the linear portion of reaction profiles during the first hour to the acid site loadings obtained from NH3 pulse chemisorption. Esterification was also explored with other common bio-oil alcohol components, anisyl alcohol, p-cresol and m-cresol for acid:alcohol molar ratios of 2:1. Water tolerance was tested by deliberately spiking a reaction with 40 mol% water relative to benzyl alcohol prior to catalyst addition. Recycle experiments were performed via hot filtration of spent catalyst in toluene and room temperature washing with methanol prior to re-use to remove residual ester. Catalyst stability was assessed via C/H/N/S elemental analysis of as-prepared and hot-filtered spent catalysts, and removal of the catalyst from the reaction medium during the initial stage of reaction and subsequent analysis of the filtrate for acetic acid conversion and ester after 5 h.
:alcohol mole ratio 2:1), and 1.28 mmol of dihexyl ether as an internal standard in 10 ml toluene. Aliquots were withdrawn periodically from the reaction mixture and analysed by off-line GC after dilution with dichloromethane using a Varian 450-GC equipped with a CP-Sil 5 CB 15 m × 0.25 mm × 0.25 μm capillary column. The impact of acid:alcohol molar ratio was investigated for a fixed 10 mmol of acetic acid, adjusting the toluene accordingly to maintain a constant reaction volume. Turnover Frequencies (TOFs) were calculated by normalising initial rates derived from the linear portion of reaction profiles during the first hour to the acid site loadings obtained from NH3 pulse chemisorption. Esterification was also explored with other common bio-oil alcohol components, anisyl alcohol, p-cresol and m-cresol for acid:alcohol molar ratios of 2:1. Water tolerance was tested by deliberately spiking a reaction with 40 mol% water relative to benzyl alcohol prior to catalyst addition. Recycle experiments were performed via hot filtration of spent catalyst in toluene and room temperature washing with methanol prior to re-use to remove residual ester. Catalyst stability was assessed via C/H/N/S elemental analysis of as-prepared and hot-filtered spent catalysts, and removal of the catalyst from the reaction medium during the initial stage of reaction and subsequent analysis of the filtrate for acetic acid conversion and ester after 5 h.
3. Results and discussion
3.1 Characterisation
Successful synthesis of the parent SBA-15 mesoporous silica support, which possesses a characteristic p6mm space group, and its retention following sulfonate grafting was first confirmed by low angle XRD and nitrogen adsorption isotherms shown in Fig. 1 and S1† respectively.33,36 XRD patterns for the PrSO3H(xML)/SBA-15 materials were essentially unchanged after the grafting procedure, with common reflections and peak intensities as a function of S coverage, evidencing hexagonally close-packed mesopore channels that were stable during the thiol functionalization and subsequent oxidation. Porosimetry showed type IV isotherms with H1 hysteresis for SBA-15 and all PrSO3H(xML)/SBA-15 samples, highlighting the presence of mesopores. Textural properties of PrSO3H/SBA-15 materials are summarised in Table 1, wherein a continual decrease in BET surface area was observed with increasing sulfonic acid loading, accompanied by a slight narrowing in the BJH mesopore diameter (Fig. 2), consistent with uniform grafting of sulfonic acid groups throughout the pore network which retain a common unit cell. | ||
| Fig. 1 Low angle powder XRD pattern of PrSO3H/SBA-15 as a function of nominal S loading. | ||
 | ||
| Fig. 2 BJH pore size distributions of PrSO3H/SBA-15 as a function of nominal S loading. | ||
| Materials | Surface areaa/m2 g−1 | BJH pore diameter/nm | Total BJH pore volume/cm3 g−1 | Unit cell parameterb/nm | Wall thicknessc/nm | Micropore volume/cc g−1 | Micropore area/m2 g−1 | Bulk S contentd/wt% | Bulk S contente/wt% |
|---|---|---|---|---|---|---|---|---|---|
| a BET. b Determined from a0 = (2d100)/√3. c Determined from a0 – pore diameter. d TGA weight loss between 400–600 °C. e Bulk S content from XRF. | |||||||||
| SBA-15 | 741 | 5.358 | 0.742 | 9.76 | 4.4 | 0.143 | 330 | — | — |
| PrSO3H(0.01ML)/SBA-15 | 675 | 5.059 | 0.771 | 9.58 | 4.52 | 0.108 | 248 | 0.27 | 0.13 |
| PrSO3H(0.1ML)/SBA-15 | 671 | 5.060 | 0.766 | 9.76 | 4.7 | 0.109 | 250 | 0.29 | 0.19 |
| PrSO3H(0.5ML)/SBA-15 | 628 | 5.060 | 0.732 | 9.80 | 4.74 | 0.099 | 226 | 0.49 | 0.82 |
| PrSO3H(1ML)/SBA-15 | 619 | 5.072 | 0.737 | 9.97 | 4.9 | 0.092 | 211 | 0.62 | 1.03 |
Thermogravimetric analysis of PrSO3H/SBA-15 revealed two regimes: loss of physisorbed water <150 °C; and decomposition of propylsulfonate groups >350 °C (Fig. S2†). Quantification of the second loss between 400–600 °C enables calculation of the bulk S content.35 As anticipated, the resulting S loading increased with the volume of MPTMS employed during synthesis (Table 1), which is confirmed by XRF analysis.
The presence of sulfonic acid functions was confirmed via DRIFTS. Fig. S3† shows DRIFT spectra of the MPTMS functionalised SBA-15 materials, and Fig. 3 those of the parent SBA-15 and PrSO3H/SBA-15 materials post-oxidation. SBA-15 exhibited bands at 700–1400 cm−1 and 3000–3800 cm−1 indicative of framework Si–O–Si and surface silanols respectively.37 Sulfonic acid grafting attenuated the silanol features, coincident with the appearance of weak new bands centred around 2900 cm−1 and 1370 cm−1, attributed to C–H and CH2–Si vibrations of the propyl backbone (Fig. S3†), whose intensities increase with increase S loading. Acidic properties of the PrSO3H/SBA-15 family were evaluated via pyridine adsorption studies and NH3 chemisorption to quantify their Brönsted/Lewis character38 and acid site densities. As expected, DRIFT spectra (Fig. S4†) of the sulfonated materials exhibited bands at 1489, 1545 and 1637 cm−1 indicative of pyridinium ions coordinated to Brönsted acidic sites. The Kubelka–Monk Brönsted peak intensity increased linearly with SO3H surface density (via XRF), in excellent correlation with the acid site density determined by NH3 titration (Fig. 4). It should be noted that the maximum sulfonic acidic density obtained in this work over SBA-15 of 0.3SO3H molecule per nm2 is comparable to that reported for MCM-41 (0.5SO3H molecule per nm2).34
 | ||
| Fig. 3 DRIFT spectra of PrSO3H/SBA-15 as a function of nominal S loading. | ||
 | ||
| Fig. 4 Evolution of surface acid properties of PrSO3H/SBA-15 as a function of S loading. | ||
3.2 Acetic acid esterification
The utility of our PrSO3H/SBA-15 family of materials for the esterification of acetic acid with benzyl alcohol (Scheme 1) was subsequently explored in toluene as a simulated bio-oil matrix. | ||
| Scheme 1 Bio-oil upgrading via acetic acid esterification with benzyl alcohol. | ||
Since the majority of reactions were conducted under excess acid, wherein complete acetic acid conversion is not possible, catalyst performance is reported with respect to either the ester yield or the maximum possible acid conversion. Under our reaction conditions, only 4% and 10% benzyl alcohol conversions were observed in blank reactions after 6 h and 24 h respectively (Fig. S5†), confirming the need for catalytic promotion at the low reaction temperatures necessary to prevent bio-oil degradation. Initial reaction conditions for acetic acid esterification catalysed by PrSO3H/SBA-15 were optimised with respect to catalyst charge and temperature (Fig. S6–7†) for the most acidic PrSO3H(1ML)/SBA-15 material. Esterification activity was approximately first order in catalyst up to a 50 mg charge, which was subsequently adopted for all reactions. Conversions increased dramatically above the background rate for temperatures ≥80 °C, reaching 100% within 6 h at 100 °C, accompanied by a selectivity to benzyl acetate of 80%, with diphenyl ether as the major by-product through acid-catalysed etherification of the alcohol. The apparent activation energy of 68 kJ mol−1 (Fig. S8†) is comparable to Amberlyst-15 sulfonic acid resins (73.3 kJ mol−1),39 but higher than that reported over HY and H-ZSM-5 zeolites (∼40 kJ mol−1),22 which operated over a significantly higher temperature range. These compares favourably with values for acetic acid esterification with methanol over sulfonic acid functionalised SBA-15 of 42–52 kJ mol−1,17,40 wherein there are no mass transfer or internal diffusion limitations for the lighter alcohol.
The impact of sulfonic acid loading on acetic acid esterification over PrSO3H/SBA-15 was subsequently explored under these optimal conditions, and the resulting reaction profiles for ester production and benzyl alcohol conversion shown in Fig. 5 and S9† respectively. Both benzyl alcohol and acetic acid (Fig. 6) conversions increased monotonically with sulfonic acid loading, with PrSO3H(1ML)/SBA-15 showing the best performance as anticipated for a Brönsted acid catalysed transformation. Diphenylether was the principal by-product from etherification. Initial rates of benzyl acetate formation shown in Fig. 6 mirror the trends in 5 h acid conversion, indicating that acid site density/strength were the predominant factors controlling esterification with the small decreases in surface area and mean pore diameter with S loading having no detrimental impact on reactivity.
 | ||
| Fig. 5 Benzyl acetate formation over PrSO3H/SBA-15 series. Reaction conditions: 50 mg catalyst, 100 °C, acid:alcohol ratio of 2:1, toluene solvent. | ||
 | ||
| Fig. 6 Acetic acid conversion and initial rate of ester production of PrSO3H/SBA-15 as a function of S loading. Reaction conditions: 50 mg catalyst, 100 °C, acid:alcohol ratio of 2:1, toluene solvent. Conversion after 5 h. | ||
Turnover Frequencies (TOFs) per acid site show a strong variation between low and high S loadings (Fig. 7), attributed to a difference in acid strength, with high sulfonic acid loadings possessing stronger acidity as previously observed for MCM-41 mesoporous silica due to steric repulsion orienting PrSO3H groups in-pore.34 The resulting TOFs of the high loading PrSO3H/SBA-15 catalysts are superior to those of microporous Hβ, HY, H-ZSM-5 zeolites (<10 h−1) and silicotungstic acid/ZrO2/SBA-15 (36 h−1):22 this is especially impressive since the latter were determined under more favourable reaction conditions. Leaching studies conducted with the PrSO3H(1ML)/SBA-15 catalyst confirmed its heterogeneous mode of action, with negligible benzyl alcohol conversion observed following catalyst removal after 30 min reaction (Fig. S10†).
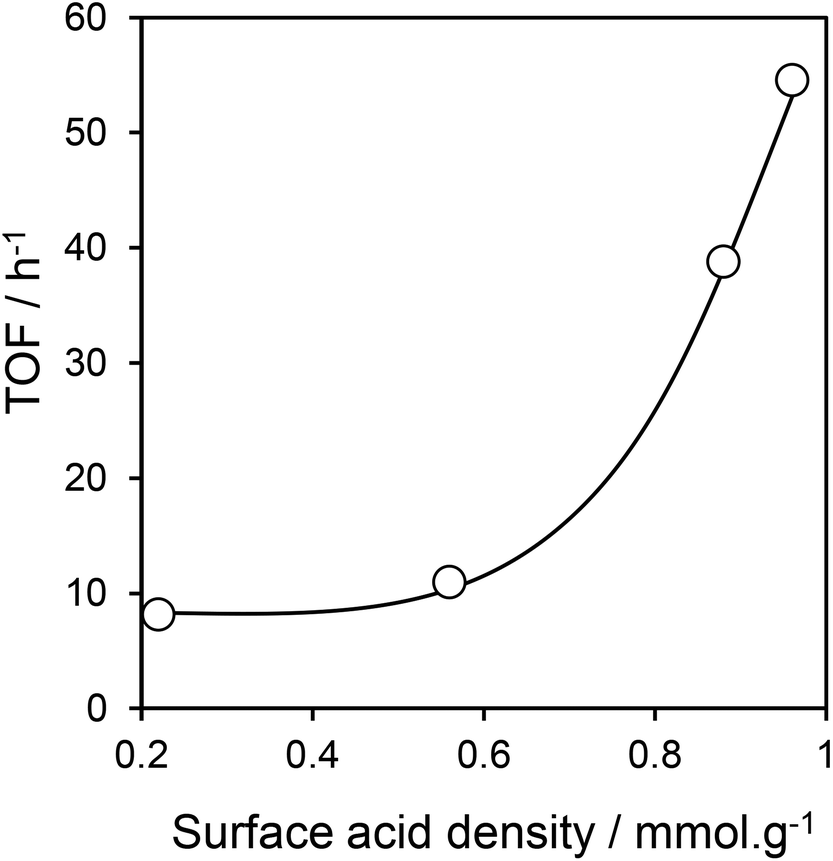 | ||
| Fig. 7 Sulfonic acid loading dependence of TOF for benzyl alcohol conversion in acetic acid esterification over PrSO3H/SBA-15. Reaction conditions: 50 mg catalyst, 100 °C, acid:alcohol ratio of 2:1, toluene solvent. | ||
An important objective of this work was to understand the impact of (simulated) bio-oil composition upon its upgrading via acetic acid esterification, and hence the performance of our most promising PrSO3H(1ML)/SBA-15 catalyst was subsequently explored as a function of acid:alcohol ratio, which varies widely in real pyrolysis bio-oils.41 The resulting impact upon acid conversion is shown in Fig. 8, revealing a significant increase in conversion under acid-rich conditions, as previously reported for zeolites,22 and indicating that benzyl alcohol is able compete effectively for adsorption sites with acetic acid, in contrast to methanol.31 This is an important observation since acetic acid is commonly in excess of phenolic components (∼6 versus 2 wt%)7 suggesting that our sulfonic acid catalysts should be efficacious against upgrading of real bio-oils via esterification under mild reaction conditions.
 | ||
| Fig. 8 Dependence of acetic acid esterification over PrSO3H(1ML)/SBA-15 on acid:alcohol ratio. Reaction conditions: 50 mg catalyst, 100 °C, toluene solvent. | ||
Bio-oils contain a range of lignin-derived phenolics, hence our PrSO3H(1ML)/SBA-15 catalyst was also screened for acetic acid esterification against representative examples, and methanol (Fig. 9). While it proved equally active towards acetic acid esterification with anisyl alcohol (4-methoxybenzyl alcohol), reactivity was far poorer employing cresols and methanol, the latter presumably reflecting the inability of methanol to compete effectively for adsorption sites in the presence of excess acid.17 This is also a reflection of the aromatic alcohols being better nucleophiles than phenol, and that lower boiling point of methanol leads to lower reaction temperatures at ambient pressure.
 | ||
| Fig. 9 Dependence of acetic acid esterification over PrSO3H(1ML)/SBA-15 on alcohol. Reaction conditions: 50 mg catalyst, 100 °C, acid:alcohol ratio of 2:1, toluene solvent. | ||
Pyrolysis bio-oils, whether derived via thermal or catalytic routes, all contain significant water (spanning 10–60 wt%42), and hence water tolerance is a key requirement for any practical bio-oil upgrading catalyst, and hence we conducted a spiking experiment in which a 40 mol% relative to benzyl alcohol was deliberately introduced to the reaction media at the start of reaction. Such a high water concentration is far in excess of that reported to strongly suppress (trans)esterification activity over heteropolyacids,43,44 sulphated zirconia-titania,45 and Amberlyst-15,46 where values around 1 mol% effectively stopped reaction. Fig. 10 shows that this high level of water had no impact on the initial rate of benzyl alcohol esterification, while overall conversion only decreased from 100 to 60% after 5 h reaction, i.e. quantitative in the amount water added, evidencing negligible catalyst deactivation, but simple displacement of the reaction equilibrium towards the reverse hydrolysis reaction as expected. Recycle experiments, and elemental analysis of the reaction mixture and isolated spent catalyst also confirmed that the PrSO3H(1ML)/SBA-15 catalyst was relatively stable to sulfur leaching under our reaction conditions, with only a small (<20%) decrease observed after three esterification cycles (Fig. S11†). In contrast, significant surface carbon accumulated over spent catalysts following each re-use, likely due to the mild regeneration treatments employed between each cycle (room temperature washing with toluene followed by methanol). This surface carbon, which likely arose from strongly adsorbed aromatic oligomers (as observed over sulfonated carbons47) has a pronounced negative impact on conversion (Fig. S12†), and we are currently exploring regeneration protocols to remove these residues.
 | ||
| Fig. 10 Impact of 40 mol% water addition on acetic acid esterification over PrSO3H(1ML)/SBA-15. Reaction conditions: 50 mg catalyst, 100 °C, acid:alcohol ratio of 2:1, toluene solvent. | ||
The excellent performance of our PrSO3H(1ML)/SBA-15 catalyst was evidenced by benchmarking against widely used commercial solid acid catalysts, specifically an Amberlyst-15 sulfonic acid resin and two USY zeolites with different Si:Al. molar ratios of 30 and 2.6. Fig. 11 shows that both zeolites offer significantly lower benzyl alcohol conversion than both sulfonic acid materials, with the poorer activity of the more acidic USY-2.6 suggesting that this may be accounted for by self-poisoning through strong adsorption of acetic acid and/or benzyl alcohol. However, despite comparable acetic acid conversion over PrSO3H(1ML)/SBA-15 and Amberlyst-15, the benzyl acetate yield is an order of magnitude lower over the latter commercial material due to preferential formation of the undesired diphenyl ether condensation product.
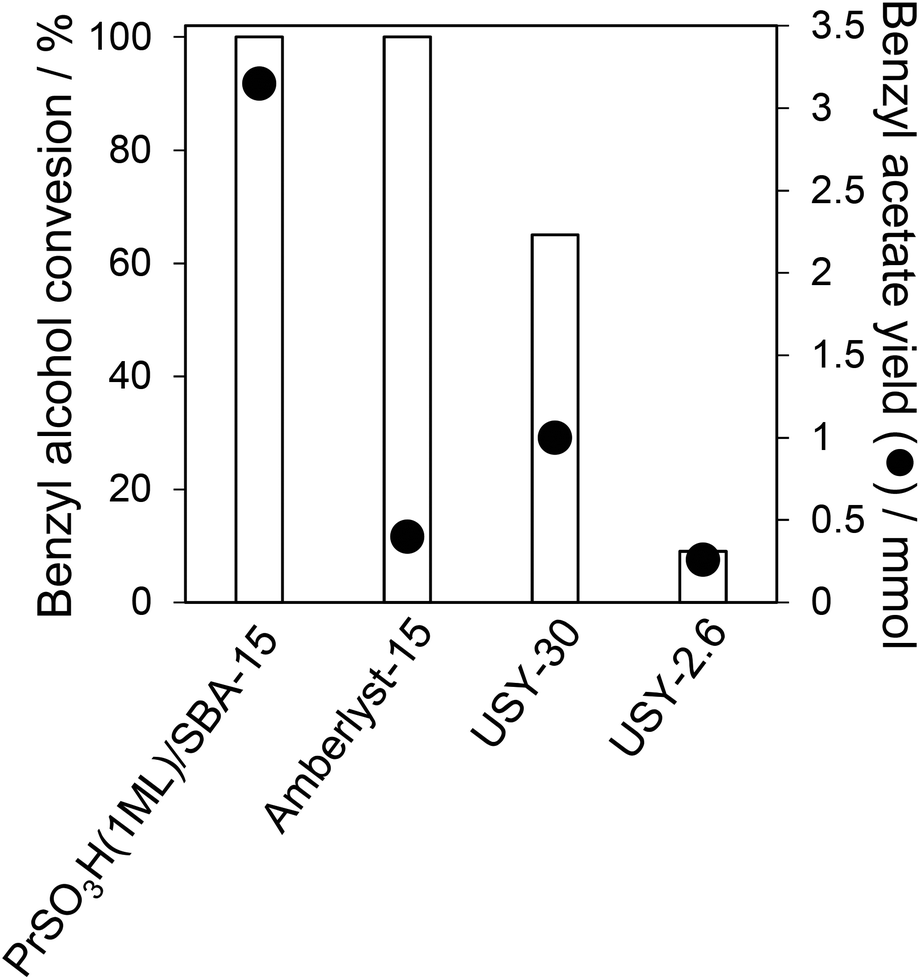 | ||
| Fig. 11 Comparison of acetic acid esterification over PrSO3H(1ML)/SBA-15 and commercial Amberlyst and USY zeolite catalysts. Reaction conditions: 50 mg catalyst, 100 °C, toluene solvent. | ||
4. Conclusions
Propyl sulfonic acid functionalised SBA-15 (PrSO3H/SBA-15) is an effective catalysts for acetic acid esterification with aromatic alcohols in toluene as a simulated bio-oil medium. Mass-normalised activities increase with S loading, reflecting and increase in sulfonic acid site densities and associated loss of surface silanols, with an concomitant rise in Turnover Frequencies (TOFs) likely associated with an increase in acid strength due to surface crowding of sulfonate groups. High loading PrSO3H/SBA-15 exhibits optimum performance under high acid:benzyl alcohol molar ratios, remaining active even in the presence of extremely high water concentrations representative of pyrolysis oils, with minimal S leaching, and appears well-suited to the upgrading of pyrolysis bio-oils. However there remains a need to develop improved catalyst regeneration protocols to remove strongly adsorbed carbon species post-reaction. Mesoporous PrSO3H/SBA-15 outperform microporous zeolites for acetic acid esterification with benzyl alcohol, and offer opportunities for further rate-enhancements through co-functionalisation with hydrophobic alkyl moieites, an approach that has proven successful in fatty acid esterification with methanol.
Acknowledgements
We thank the EPSRC for financial support (EP/K000616/2, EP/G007594/4 and EP/K014749/1) and the award of a Leadership Fellowship to AFL. KW thanks the Royal Society for an Industry Fellowship. Support from the European Union Seventh Framework Programme (FP7/2007–2013) under grant agreement no. 604307 is also acknowledged.Notes and references
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS PubMed.
- A. F. Lee and K. Wilson, Catal. Today, 2015, 242, 3–18 CrossRef CAS.
- A. F. Lee, J. A. Bennett, J. C. Manayil and K. Wilson, Chem. Soc. Rev., 2014, 43, 7887–7916 RSC.
- L. Ciddor, J. A. Bennett, J. A. Hunns, K. Wilson and A. F. Lee, J. Chem. Technol. Biotechnol., 2015, 90, 780–795 CrossRef CAS.
- S. Zhang, Y. Yan, T. Li and Z. Ren, Bioresour. Technol., 2005, 96, 545–550 CrossRef CAS PubMed.
- A. Alcala, A. V. Bridgwater, A. Alcala and A. V. Bridgwater, Blends Biodiesel Pyrolysis Oil, 2014, 109, 417–426 Search PubMed.
- C. A. Mullen, A. A. Boateng, N. M. Goldberg, I. M. Lima, D. A. Laird and K. B. Hicks, Biomass Bioenergy, 2010, 34, 67–74 CrossRef CAS.
- D. Mohan, C. U. Pittman and P. H. Steele, Energy Fuels, 2006, 20, 848–889 CrossRef CAS.
- Q. Zhang, J. Chang, T. Wang and Y. Xu, Energy Convers. Manage., 2007, 48, 87–92 CrossRef CAS.
- C. A. Mullen and A. A. Boateng, Energy Fuels, 2008, 22, 2104–2109 CrossRef CAS.
- P. M. Mortensen, J. D. Grunwaldt, P. A. Jensen, K. G. Knudsen and A. D. Jensen, Appl. Catal., A, 2011, 407, 1–19 CrossRef CAS.
- A. V. Bridgwater, Biomass Bioenergy, 2012, 38, 68–94 CrossRef CAS.
- A. H. Zacher, M. V. Olarte, D. M. Santosa, D. C. Elliott and S. B. Jones, Green Chem., 2014, 16, 491–515 RSC.
- M. Milina, S. Mitchell and J. Pérez-Ramírez, Catal. Today, 2014, 235, 176–183 CrossRef CAS.
- I. Graca, J. D. Comparot, S. Laforge, P. Magnoux, J. M. Lopes, M. F. Ribeiro and F. R. Ribeiro, Energy Fuels, 2009, 23, 4224–4230 CrossRef CAS.
- Z. Q. Ma and J. A. van Bokhoven, ChemCatChem, 2012, 4, 2036–2044 CrossRef CAS.
- S. Miao and B. H. Shanks, Appl. Catal., A, 2009, 359, 113–120 CrossRef CAS.
- D. P. Sawant, A. Vinu, J. Justus, P. Srinivasu and S. B. Halligudi, J. Mol. Catal. A: Chem., 2007, 276, 150–157 CrossRef CAS.
- J. Bedard, H. Chiang and A. Bhan, J. Catal., 2012, 290, 210–219 CrossRef CAS.
- F. H. Mahfud, I. Melián-Cabrera, R. Manurung and H. J. Heeres, Process Saf. Environ. Prot., 2007, 85, 466–472 CrossRef CAS.
- M. J. Climent, A. Corma and S. Iborra, Chem. Rev., 2011, 111, 1072–1133 CrossRef CAS PubMed.
- S. R. Kirumakki, N. Nagaraju and S. Narayanan, Appl. Catal., A, 2004, 273, 1–9 CrossRef CAS.
- S. H. Ali and S. Q. Merchant, Ind. Eng. Chem. Res., 2009, 48, 2519–2532 CrossRef CAS.
- J. D'Souza and N. Nagaraju, Indian J. Chem. Technol., 2006, 13, 605–613 Search PubMed.
- W.-J. Liu, K. Tian, H. Jiang and H.-Q. Yu, Sci. Rep., 2013, 3 Search PubMed.
- K. Wilson, A. F. Lee, D. J. Macquarrie and J. H. Clark, Appl. Catal., A, 2002, 228, 127–133 CrossRef CAS.
- J. A. Melero, G. D. Stucky, R. van Grieken and G. Morales, J. Mater. Chem., 2002, 12, 1664–1670 RSC.
- S. Shylesh, S. Sharma, S. P. Mirajkar and A. P. Singh, J. Mol. Catal. A: Chem., 2004, 212, 219–228 CrossRef CAS.
- A. F. Lee and K. Wilson, in Handbook of Green Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2010 Search PubMed.
- J. A. Melero, L. F. Bautista, G. Morales, J. Iglesias and R. Sánchez-Vázquez, Chem. Eng. J., 2010, 161, 323–331 CrossRef CAS.
- S. Miao and B. H. Shanks, J. Catal., 2011, 279, 136–143 CrossRef CAS.
- N. Lohitharn and B. H. Shanks, Catal. Commun., 2009, 11, 96–99 CrossRef CAS.
- D. Y. Zhao, J. L. Feng, Q. S. Huo, N. Melosh, G. H. Fredrickson, B. F. Chmelka and G. D. Stucky, Science, 1998, 279, 548–552 CrossRef CAS PubMed.
- J.-P. Dacquin, H. E. Cross, D. R. Brown, T. Duren, J. J. Williams, A. F. Lee and K. Wilson, Green Chem., 2010, 12, 1383–1391 Search PubMed.
- C. Pirez, J.-M. Caderon, J.-P. Dacquin, A. F. Lee and K. Wilson, ACS Catal., 2012, 2, 1607–1614 CrossRef CAS.
- D. Y. Zhao, Q. S. Huo, J. L. Feng, B. F. Chmelka and G. D. Stucky, J. Am. Chem. Soc., 1998, 120, 6024–6036 CrossRef CAS.
- C. Pirez, K. Wilson and A. F. Lee, Green Chem., 2014, 16, 197–202 RSC.
- A. Osatiashtiani, A. F. Lee, D. R. Brown, J. A. Melero, G. Morales and K. Wilson, Catal. Sci. Technol., 2014, 4, 333–342 CAS.
- R. Roy and S. Bhatia, J. Chem. Technol. Biotechnol., 1987, 37, 1–10 CrossRef CAS.
- Y. Liu, E. Lotero and J. G. Goodwin Jr., J. Catal., 2006, 242, 278–286 CrossRef CAS.
- D. Carpenter, T. L. Westover, S. Czernik and W. Jablonski, Green Chem., 2014, 16, 384–406 RSC.
- K. Smets, P. Adriaensens, J. Vandewijngaarden, M. Stals, T. Cornelissen, S. Schreurs, R. Carleer and J. Yperman, J. Anal. Appl. Pyrolysis, 2011, 90, 100–105 CrossRef CAS.
- F. Chai, F. Cao, F. Zhai, Y. Chen, X. Wang and Z. Su, Adv. Synth. Catal., 2007, 349, 1057–1065 CrossRef CAS.
- X. Duan, Y. Liu, Q. Zhao, X. Wang and S. Li, RSC Adv., 2013, 3, 13748–13755 RSC.
- N. Kaur and A. Ali, Renewable Energy, 2015, 81, 421–431 CrossRef CAS.
- J.-Y. Park, Z.-M. Wang, D.-K. Kim and J.-S. Lee, Renewable Energy, 2010, 35, 614–618 CrossRef CAS.
- J. A. Melero, R. van Grieken and G. Morales, Chem. Rev., 2006, 106, 3790–3812 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: DRIFT spectra, porosimetry data, TGA, Pyridine adsorption spectra, esterification reaction results. See DOI: 10.1039/c5gc01889g |
| This journal is © The Royal Society of Chemistry 2016 |