DOI:
10.1039/C5GC01694K
(Paper)
Green Chem., 2016,
18, 760-770
Bio-based polycarbonate from limonene oxide and CO2 with high molecular weight, excellent thermal resistance, hardness and transparency†
Received
23rd July 2015
, Accepted 8th September 2015
First published on 8th September 2015
Abstract
Completely bio-based, high molecular weight (>100 kDa) poly(limonene carbonate) (PLimC) with attractive thermal (glass transition temperature, Tg = 130 °C) and optical properties (transmission 94%, haze 0.75%) was prepared by coupling bio-based limonene oxide (LO) and carbon dioxide (CO2) using a β-diiminate zinc catalyst (bdi)Zn(OAc). The molar mass of the polymer could be controlled by changing the ratio of catalyst and monomer, keeping molar mass dispersities low. The synthesis of the LO with very high content (>85%) of trans-isomer and absence of impurities with hydroxyl functionality was also established, which was necessary to obtain a high molar mass of polymer and almost quantitative conversion of epoxide during polymerisation. The upscaled syntheses of both the monomer and the polymer (>1 kg per batch) were readily realised, suggesting an easy transfer to pilot plant scale. The polymerisation kinetics were studied suggesting a second order dependence on LO concentration, wherefrom a mechanism is proposed with an alternating insertion of LO and CO2. The effect of chain-ends on the thermal stability of PLimC was studied with a thermal desorption unit coupled with gas chromatography-mass spectrometry (GC-MS) experiments. PLimC with thermal stability as high as 240 °C could be achieved using appropriate end-capping agents. PLimC is characterised by excellent transparency and hardness.
Introduction
The conventional polycarbonate (PC) made by the condensation of bisphenol-A (BPA) and phosgene is an amorphous polymer with high impact strength, toughness, heat resistance and transparency, which finds applications as medical, automotive, optical and electronic devices.1 The drawback of BPA-PC is found in the choice of monomers, with phosgene being a highly toxic volatile compound and BPA raising concerns regarding negative health effects due to leaching out of the polymer when in contact with food.2 Additionally, both monomers are petro-based compounds and, recently, efforts have been made to replace them, at least partially, with bio-based diols, e.g. isosorbide derived from carbohydrate,3,4L-tyrosyine5 or terpene derivatives.6 An alternative route to the synthesis of PCs is the reaction of epoxides with CO2.7 This route avoids the use of poisonous phosgene by replacement with CO2, which is the most abundant greenhouse gas, far less hazardous and inherently bio-based.8 Inoue et al. published pioneering work in 1969 on the synthesis of aliphatic polycarbonates by coupling CO2 and epoxides with a zinc catalyst.9,10 Since then, a lot of research has been directed towards the copolymerisation of CO2 and the commodity chemical propylene oxide (PO) to yield poly(propylene carbonate), which is a low Tg (∼40 °C) amorphous thermoplastic that is produced on an industrial scale.11–13 The softening temperature of this material obviously limits the range of applications, thus, the use of bulkier epoxides resulting in higher dimensional stability at elevated temperature is desirable. Cyclohexene oxide is such an example for the coupling with CO2 that gives a PC with a Tg at around 120 °C, but, similar to PO, it is a petro-based monomer and its brittle nature results in poor mechanical properties.14 The PC produced in this way is always partially bio-based and can become fully bio-based depending upon the source of epoxide. In order to produce 100% sustainable PC, the choice of epoxide is crucial and, to the best of our knowledge, only epoxidised fatty acids,15 1,4-cyclohexadiene16,17 and limonene have been investigated.18 Limonene is an abundant alicyclic terpene, found in the peel of many citrus fruits and is the main component of orange oil, resulting in a capacity of more than 520![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tons a−1 (estimated from 70 Mt a−1 oranges produced) of which 70 kt are extracted each year.19,20 This available renewable non-food resource is a great choice for sustainable chemistry and the oxidation product LO is used as a monomer for direct cationic polymerisation,21 as a dioxide for isocyanate-free synthesis of polyurethanes22 or for copolymerisation with CO2.18,23,24 Coates et al. discovered an efficient single-site β-diiminate (bdi) zinc catalyst for copolymerisation of CO2 and LO to produce poly(limonene carbonate) with promising Tg (110 °C).18 The molar mass reported for the polymer was limited to 25 kDa and less material characterisation was reported. The polymerisation is stereoselective, incorporating only trans-LO in the polymer. Therefore, the commercial mixture of LO with an approximate content of at least 45% of the cis stereoisomer is rather uneconomical for this reaction because there is a huge amount of unreacted high boiling epoxide in the reaction mixture that has to be extracted, not to mention the waste of valuable bio resources. Later, the use of an Al(III) catalyst has proven to incorporate both isomers (with preference for trans-LO) into the polymer chain, but molar masses were still less than 11 kDa and monomer conversion was limited to <75%.24 The present work provides high molar mass (>100 kDa) PLimC in almost quantitative monomer conversion (>90%) for the first time, using a bdi-zinc catalyst with very attractive material properties that might challenge the predominance of BPA-PC by offering a bio-based, non-food, high-Tg alternative. The use of LO with a very high content (>95%) of trans-isomer and the absence of impurities with hydroxyl functionality in the monomer were critical points for obtaining high molar mass and almost quantitative conversion of monomer during polymerisation. Both the monomer and polymer synthesis were readily scaled-up to ∼1.2 kg per batch. An in-depth study on the kinetics of polymerisation was performed to elucidate the mechanism of the alternating insertion of LO and CO2.
000 tons a−1 (estimated from 70 Mt a−1 oranges produced) of which 70 kt are extracted each year.19,20 This available renewable non-food resource is a great choice for sustainable chemistry and the oxidation product LO is used as a monomer for direct cationic polymerisation,21 as a dioxide for isocyanate-free synthesis of polyurethanes22 or for copolymerisation with CO2.18,23,24 Coates et al. discovered an efficient single-site β-diiminate (bdi) zinc catalyst for copolymerisation of CO2 and LO to produce poly(limonene carbonate) with promising Tg (110 °C).18 The molar mass reported for the polymer was limited to 25 kDa and less material characterisation was reported. The polymerisation is stereoselective, incorporating only trans-LO in the polymer. Therefore, the commercial mixture of LO with an approximate content of at least 45% of the cis stereoisomer is rather uneconomical for this reaction because there is a huge amount of unreacted high boiling epoxide in the reaction mixture that has to be extracted, not to mention the waste of valuable bio resources. Later, the use of an Al(III) catalyst has proven to incorporate both isomers (with preference for trans-LO) into the polymer chain, but molar masses were still less than 11 kDa and monomer conversion was limited to <75%.24 The present work provides high molar mass (>100 kDa) PLimC in almost quantitative monomer conversion (>90%) for the first time, using a bdi-zinc catalyst with very attractive material properties that might challenge the predominance of BPA-PC by offering a bio-based, non-food, high-Tg alternative. The use of LO with a very high content (>95%) of trans-isomer and the absence of impurities with hydroxyl functionality in the monomer were critical points for obtaining high molar mass and almost quantitative conversion of monomer during polymerisation. Both the monomer and polymer synthesis were readily scaled-up to ∼1.2 kg per batch. An in-depth study on the kinetics of polymerisation was performed to elucidate the mechanism of the alternating insertion of LO and CO2.
Results and discussion
Synthesis of limonene oxide
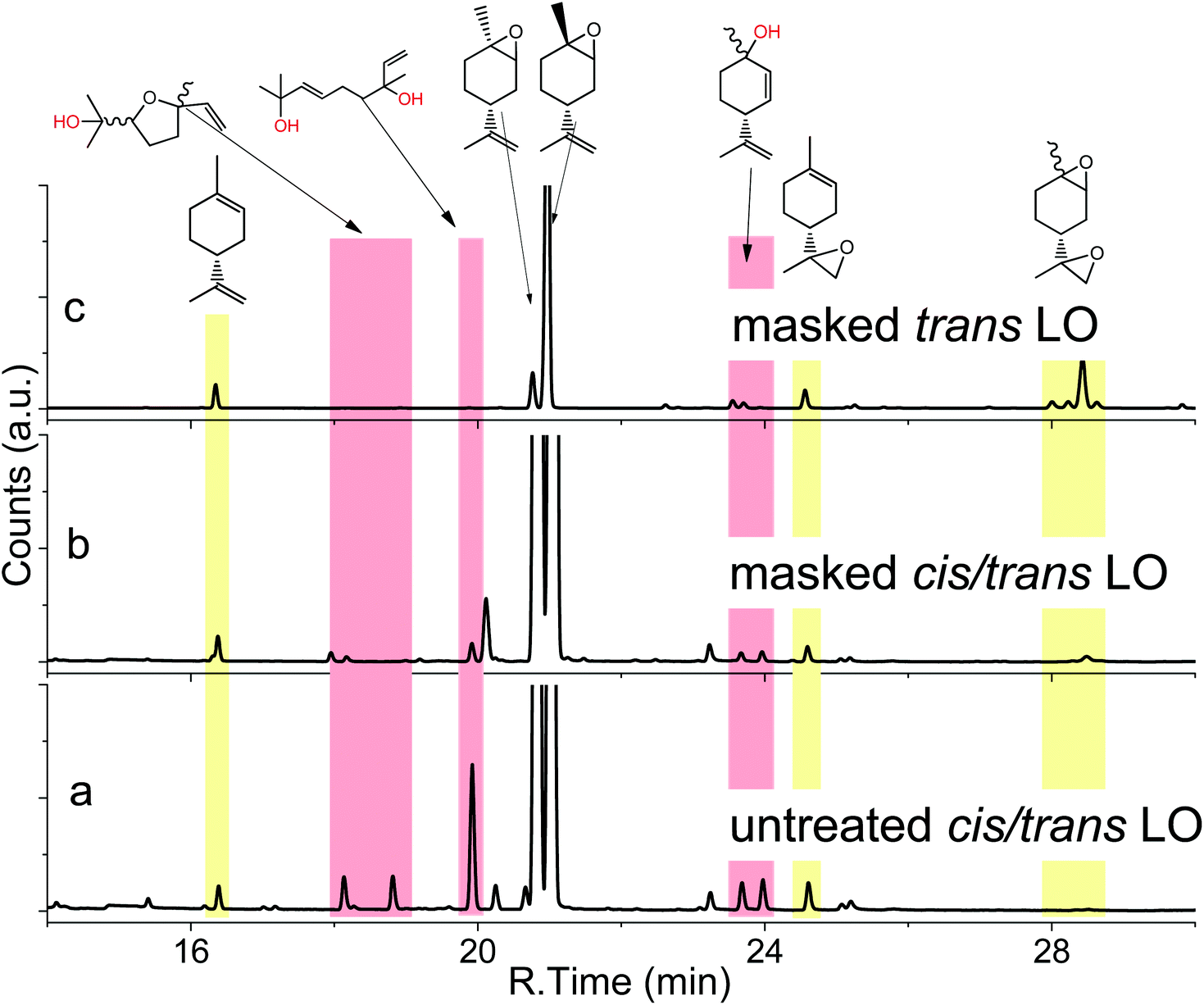
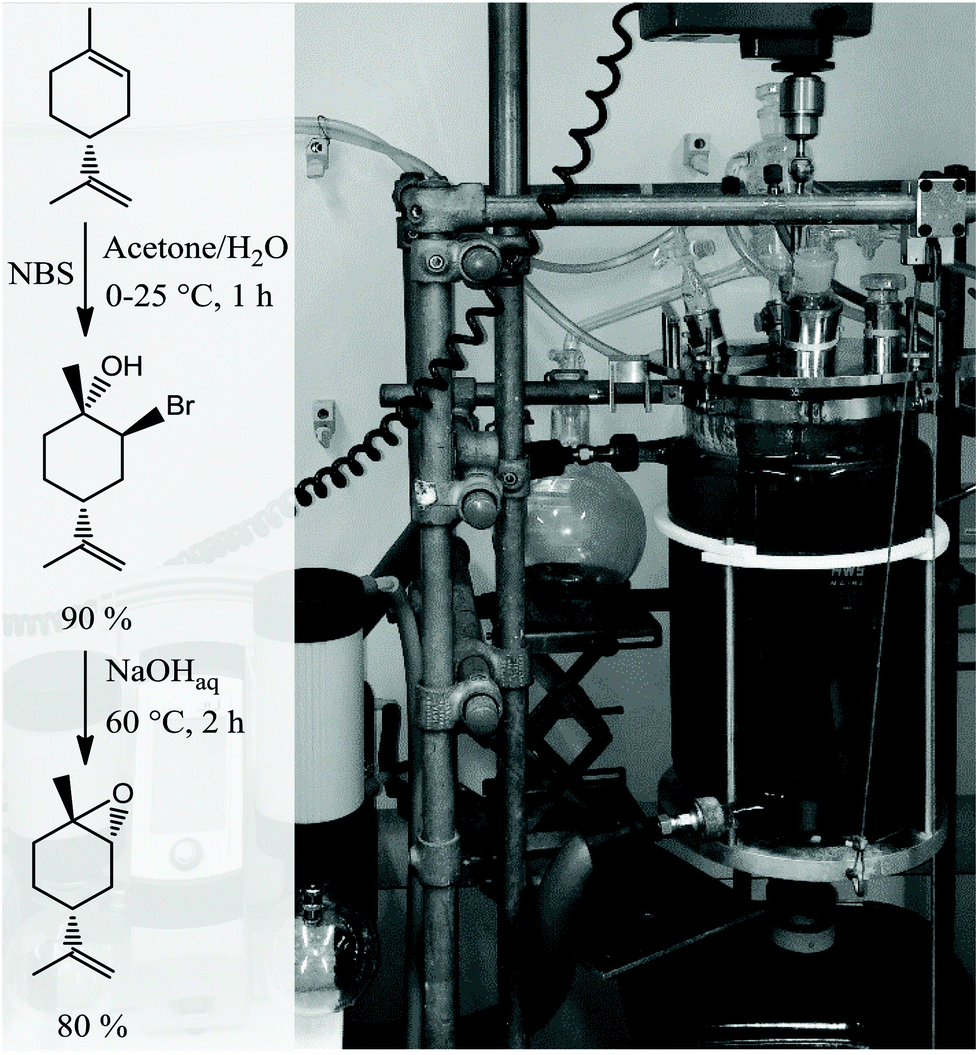
Coates et al.18 found in 2004 that the copolymerisation of LO and CO2 with [(bdi)Zn(OAc)] is highly stereoselective with only the trans isomer being incorporated into the polymer. It would be desirable to start with LO containing high amounts of trans isomer for the reaction with CO2 to create an efficient reaction in terms of yield. Moreover, the commercial monomer contains large amounts of impurities, including hydroxyl functionalised molecules with high boiling points (170–200 °C), as identified by GC-MS (Fig. 1a). The hydroxyl acts as a chain transfer agent (CTA),25 even in very small amounts in the metal catalysed polymerisation of CO2 and LO, and provides only low molar mass polymers of around 25 kg mol−1.18 In order to overcome these two major problems during copolymerisation, the monomer with a majority of trans isomer was synthesised and hydroxyl groups of impurities are masked in the present work. A slightly modified procedure developed by Gurudutt et al. was used for the synthesis of LO to give about 83% of the trans isomer (Scheme 1).26 The monomer contained only 7% cis isomer and 10% other impurities coming from starting material and side reactions during the formation of bromohydrin (exo-double bond was also or exclusively brominated, respectively), as proved by GC (Fig. 1c).
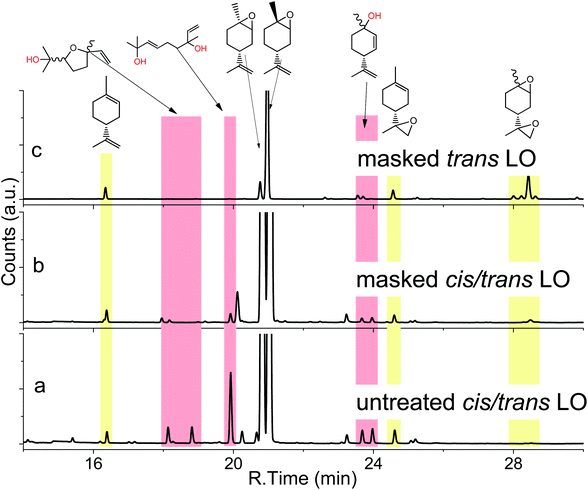 |
| | Fig. 1 GC spectra of a commercial mixture of cis- and trans-LO (a), the same mixture after masking of the hydroxy impurities (b) and the masked product from stereoselective synthesis of trans-LO (c) with structures of identified hydroxyl impurities (by GC-MS). | |
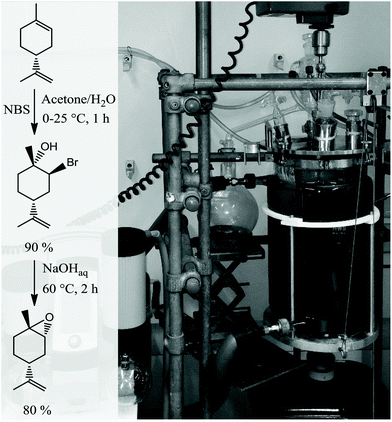 |
| | Scheme 1 Stereoselective synthesis of trans-LO via the corresponding trans-bromohydrin that is subsequently ring-closed by the addition of a base at elevated temperature (left) and 10 L reactor employed for monomer synthesis (right). | |
The synthetic route involves the endo-cyclic bromohydrin (regio- and stereoselective for limonene) in aqueous acetone with N-bromosuccinimide (NBS) as the bromine source. This bromohydrin is readily converted to the corresponding epoxide in the presence of aqueous sodium hydroxide. The monomer synthesis was successfully scaled up to 1.3 kg of LO per batch by transferring the reaction into a 10 L double-walled glass reactor with stirrer and thermostat to control the reaction temperature between 0–60 °C (Scheme 1). All organic solvents employed in the process were recovered by vacuum distillation containing almost no limonene by-products (high boiling liquids). Thus, the solvents can be easily recycled in the process without prior purification which gives this synthesis a sustainable character. Both the commercial mixture and the stereoselective synthesised LO in this work contain impurities with hydroxyl groups. Although the latter produces fewer of these CTAs, they are still observable in the GC spectra (Fig. 1a and c). The impurities were not removable by simple distillation and chromatographic methods in an efficient way. Therefore, all hydroxyls were masked by O-methylation. Here the Williamson ether synthesis has proven to be the most efficient reaction by using sodium hydride (NaH) as a deprotonating agent and methyl iodide (MeI) as an O-methylating moiety (Scheme 2). A GC spectrum of the commercial LO mixture after such a treatment is shown in Fig. 1b, where the signals assigned to the hydroxyl impurities have disappeared completely after the masking reaction (or partially depending on reaction time and steric demand of the molecule) Other masking reactions, such as silylation with trimethylsilyl chloride and catalytic amounts of Mg, gave some side reactions with the oxirane, while the use of benzyl bromide instead of MeI slowed down the ether synthesis significantly, i.e. seems to be too hindered to react with the bulky molecules carrying hydroxyl groups shown in Fig. 1. Another great advantage of this epoxy non-destructive reaction is the simultaneous use of NaH as a drying agent for the monomer. The methylated mixture was then fractionally distilled to give an LO monomer with approximately 85% trans isomer and mainly cis-LO as a side product. This treatment might also be applicable to other epoxides (or other aprotic monomers) where hydroxyl impurities are disturbing the polymerisation.
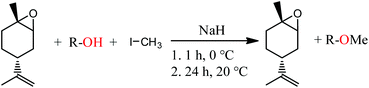 |
| | Scheme 2
O-Methylation of OH-impurities with sodium hydride and methyl iodide by first stirring it for 1 h at 0 °C followed by 24 h at 20 °C. | |
Synthesis of poly(limonene carbonate)
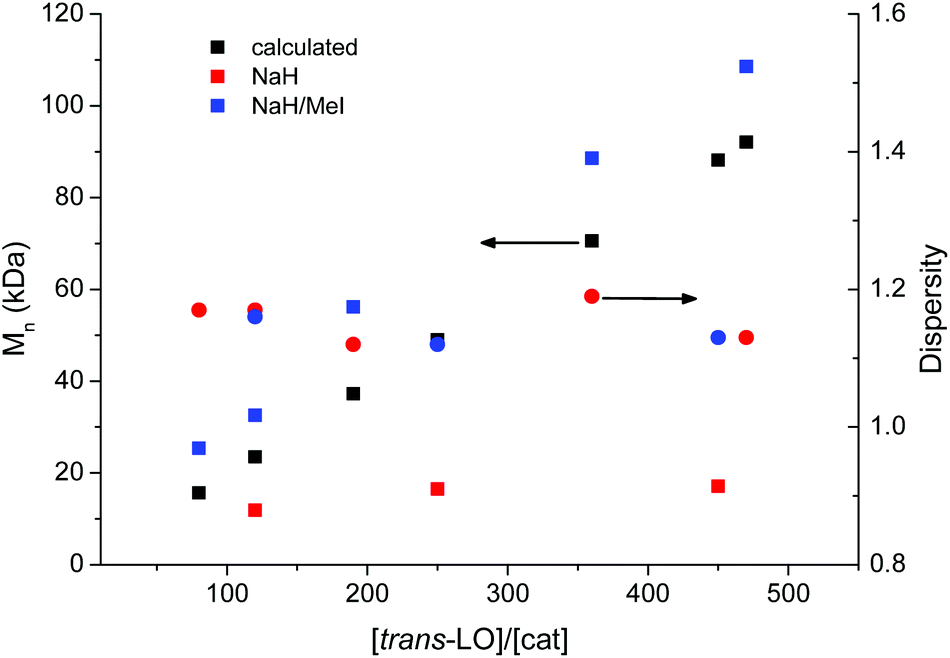
An inherent problem during the copolymerisation of LO and CO2 (see Scheme 3, ESI Fig. S1 and S2† for NMR spectra) to produce PLimC was the limited molecular weight obtained even with very low catalyst concentrations, dried epoxide (Table 1, entries 1–3) and high conversions (>90%). We expected longer chains at lower catalyst concentrations for this supposedly living catalytic system,27 but only slight changes in the molecular weight could be observed when the ratio monomer/catalyst was increased (see Fig. 2, red squares). The highest Mn received with a dried sample (over CaH2 or NaH) of trans-LO was 17.1 kDa (entry 1), while Đ was kept between 1.12–1.16 (Fig. 2, red circles). The low molecular weight obtained was supposedly due to the presence of impurities with hydroxyl groups in the monomer. After the treatment of the dried epoxide with iodomethane (masking agent, Scheme 2), the final copolymer had the expected high molecular weight corresponding to the amount of catalyst added (Table 1, entries 4–8 and Fig. 2, blue squares). In this way, we were able to produce PLimC with molar masses up to 109 kDa, which is, to the best of our knowledge, the highest reported Mn for PLimC while keeping Đ low (1.10–1.19, Fig. 2, blue circles).
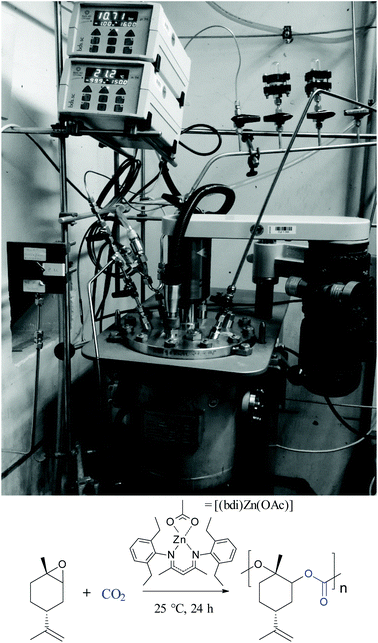 |
| | Scheme 3 Copolymerisation of cis/trans-LO and CO2 in the presence of a β-diiminate zinc complex and the corresponding 10 L high-pressure reactor used for the production of PLimC on the kilogram scale. | |
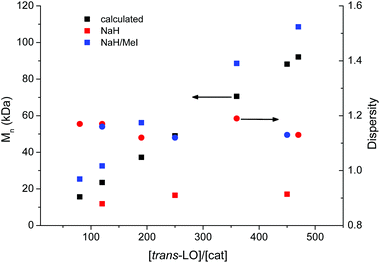 |
| | Fig. 2 Dependency of the molecular weight (determined by CHCl3-GPC) on the amount of catalyst loading after NaH (red squares) and after NaH/MeI- (blue squares) treatment, in comparison with the theoretical values (filled squares) for an expected immortal catalytic system. The dispersity of the experimental values are represented by the circles, whereas red corresponds to NaH treatment and blue to the NaH/MeI treatment. | |
Table 1 Copolymerisation of trans-LO and CO2 after different treatments of the epoxide with corresponding gel permeation chromatography (GPC) data
| Entry |
Treatment |
[trans-LO]/[cat] |
Conv.a [%] |
M
n,calc. [kDa] |
M
n,GPCb [kDa] |
Đ
|
|
Conversion relative to trans-LO in mixture.
Molecular weights and dispersities were determined by CHCl3 GPC calibrated with polystyrene standards.
|
| 1 |
NaH |
450 |
98 |
88.2 |
17.1 |
1.13 |
| 2 |
NaH |
250 |
98 |
49.0 |
16.5 |
1.12 |
| 3 |
NaH |
120 |
95 |
23.5 |
11.9 |
1.16 |
| 4 |
NaH/MeI |
470 |
85 |
92.2 |
108.6 |
1.13 |
| 5 |
NaH/MeI |
360 |
98 |
70.6 |
88.6 |
1.19 |
| 6 |
NaH/MeI |
190 |
93 |
37.2 |
56.2 |
1.13 |
| 7 |
NaH/MeI |
120 |
93 |
23.5 |
32.6 |
1.15 |
| 8 |
NaH/MeI |
80 |
98 |
15.7 |
25.4 |
1.17 |
The linear increase of the molecular weight with a growing LO/catalyst ratio is now in very good agreement with the data calculated (Fig. 2, black squares). It is self-explanatory that the molecular weight for almost any application should be as high as possible to maintain a certain mechanical strength. By masking the hydroxyl impurities in the monomer mixture, we are now able to adjust the molecular weight to the value desired, hence, opening the door to a new world of applications for this completely bio-based material (for full structural characterisation by 1H and 13C NMR, see Fig. S1 and S2, respectively, in ESI† which match well with literature).18 After gaining full control over the copolymerisation, i.e. the molecular weight of the polymer and stereoselective synthesis of the monomer, the next challenge was to upscale the polymerisation reaction. For this, a monomer/catalyst solution was transferred into an evacuated high-pressure 10 L stainless steel reactor (Scheme 2) via cannula, where it is diluted to a 33 vol% solution with toluene via steel tubing connected to a toluene still.
After pressurising the reactor with CO2, the mixture is stirred for 72 h to produce up to 1.2 kg PLimC, which can be used for further processing.
Catalyst removal with immobilised EDTA
The scale up of PLimC raises the question how the amount of organic solvents can be avoided or at least minimised. The work-up of crude PLimC is usually conducted by repeated precipitation of the polymer (dissolved in chloroform or toluene) in methanol to remove residual catalyst. On the kilogram scale, this technique proves very cumbersome and all but eco-friendly. Therefore, an efficient method of inactivating the Zn(II) metal towards PLimC was developed. One of the most common chelating ligands was used for the improvement of the thermal stability of the product: ethylenediaminetetraacetic acid (EDTA). The decomposition temperature was increased dramatically simply by adding the fully protonated (H4EDTA) form of the ligand to a crude polymer solution.
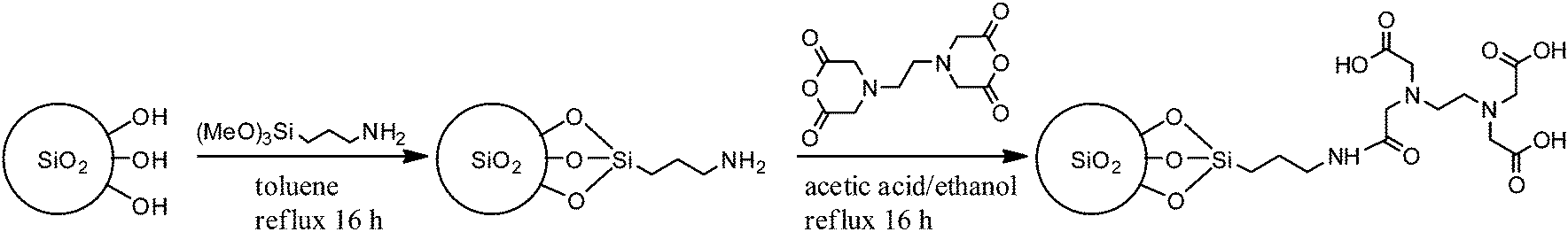
The only drawback of this method comes from the bad solubility of EDTA in CHCl3 or toluene, hence, giving a suspension that is difficult to separate by filtration (the EDTA particles are too small). In order to not only inactivate the metal, but also to separate the inactivated complex (and excess EDTA) from the mixture, the use of immobilised EDTA was a probable solution that also supports the upscaling process, where a packed bed for catalyst removal is mandatory. A method to immobilise EDTA on silica was chosen because of its simplicity and because no swelling is necessary to enter the pores.28 Firstly, the silica is functionalised with 10% primary amine and a propyl spacer then this amine reacts readily with EDTA anhydride to give an amide-coupled EDTA on silica to give EDTA silica (Scheme 4). The reaction was followed by infrared spectroscopy and thermogravimetric analysis (TGA) (see Fig. S4 and S5†). A very efficient procedure was developed to thermally stabilise PLimC without even precipitating the polymer once. To do this, a freshly prepared reaction mixture in toluene was further diluted with toluene and an end-capping agent acetic anhydride (20 wt% with respect to polymer). Then the solution was subdivided into three vessels for treatment with either no, 1 or 10 wt% EDTA silica, respectively.
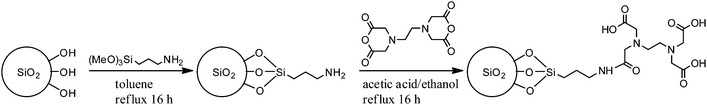 |
| | Scheme 4 Synthesis of EDTA silica by primary amine functionalisation of silica with 3-aminopropyltrimethoxysilane and subsequent condensation with EDTA anhydride. | |
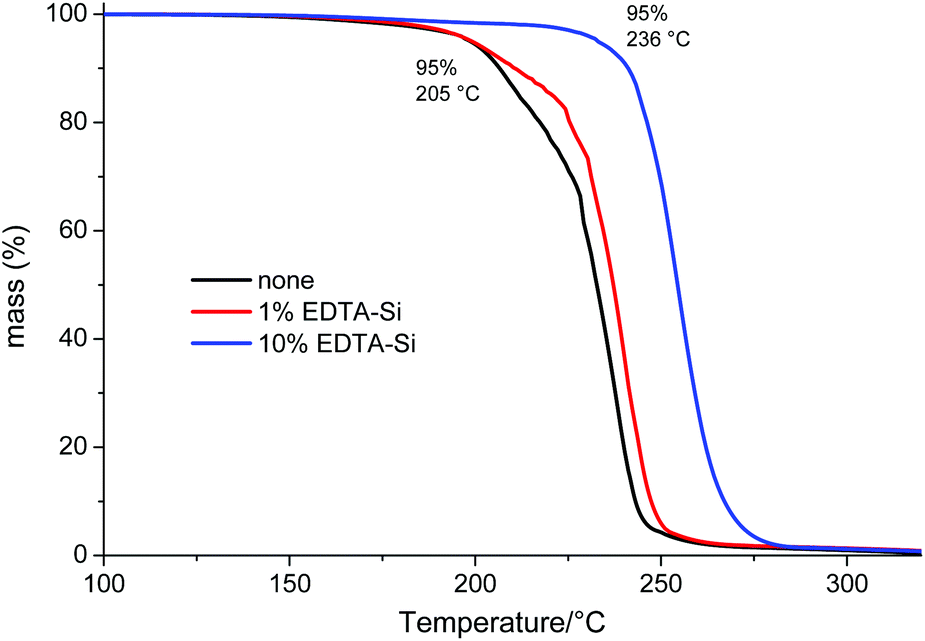
The mixtures were stirred for 2 h, filtered to remove silica particles and eventually cast into petri dishes to yield films of approximately 200 μm thickness. These films were dried in vacuo at 145 °C for 30 min to remove high boiling volatiles above the polymers Tg and then analysed by TGA (Fig. 3). The effect of EDTA silica is well illustrated in these thermograms, since obviously without any catalyst removal, the 5% decomposition temperature lies only at 205 °C (end-capping conditions were the same for all three samples). There is a slight increase of T5% with 1% EDTA silica, but still well below 210 °C, whereas with 10% of the immobilised chelating ligand, the thermal stability has risen to 236 °C (close to the all-time maximum for PLimC of 241 °C). Hence, with 10% of that silica, no precipitation of the polymer is needed at all. The reason why 1 wt% is not enough arises simply from the fact that only 10% (or rather less, because immobilisation is hardly quantitative) of the 1% EDTA silica bears EDTA groups, i.e. there is less than 0.2 mol% EDTA in the mixture, which translates to twice as many moles of Zn(II) compared to EDTA in that sample (having used 0.35 mol.% of catalyst).
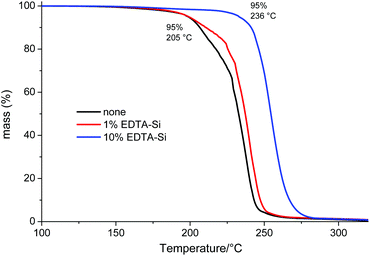 |
| | Fig. 3 TGA of cast films of a crude PLimC solution (in toluene) treated with 20 wt% acetic anhydride and no (black), 1% (red) or 10% (blue) EDTA silica. | |
Kinetic studies and mechanistic investigations of the copolymerisation of LO and CO2
Upscaling the polymer production eventually leads to a re-evaluation of the catalytic system involved. A homogeneous catalyst might be most convenient for gram synthesis, but catalyst removal in up-scaled processes becomes tedious (see section above) and solvent intensive. Therefore, a heterogeneous catalyst is desirable that can remain in the polymer and that, most ideally, works in an aqueous environment to create a completely sustainable synthesis of polycarbonate. For the design of the latter, a deeper understanding of the mechanism of insertion of LO and CO2 into the Zn–O bond is essential and the kinetic studies presented here can help to elucidate the details of transition states and active species. In order to establish the rate law for the copolymerisation of trans-LO and CO2, the reaction order in monomer, CO2 and catalyst were determined. Utilising in situ attenuated total reflectance infrared (ATR IR) spectrometry, the reaction progress was observed by monitoring the increase of the carbonyl vibration stretching-mode of PLimC at 1749 cm−1. All experiments were performed at 20 °C.18 The kinetic equation can be described by −d[LO]/dt = k·[CO2]a·[LO]b·[Zn]c, where k is the apparent rate constant and [LO], [CO2] and [Zn] are the concentrations of LO, CO2 and the catalyst, respectively. Finally, a, b and c are the orders of [LO], [CO2] and [Zn], respectively. To ascertain the catalyst order, the concentration of (bdi)ZnOAc was varied from 12.3–32.9 mM at otherwise constant reaction conditions at room temperature and 10 bar CO2 pressure. The reaction rates kobs were determined from the initial slopes. It has to be mentioned that an induction period of about 10–20 min was observed for all copolymerisation experiments. The double logarithmic plot of kobs against [Zn] showed that the copolymerisation reaction of LO was nearly first order in catalyst (Fig. S6†). The measured kobs rate constants (a.u. s−1) can be converted into TOF values (h−1) (Table S1, Fig. S6† bottom). At 20 °C, 11 bar CO2 and 0.4 mol% catalyst, an in situ determined TOF of 70 h−1 was observed, which is the highest reported TOF for the formation of PLimC in literature until now.18 The reaction order in CO2 was determined to be zero by varying the CO2 pressure from 5–20 bar with an otherwise constant composition of the reaction mixture (Fig. S7†). All observed rate constants kobs are in the same order of magnitude around 7.4 × 10−5 a.u. s−1 (Table S2†). In order to assign the reaction order in LO, the concentration of LO is normally varied utilising an additional solvent, such as toluene.29–31 An experiment of 2.5 mL toluene and 2.5 mL LO at a catalyst loading of 0.4 mol% at 10 bar CO2 pressure showed an induction period of nearly 1 h. By changing the volume of toluene to 3.0 mL, the induction period was increased to about 2 h. Therefore, rate calculation does not start before the carbonyl band of the polymer begins to rise (Fig. S8†). Experiments with limonene concentrations of 2.44–6.10 M were conducted, whereby plotting of lnkobs against lnc(LO) resulted in an order of two in LO. Increasing the amount of toluene, the rate constants kobs and activities are decreasing (Table S3†). As the induction process might still have some influence on the linear section of the reaction curve, an additional method for determination of the reaction order for the epoxide was established. Accordingly, an experiment with a catalyst loading of 0.4 mol%, 11 bar CO2 and 2.5 mL toluene (total volume 5.0 mL) was performed (Fig. S10c†). The concentration of the catalyst and the CO2 pressure during the entire copolymerisation reaction remained practically unchanged, whereas the concentrations of LO and PLimC were varied. The reaction rate kobs can now be calculated for different epoxide concentrations out of the derivation of the curve measured at different conversions (Fig. S10a, b and eqn (S2)†). The intensity of the carbonyl band of the polymer was correlated to the concentration of the polymer in the mixture by an independent multipoint calibration and, as a consequence, to the concentration of LO, as shown in Table S4.† Deriving the polynomial copolymerisation curve, the slope of the tangents, directly related to the reaction rate, at 20, 30, 40, 60 and 80% conversion can be calculated. After double logarithmic plotting of the LO concentration against the corresponding reaction rates, the order in epoxide was again determined to be two (Fig. S10d†). Hence, the second method to establish the order in monomer confirms the first result, wherefore the overall rate equation can be written as −d[LO]/dt = k·[CO2]0·[LO]2·[Zn]1. The group of Coates et al. investigated the reaction order for the copolymerisation of cyclohexene oxide and CO2 with the same (bdi)ZnOAc catalyst in 2003.29 They found an overall rate equation of −d[CHO]/dt = k·[CO2]0·[CHO]1·[Zn]1, whereby the reaction order of one in cyclohexene oxide deviates from the reaction order of two in LO. Comparing cyclohexene oxide and limonene oxide, the steric demand of the latter attracts attention. The following pathway is proposed bearing all the reaction orders measured in mind (Fig. 4).
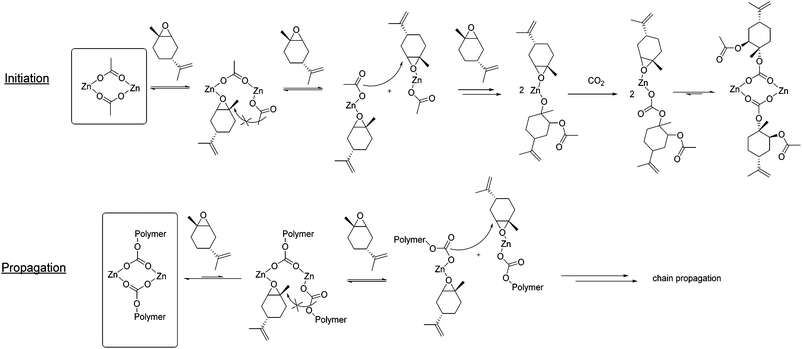 |
| | Fig. 4 Postulated mechanism for copolymerisation of CO2 and LO with catalyst (bdi)ZnOAc. | |
In a first step, the dimeric catalyst coordinates one LO. 1H NMR measurements showed that the catalyst exists preferentially in its dimeric state,27,29 even in strong coordinating and polar solvents such as tetrahydrofurane (Fig. S11 and S12†). Due to the steric demand of the monomer, the acetate group of the other zinc centre probably cannot attack the epoxide. After a second LO is coordinated to the second zinc centre the monomeric catalyst can rearrange to enable the nucleophilic attack of the initiating group. Carbon dioxide is inserted and a dimeric state is formed again, as the equilibrium lies on the dimeric side. In order to enable chain propagation, again two LO molecules have to be coordinated to the catalyst in order to separate the two zinc centres.
End-capping of PLimC with anhydrides and silicates
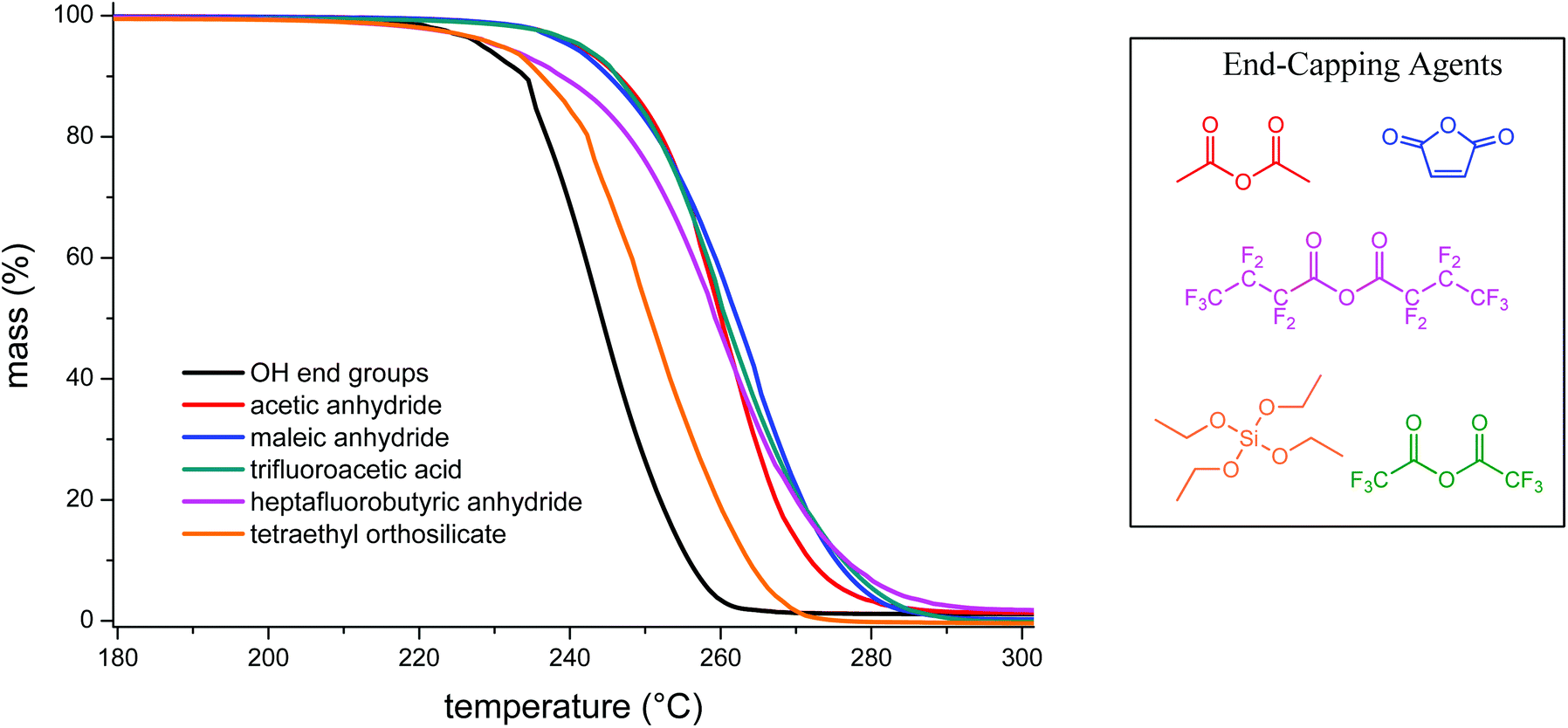
High molecular weight PLimC has a glass transition temperature of 130 °C (for DSC trace see Fig. S3†). This is, to the best of our knowledge, the highest Tg reported for an aliphatic PC and comes relatively close to commercial BPA-PC that has a Tg of 145 °C. On the other hand, the thermal stability of aliphatic polycarbonates is possibly the greatest obstacle when it comes to commercialisation of this polymer class for a broad range of applications.32,33 Poly(propylene carbonate) has a Tg at around 40 °C and a decomposition temperature of 200 °C seems acceptable.32 However, this leaves a very narrow processing window for PLimC with its glass transition at 130 °C. To overcome this limitation of a 5% decomposition temperature (T5%) for a catalyst-free polymer at 225 °C (Table 2, entry 1 and Fig. 5), an attempt was made to end-cap the polymer that is presumably terminated by OH groups when precipitated in methanol. Four different anhydrides: acetic anhydride, maleic anhydride, trifluoroacetic anhydride, heptafluorobutyric anhydride and tetraethyl orthosilicate were chosen as end capping agents. With acetic, maleic and trifluoroacetic anhydride, an increase of 15 °C in T5% and of 16–20 °C for the maximum decomposition temperature (highest decomposition rate, Tmax) could be observed (Table 2, entries 2–4). Capping the polymer with fluorinated butyric anhydride and silicate only slightly increased the onset of decomposition of PLimC (Table 2, entries 5 and 6), while the first at least increased Tmax significantly.
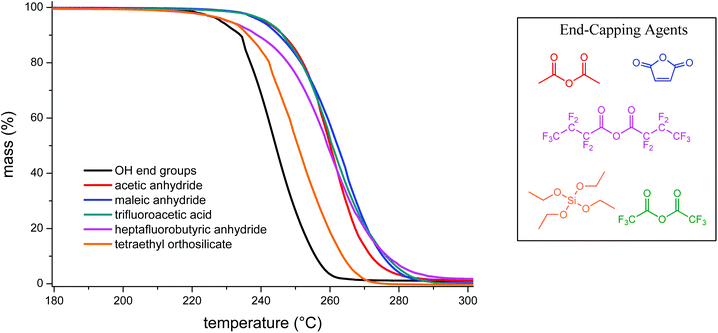 |
| | Fig. 5 TGA of PLimC treated with various end-capping agents compared to an untreated sample (black curve). | |
Table 2 Thermal degradation properties of pure and end-capped PLimC
| Entry |
End-capper |
T
5%a [°C] |
T
maxa [°C] |
|
Measured at 10 K min−1.
|
| 1 |
— |
225 |
244 |
| 2 |
Acetic anhydride |
241 |
260 |
| 3 |
Maleic anhydride |
240 |
265 |
| 4 |
Trifluoroacetic anhydride |
242 |
260 |
| 5 |
Heptafluorobutyric anhydride |
231 |
261 |
| 6 |
Tetraethyl orthosilicate |
231 |
252 |
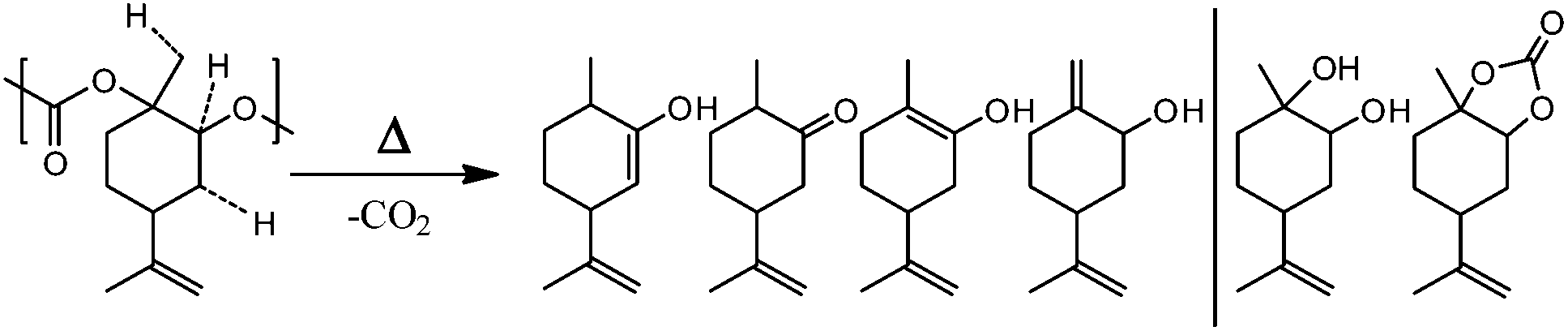
This is a hint of only partial end-capping that can result from bad solubility of the end-capping agent (fluorinated butyric anhydride) or low reactivity under the conditions chosen (orthosilicate). End-capping can only slightly improve the thermal stability of PLimC, therefore, the thermal degradation was studied in more detail to find a possible alternative technique of stabilising the polymer at elevated temperatures. To understand the mechanisms involved in the degradation of the PC, GC MS coupled with a thermal desorption unit was employed. Here, the sample is heated to the temperature desired, while its decomposition products are condensed in another chamber for a defined time, where they are eventually injected onto the column. The spectra and therewith potentially identified products from a decomposition of PLimC for 20 °C min at 230 °C are presented in Fig. 6. The spectra of PLimC with and without end-capping look very similar and suggest decomposition via abstraction of CO2, probably by nucleophilic attack at an acidic hydrogen and a subsequent chain reaction. Though interestingly, only the spectrum of maleic anhydride end-capped PLimC shows a signal at 13.2 min, which is tentatively assigned to limonene carbonate which seems to be produced in significant amounts when the polymer is capped with maleic acid. The great variety of decomposition products is also illustrated in Scheme 5, where the ‘most labile’, i.e. acidic protons, are indicated by dashed lines. Those protons might be prone to nucleophilic attack (any nucleophile that is basic enough) and subsequent unzipping reactions then produce the products identified via abstraction of CO2. The diol might evolve by a different mechanism where no CO2, but rather CO abstraction is involved. The limonene carbonate mentioned above was possibly the backbiting product of maleic anhydride that activates a new pathway of decomposition to form the cyclic carbonate. This seems unreasonable for an end-capped product that should increase the thermal stability of the polymer, but at least the labile proton of a hydroxyl end group is protected against abstraction and TGA has proven the concept to be successful.
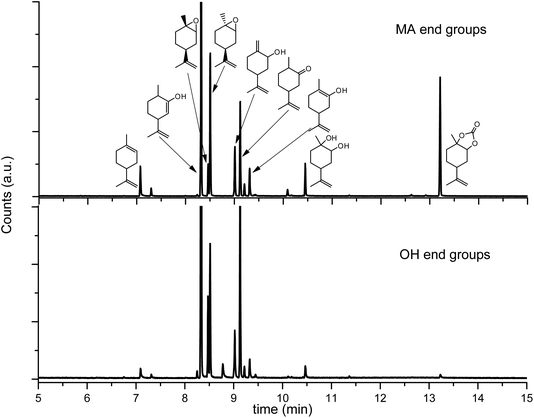 |
| | Fig. 6 GC spectra of the products evolved by heating samples of pure (bottom) and end-capped (top) PLimC for 20 min at 230 °C. The assignment of signals in the top spectrum results from mass spectroscopy analysis. | |
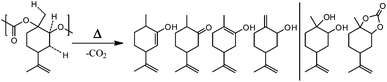 |
| | Scheme 5 Thermal decomposition products of PLimC proposed by GC MS studies. The dashed lines on PLimC indicate ‘labile’ protons and right-hand side on the product side are decomposition products where no simple CO2 abstraction mechanism has taken place. | |
Properties of PLimC
The material PLimC possesses a variety of very interesting properties that make it so worthwhile to work with. They are summarised in Table 3 with the method of determination in the right hand column. The carbonate content in the backbone is greater than 99% for a polymer with 52.4 kDa and a dispersity of 1.10. Such an alicyclic PC has a density as low as 1.08 g cm−3, which resembles that of PP or PE.
Table 3 Collection of general, mechanical, thermal and optical properties of PLimC
| |
Property |
Value |
Unit |
Method |
|
Measured at 10 K min−1.
Thickness of sample 0.24 mm.
Test conducted according to ISO 15184 with BYK Pencil Hardness Tester and Derwent Graphic pencils.
|
|
General
|
Density |
1.08 |
g cm−3 |
Balance (Archimedes) |
| Carbonate content |
99+ |
% |
1H-NMR |
| Contact angle to water |
93 |
° |
Drop shape analyser |
|
M
n
|
53.4 |
kDa |
GPC (CHCl3) |
|
Đ
|
1.10 |
— |
GPC (CHCl3) |
|
Mechanical
|
E-Modulus |
0.95 |
GPa |
Tensile tester |
|
σ
m
|
55 |
MPa |
Tensile tester |
| Elongation at break |
15 |
% |
Tensile tester |
| Pencil hardness |
B |
|
Pencil hardness testerc |
|
Thermal
|
T
g
|
130 |
°C |
DSC |
|
T
m
|
— |
°C |
DSC |
|
T
5%
|
240 |
°C |
TGA |
|
T
max
|
265 |
°C |
TGA |
|
Optical
|
Transmission |
94 |
% |
Hazemeter |
| Haze |
0.75 |
% |
Hazemeter |
| Clarity |
99.8 |
% |
Hazemeter |
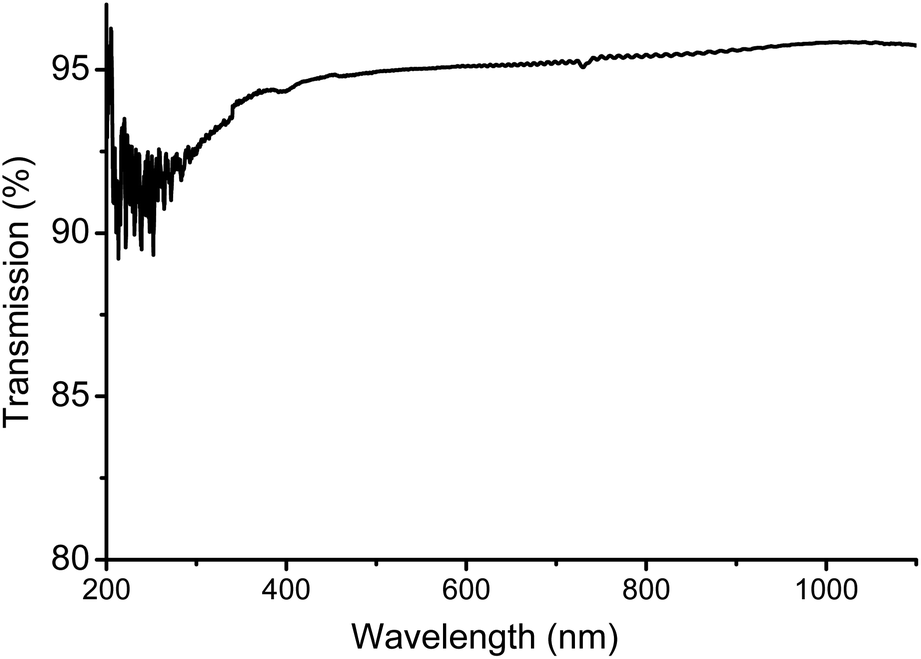
The tensile properties were tested on dog-bone shaped specimen produced by hot-pressing – which had no effect on the molecular weight of the polymer – and suggest a mechanical behaviour ranging between PS and BPA-PC. Though it should be noted that injection moulded samples might improve upon tensile testing. The hardness and scratch resistance was evaluated with respect to pencil hardness. The test method proved to be independent of film thickness (films of 50–400 μm were tested), as scratching is only superficial. A pencil of hardness B left no scratch behind on the surface of a PLimC film. This is already a significant improvement compared to BPA-PC (Makrolon 2800® Bayer; test with same set-up) that could only resist 8B or softer. Acrylics (ZK50 Plexiglas® Evonik), on the other hand, are resistant to pencils as hard as 5H, but suffer from a very high brittleness. Thermally, this polymer is also surprising with (to the best of our knowledge) the highest reported Tg for an aliphatic PC of 130 °C. This thermal resistance makes it an interesting choice wherever contact with boiling water is likely. The amorphous polymer with its alicyclic structure suggests very attractive optical properties and, indeed, films, produced by solvent casting, tested on a hazemeter gave values for transmission, haze and clarity of 94%, 0.75% and 99.8%, respectively. These results were independent of film thickness (tested films of 50–400 μm thickness) and we would ascribe the missing 6% of transmission to 3% reflection on each of the interfaces (air/polymer–polymer/air). Upon reversion, this observation highlights the very low small and wide angle scattering, i.e. the high optical purity of PLimC. An ultraviolet visible (UV/Vis) spectrum of such a film (with a thickness of 240 μm) is also shown in Fig. 7. There is almost no absorbance of light in the visible region, thus, making it a perfect choice for applications where high transparency is necessary, e.g. packaging material or optical lenses.
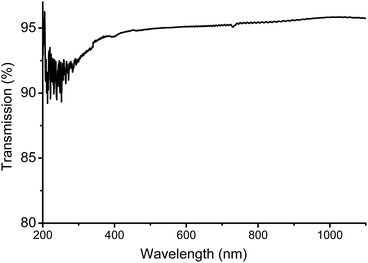 |
| | Fig. 7 UV/Vis spectrum of a 240 μm thick film of PLimC produced by solvent casting. | |
Conclusions
In conclusion, we would like to present a 100% bio-based high molecular weight PLimC (>100 kDa) where we can tune the molecular weight over a wide range. Very crucial were the successful conversion of LO to PLimC by regio- and stereoselective high-yield epoxidation of limonene and the quantitative masking of hydroxyl impurities in the LO mixture. Future efforts are directed towards the search of a less harmful masking agent than iodomethane, though. The production of LO and PLimC was scaled up to kilograms per batch, while reducing the amount of organic solvents employed by a factor of 10 compared to standard precipitation methods. Kinetic studies suggested a second order dependence on LO concentration and indicate an alternating insertion of CO2 and LO. PLimC has a range of very attractive properties, e.g. a Tg as high as 130 °C, which is close to technical BPA-based PC. The excellent transparency of PLimC in combination with good hardness, both better than BPA-PC, makes it a highly promising green coating material, which will be the topic of upcoming work. Furthermore the biodegradability of PLimC in comparison with other polycarbonates and polyesters will be investigated in the future.
Experimental
Materials
R-Limonene (97%), acetyl acetone (99%), 2,4-diethylaniline (99%), N-bromosuccinimide (97%), sodium hydride (60% dispersion in mineral oil), iodomethane (99%, stabilised with silver), ethylenediaminetetraacetic acid (EDTA, 98.5%), acetic acid (99%) and acetic anhydride (99%) were used as received. R-LO (97%) was dried over NaH and distilled. Carbon dioxide (5.0, Linde Gase) was dried by passing through a column packed with a molecular sieve of 3 Å. The catalyst [(bdi)Zn(μ-OAc)]34 and EDTA-silica28 were synthesised according to literature procedures.
Methods
NMR spectra were recorded on a Bruker AMX-300 operating at 300 MHz. Chemical shifts δ are indicated in parts per million (ppm) with respect to residual solvent signals. Thermogravimetric analysis (TGA) was performed on a Netzsch TG 209 F1 Libra and differential scanning calorimetry (DSC) on a Mettler Toledo DSC 821c, both at a heating rate of 10 K min−1 under N2 atmosphere. In situ IR measurements were carried out under an argon atmosphere using an ATR IR Mettler Toledo system. IR spectra of solids were obtained with a Digilab Excalibur FTS-3000 equipped with an ATR unit. Gas chromatography spectra were recorded on a Shimadzu QP-5050 with N2 as the carrier gas. GC-MS spectra were recorded on an Agilent 5977A MSD with He as the carrier gas. Relative molecular weights and dispersities were determined by GPC on an Agilent 1200 system with chloroform as the eluent and polystyrene as the calibration standard. A Hazemeter BykGardner Haze-Gard Plus and a UV/Vis spectrometer V-670 (JASCO) were employed for the testing of optical properties of solvent cast PLimC films having a thickness between 100 and 400 μm. A Zwick/Roell Z0.5 with testXpert II software and 5 mm min−1 test speed were used for tensile testing of heat pressed PLimC specimen having a thickness of 300 μm. A BYK Pencil Hardness Tester and Derwent Graphic pencils were used to determine pencil hardness.
Synthetic procedures
Synthesis of trans-limonene oxide.
A modified procedure from Gurudutt et al.26 was used. A volume of 1.2 L (8.8 mol) limonene, 1 L water and 4.5 L acetone were filled into a 10 L temperature-controlled reactor. The mixture was cooled down to 0 °C and 9.2 mol N-bromosuccinimide was added within 30 min. The solvent was evaporated at 50 °C and the resulting organic phase was diluted with 3 L diethyl ether. After removing the aqueous phase, the organic phase was washed with water once before the ether was removed at 50 °C. The crude bromohydrin was converted directly to the epoxide with 2 L of a 6 M NaOH solution at 60 °C for 1.5 h. The alkaline solution was removed and the crude product diluted with 3 L diethyl ether before it was washed with 1.5 L of a saturated sodium bicarbonate solution and, subsequently, 1.5 L of water. After evaporating the solvent, 1.4 L of the crude product was obtained as a yellow viscous liquid. Gas chromatography analysis revealed a trans-LO content of 60%, which was isolated by fractional distillation at 80 °C and dynamic vacuum. The final product consisted of 5% limonene, 9% cis-LO and 85% trans-LO and 1% by-products (determined by GC analysis).
Masking of hydroxyl impurities in limonene oxide.
All manipulations were carried out under inert atmosphere. The crude oxirane was treated with 0.16 eq. of NaH (60% dispersion in mineral oil) at 0 °C. After stirring for 60 min, 0.08 eq. of iodomethane was added and the mixture was kept at 0 °C for another hour. The process of the reaction was monitored via GC and after 24 h, no more hydroxyl impurities could be detected and the mixture was fractionally distilled at 80 °C in a dynamic vacuum. The main fraction was collected at 5 mbar (∼66%, containing 85% trans-limonene oxide).
Synthesis of poly(limonene carbonate).
An in vacuo pre-dried 120 mL autoclave at 90 °C was charged in a glove box with 30 mmol of epoxide, 3 mL toluene and 0.4 mmol catalyst. The closed reactor was stirred for 15 min to dissolve the catalyst before it was evacuated and, subsequently, pressurised with 10 bar CO2. The reactor was opened after an adequate time and the viscous mixture diluted with toluene before 20 wt% (relative to LO) of acetic anhydride was added. After 1 h, the solution was precipitated in methanol and the product dried in vacuo. The product was characterised by 1H and 13C NMR spectroscopy (ESI†).
Purification of PLimC with immobilised EDTA.
An amount of 20 wt% acetic anhydride and 10 wt% EDTA silica were added to a 0.2 g mL−1 solution of crude PLimC in toluene and stirred for 3 h at room temperature. The suspension was filtered through a G3 glass frit to remove silica particles. The viscous polymer solution was cast on a glass plate after concentration and dried in vacuo at 50–120 °C for 12 h.
Acknowledgements
We would like to thank DFG for funding, M. Langner for the thermal desorption unit GC-MS measurements and R. Agarwal for initial discussions.
Notes and references
-
D. G. LeGrand and J. T. Bendler, Handbook of Polycarbonate Science and Technology, Marcel Dekker, Inc., 2000 Search PubMed
 .
.
-
J. Aungst, Bisphenol A (BPA): Use in Food Contact Application, U.S. Food and Drug Administration, 2014 Search PubMed .
- M. Yokoe, K. Aoi and M. Okada, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 3909–3919 CrossRef CAS .
- S. Chatti, H. R. Kricheldorf and G. Schwarz, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 3616–3628 CrossRef CAS .
- H. Xia, Z. Y. Suo, G. J. Qiang and C. J. Chang, J. Appl. Polym. Sci., 2008, 110, 2168–2178 CrossRef CAS .
- Y. Xin and H. Uyama, J. Polym. Res., 2012, 19, 15 CrossRef .
- S. Klaus, M. W. Lehenmeier, C. E. Anderson and B. Rieger, Coord. Chem. Rev., 2011, 255, 1460–1479 CrossRef CAS .
-
B. M. Bhanage and M. Arai, Transformation and Utilization of Carbon Dioxide, Springer, Berlin, Heidelberg, 2014 Search PubMed .
- S. Inoue, H. Koinuma and T. Tsuruta, Die Makromol. Chemie, 1969, 130, 210–220 CrossRef CAS .
- S. Inoue, H. Koinuma and T. Tsuruta, Polym. Lett., 1969, 7, 287–292 CrossRef CAS .
- N. Takeda and S. Inoue, Die Makromol. Chemie, 1978, 179, 1377–1381 CrossRef CAS .
- S. J. Wang, L. C. Du, X. S. Zhao, Y. Z. Meng and S. C. Tjong, J. Appl. Polym. Sci., 2002, 85, 2327–2334 CrossRef CAS .
-
G. A. Luinstra and E. Borchardt, in Synthetic Biodegradable Polymers, Springer, Berlin/Heidelberg, 2012, pp. 29–48 Search PubMed .
- C. Koning, J. Wildeson, R. Parton, B. Plum, P. Steeman and D. Darensbourg, Polymer, 2001, 42, 3995–4004 CrossRef CAS .
- Y. Zhang, X. Zhang, R. Wei, B. Du, Z. Fan and G. Qi, RSC Adv., 2014, 4, 36183–36188 RSC .
- D. J. Darensbourg, W. Chung, C. J. Arp, F. Tsai and S. J. Kyran, Macromolecules, 2014, 47, 7347–7353 CrossRef CAS .
- M. Winkler, C. Romain, M. A. R. Meier and C. K. Williams, Green Chem., 2015, 17, 300–306 RSC .
- C. M. Byrne, S. D. Allen, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2004, 126, 11404–11405 CrossRef CAS PubMed .
-
P. Liu, I. Boto, R. Kortbech-Olesen, B. Vrolijk and P. Pilkauskas, Citrus Fruit Fresh and Processed, Food and Agriculture Organization of the United Nations, 2012 Search PubMed .
- R. Ciriminna, M. Lomeli-Rodriguez, P. Demma Carà, J. A. Lopez-Sanchez and M. Pagliaro, Chem. Commun., 2014, 50, 15288–15296 RSC .
- H. J. Park, C. Y. Ryu and J. V. Crivello, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 109–117 CrossRef CAS .
- M. Bähr, A. Bitto and R. Mülhaupt, Green Chem., 2012, 14, 1447 RSC .
- F. Auriemma, C. De Rosa, M. R. Di Caprio, R. Di Girolamo, C. W. Ellis and G. W. Coates, Angew. Chem., Int. Ed., 2015, 54, 1215–1218 CrossRef CAS PubMed .
- L. Peña Carrodeguas, J. González-Fabra, F. Castro-Gómez, C. Bo and A. W. Kleij, Chem. – Eur. J., 2015, 21, 6115–6122 CrossRef PubMed .
- M. Helou, O. Miserque, J.-M. Brusson, J.-F. Carpentier and S. M. Guillaume, Chemistry, 2008, 14, 8772–8775 CrossRef CAS PubMed .
- K. N. Gurudutt, S. Rao and P. Srinivas, Flavour Fragrance J., 1992, 7, 343–345 CrossRef CAS .
- M. Cheng, D. R. Moore, J. J. Reczek, B. M. Chamberlain, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2001, 123, 8738–8749 CrossRef CAS PubMed .
- R. Kumar, M. A. Barakat, Y. A. Daza, H. L. Woodcock and J. N. Kuhn, J. Colloid Interface Sci., 2013, 408, 200–205 CrossRef CAS PubMed .
- D. R. Moore, M. Cheng, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2003, 125, 11911–11924 CrossRef CAS PubMed .
- F. Jutz, A. Buchard, M. R. Kember, S. B. Fredriksen and C. K. Williams, J. Am. Chem. Soc., 2011, 133, 17395–17405 Search PubMed .
- M. W. Lehenmeier, S. Kissling, P. T. Altenbuchner, C. Bruckmeier, P. Deglmann, A. K. Brym and B. Rieger, Angew. Chem., Int. Ed., 2013, 52, 9821–9826 CrossRef CAS PubMed .
- G. A. Luinstra, Polym. Rev., 2008, 48, 192–219 CrossRef CAS .
- S. D. Thorat, P. J. Phillips, V. Semenov and A. Gakh, J. Appl. Polym. Sci., 2003, 89, 1163–1176 CrossRef CAS .
- S. D. Allen, D. R. Moore, E. B. Lobkovsky and G. W. Coates, J. Organomet. Chem., 2003, 683, 137–148 CrossRef CAS .
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5gc01694k |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence