
Highly selective hydrogenation and hydrogenolysis using a copper-doped porous metal oxide catalyst†
Laurene
Petitjean
a,
Raphael
Gagne
b,
Evan S.
Beach
 a,
Dequan
Xiao
*b and
Paul T.
Anastas
*a
a,
Dequan
Xiao
*b and
Paul T.
Anastas
*a
aYale University, Forestry & Environmental Studies, New Haven, Connecticut, USA. E-mail: paul.anastas@yale.edu
bUniversity of New Haven, New Haven, Connecticut, USA
First published on 6th October 2015
Abstract
A copper-doped porous metal oxide catalyst in combination with hydrogen shows selective and quantitative hydrogenolysis of benzyl ketones and aldehydes, and hydrogenation of alkenes. The approach provides an alternative to noble-metal catalysed reductions and stoichiometric Wolff-Kishner and Clemmensen methods.
Sustainability has emerged as a global concern and has prompted chemists to develop procedures that minimize impact on the environment. Catalysis is the basis for many improvements in sustainable chemical transformations, facilitating the use of reduced energy and material inputs for processes that society requires.1 Heterogeneous catalysis, in particular, provides many advantages such as increased catalyst stability and lifetime, as well as ease of catalyst separation from the product mixture.2 Additionally, methods based on supported heterogeneous catalysts show superior applicability for industrial scale-up.3 Supply vulnerabilities and depletion of natural resources have increased the attractiveness of catalysts based on earth-abundant metals.1
The hydrogenation of unsaturated functional groups such as carbon–carbon double bonds and the hydrogenolysis of carbon–oxygen bonds are both important reactions in synthetic chemistry4 particularly in the liquid fuels sector5 where catalytic methods are used to reduce oxygen content and improve hydrogen/carbon ratios.6 Reductions of alkenes are typically conducted with high selectivity using noble metal catalysts that are active under mild conditions.3 For example, hydrogenation of the propene moiety in eugenol can be performed using Pd/C in combination with stoichiometric triethylsilane and an acid quench.7 One of the earliest reports of eugenol reduction used an insoluble rhodium catalyst in water.8 Even with noble metal catalysts, selectivity can frequently be difficult to achieve. The reduction of eugenol with a heterogeneous Pt/γ-Al2O3 catalyst in combination with 0.14 MPa of N2/H2 stream (90/10) at 300 °C yielded dozens of products in addition to guaiacol and n-propylguaiacol.9 Hydrogenolysis of ketones or aldehydes from aryl-substituted compounds is even more difficult to effect selectively. Catalytic methods commonly use noble metals and typically require forcing conditions.3 A recent report of selective catalytic vanillin hydrogenolysis utilizes Au on carbon nanotubes,10 while another employs Pd nanoparticles supported on mesoporous N-doped carbon to provide creosol.11 Various other supported Pd catalysts have been used but show lower selectivity in the reduction of vanillin to creosol,12–14 yielding mixtures of creosol and vanillyl alcohol.
Alternatively, selective but stoichiometric methods such as Wolff-Kishner15 or Clemmensen16 conditions are extensively utilized for carbonyl removal. They historically employ toxic reagents such as hydrazine and mercury and generate hazardous waste. Recently, methods have been developed to avoid use of noble metals or the above stoichiometric reactions for hydrogenation and hydrogenolysis. Both Ni nanoparticles17 and a Ni/Al alloy catalyst18 are capable of hydrogenating eugenol to propyl-guaiacol. Unfortunately, the catalysts’ synthesis either employs several equivalents of toxic reagent or is energy intensive. Thus, easily synthesized green catalysts based on earth abundant elements could provide promising solutions for the large-scale applications of catalytic hydrogenation and hydrogenolysis.
A general challenge for all catalytic methods of hydrogenation or hydrogenolysis, particularly by those based on earth abundant elements, is improving selectivity. For example, using a CoMo/Al2O3 catalyst to reduce vanillin at 300 °C and 5 MPa H2 showed poor conversion and provided mixtures of over four compounds including creosol.19 Copper has the advantages of being an earth-abundant metal and having low tendency to catalyze arene hydrogenation, preventing over-reduction and thus improving selectivity.3 Recently, Kong et al. reported the use of copper-doped HZSM-5 zeolite for hydrogenolysis of aryl aldehydes and ketones.20 Porous metal oxides (PMOs), derived from hydrotalcite-like precursors of general formula Mg6Al2CO3(OH)16·4H2O, are promising catalysts for a wide range of applications. This is due to their high potential for tunability through altering the M2+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :M3+ ratio and metal dopants. Other advantages include high surface area, stability against sintering, simplicity of preparation, and ease of handling.21,22 Thus, doping copper into hydrotalcite-derived compounds can be a promising strategy for a wide range of reduction methods. For example, Kaneda et al. successfully utilised a copper-nanoparticle catalyst synthesized from Cu–Al hydrotalcite to effect the quantitative hydrogenolysis of glycerol to 1,2-propanediol.23
:M3+ ratio and metal dopants. Other advantages include high surface area, stability against sintering, simplicity of preparation, and ease of handling.21,22 Thus, doping copper into hydrotalcite-derived compounds can be a promising strategy for a wide range of reduction methods. For example, Kaneda et al. successfully utilised a copper-nanoparticle catalyst synthesized from Cu–Al hydrotalcite to effect the quantitative hydrogenolysis of glycerol to 1,2-propanediol.23
In this communication, the reactivity and selectivity of copper-doped PMO (Cu-PMO) is evaluated. Our previous work with the catalyst suggested it was capable of very selective transformations.24 This work clarifies the scope of the reactivity towards various C–C and C–O bond configurations. Density functional theory (DFT) calculations were performed to evaluate the thermodynamic bias of each reaction at relevant pressures. The computational results are integrated with experimental data from Cu-PMO catalysed reductions to show improvements in efficiency and selectivity provided by the catalyst.
Cu-PMO is synthesized by co-precipitation of Cu, Mg and Al nitrate salts in aqueous media. Copper constitutes 20 mol% of M2+, with M2+:M3+ kept at 3:1. Elemental analyses proved that the metals are incorporated in the anticipated amounts, furnishing a catalyst with metal ratios of Cu0.57Mg2.25Al1.00 (see ESI†). XRPD measurements indicate that Cu-PMO changes from a hydrotalcite-like structure to become an amorphous material after calcination in air for 24 hours at 460 °C. Cu-PMO was previously reported to have a surface area of ∼137 m2 g−1.25
The Gibbs free energy of different reaction pathways was determined using the high-performance computational chemistry software NWChem.26 The structures were built in .xyz format using the model-building program Avogadro.27 Initial molecular geometries were then optimized using density functional theory at the B3LYP/6-31g* level. The optimized structures were subjected to thermochemistry analysis based on vibrational frequency calculations and solvation energy calculations using the COSMO solvation model.28 The output of the vibrational frequency calculations provided the zero-point correction to energy (E1), thermal correction to enthalpy (H) and total entropy (S). The solvation calculation provided the total density function theory (DFT) energy (E0) and the electrostatic solvation energy (Es). Eqn (1) was used to determine the change in Gibbs free energy (dG).
| dG = H − TS | (1) |
This information was then used to compute Gibbs free energy (G) of each structure in gas phase by eqn (2).
| G = dG + E0 + E1 | (2) |
For specific hydrogenation or hydrogenolysis reactions, we followed the Born–Haber cycle to compute the reaction Gibbs energy in solutions (see ESI†). R is the organic molecule prior to hydrogenation, H2 is molecular hydrogen, and RH2 is the organic molecule after hydrogenation. In the notations of ΔG terms, ‘g’ denotes gas phase, ‘solu’ denotes solution, and ‘s’ denotes solvation.
The reaction Gibbs free energy in gas phase was first computed using eqn (3):
| ΔGg = G(RH2,g) − G(R,g) − G(H2,g) | (3) |
Then, the reaction Gibbs free energy in solution was computed by eqn (4):
| ΔGsolu = ΔGg + ΔGs,RH2− ΔGs,R − ΔGs,H2 | (4) |
To examine the chemoselectivity of eugenol reduction, DFT calculations were performed to evaluate the thermodynamic feasiblity of potential products at varying pressures of hydrogen (Fig. 1). At reaction conditions over 1 MPa of hydrogen pressure at 180 °C, several pathways are calculated to be thermodynamically favourable: hydrogenation of the alkene, as well as hydrogenolysis of the methoxy–aryl bonds. Interestingly, the most thermodynamically favorable product at H2 pressures of 0.1–6 MPa is predicted to be the catechol resulting from aryl–ether bond cleavage (path 2, Fig. 1). This is potentially due to the added entropy gain from methane release after bond cleavage. However, experiments with Cu-PMO did not yield catechol product, suggesting that the production of catechol is subjected to the kinetic control of the catalysis.
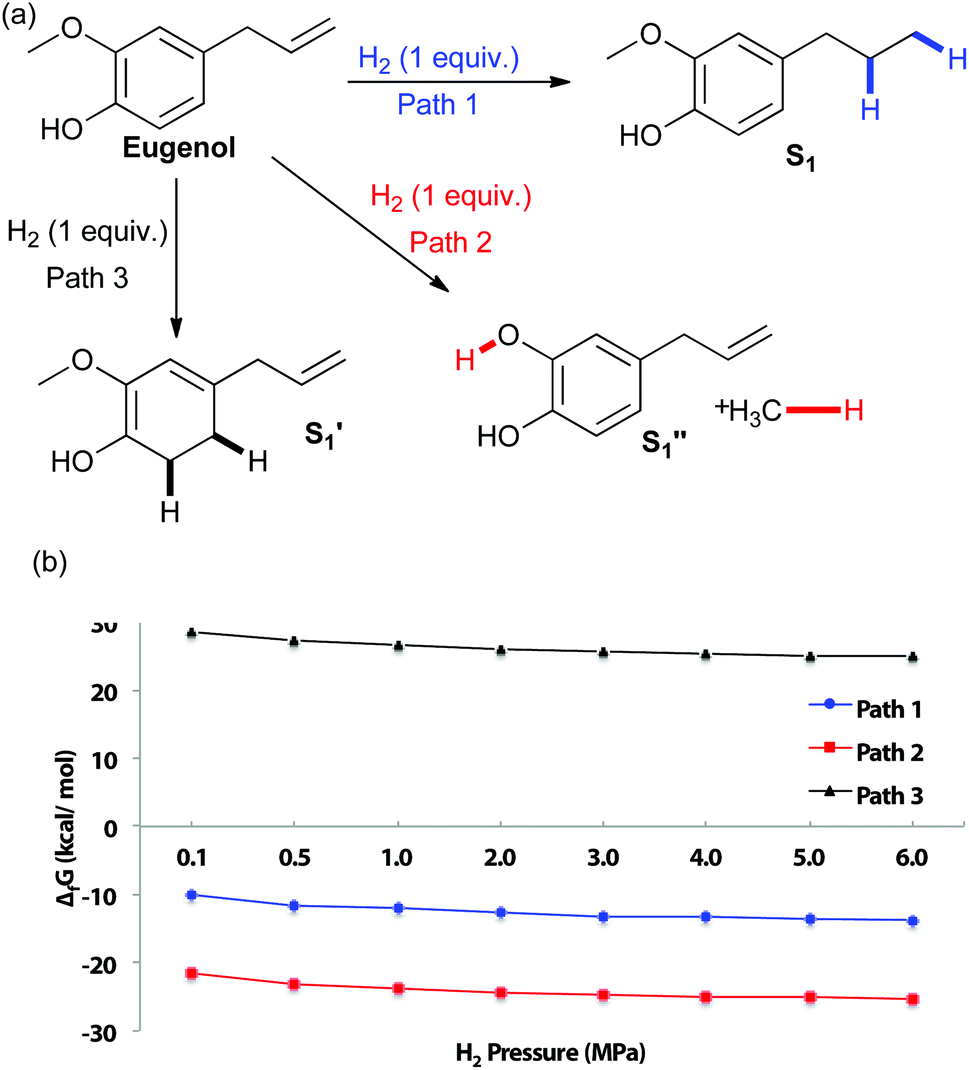 | ||
| Fig. 1 (a) Potential pathways of eugenol reduction (b) changes in Gibbs free energy with varying H2 pressure at 180 °C. | ||
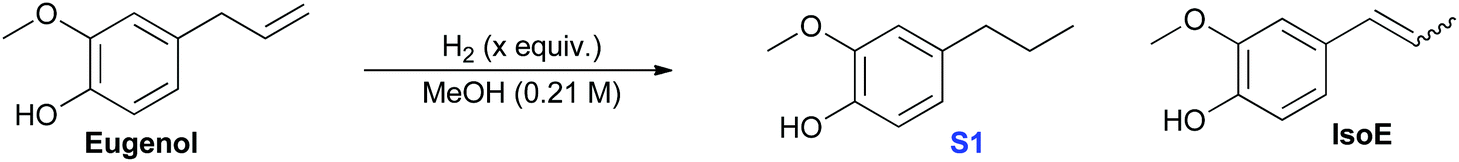
The hydrogenation of the propene group occurs under our reaction conditions, as it is thermodynamically allowed at various H2 pressure (see Fig. 1b, path 1). Experiments also found that the hydrogenation of propene is under the control of reaction kinetics (Table 1). Product S1 is obtained quantitatively after stirring for 18 h in a sealed Parr Reactor at 180 °C with an initial pressure of 4 MPa of hydrogen (Table 1, entry 1). The efficiency was excellent at temperatures as low as 100 °C, but dropped with further decrease in temperature (Table 1, entries 2–4). Optimal conditions appear to be around 3 h reaction at 100 °C (Table 1, entry 5). Lowering hydrogen pressure slows the reaction, yet quantitative yields of S1 can be obtained in only 4 h at 100 °C and 1 MPa of hydrogen (Table 1, comparing entries 5, 7 and 8). The control experiments suggest that hydrogenation of eugenol is kinetically controlled. Only trace reactivity was observed even after 18 h at 180 °C with 4 MPa of hydrogen (Table 1, entry 10).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Catalyst (mol%) | Temp (°C) | Time (h) | H2c (MPa) | Conversionb (%) | YieldbS1 (%) | Yieldb IsoE (%) |
| a All reactions were carried out in a high pressure 100 mL Parr Reactor using Eugenol (6.456 mmol) in MeOH (30 mL). b Conversion and Yield determined by 1H NMR Spectroscopy using DMF or Dodecane as internal standards. c Pressure as added at room temperature; tr = trace. | |||||||
| (1) | Cu-PMO (11) | 180 | 18 | 4 | 100 | >95 | 0 |
| (2) | Cu-PMO (11) | 100 | 18 | 4 | 100 | >95 | 0 |
| (3) | Cu-PMO (11) | 60 | 18 | 4 | 53 | 39 | 0 |
| (4) | Cu-PMO (11) | 22 | 18 | 4 | 0 | 0 | 0 |
| (5) | Cu-PMO (11) | 100 | 3 | 4 | 100 | >95 | 0 |
| (6) | Cu-PMO (11) | 70 | 3 | 4 | <5 | tr | 0 |
| (7) | Cu-PMO (11) | 100 | 3 | 1 | 40 | 40 | 0 |
| (8) | Cu-PMO (11) | 100 | 4 | 1 | 100 | >95 | 0 |
| (9) | PMO (250 mg) | 180 | 18 | 4 | 57.5 | 35 | 18 |
| (10) | — | 180 | 18 | 4 | 26 | 15 | 0 |
| (11) | Cu(OAc)2·H2O (0.3) | 180 | 18 | 4 | 90 | 60 | 0 |
| (12) | PMO (250 mg) | 100 | 4 | 1 | 0 | 0 | 0 |
| (13) | — | 100 | 4 | 1 | <5 | tr | 0 |
| (14) | Cu(OAc)2·H2O (0.3) | 100 | 4 | 1 | 15 | tr | 0 |
The phase of the reaction mixture could play an important role in the efficiency and selectivity of reduction, by altering the mechanism of catalysis.29,30 In the present system, methanol remains in the liquid phase throughout the reaction under all conditions reported.31 The system pressure increased as the temperature approached the set point, typically reaching 1.4 MPa at 100 °C and 5.9 MPa at 180 °C. Accordingly the density varies in the early stages of the reaction. For a transformation performed at 180 °C and 4 MPa, the hydrogen pressure is introduced at room temperature and a density of 790.5 g mL−1 is expected for methanol.31 Once the set temperature is reached, the pressure has increased and the density of methanol is calculated to be 608.6 g mL−1.31 For the milder reaction conditions, the effect is lower; at the start of the reaction, a density of 787.6 g mL−1 is expected at room temperature and 1 MPa H2. A lower experimental density of methanol at 712.6 g mL−1 can be reached with 100 °C and 1.4 MPa H2. A lower solvent density may facilitate hydrogen solvation and increase the reaction rate. Changes in solvent density could also alter solvent polarity, in turn affecting reduction efficiency and selectivity.32
Cu-PMO is able to overcome the transition state barrier associated with hydrogenation of eugenol. Interestingly, the Cu-free porous-metal oxide (PMO) material derived from Mg/Al hydrotalcite is also active for eugenol hydrogenation (Table 1, entry 9). This control indicates that Cu is essential for reaction efficiency as well as selectivity, since the PMO-promoted hydrogenation of eugenol yields isoeugenol in 15% yield (2:1 ratio trans![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) :cis). Eugenol isomerization is known to be catalyzed by hydrotalcite-like compounds due to their solid base character.33 With hydrotalcite-like compounds, reduced reactivity for isomerization is observed if the catalyst is calcined to a PMO, or when polar solvents are utilized.34 In the present case, a calcined catalyst is utilized in polar methanol, yet isomerization is still observed. This suggests that the rate of PMO-catalysed hydrogenation of both eugenol and isoeugenol are low enough to allow isoeugenol to be observed as a co-product.
:cis). Eugenol isomerization is known to be catalyzed by hydrotalcite-like compounds due to their solid base character.33 With hydrotalcite-like compounds, reduced reactivity for isomerization is observed if the catalyst is calcined to a PMO, or when polar solvents are utilized.34 In the present case, a calcined catalyst is utilized in polar methanol, yet isomerization is still observed. This suggests that the rate of PMO-catalysed hydrogenation of both eugenol and isoeugenol are low enough to allow isoeugenol to be observed as a co-product.
The use of a homogeneous copper catalyst for eugenol hydrogenation is not as effective as Cu-PMO (Table 1, entry 11). The Cu-PMO loading (11 mol%) furnishes 0.3 mol% of Cu which is identical to the absolute amount of Cu in the Cu(OAc)2 experiment, yet Cu-PMO performs significantly better. Control experiments with milder conditions were performed (Table 1, entries 12–14) and it is evident that the Cu-PMO structure and composition are essential for overcoming the transition state energy barrier leading to the reduction product S1.
In an effort to explore the applicability of our method towards C–O bonds, DFT calculations of vanillin reduction products thermodynamic stability, at varying hydrogen pressure, were first performed (Fig. 2). As for eugenol, the most thermodynamically favored product is predicted to be that from cleavage of the aryl–methoxy moiety, due to entropy gain. For analogous reasons, formation of creosol also displays negative Gibbs free energy at all studied hydrogen pressures. Hydrogenation of the aromatic unit is calculated to be particularly disfavored. It is more difficult to obtain the product of aromatic hydrogenation for vanillin than eugenol, correlating with the increased electron-donating ability of the propene unit versus the aldehyde (as also indicated by a pKa of 10.19 for eugenol and 7.38 for vanillin).35 Hydrogenation of the aldehyde to the corresponding benzylic alcohol also displays a positive change in Gibbs free energy, most likely because the conjugation of the aldehyde to the aromatic unit makes it more difficult to reduce.
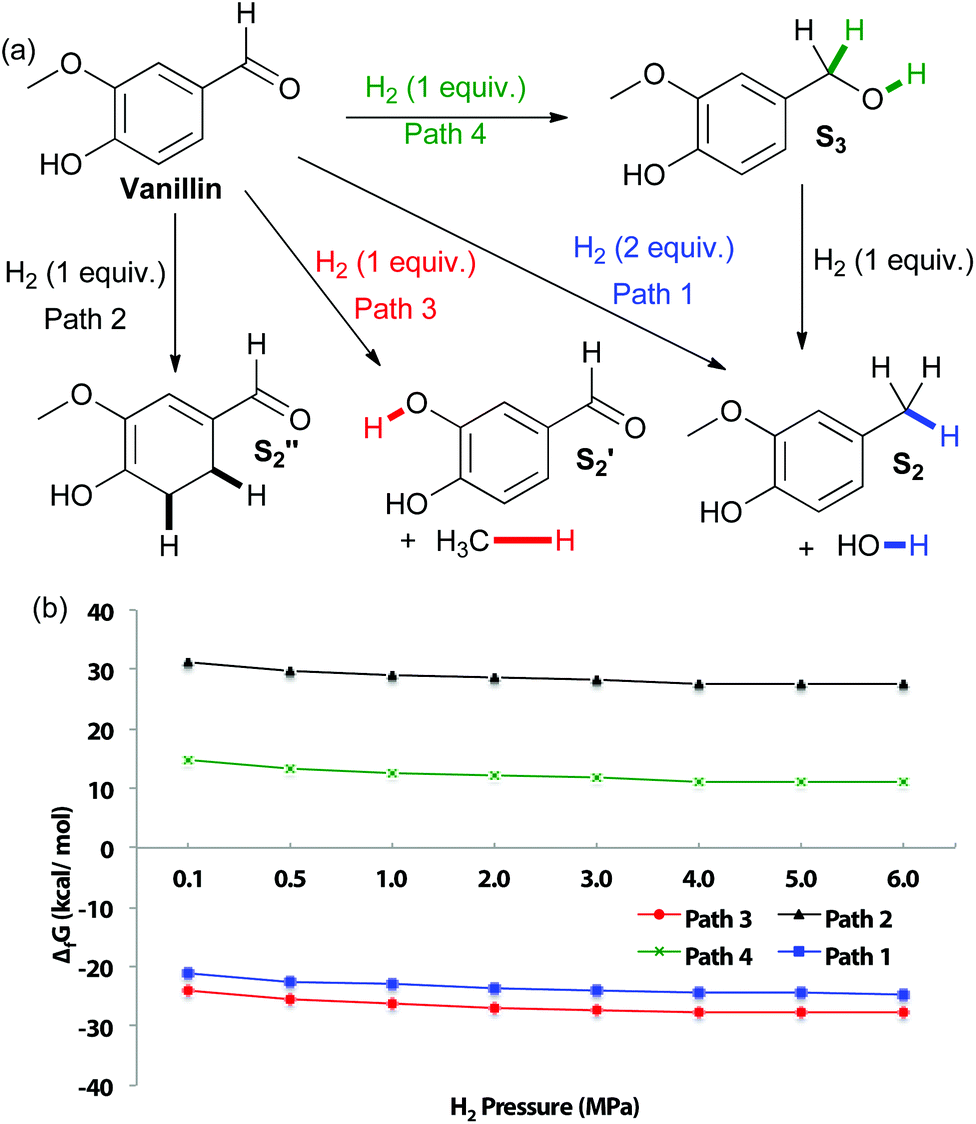 | ||
| Fig. 2 (a) Potential pathways of vanillin reduction (b) changes in Gibbs free energy with varying H2 pressure at 180 °C. | ||
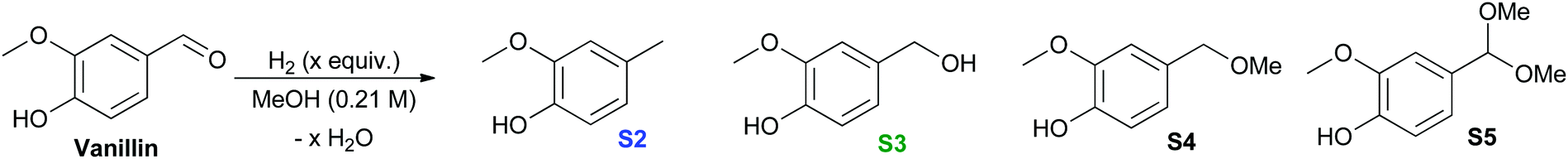
It is interesting that experimentally, Cu-PMO does not favor cleavage of the methoxy bond that is predicted to be the most thermodynamically favored pathway (Table 2), implicating highly selective kinetic control by the Cu-PMO catalyst. Many other systems have shown similar, although less pronounced, selectivity.10–14,36–38 At 4 MPa of hydrogen and 180 °C for 18 h, full hydrogenolysis of vanillin to creosol (S2) was observed (Table 2, entry 1). Interestingly, the catalyst seems to be required for hydrogenolysis, as a different product distribution is seen in its absence (Table 2, entries 2 and 3). S4 is obtained in 15–19% yield with poor mass balance using Cu-free PMO or no catalyst. Using homogeneous copper acetate, conversion of vanillin and formation of S4 is suppressed compared to the same reaction with no catalyst (Table 2, entries 3 & 4). Importantly, no creosol was observed with copper acetate, suggesting that both the Cu loading (overall composition) and structure of Cu-PMO are necessary for selective conversion to S2. At lower temperature, lower hydrogen pressure and shorter time, Cu-PMO yields a different product distribution, mainly S3 (Table 2, entry 5). This result suggests that S3 may be an intermediate in the formation of S2, as expected. This was confirmed by the direct quantitative reduction of S3 to S2 using Cu-PMO at 180 °C and 4 MPa H2 (see ESI†). If Cu is excluded from the reaction at lower temperature and pressure, no reduction of vanillin is observed (Table 2, entries 6 and 7). Instead, acetal S5 is obtained which probably results from addition of methanol to the aldehyde, followed by elimination of water and addition of a second equivalent of methanol. The observation of product S5 is significant since acetal formation is typically effected by acid catalysis, yet there is no explicit source of acid in the present conditions.39 The catalysis provided by PMO or Cu(OAc)2 is not sufficient to overcome transition state barriers for hydrogenolysis of vanillin. Overall, the vanillin studies again lead to the conclusion that the Cu-PMO structure and copper loading are essential for efficiency and selectivity.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst (mol%) | Temp (°C) | Time (h) | H2c (MPa) |
Conversionb (%) | YieldbS2:S3:S4:S5 (%) |
| a All reactions were carried out in a high pressure 100 mL Parr Reactor using vanillin (6.572 mmol) in MeOH (31.3 mL). b Conversion and Yield determined by 1H NMR Spectroscopy using Dodecane or DMF as internal standard isolated yields in parentheses. c Pressure as measured at room temperature; tr = trace. | ||||||
| (1) | Cu-PMO (11) | 180 | 18 | 4 | 100 | 90:0:0:0 |
| (2) | PMO (250 mg) | 180 | 18 | 4 | 75 | tr:tr:15:0 |
| (3) | — | 180 | 18 | 4 | 77 | tr:0:(19):0 |
| (4) | Cu(OAc)2·H2O (0.3) | 180 | 18 | 4 | 45 | 0:0:tr:0 |
| (5) | Cu-PMO (11) | 100 | 4 | 1 | 52 | tr:(36):tr:tr |
| (6) | PMO (250 mg) | 100 | 4 | 1 | 15 | 0:0:0:15 |
| (7) | — | 100 | 4 | 1 | 40 | 0:0:0:23 |
| (8) | Cu(OAc)2·H2O (0.3) | 100 | 4 | 1 | 0 | 0:0:0:0 |
The effect of increased steric hindrance and a more electron rich reduction centre was probed by studying acetovanillone (Fig. 3). Calculations of the change in Gibbs free energy with varying pressure at 180 °C indicate that both hydrogenolysis of the aryl ketone and cleavage of the aryl ether are thermodynamically favored. Although fission of the phenolic ring appears more thermodynamically favourable than hydrogenolysis of the ketone, the catalyst biases selectivity so that solely the ethyl-substituted phenol is obtained experimentally (Table 3). Indeed, no conversion of acetovanillone was seen except when using Cu-PMO at 180 °C, 4 MPa of hydrogen for 18 h (Table 3, entry 1) which effected selective and efficient hydrogenolysis of the ketone, yielding S6 quantitatively.
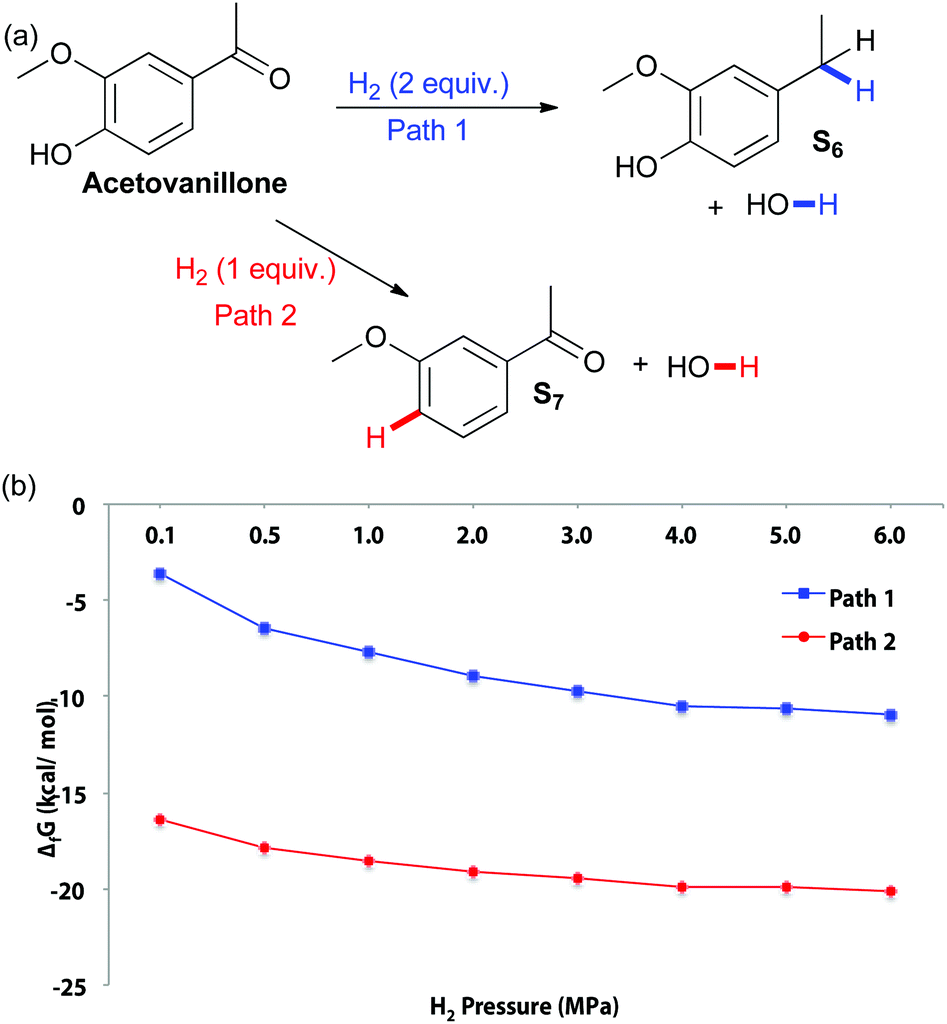 | ||
| Fig. 3 (a) Potential pathways of acetovanillone reduction (b) changes in Gibbs free energy with varying H2 pressure at 180 °C. | ||
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst (mol%) | Temp (°C) | Time (h) | H2d (MPa) | Conversionb (%) | YieldbS6 (%) |
| a All reactions were carried out in a high pressure 100 mL Parr Reactor using acetovanillone (6.456 mmol) in MeOH (30 mL). b Conversion and Yield determined by 1H NMR Spectroscopy using DMF as internal standard. c Conversion determined by recovery of starting material. d Pressure as measured at room temperature; nd = not determined. | ||||||
| (1) | Cu-PMO (11) | 180 | 18 | 40 | 100 | >95 |
| (2) | PMO (250 mg) | 180 | 18 | 40 | 0c | 0 |
| (3) | — | 180 | 18 | 40 | 0 | 0 |
| (4) | Cu(OAc)2·H2O (0.3) | 180 | 18 | 40 | 0 | 0 |
| (5) | Cu-PMO (11) | 100 | 4 | 10 | <5 | nd. |
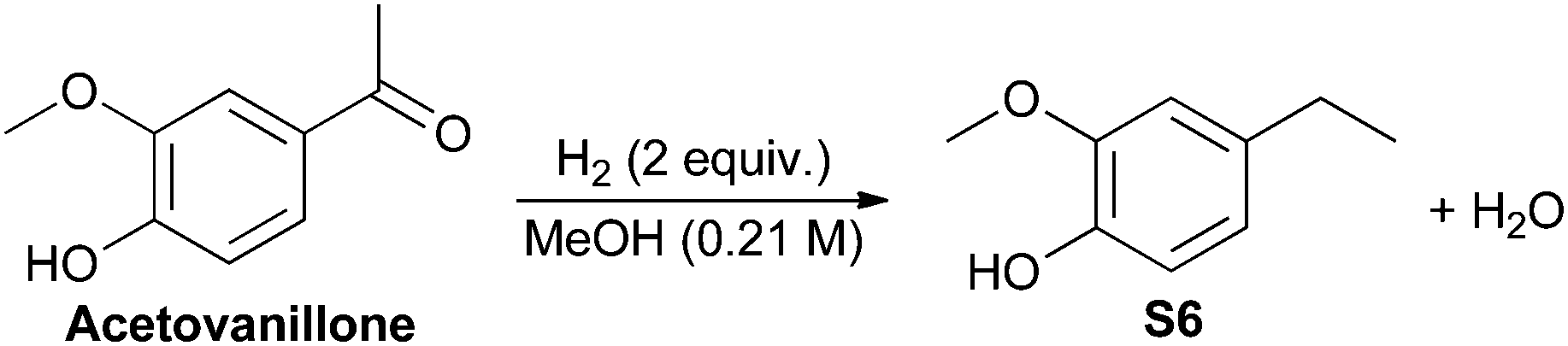
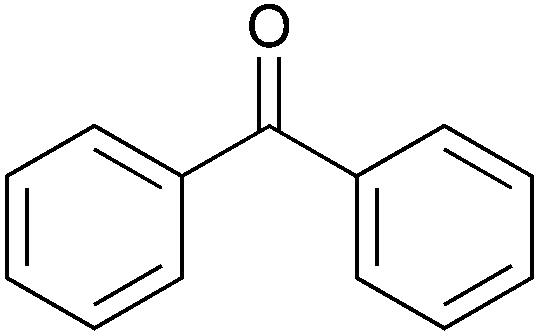
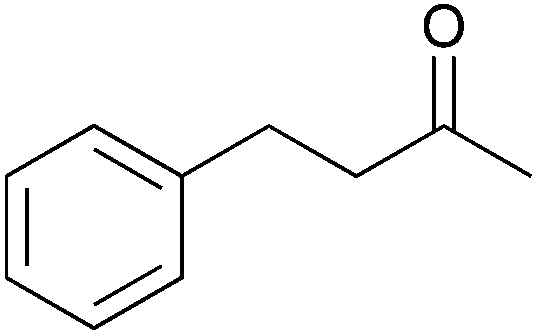
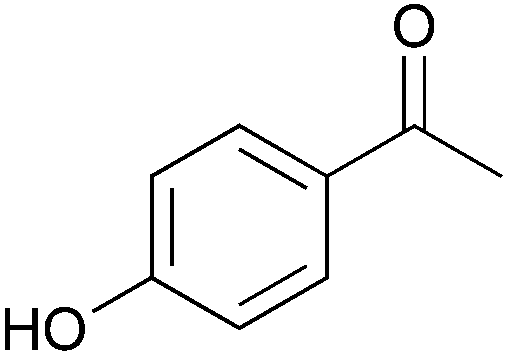
To investigate the robustness, selectivity and utility of Cu-PMO, several other ketones were investigated (Table 4). Benzyl ketones are very well tolerated, as evidenced by the quantitative hydrogenolysis of 2-acetonaphthone, 4′-hydroxyacetophenone and benzophenone. In contrast, the aliphatic ketone benzylacetone furnishes the corresponding alcohol quantitatively under the same conditions. Control experiments attribute both reactivity and selectivity to Cu-PMO.
|
|
||
|---|---|---|
| Substrate | Yieldb (%) condition A P1:P2 |
Yieldb (%) condition B P1:P2 |
| a All reactions were carried out in a high pressure 100 mL Parr Reactor using 6.456 mmol substrate. b Conversion and Yield determined by 1H NMR Spectroscopy using DMF as internal standard. c Pressure as measured at room temperature; nd = not determined. | ||
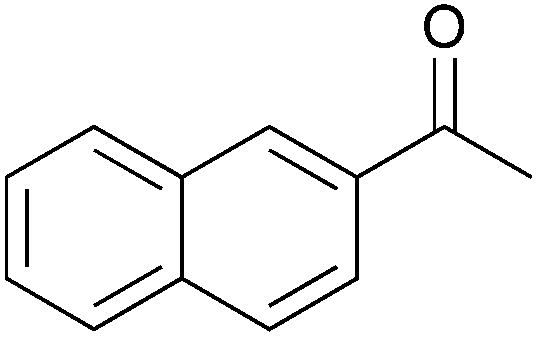
|
0:>95 |
0:<5 |
|
0:>95 |
13:<15 |
|
>95:0 |
0:0 |
|
0:>95 |
0:0 |
Even though the hydrogenolysis of methoxy–aryl bonds or phenol groups are also thermodynamically allowed, our Cu-PMO catalyst has a high selectivity (with mostly >95% yields) for the hydrogenation or hydrogenolysis of carbonyl groups and C–C double bonds, indicating strong kinetic control of the catalysis. Many other catalytic systems have shown similar product distributions but with lower selectivity.10–14,36–38
Moreover, our Cu-PMO catalyst has the advantage of being composed entirely of earth-abundant materials and of operating at very low loadings of Cu (0.3 mol%). Compared to other earth-abundant metal catalysts,20,21 Cu-PMO is resilient to phenolic units and is able to accommodate electron-rich and sterically hindered substrates.
Recycling experiments of eugenol hydrogenation (see ESI†) showed that it was possible to recycle the catalyst up to 11 times before noticing a decrease in activity. Analyses by ICP-OES of the spent catalyst revealed that the original metal ratio is retained after reaction. SEM and TEM images of Cu-PMO before and after reaction show little changes in the aggregation pattern and structure of the catalyst. XRPD pattern of spent Cu-PMO shows it is still amorphous after reaction. XPS investigations of recovered Cu-PMO versus fresh catalyst indicate some reduction of Cu(II) to Cu(I) and possibly Cu(0) after reaction (see ESI†).
In summary, we have developed a very selective method for hydrogenolysis of benzyl ketones and aldehydes as a greener alternative to Wolff-Kishner and Clemmensen conditions or noble-metal catalysed reductions. Additionally, our method allows selective reductions of alkenes. Ongoing investigations in our laboratory aim to extend the utility of the Cu-PMO system and elucidate its mechanism of reduction.
Research at University of New Haven is supported by the new faculty start-up fund and 2014 summer research grant and research fellowship of the University of New Haven. The Center for Green Chemistry and Green Engineering at Yale U. thanks the Yale School of Forestry and Environmental Studies for its support. We thank Amanda Lounsbury for the TEM images of Cu-PMO.
Notes and references
- P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, Oxford, 1998 Search PubMed.
- R. A. Sheldon, I. Arends and U. Hanefeld, Green Chemistry and Catalysis, WILEY-VCH, Weinheim, Germany, 2007 Search PubMed.
- S. Nishimura, Handbook of Heterogeneous Catalytic Hydrogenation for Organic Synthesis, John Wiley & Sons, Inc., United States of America, 2001 Search PubMed.
- M. R. Arnold, Ind. Eng. Chem., 1956, 48, 1629–1642 CrossRef CAS.
- D. D. Laskar, B. Yang, H. Wang and J. Lee, Biofuels, Bioprod. Biorefin., 2013, 7, 602–626 CrossRef CAS.
- C. Zhang, J. Xing, L. Song, H. Xin, S. Lin, L. Xing and X. Li, Catal. Today, 2014, 234, 145–152 CrossRef CAS.
- S. Tuokko and P. M. Pihko, Org. Process Res. Dev., 2014, 18, 1740–1751 CrossRef CAS.
- C. Larpent, R. Dabard and H. Patin, Tetrahedron Lett., 1987, 28, 2507–2510 CrossRef CAS.
- T. Nimmanwudipong, R. C. Runnebaum, S. E. Ebeler, D. E. Block and B. C. Gates, Catal. Lett., 2012, 142, 151–160 CrossRef CAS.
- X. Yang, Y. Liang, X. Zhao, Y. Song, L. Hu, X. Wang, Z. Wang and J. Qiu, RSC Adv., 2014, 4, 31932–31936 RSC.
- X. Xu, Y. Li, Y. Gong, P. Zhang, H. Li and Y. Wang, J. Am. Chem. Soc., 2012, 134, 16987–16990 CrossRef CAS PubMed.
- Z. Zhu, H. Tan, J. Wang, S. Yu and K. Zhou, Green Chem., 2014, 16, 2636–2643 RSC.
- A. Modvig, T. L. Andersen, R. H. Taaning, A. T. Lindhardt and T. Skrydstrup, J. Org. Chem., 2014, 79, 5861–5868 CrossRef CAS PubMed.
- Q. Wang, Y. Yang, Y. Li, W. Yu and Z. J. Hou, Tetrahedron, 2006, 62, 6107–6112 CrossRef CAS.
- L. Wolff, Justus Liebigs Ann. Chem., 1912, 394, 86–108 CrossRef.
- E. Clemmensen, Ber. Dtsch. Chem. Ges., 1913, 46, 1837–1843 CrossRef.
- F. Alonso, P. Riente and M. Yus, Tetrahedron, 2009, 65, 10637–10643 CrossRef CAS.
- J. Petro, L. Hegedus and I. E. Sajo, Appl. Catal., A, 2006, 308, 50–55 CrossRef CAS.
- A. L. Jongerius, R. Jastrzebski, P. C. A. Bruijnincx and B. M. Weckhuysen, J. Catal., 2012, 285, 315–323 CrossRef CAS.
- X. Kong and L. Chen, Catal. Commun., 2014, 57, 45–49 CrossRef CAS.
- M. R. Sturgeon, M. H. O'Brien, P. N. Ciesielski, R. Katahira, J. S. Kruger, S. C. Chmely, J. Hamlin, K. Lawrence, G. B. Hunsinger, T. D. Foust, R. M. Baldwin, M. J. Biddy and G. T. Beckham, Green Chem., 2014, 16, 824–835 RSC.
- D. P. Debecker, E. M. Gaigneaux and G. Busca, Chem. – Eur. J., 2009, 15, 3920–3935 CrossRef CAS PubMed.
- T. Mizugaki, R. Arundhathi, T. Mitsudome, K. Jitsukawa and K. Kaneda, Chem. Lett., 2013, 42, 729–731 Search PubMed.
- K. Barta, G. R. Warner, E. S. Beach and P. T. Anastas, Green Chem., 2014, 16, 191–196 RSC.
- G. S. Macala, T. D. Matson, C. L. Johnson, R. S. Lewis, A. V. Iretskii and P. C. Ford, ChemSusChem, 2009, 2, 215–217 CrossRef CAS PubMed.
- M. Valiev, E. J. Bylaska, N. Govind, K. Kowalski, T. P. Straatsma, H. J. J. Van Dam, D. Wang, J. Nieplocha, E. Apra, T. L. Windus and W. A. de Jong, Comput. Phys. Commun., 2010, 181, 1477–1489 CrossRef CAS.
- M. Hanwell, D. Curtis, D. Lonie, T. Vandermeersch, E. Zurek and G. Hutchison, J. Cheminf., 2012, 4, 17 CAS.
- A. Klamt and G. Schuurmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805, 10.1039/P29930000799.
- H. Wang, A. Sapi, C. M. Thompson, F. Liu, D. Zherebetskyy, J. M. Krier, L. M. Carl, X. Cai, L.-W. Wang and G. A. Somorjai, J. Am. Chem. Soc., 2014, 136, 10515–10520 CrossRef CAS PubMed.
- U. K. Singh and M. A. Vannice, Appl. Catal., A, 2001, 213, 1–24 CrossRef CAS.
- I. H. Bell, J. Wronski, S. Quoilin and V. Lemort, Ind. Eng. Chem. Res., 2014, 53, 2498–2508 CrossRef CAS PubMed.
- C. Reichardt and T. Welton, Solvents and Solvent Effects in Organic Chemistry, Wiley-VCH, Weinheim, Germany, 4th edn, 2011 Search PubMed.
- C. M. Jinesh, C. A. Antonyraj and S. Kannan, Catal. Today, 2009, 141, 176–181 CrossRef CAS.
- D. Kishore and S. Kannan, Appl. Catal., A, 2004, 270, 227–235 CrossRef CAS.
- N. V. Shorina, D. S. Kosyakov and K. G. Bogolitsyn, Russ. J. Appl. Chem., 2005, 78, 125–129 CrossRef CAS.
- X. Yang, Y. Liang, Y. Cheng, W. Song, X. Wang, Z. Wang and J. Qiu, Catal. Commun., 2014, 47, 28–31 CrossRef CAS.
- F. H. Mahfud, S. Bussemaker, B. J. Kooi, G. H. Ten Brink and H. J. Heeres, J. Mol. Catal. A: Chem., 2007, 277, 127–136 CrossRef CAS.
- M. B. Ezhova, A. Z. Lu, B. R. James and T. Q. Hu, Chem. Ind., 2005, 104, 135–143 CAS.
- P. G. M. Wuts and T. W. Greene, Greene's Protective Groups in Organic Synthesis, John Wiley & Sons, Inc., Hoboken, New Jersey, 4th edn, 2007 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: See DOI: 10.1039/c5gc01464f |
| This journal is © The Royal Society of Chemistry 2016 |