
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cajanus cajan – a source of PPARγ activators leading to anti-inflammatory and cytotoxic effects†
Roswitha
Schuster
a,
Wolfgang
Holzer
b,
Hannes
Doerfler
c,
Wolfram
Weckwerth
cd,
Helmut
Viernstein
a,
Siriporn
Okonogi
e and
Monika
Mueller
*a
aDepartment of Pharmaceutical Technology and Biopharmaceutics, University of Vienna, Althanstrasse 14, A-1090 Vienna, Austria. E-mail: monika.mueller@univie.ac.at; Tel: +43 1 4277 55414
bDepartment of Pharmaceutical Chemistry – Division of Drug Synthesis, University of Vienna, Althanstrasse 14, A-1090 Vienna, Austria
cDepartment of Ecogenomics and Systems Biology, University of Vienna, Althanstraße 14, A-1090 Vienna, Austria
dVienna Metabolomics Center (VIME), University of Vienna, Althanstraße 14, A-1090 Vienna, Austria
eDepartment of Pharmaceutical Sciences, Faculty of Pharmacy, Chiang Mai University, Chiang Mai 50200, Thailand
First published on 6th September 2016
Abstract
Cajanus cajan is an important legume crop in the human diet in many parts of the world. Due to its pharmacological properties, C. cajan is, moreover, used in traditional medicine for treating skin diseases, diabetes, inflammatory disorders and various other dysfunctions. In this study, we focused on the role of peroxisome proliferator-activated receptor gamma (PPARγ) as a potential therapeutic target of Cajanus cajan and its main compounds for the treatment of cancer, inflammation and inflammation-related disorders. The anti-inflammatory potential of C. cajan and its bioactive compounds and their cytotoxicity on the human cervical adenocarcinoma cell line HeLa, the human colorectal adenocarcinoma cell line CaCo-2 and the human breast adenocarcinoma cell line MCF-7 were elucidated. C. cajan and its compounds exerted significant anti-inflammatory activity on lipopolysaccharide-stimulated macrophages, showed good cytotoxic effects on the 3 different cancer cell lines and proved PPARγ activity in vitro. The main active compounds were orientin, pinostrobin and vitexin. Cajaninstilbene acid and pinosylvin monomethylether were identified as novel PPARγ activators. Based on these data, C. cajan provides excellent beneficial medicinal attributes and may be used as a potential food or a pharmaceutical supplement.
1. Introduction
Cajanus cajan (L.) Millsp. (syn.: pigeon pea, congo pea, red gram, non-eye pea, gunga pea, etc., Fabaceae) is one of the major grain legume crops of tropical and subtropical areas, with special significance in India, East Africa and Central America.1 In these countries, the seeds of the shrub, which are very rich in proteins, carbohydrates, minerals, vitamins and essential amino acids, are processed into meal or consumed as a whole, whereas the leaves are eaten fresh in salads or as a streamed or blanched vegetable.2–4 For example, in India, Cajanus cajan is primarily consumed as so called dhal (decorticated split cotyledons).1Besides its importance in nutrition, the plant is very popular in traditional medicine, where it is used as an infusion or tea preparation against various skin diseases, including bedsores, oral ulcers and measles, as well as urinary tract infections, menstrual disorders, genital irritations, hepatitis, diabetes, dysentery etc.5,6 Conventional treatments of these diseases (i.e. NSAIDs, glucocorticoids) exert excellent curative effects, but show several serious side effects in long term use including ulcers, edema and weight gain. For this reason, the interest in natural products has increased rapidly in recent years.
Extracts of C. cajan have been found to exert anti-inflammatory, antioxidative, antibacterial, anti-carcinogenic, hypoglycemic and hypocholesterolemic effects.6–12 The main compounds are classified into flavonoids (orientin, vitexin, pinostrobin) and stilbenes (cajaninstilbene acid, longistilin A and C).11,12 To the best of our knowledge, there is no previously reported study of C. cajan and its effect on peroxisome proliferator-activated receptors (PPARs), a group of transcription factors which modulate the expression of specific target genes when activated by an endogenous or exogenous ligand.13 In this way, PPARs play essential roles in inflammation, lipid metabolism, cell proliferation and differentiation, cancer, obesity and energy homeostasis.14
In the continuous search for natural remedies, we focus on the role of PPARγ as a potential therapeutic target of C. cajan leaves and their isolated compounds for the prevention and treatment of inflammation, inflammation-related disorders and cancer. We also elucidate the anti-inflammatory potential of C. cajan and its bioactive compounds and their cytotoxicity on the three different cancer cell lines (HeLa, CaCo-2, MCF-7). Furthermore, the main active compounds of C. cajan are isolated and identified.
2. Materials and methods
2.1. Chemicals and reagents
Absolute ethanol, formic acid, disodium hydrogen phosphate, hydrochloric acid, potassium chloride, sodium chloride, sodium dodecyl sulfate (SDS) and Tween 20 were obtained from Merck (Darmstadt, Germany). Bovine serum albumin (BSA), potassium dihydrogen phosphate and trifluoroacetic acid were purchased from Sigma-Aldrich (St Louis, MO, USA). Acetonitrile (HPLC grade) was purchased from Promochem (Wesel, Germany). Macrophage RAW 264.7, HeLa, CaCo-2 and MCF-7 cells were obtained from American Type Culture Collection (ATCC-TIB-71, ATCC-CCL-2, ATCC-HTB-37, ATCC-HTB-22). Fibroblast NIH-3T3 cells were purchased from the German Collection of Microorganisms and Cell Cultures (GSMZ, Ac. No. 59). One Shot® TOP10 Chemically Competent E. coli, Dulbecco's minimum essential medium (DMEM), fetal bovine serum (FBS), L-glutamine and a penicillin/streptomycin mixture were purchased from Life Technologies (Carlsbad, CA, USA). Rabbit polyclonal anti-iNOS was purchased from Santa Cruz Biotechnology (Dallas, TX, USA). Goat Anti-Rabbit IgG, DyLight™ 800, was obtained from Thermo Fisher Scientific (Rockford, IL, USA). Lipopolysaccharide from E. coli (LPS), thiazolyl blue tetrazolium bromide (MTT) and rosiglitazone were obtained from Sigma-Aldrich (Vienna, Austria), SuperFect and a Qiagen Plasmid Maxi kit were purchased from Qiagen (Germantown, MD, USA). An Enzyme Linked Immunosorbent Assay (ELISA) kit was obtained from eBioscience (San Diego, CA, USA). A Dual Glo Luciferase Assay System was obtained from Promega (Madison, WI, USA).2.2. Preparation of plant extracts
Fresh leaves of C. cajan were collected from the area of the Chiang Mai province, Thailand. The plant voucher specimens (no. 023173 and 023174) were deposited at the Herbarium of the Faculty of Pharmacy, Chiang Mai University (CMU) and authenticated by the CMU staff botanist. After washing, the leaves were cut into small pieces, dried in a circulating oven at 55 °C and ground into a fine powder which was macerated with 50% ethanol for 24 h at room temperature. The mixture was filtered (Whatman no. 1, Sigma-Aldrich) and the solvent was evaporated in a rotary evaporator at 40 °C. The remaining water was removed by freeze-drying in a Heto Power Dry LL3000 freeze-dryer (Thermo Scientific, Waltham, MA, USA) and the crude extracts were stored at room temperature in the dark. For further investigations the residues were freshly dissolved in 50% ethanol at a concentration of 100 mg ml−1.2.3. Separation by preparative high performance liquid chromatography (HPLC)
An extract of C. cajan was separated by preparative HPLC using a Thermo Scientific Ultimate 3000 system connected to a Hypersil GOLD™ column (5 μm, 21.2 mm × 250 mm, Thermo Scientific). The solvent gradient was set as follows: 0.5% B (0 min), 10% B (5 min), 17% B (14 min), 44% B (16 min), 50% B (20 min), 90% B (30 min), 99.5% B (32 min), 99.5% B (33 min), 0.5% B (34 min), 0.5% B (36 min), with a mobile phase, consisting of eluent A (formic acid/acetonitrile/water 0.1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5:94.9 v/v/v) and eluent B (formic acid/acetonitrile 0.1:99.9 v/v), a flow rate of 15 ml min−1 and UV detection at 280 nm. In repeated preparative HPLC separations, the fractions were collected and pooled and the solvent was evaporated. The remaining water was removed by freeze-drying (Heto Power Dry LL3000). The dry extracts were stored at room temperature until further use.
:5:94.9 v/v/v) and eluent B (formic acid/acetonitrile 0.1:99.9 v/v), a flow rate of 15 ml min−1 and UV detection at 280 nm. In repeated preparative HPLC separations, the fractions were collected and pooled and the solvent was evaporated. The remaining water was removed by freeze-drying (Heto Power Dry LL3000). The dry extracts were stored at room temperature until further use.
2.4. Characterization of C. cajan and its isolated compounds by HPLC-UV
Characterization of the plant extract and the isolated fractions was performed by analytical HPLC using a Thermo Scientific Ultimate 3000 system connected to a Kinetex™ C-18 column (5 μm, 100 Å, 150 × 4.6 mm, Phenomenex) and a PDA-100 Photodiode Array Detector. The elution programme was set as follows: 0% B (0 min), 10% B (1 min), 21% B (11 min), 28% B (12 min), 32% B (16 min), 36% B (17 min), 44% B (21 min), 48% B (22 min), 75% B (26 min), 100% B (27 min), 100% B (29 min), 0% B (29.1 min), 0% B (32 min), with a mobile phase consisting of eluent A (trifluoroacetic acid/acetonitrile/water 0.1:5:94.9 v/v/v) and eluent B (trifluoroacetic acid/acetonitrile 0.1:99.9 v/v), using a flow rate of 0.8 ml min−1 and UV detection at 280 nm. The extracts were analyzed at a concentration of 1 mg ml−1. The absorption maxima of the single compounds were determined by performing a 3D scan using different wavelengths which are described for these components in the literature.11 For further investigations we chose 280 nm, which was found to be the (or near the) absorption maximum for the majority of compounds of C. cajan.
2.5. Identification by mass spectrometry
Samples were prepared in a methanolic solution with 0.01% formic acid and subsequently analyzed on an Orbitrap Elite mass spectrometer (Thermo Scientific) via a heated electrospray ionisation (HESI) source in positive ([M + H+]+) mode facilitating a flow rate of 5 μl min−1. Full-scan (FS) data were recorded at a resolution of 120000 in a mass window of 100–500 m/z. Selected precursor ions underwent MS2 and MS3 fragmentation (collision-induced dissociation, CID) at a normalized collision energy of 50 with an isolation width of 1. Both FS and MSn scan events were recorded in the Fourier transformation (FT) mode of the mass spectrometer, yielding accurate m/z features of 1 ppm or lower.
2.6. Identification by nuclear magnetic resonance (NMR)
1H and 13C NMR spectra were recorded with a Bruker Avance III 400 spectrometer (400 MHz for 1H, 100 MHz for 13C) at 297 K using a “directly” detecting broadband observe (BBFO) probe. The center of the solvent signal was used as an internal standard which was related to tetramethylsilane with δ 2.49 ppm (1H not completely deuterated DMSO as in purity in DMSO-d6) and δ 39.50 ppm (13C in DMSO-d6). Full and unambiguous assignment of all resonances was achieved by the combined application of standard NMR techniques, such as an attached proton test (APT), heteronuclear single quantum coherence spectroscopy (HSQC), heteronuclear multiple bond correlation (HMBC), correlated spectroscopy (COSY) and nuclear Overhauser enhancement spectroscopy (NOESY) experiments.152.7. Assay of anti-inflammatory activity
To determine anti-inflammatory activity, an assay with RAW 264.7 cells was performed as described by Mueller et al.16 Macrophages were cultured in DMEM containing 10% FBS, 100 U ml−1 penicillin/streptomycin and 4 mM L-glutamine at 37 °C, 5% CO2 and 95% relative humidity. The cells were seeded into 24 well plates at a density of 2 × 106 cells per ml and incubated for 24 h. The samples were added on the following day and further incubated for 3 h.To stimulate cytokine production, the cells were treated with LPS at a final concentration of 1 μg mL−1. Wells without LPS and test substance addition served as the negative control, and wells with LPS but without test substance addition served as the positive control. The plate was once more incubated at 37 °C for 24 h. 500 μl of cell supernatants were removed, centrifuged at 12500 rpm to remove any cell residues and stored at −20 °C until performing ELISA.
The metabolic activity and thus the viability of the remaining cells were assessed using the colorimetric MTT assay based on the reduction of yellow MTT to purple formazan by living cells. 50 μl of MTT solution (5 mg ml−1 in 1× phosphate-buffered saline, PBS) was added to each well of the remaining cells and the plate was incubated for 2 h under normal culture conditions. Then, the supernatant was removed and the cells were lysed with 500 μl of lysis buffer (10% SDS in 0.01 N HCl). The absorption of the lysed cells was measured at 570 nm and corrected by a reference wavelength of 690 nm using a microplate reader (Infinite M200, Tecan, Austria).
The amount of secreted cytokines (IL-6, IL-10 and TNF-α) in the cell supernatants was quantified using the ELISA assay, according to the manufacturer's protocol (eBioscience). The concentration of cytokines of the positive control (only LPS) was defined as 100%. The variation of cell density was reduced by normalizing cytokine production to cell viability. The results from the test substances were calculated as a percentage of the positive control.
2.8. Western blot analysis
For western blot analysis, cultured cells were harvested, lysed and separated by SDS gel electrophoresis. After being transferred to a nitrocellulose membrane, the membrane was blocked in PBS containing 3% BSA overnight at 4 °C. On the next day, the membrane was washed with PBS containing 0.1% Tween 20 and preincubated with a primary antibody (anti-iNOS antibody) for 2 h at room temperature before incubation with the appropriate secondary antibody (DyLight conjugated anti-rabbit antibody). The blots were developed using an Odyssee®CLx Infrared Imaging System (LI-COR Biosciences, Vienna, Austria).2.9. Assay of PPAR transactivation
PPARγ activation by C. cajan and its isolated fractions was tested using a chimeric GAL-4 system in which the DNA binding domain of a chimeric GAL-4 was expressed as a fusion protein with the ligand binding domain of a human PPAR according to Mueller & Jungbauer.17 Simultaneously, a luciferase reporter plasmid was expressed which contained a firefly luciferase gene. In order to control transfection efficiency and to equalize variations in cell viability and sample preparation, a plasmid which encodes the renilla luciferase gene was used.According to the manufacturer's protocol, One Shot® TOP10 Chemically Competent E. coli was transformed for replication and cloning of the required plasmids. The bacterial strains were inoculated into LB-medium and cultured overnight under normal conditions. A Qiagen Plasmid Maxi Prep kit was used for the purification of the plasmid DNA from the bacterial pellet in conformity with the manufacturer's protocol.
For the transactivation assay, NIH-3T3 cells were seeded at a density of 2 × 106 cells per ml into 96 well microplates and incubated for 24 h. Subsequently, the cells were co-transfected with 300 ng pFR_Luc reporter plasmid, 60 ng pGAL4-hPPARγ-LBD expression plasmid and 30 ng pRL-TK control plasmid, using the SuperFect transfection reagent according to the manufacturer's instructions. After transfection, the standard rosiglitazone, a strong PPARγ activator, the plant extracts and pure compounds were added and incubated for another 24 h under normal growth conditions. On the next day, a luciferase assay was performed according to the manufacturer's protocol, using a Dual Glo® Luciferase Assay System. The renilla and firefly luminescences were recorded using a Genious Pro plate reader (Tecan), and the ratio of the luminescence from the experimental reporter (firefly) to that from the control reporter (renilla) was calculated and normalized to the ratio of the standard whose efficiency was defined as 100%.
2.10. Assay of cytotoxicity to cancer cell lines
An MTT-assay was performed to determine the cell viability and thus potential cytotoxicity of the substances towards three different cancer cell lines (HeLa, CaCo-2, MCF-7). Cells were seeded into 96 well plates at a density of 2 × 106 cells per ml and incubated for 24 h under normal culture conditions. On the next day, the cells were incubated with the test substances for another 24 h. Then, the wells were loaded with 10 μl MTT solution, and after 2 h of incubation, the cells were lysed with 100 μl of lysis buffer. The absorption was measured at 570 nm with a reference wavelength of 690 nm using an Infinite M200 microplate reader. The amount of cells of the positive control (cells only incubated with DMEM) was set as 100%. The results from the test substances were expressed as a percentage of the positive control.2.11. Statistical calculations
The anti-inflammatory assay, western blot analysis, the assay of PPAR transactivation and the assay of cytotoxicity to cancer cell lines were performed in triplicate on individual days. The results are expressed as means ± standard deviation. IC50 was determined using a logistic dose–response model of the Table Curve 2D software (Systat Software, San Jose, CA, USA).3. Results
3.1. Preparation and separation of a plant extract
The 50% ethanolic extract of C. cajan was an ochre powder. The recorded chromatogram of the preparative HPLC showed 10 main peaks at a wavelength of 280 nm (Fig. 1). Fraction 1 showed the highest yield of the extract, namely 20%, fractions 2–7 showed low yields of 1–4% and fractions 8–10 showed the least yields with a maximum of 0.4%. All fractions showed a purity of at least 75%, except fractions 3, 4 and 7 with a maximum of 50% (Table 1).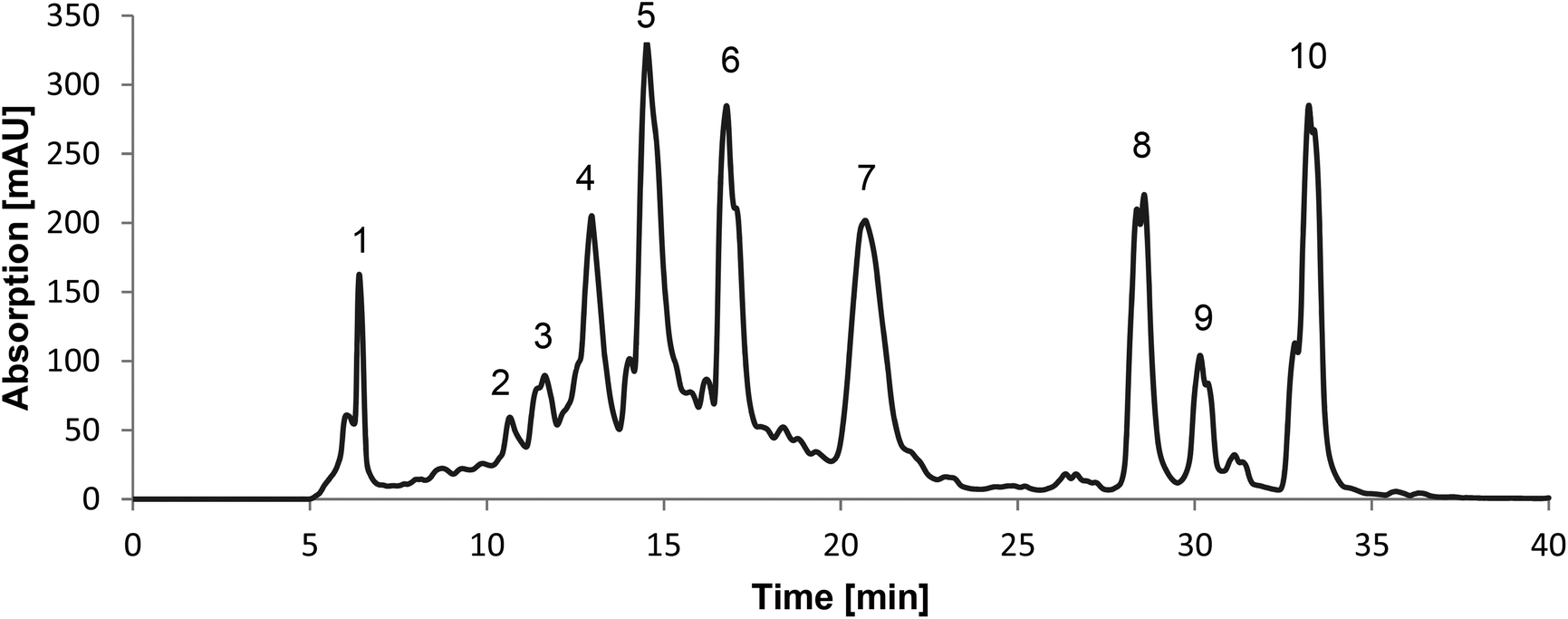 | ||
| Fig. 1 Preparative HPLC chromatogram of C. cajan showing 10 main peaks at a wavelength of 280 nm. Fractions 1–10 were collected as labeled at retention times [min] 6.4 (F1), 10.7 (F2), 11.6 (F3), 13 (F4), 14.5 (F5), 16.8 (F6), 20.7 (F7), 28.6 (F8), 30.2 (F9) and 33 (F10). | ||
| Fraction | Yield [%] | Purity [%] | Identification | Mass accuracy (ppm) | Product ions | |||
|---|---|---|---|---|---|---|---|---|
| HPLC (RT; min) | MS ([M + H+]+) | NMR | Compound | |||||
| 1 | 20.0 | >80 | — | |||||
| 2 | 0.68 | >75 | 10.5 | 205.1 | Tryptophan | −0.10 | 188 | |
| 3 | 0.64 | >45 | — | |||||
| 4 | 1.93 | >50 | — | |||||
| 5 | 2.22 | >75 | 9.8 | 449.1 | Orientin | 0.53 | 431, 413, 383 | |
| 6 | 1.36 | >80 | 11.8 | 433.1 | Vitexin | 0.48 | 415, 397, 367 | |
| 7 | 4.37 | >10 | — | |||||
| 8 | 0.32 | >90 | 227.1 | × | Pinosylvin monomethylether | −0.36 | 149, 143, 117 | |
| 9 | 0.18 | >90 | 25 | 271.1 | Pinostrobin | 0.76 | 167, 131, 173 | |
| 10 | 0.38 | >90 | 339.2 | × | Cajaninstilbene acid | −0.25 | 331, 313, 239 | |
3.2. Identification of the main compounds
Fractions 2, 5, 6 and 9 were identified as tryptophan, orientin, vitexin and pinostrobin by comparing the retention times with standards. Tryptophan (fraction 2) was not further analysed because there are already many studies on the bioactivity of this compound.18,19The NMR spectroscopic analysis (in DMSO-d6) of fraction 10 revealed the presence of a single product. The combined application of standard 2D-NMR experiments (HSQC, HMBC, NOESY, COSY) enabled us to unequivocally establish the structure of 2-hydroxy-4-methoxy-3-(3-methyl-2-buten-1-yl)-6-[(E)-2-phenylvinyl] benzoic acid (cajaninstilbene acid, Fig. 2A) as well as the full assignment of all 1H and 13C resonances as in ESI 1A.†
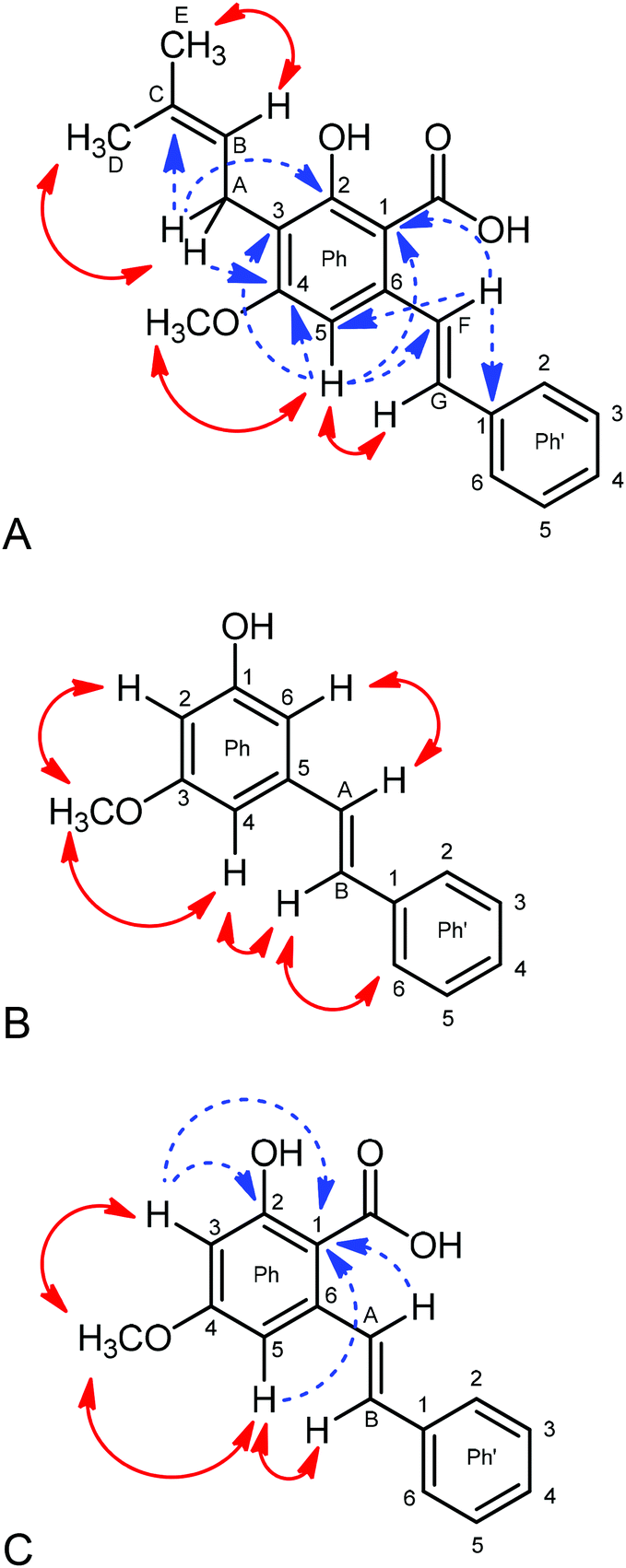 | ||
| Fig. 2 (A) Crucial NOEs (double headed arrows) and HMBC correlations (dashed arrows) detected in the NMR spectra of cajaninstilbene acid; (B) NOEs (double headed arrows) used in the structural elucidation of pinosylvin monomethyl ether; and (C) crucial NOEs (double headed arrows) and HMBC correlations (dashed arrows) detected in the NMR spectra of 4-O-methylpinosylvic acid. | ||
Fig. 2A shows the crucial NOEs (double headed arrows) and HMBC correlations (dashed arrows) used for this purpose. An unambiguous discrimination of the terminal methyl groups of the side chain attached at C-3 was possible by considering NOEs between alkene-H (HB) and MeE, whereas the methylene proton HA shows such a through-space interaction with MeD. The trans-position of the alkene protons HF and HG of the styryl moiety follows from a vicinal coupling constant of 16.1 Hz. A considerably larger NOE between H-5 and HG compared to that between H-5 and HF suggests that the rotameric form proved (proximity of H-3 and HG) to be favored in solution. The 1H and 13C chemical shifts found by us are in good agreement with those reported by Green et al.20 and Ji et al.,21 albeit these data originate from recordings in CDCl3.
The 1H-NMR of fraction 8 exhibited two signal sets arising from two structurally related compounds in the ratio of 1:0.4. The major component turned out to consist of a trisubstituted benzene ring with a methoxy, a hydroxy and a styryl moiety attached in the meta position, respectively, and thus to be a pinosylvin monomethyl ether (S.1B†). The two alkene protons HA and HB establish an AB-system with a vicinal coupling of 16.4 Hz indicating trans-position at the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond. Protons H-2 and H-4 can be discriminated from H-6 via NOEs to the methoxy protons; H-4 is unequivocally identified via NOEs to HA and HB (Fig. 2B).
C double bond. Protons H-2 and H-4 can be discriminated from H-6 via NOEs to the methoxy protons; H-4 is unequivocally identified via NOEs to HA and HB (Fig. 2B).
A plethora of HMBC correlations unambiguously confirm the structure and permit all assignments. HMBC correlations: OCH3 → C-3; H-2 → C-1, C-3, C-4, C-6; H-4 → C-2, C-6, CA; H-6 → C-1, C-2, C-4, CA; HA → C-4, C-5, C-6, C-1 of Ph′; HB → C-5, C-2.6 of Ph′. The NMR data are in good agreement with those given by Ngo & Brown.22
The minor component of fraction 8 was identified as 2-hydroxy-4-methoxy-6-[(E)-2-phenylvinyl] benzoic acid (4-O-methylpinosylvic acid, Fig. 2C). Only two protons (H-3 and H-5) are attached to the central benzene ring which must be arranged in meta-position due to a coupling constant 4J = 2.4 Hz. Both resonances receive strong NOEs from the OCH3 protons, thus indicating attachment of the methoxy group between H-3 and H-5 (4-position). Similarly to the main component, H-3 and H-5 can be easily distinguished by the fact that, in contrast to H-3, H-5 shows NOEs to the alkene protons HA and HB. This proves the spatial closeness of H-5 and the styryl substituent, the latter consequently attached at position 6 at the central benzene ring and its alkene protons HA and HB being in trans-position due to a coupling constant of 16.3 Hz (S.1C†).
Finally, the position of the hydroxy and the carboxy substituent can be determined via HMBC correlations. C-1 (108.07 ppm) is correlated to H-3, H-5 and HA, whereas C-2 (162.52 ppm) is only correlated to H-3, thus clearly evidencing that OH is attached to C-2 and COOH to C-1. Our NMR data for this compound are closely similar to those reported by Ohwaki et al.23 and Mitra et al.24
Furthermore, the recorded mass-to-charge ratios of fractions 2, 5, 6, 8, 9 and 10 by high mass accuracy MS were confirmed to be the identified compounds (Table 1). Analytical HPLC showed that fractions 3, 4 and 7 were not pure enough to identify them as single substances.
3.3. PPARγ activation
C. cajan and fraction 10 exhibited the highest transactivation efficiency with at least 40% efficiency in comparison with rosiglitazone (Table 2 and Fig. 3), followed by fraction 8 with almost 30%. Fractions 4 and 9 had good effects with about 15%. Fractions 3 and 5 showed moderate effects; fractions 1, 6 and 7 had no effect on transactivation efficiency. Furthermore, IC50 was determined for C. cajan (60 μg ml−1), fraction 8 (15 μg ml−1) and fraction 10 (14 μg ml−1). According to Mueller & Jungbauer, the IC50 value for rosiglitazone was determined to be 0.21 μg ml−1.17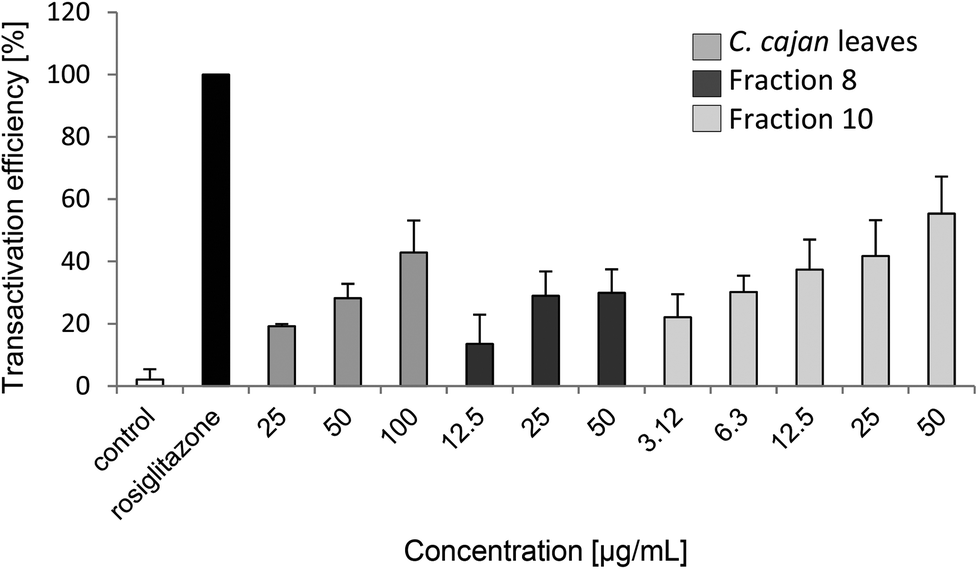 | ||
| Fig. 3 Concentration dependent transactivation efficiency [%] of C. cajan leaves, fraction 8 and fraction 10, using a chimeric GAL-4 system. For a positive control, cells were treated with rosiglitazone, a strong PPARγ activator; for a negative control, cells were not treated with any substance. | ||
| Substance | Concentration | Transactivation efficiency [%] | Secretion of [%] | Expression of iNOS [%] | Viability of [%] | IC50 [μg ml−1] | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IL-6 | TNF-α | IL-10 | HeLa | CaCo-2 | MCF-7 | For IL-6 reduction | For cytotoxicity to | ||||||
| HeLa | CaCo-2 | MCF-7 | |||||||||||
| Control | 2 ± 3 | 12 ± 1 | 15 ± 7 | 11 ± 6 | 0 | 100 | 100 | 100 | |||||
| Rosiglitazone | 10−5 M | 100 | |||||||||||
| LPS | 100 | 100 | 100 | 100 | |||||||||
| C. cajan leaves | 100 μg mL−1 | 43 ± 10 | 40 ± 5 | 84 ± 7 | 113 ± 10 | 33 ± 4 | 71 ± 9 | 79 ± 10 | 47 ± 11 | 18 | >80 | >80 | 52 |
| Fraction 1 | 100 μg mL−1 | No effect | 64 ± 3 | 92 ± 6 | 105 ± 3 | 98 ± 2 | 55 ± 13 | 51 ± 11 | 55 ± 6 | >80 | >80 | >80 | >80 |
| Fraction 3 | 100 μg mL−1 | 3 ± 0 | 40 ± 5 | 92 ± 12 | 77 ± 11 | 32 | 55 ± 15 | 43 ± 13 | 88 ± 13 | ||||
| Fraction 4 | 100 μg mL−1 | 13 ± 9 | 62 ± 5 | 95 ± 14 | 87 ± 9 | 60 ± 3 | 75 ± 10 | 53 ± 12 | 74 ± 14 | ||||
| Fraction 5 | 100 μg mL−1 | 7 ± 4 | 47 ± 3 | 83 ± 3 | 78 ± 13 | 87 ± 6 | 68 ± 17 | 55 ± 2 | 63 ± 12 | 40 | >80 | >80 | >80 |
| Fraction 6 | 100 μg mL−1 | No effect | 69 ± 4 | 112 ± 12 | 88 ± 8 | 75 ± 2 | 66 ± 6 | 52 ± 7 | 113 ± 6 | >80 | >80 | 72 | No effect |
| Fraction 7 | 100 μg mL−1 | No effect | 55 ± 2 | 108 ± 18 | 87 ± 13 | 25 ± 2 | 96 ± 18 | 122 ± 3 | 75 ± 32 | ||||
| Fraction 8 | 50 μg ml−1 | 30 ± 8 | 82 ± 4 | 91 ± 14 | 119 ± 5 | 0 | 67 ± 3 | 51 ± 8 | 59 ± 11 | >80 | >80 | 65 | >80 |
| Fraction 9 | 100 μg mL−1 | 16 ± 7 | Cytotoxic | 49 ± 10 | 48 ± 10 | 55 ± 3 | >80 | 62 | 54 | 42 | |||
| 50 μg mL−1 | 79 ± 4 | 73 ± 10 | 98 ± 12 | 16 ± 1 | |||||||||
| Fraction 10 | 100 μg mL−1 | 37 ± 4 | 103 ± 9 | 73 ± 7 | 0 | 16 ± 3 | 20 ± 6 | 54 ± 12 | 48 | 39 | 32 | 65 | |
| 50 μg mL−1 | 55 ± 12 | ||||||||||||
3.4. Anti-inflammatory effect
The concentrations of secreted IL-6, IL-10 and TNF-α in response to treatment with C. cajan and its isolated compounds in LPS-stimulated macrophages are summarized in Table 2. Secretion of the pro-inflammatory cytokine IL-6 was significantly reduced by at least 20% upon adding fractions 6, 8 and 9, 40% upon adding fractions 1, 4, 5 and 7 and more than 60% upon incubation with the extract of C. cajan and fractions 3 and 10. Fraction 10 also showed significant inhibition of IL-6 secretion at concentrations of 50 or 25 μg ml−1 (Fig. 4). No significant effect on IL-10 secretion was found. A slight reduction of TNF-α secretion was found for fractions 5 and 9 (Table 2). IC50 for the reduction of IL-6 ranged from 6 to >80 μg ml−1 (Table 2). As a positive control, cells were incubated with dexamethasone, an anti-inflammatory hormone. A significant reduction of IL-6 was observed upon incubating the cells with 5 μM cortisol.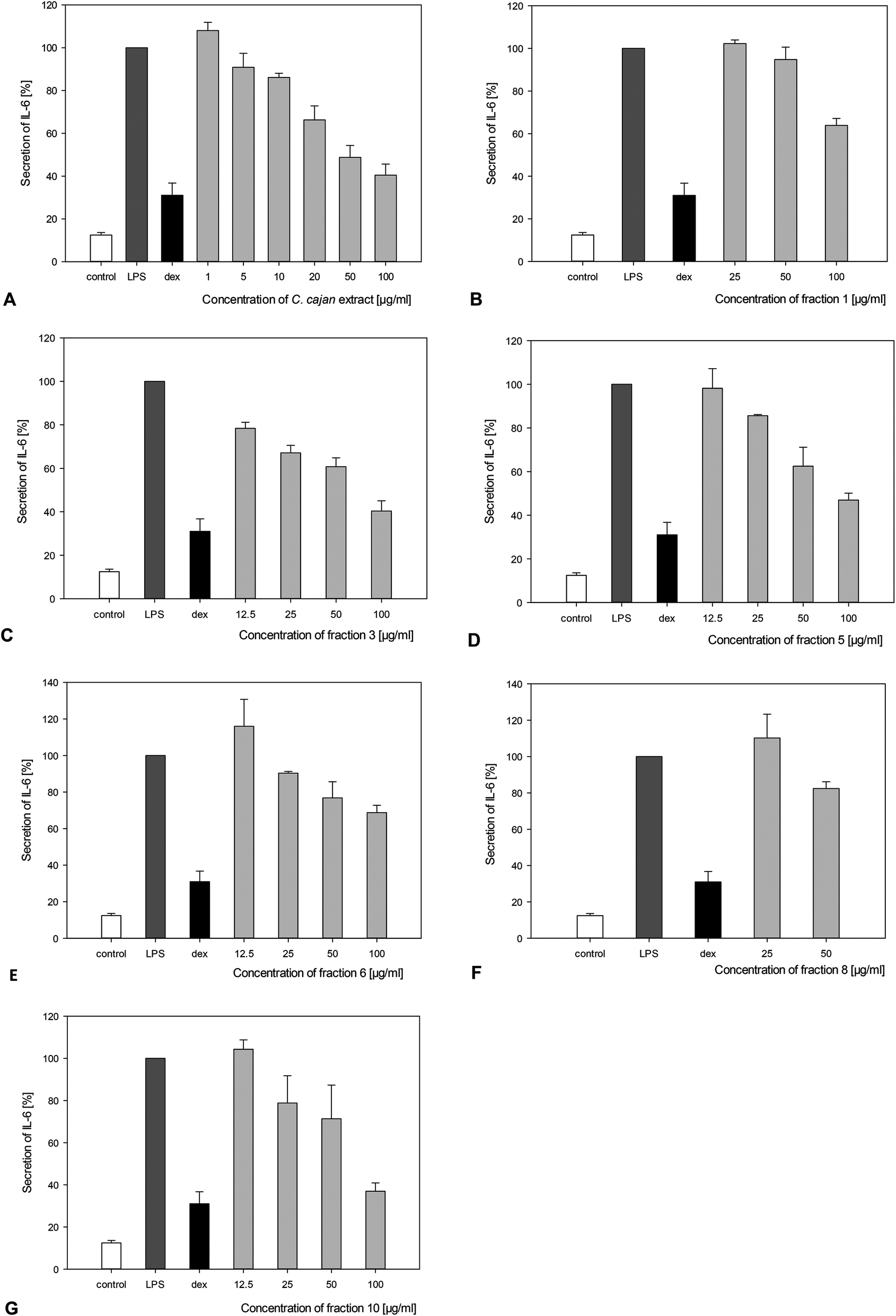 | ||
| Fig. 4 Concentration dependent reducing effect of the C. cajan leave extract and fractions 1, 3, 5, 6, 8 and 10 (A–G) on the IL-6 secretion of LPS-stimulated macrophages, indicated by the amounts of secreted cytokines, as determined by ELISA. For a positive control, cells were only treated with LPS (LPS); for a negative control, cells were not treated with any substance (control). Simultaneously, 5 μM dexamethasone (dex), an anti-inflammatory hormone, was used as a control substance. | ||
The C. cajan extract and several fractions exerted a significant reducing effect on the expression of iNOS (Table 2). In fact, C. cajan and fractions 1, 3–7 and 10 were tested at a final concentration of 100 μg ml−1, due to their cytotoxic effects on the macrophages (at a concentration of 100 μg ml−1), and fractions 8 and 9 were tested at a final concentration of 50 μg ml−1. Fraction 1 had no effect on the expression of iNOS, and fractions 4, 5 and 6 reduced the expression of iNOS by more than 20%. C. cajan and fractions 3, 7 and 9 decreased the expression of iNOS by more than 70%. Notably, fractions 8 and 10 completely inhibited iNOS expression.
3.5. Cytotoxicity to cancer cell lines
The C. cajan extract and its isolated compounds show diverse cytotoxic effects on the three cancer cell lines (Table 2 and S.2†). Treating the cell line HeLa with C. cajan, fractions 4, 5, 6 and 8 decreased the metabolic activity of cells by at least 25%, and incubating the cells with fractions 1, 3 and 9 led to a reduction in cell viability of at least 50%. Notably, fraction 10 showed the highest decrease of cell viability by about 85%. Incubating the cancer cell line CaCo-2 with C. cajan led to a reduction of metabolic activity by about 20%. Fractions 1, 3, 4, 5, 6, 8, and 9 lowered the cell viability by at least 50%, and fraction 10 minimized the cell viability significantly by at least 80%. Fractions 4 and 7 decreased the cell viability of the cancer cell line MCF-7 by at least 25% and C. cajan and fractions 1, 5, 8, 9 and 10 by at least 40%. Fraction 7 showed no effect on HeLa and CaCo-2 and fractions 3 and 6 had no significant influence on MCF-7. IC50 was determined for each cancer cell line, ranging from 39 to >80 μg ml−1 for HeLa, from 32 to >80 μg ml−1 for CaCo-2 and from 42 to >80 μg ml−1 for MCF-7 (Table 2).4. Discussion
Since ancient times, plants and plant derived formulations have been used in traditional medicine for treating various diseases. In recent years, the beneficial pharmacological effects of spices, herbals, fruits, vegetables, etc. have been demonstrated and the interest in so called functional foods has been constantly increasing.It is well known that a wide variety of phytochemicals in plants, spices and herbals (for example, isoflavones, pomegranate and liquorice components, pepper, ginger) can activate PPARs and show anti-inflammatory and anti-diabetic effects.16,17,25 In traditional medicine, C. cajan is, amongst others, used to treat diabetes and, accordingly, scientific studies have reported its anti-diabetic potential in diabetic mice.6,26 In order to explain the mechanism of the anti-diabetic and anti-inflammatory effects of C. cajan and its main compounds, the plant was tested for its PPARγ activity. We therefore isolated 10 compounds from the extract and identified 6 of them, namely tryptophan (2), orientin (5), vitexin (6), pinostrobin (9) and cajaninstilbene acid (10). The NMR spectrum of fraction 8 showed a major (pinosylvin monomethylether) and a minor (4-O-methylpinosylvic acid) component. The presence of the two components could be attributed to a decarboxylation process by heat generation during the measurement. Furthermore, the main component may be present in a stable form in the plant and disintegrate into a fission product during sample preparation. The identified components have all been described previously and various studies on the bioactivity of the main compounds have been published.11,12,27–29 Previously, Huang et al.30 reported an inhibition of the activation of the NF-κB and MAPK pathways by cajaninstilbene acid, assuming that this compound enhances the activity of PPARγ. This study revealed that cajaninstilbene acid and pinosylvin monomethylether, two isolated compounds from C. cajan, exhibit high transactivation efficiency in PPARγ in vitro.
Besides, previous studies have proved the anti-inflammatory activity of C. cajan leaves as indicated by a decrease of production of pro-inflammatory cytokines such as IL-1β and TNF-α in a LPS-stimulated macrophage model, but there is only a little information on the effect of its main compounds on the IL-6 and IL-10 secretion.12,27,31 In this study we have shown the anti-inflammatory effect of C. cajan leaves and its 10 main compounds as indicated by a significant decrease of IL-6 secretion by most of the main fractions, including cajaninstilbene acid, orientin, and fractions 3 and 7. The secretion of TNF-α was only slightly reduced by two fractions, namely orientin and pinostrobin; the secretion of IL-10 was not affected at all.
Since ancient times, various cultures have used plants or plant-derived formulations (for example, traditional Chinese medicine or Ayurveda) in the treatment of inflammatory disorders and related diseases.32,33 These data indicate that C. cajan, an important plant in human nutrition in many parts of the world, has a beneficial effect on inflammation, the metabolic syndrome and related disorders.
Up to now, there has been no study on the cytotoxicity of the 10 main compounds of C. cajan leaves on the three cancer cell lines HeLa, CaCo-2 and MCF-7. Only pinostrobin was found to have a cytotoxic effect on MCF-7.34 In this study, cajaninstilbene acid exerted the most significant effect on all the three cell lines, followed by pinosylvin monomethylether. The remaining fractions have some cytotoxic potential on each cell line. These data prove that even natural products have cytotoxic effects but in order to make a concrete statement about the cytotoxic effect on tumors and on the impact on the human body, in vivo studies would be required.
5. Conclusion
This study revealed that C. cajan leaves and their isolated compounds exhibit significant anti-inflammatory and cytotoxic effects as well as PPARγ activity in vitro. Cajaninstilbene acid and pinosylvin monomethylether were identified as new PPARγ activators. These results confirm the popular use of this plant in the traditional treatment of inflammatory skin diseases and related disorders and may also lead to a better understanding of the toxic effects on different cancers. This work demonstrates that C. cajan and its compounds are potential candidates for food or pharmaceutical supplements in the treatment of inflammation and related disorders.Funding
The research reported in this publication was supported by a short term grant from the ASEAN-European Academic University Network (ASEA-UNINET).References
- U. Singh, R. Jambunathan, K. Saxena and N. Subrahmanyam, J. Sci. Food Agric., 1990, 50, 201–209 CrossRef.
- J. F. Morton, HortScience, 1976, 11, 11–19 Search PubMed.
- D. K. Salunkhe, J. K. Chavan, S. S. Kadam and N. R. Reddy, Crit. Rev. Food Sci. Nutr., 1986, 23, 103–145 CrossRef CAS PubMed.
- K. B. Saxena, Genet. Resour., Chromosome Eng., Crop Improv., 2005, 1, 85–115 Search PubMed.
- D. K. Abbiw, Useful plants of Ghana: West African uses of wild and cultivated plants, Intermediate Technology Publications and The Royal Botanic Gardens, 1990 Search PubMed.
- T. Amalraj and S. Ignacimuthu, Indian J. Exp. Biol., 1998, 36, 1032–1033 CAS.
- J. K. Grover, S. Yadav and V. Vats, J. Ethnopharmacol., 2002, 81, 81–100 CrossRef CAS PubMed.
- Y. Kong, Y. J. Fu, Y. G. Zu, F. R. Chang, Y. H. Chen, X. L. Liu, J. Stelten and H. M. Schiebel, Food Chem., 2010, 121, 1150–1155 CrossRef CAS.
- Q. F. Luo, L. Sun, J. Y. Si and D. H. Chen, Phytomedicine, 2008, 15, 932–939 CrossRef CAS PubMed.
- M. Luo, X. Liu, Y. Zu, Y. Fu, S. Zhang, L. Yao and T. Efferth, Chem.-Biol. Interact., 2010, 188, 151–160 CrossRef CAS PubMed.
- N. Wu, K. Fu, Y. J. Fu, Y. G. Zu, F. R. Chang, Y. H. Chen, X. L. Liu, Y. Kong, W. Liu and C. B. Gu, Molecules, 2009, 14, 1032–1043 CrossRef CAS PubMed.
- N. K. Patel and K. K. Bhutani, Phytomedicine, 2014, 21, 946–953 CrossRef CAS PubMed.
- L. Michalik, J. Auwerx, J. P. Berger, V. K. Chatterjee, C. K. Glass, F. J. Gonzalez, P. A. Grimaldi, T. Kadowaki, M. A. Lazar and S. O'Rahilly, Pharmacol. Rev., 2006, 58, 726–741 CrossRef CAS PubMed.
- K. L. Houseknecht, B. M. Cole and P. J. Steele, Domest. Anim. Endocrinol., 2002, 22, 1–23 CrossRef CAS PubMed.
- S. Braun, H. O. Kalinowski, S. Berger, L. Ernst and K. Ibrom, Angew. Chem., German Ed., 1999, 111, 1227–1227 Search PubMed.
- M. Mueller, S. Hobiger and A. Jungbauer, Food Chem., 2010, 122, 987–996 CrossRef CAS.
- M. Mueller and A. Jungbauer, Food Chem., 2009, 117, 660–667 CrossRef CAS.
- A. L. Mellor, J. Sivakumar, P. Chandler, K. Smith, H. Molina, D. Mao and D. H. Munn, Nat. Immunol., 2001, 2, 64–68 CrossRef CAS PubMed.
- S. Löb, A. Königsrainer, D. Zieker, B. L. D. M. Brücher, H. G. Rammensee, G. Opelz and P. Terness, Cancer Immunol. Immunother., 2009, 58, 153–157 CrossRef PubMed.
- P. W. C. Green, P. C. Stevenson, M. S. J. Simmonds and H. C. Sharma, J. Chem. Ecol., 2003, 29, 811–821 CrossRef CAS PubMed.
- X. Ji, J. Jin, G. Zheng and Z. Li, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2013, 69, o91–o91 CAS.
- K. S. Ngo and G. D. Brown, Phytochemicals, 1998, 47, 1117–1123 CrossRef CAS.
- Y. Ohwaki, J. I. Ogino and K. Shibano, Soil Sci. Plant Nutr., 1993, 39, 55–61 CrossRef CAS.
- P. Mitra, B. Shome, S. R. De, A. Sarkar and D. Mal, Org. Biomol. Chem., 2012, 10, 2742–2752 CAS.
- M. Mueller and A. Jungbauer, Menopause, 2008, 15, 1120–1131 CrossRef PubMed.
- A. M. Espósito, A. Diaz, I. De Gracia, R. De Tello and M. P. Gupta, Rev. Med. Panama, 1991, 16, 39–45 Search PubMed.
- Y. S. Lai, W. H. Hsu, J. J. Huang and S. C. Wu, Food Funct., 2012, 3, 1294–1301 CAS.
- L. Liang, M. Luo, Y. Fu, Y. Zu, W. Wang, C. Gu, C. Zhao, C. Li and T. Efferth, Toxicol. Lett., 2013, 219, 254–261 CrossRef CAS PubMed.
- D. M. Zhang, Y. Li, W. San Cheang, C. W. Lau, S. M. Lin, Q. L. Zhang, N. Yao, Y. Wang, X. Wu and Y. Huang, PLoS One, 2012, 7, e47030 CAS.
- M. Y. Huang, J. Lin, K. Lu, H. G. Xu, Z. Z. Geng, P. H. Sun and W. M. Chen, J. Agric. Food Chem., 2016, 64, 2893–2900 CrossRef CAS PubMed.
- W. Lee, S. K. Ku and J. S. Bae, Vasc. Pharmacol., 2014, 62, 3–14 CrossRef CAS PubMed.
- Q. Wang, H. Kuang, Y. Su, Y. Sun, J. Feng, R. Guo and K. Chan, J. Ethnopharmacol., 2013, 146, 9–39 CrossRef CAS PubMed.
- A. Nelly, D. D. Annick and D. Frederic, J. Ethnopharmacol., 2008, 120, 315–334 CrossRef PubMed.
- J. S. Ashidi, P. J. Houghton, P. J. Hylands and T. Efferth, J. Ethnopharmacol., 2010, 128, 501–512 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6fo00689b |
| This journal is © The Royal Society of Chemistry 2016 |