
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Heat-killed and live Lactobacillus reuteri GMNL-263 exhibit similar effects on improving metabolic functions in high-fat diet-induced obese rats
Feng-Ching
Hsieh
a,
Cheng-Che E.
Lan
b,
Tsui-Yin
Huang
c,
Kuan-Wei
Chen
c,
Chee-Yin
Chai
d,
Wan-Tzu
Chen
d,
Ai-Hui
Fang
e,
Yi-Hsing
Chen†
cf and
Ching-Shuang
Wu†
*ag
aDepartment of Medical Laboratory Science and Biotechnology, Kaohsiung Medical University, Kaohsiung, Taiwan. E-mail: m785034@kmu.edu.tw; Fax: +886-7-3113449; Tel: +886-7-3121101 ext. 2354
bDepartment of Dermatology, College of Medicine, Kaohsiung Medical University, Kaohsiung, Taiwan
cDepartment of Research and Development, GenMont Biotech. Inc., Tainan, Taiwan
dDepartment of Pathology, Kaohsiung Medical University Hospital, Kaohsiung, Taiwan
eDepartment of Microbiology, College of Medicine, Kaohsiung Medical University, Kaohsiung, Taiwan
fInstitute of Biomedical Science, National Chung Hsing University, Taichung, Taiwan
gInstitute of Biomedical Sciences, National Sun Yat-Sen University, Kaohsiung, Taiwan
First published on 26th April 2016
Abstract
Our objective was to investigate and compare the effects of heat-killed (HK) and live Lactobacillus reuteri GMNL-263 (Lr263) on insulin resistance and its related complications in high-fat diet (HFD)-induced rats. Male Sprague-Dawley rats were fed with a HFD with either HK or live Lr263 for 12 weeks. The increases in the weight gain, serum glucose, insulin, and lipid profiles in the serum and liver observed in the HFD group were significantly reduced after HK or live Lr263 administration. Feeding HK or live Lr263 reversed the decreased number of probiotic bacteria and increased the number of pathogenic bacteria induced by high-fat treatment. The decreased intestinal barrier in the HFD group was markedly reversed by HK or live Lr263 treatments. The elevations of pro-inflammatory associated gene expressions in both adipose and hepatic tissues by high-fat administration were markedly decreased by HK or live Lr263 treatments. The increased macrophage infiltration noticed in adipose tissue after high-fat treatment was effectively suppressed by HK or live Lr263 consumption. The insulin resistance associated gene expressions in both adipose and hepatic tissues, which were downregulated in the HFD group, were markedly enhanced after HK or live Lr263 administration. HK or live Lr263 consumption significantly decreased hepatic lipogenic gene expressions stimulated by high-fat treatment. Administration of HK or live Lr263 significantly reduced hepatic oil red O staining and ameliorated the hepatic steatosis observed in high-fat treated rats. Our data suggested that similar to live Lr263, HK Lr263 exerted significant effects on attenuating obesity-induced metabolic abnormalities by reducing insulin resistance and hepatic steatosis formation.
1. Introduction
The global prevalence of obesity and obesity-related illnesses is increasing at an overwhelming rate. Between the years 1995 and 2000, the number of diagnosed cases of obesity increased by approximately 50%, exceeding 300 million persons aged 15 or older worldwide.1 Obesity is generally recognized as an increasingly important cause of childhood and adolescent morbidity worldwide and is a contributor to chronic diseases such as insulin resistance, non-alcoholic fatty liver disease (NAFLD) and coronary heart disease (CHD).2–4 Obesity is associated with increased adipose tissue macrophage (ATM) infiltration in rodents and humans.5,6 Previous studies from many rodent and human models suggested that increased ATM infiltration secreting several proinflammatory cytokines, including tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), and interleukin-1β (IL-1β), was responsible for obesity-associated inflammation and obesity-induced insulin resistance.7–9Recent evidence indicated that the intestinal microbiota played a crucial role in obesity.10 Moreover, a recently proposed hypothesis indicated that the gut flora could be implicated in metabolic diseases associated with obesity.11,12 Therefore, the intentional manipulation of community structure of gut microbiota may be a potential strategy to treat obesity. Accumulating results have demonstrated that probiotics conferring health benefits, either by manipulation of the intestinal microbiota or by affecting the host, have been proven to ameliorate metabolic and infectious diseases.13 Among these various probiotics, Lactobacilli strains have been reported to promote beneficial effects via anti-inflammatory actions and stabilizing the intestinal barrier, which might attenuate hepatic pathologies.14 Live probiotics such as Lactobacilli spp. and Bifidobacteria spp. colonizing in the host intestines were generally considered to be one of the important underlying mechanisms improving intestinal microbiota and enhancing the immunity of the host against various diseases.15 However, administering live probiotics to immunocompromised patients may be risky. The unwanted adverse effects of probiotics can be tolerated by healthy individuals, but may be risky to the patients with underlying conditions predisposing them to infection.16 Growing evidence has demonstrated that even heat-killed probiotics may also confer beneficial effects on the host.17 Compared to live probiotics, heat-killed probiotics may be advantageous for industrial production and consumer usage. More specifically, heat-killed probiotics are easier and more convenient to handle and distribute. Furthermore, heat-killed probiotics have the advantages of allowing a longer product shelf life and easier storage and transport. Therefore, additional scientific studies on the potent health-promoting effects of heat-killed probiotics will help to promote their academic and commercial usage.18
Lactobacillus reuteri GMNL-263 (Lr263), firstly isolated by us, is a representative probiotic bacteria that is commercially available as a healthy food in Taiwan. Our previous study demonstrated that administration of Lr263 in high-fructose fed rats significantly reduced insulin resistance as well as hepatic steatosis formation.19 However, the effects of heat-killed Lr263 on insulin resistance and its related complications have remained undetermined. We proposed that heat-killed Lr263 may exert similar beneficial effects as the live Lr263 did in vivo on preventing obesity associated insulin resistance and its related complications. Thus, the present study was conducted to investigate and compare the effects of heat-killed and live Lr263 on insulin resistance and the associated metabolic complications in high-fat diet (HFD)-fed rats.
2. Materials and methods
2.1. Preparation of Lactobacillus reuteri GMNL-263 (Lr263)
Lr263 was obtained from the culture collection of the GenMont Biotech Incorporation (Taiwan). Lactobacilli microorganisms for oral administration were prepared from overnight cultures of Lactobacilli and subcultured in Man Rogosa Sharpe (MRS) broth (Difco Laboratories, MI, USA) at 37 °C. Cultured bacterial cells were collected by centrifugation at 7000g and washed with sterile 0.85% NaCl. After washing, the collected bacteria were killed by heat treatment at 121 °C for 5 min in sterile 0.85% NaCl. The heat-killed bacteria were lyophilized and then kept at −80 °C until use. The bacterial cell count in the lyophilized powder was greater than 1011 cells per gram. The lyophilized bacteria powder was dissolved with RO water when orally administered to the rats.2.2. Animal model
Male Sprague-Dawley rats (BioLASCO Taiwan Co., Ltd, Yi-Lan, Taiwan) were housed in plastic cages in an animal room maintained with a 12 h light–dark cycle at 24 ± 1 °C and 50% humidity. Animals were allowed free access to water and food throughout the study. All of the experimental procedures described were approved by the GenMont Biotech Inc. Institutional Animal Care and Use Committee (IACUC) (approval no. GMN-20121201) and conducted according to the guidelines laid down by the IACUC. All surgeries were performed under sodium pentobarbital anesthesia, and all efforts were made to minimize suffering.2.3. Experimental design
Animals used in the study were divided into (1) the control group (control; n = 6) that consumed a standard diet, and was fed with RO water by an oral gavage every day; (2) the control + heat-killed Lr263 (HK Lr263) group (control + HK Lr263; n = 6) that consumed a standard diet, and was fed with HK Lr263 (2 × 109 cells per rat) by an oral gavage every day; (3) the control + live Lr263 group (control + live Lr263; n = 6) that consumed a standard diet, and was fed with live Lr263 (2 × 109 cells per rat) by an oral gavage every day; (4) the high-fat diet (HFD) group (n = 6) that consumed a high-fat diet (a diet containing 59.3% energy from fat), and was fed with RO water by an oral gavage every day; (5) the HFD + HK Lr263 group (n = 6) that consumed a high-fat diet, and was fed with HK Lr263 (2 × 109 cells per rat) by an oral gavage every day; (6) the HFD + live Lr263 group (n = 6) that consumed a high-fat diet, and was fed with live Lr263 (2 × 109 cells per rat) by an oral gavage every day. The oral gavage was performed between 9:00 AM and 11:00 AM every day. The components of the standard diet and the high-fat diet are listed in Table 1. At the end of the experimental period (12 weeks), rats were fasted overnight before sacrificing. The body weight of each rat was measured at pre-test, weekly thereafter, and at sacrifices.| Components (g kg−1) | Standard diet | High-fat diet |
|---|---|---|
| N.D.: none detectable. | ||
| Casein | 200 | 232 |
| L-Cystine | 3.0 | 3.0 |
| D,L-Methionine | N.D. | 3.5 |
| Corn starch | 397.48 | 137 |
| Maltodextrin | 132 | 150 |
| Sucrose | 100 | 162.58 |
| Cellulose | 50 | 50 |
| Cholesterol | N.D. | 1.9 |
| Mineral mix (AIN-93) | 35 | 40.60 |
| Calcium phosphate dibasic | N.D. | 4.64 |
| Vitamin mix (AIN-93) | 10 | 16.24 |
| Choline bitartrate | 2.5 | 5 |
| tert-Butylhydroquinone | 0.014 | 0.04 |
| Soybean oil | 70 | 40 |
| Lard | N.D. | 153.5 |
2.4. Blood and tissue sample collection
At the end of the experiments, the animals were sacrificed by CO2 anesthesia in a fasting state for at least 8 h. The blood samples were collected by cardiac puncture and centrifuged at 4000g for 10 min at 4 °C, and then stored at −80 °C. Liver and adipose tissues were weighed and washed with ice-cold saline and then stored at −80 °C until use.2.5. Biochemical parameters analysis
Serum biochemical parameters including triglyceride (TG), low density lipoprotein (LDL), cholesterol, aspartate aminotransferase (AST), and alanine aminotransferase (ALT) were analyzed using an automatic clinical analyzer (Hitachi High-Technologies Corporation, Tokyo, Japan). Serum glucose concentrations were determined using a glucometer (TaiDoc Technology Co., Taipei, Taiwan). Serum levels of insulin were measured using a commercial rat insulin ELISA kit (Millipore, MA, USA) according to the manufacturer's instructions. Homeostasis model assessment (HOMA) was computed with the formula: HOMA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) = (FPI × FPG)/22.5, where FPI is fasting plasma insulin and FPG is fasting plasma glucose.20
= (FPI × FPG)/22.5, where FPI is fasting plasma insulin and FPG is fasting plasma glucose.20
2.6. Oral glucose tolerance test (OGTT)
An oral glucose tolerance test was performed after 11 weeks treatment in this study. The diets were removed from animal cages for 12 h before the administration of an oral glucose load (2 g per kg of body weight). Blood samples were collected from the tail vein at 0, 30, 60, 90, and 120 min after glucose administration. Glucose concentrations were determined by using a glucometer (TaiDoc Technology Co., Taipei, Taiwan). The total glucose areas under the curve (AUCglucose) between 0 and 120 min represented the magnitude of the glucose response and were calculated as described previously.192.7. Determination of transepithelial electrical resistance (TEER)
The Ussing chamber (World Precision Instruments Inc., FL, USA) was used to measure the TEER and Isc of fresh colonic tissues.21 Colon segments were immediately excised postmortem and placed in a fresh Ussing chamber buffer (116.0 mM NaCl, 5.4 nM KCl, 0.4 mM MgCl2, 1.8 mM CaCl2, 5.5 mM glucose, 26.0 mM NaHCO3, and 0.9 mM NaH2PO4 at pH 7.4). Tissue segments were then opened along the mesenteric border, cut into a flat sheet, and rinsed free of luminal contents with phosphate buffered saline (PBS; Gibco Invitrogen, Carlsbad, CA, USA) prior to being vertically mounted in the Ussing chamber. The chamber exposed tissues to 10 mL of circulating oxygenated buffer at 37 °C on both the mucosal and serosal sides. The serosal bathing solution contained 10.0 mM glucose as a source of energy, which was osmotically balanced by mannitol (10.0 mM) on the mucosal side. The chamber was connected to matched voltage and current electrodes (EKV and EKC, resp.; World Precision Instruments Inc.) through a KCl saturated agar bridge to monitor the potential differences across the tissues. Following equilibration for 30 min, the Isc, an index of net active ion passage, was recorded continually in the voltage clamp mode at 0 potential difference. A voltage pulse of 2 mV was imposed for 3 s at 60 second intervals across the colon tissues in order to estimate the TEER (expressed as Ω cm2), as a surface area normalized ratio of imposed voltage pulse to the observed deflection in the resultant circuit current, according to the Ohm's law formula:where A is an exposed window surface area of 1 cm2.
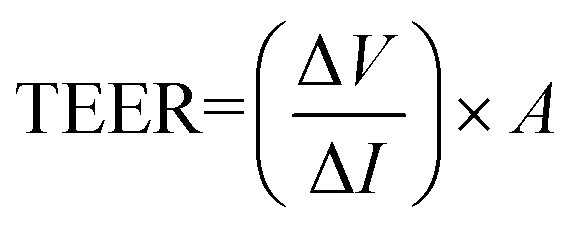
2.8. DNA extraction from fecal samples
Fecal samples were collected after sacrificing and immediately stored at −80 °C until analyzed. DNA extraction from 200 mg of stools was performed using a QIAamp DNA stool Mini kit (Qiagen, Hilden, Germany) following the manufacturer's instructions. The DNA concentration was determined by measuring the absorbance at 260 nm, and the purity was estimated by determining the A260/A280 ratio with a Nanodrop spectrophotometer (Nanodrop Technologies, Wilmington, DE).2.9. RNA isolation from adipose and liver tissues
Total RNA was extracted from adipose and liver tissues using an RNeasy Mini kit (Qiagen). RNA concentrations were determined spectrophotometrically, and 0.25 mg total RNA was reverse transcribed using a SuperScript® II Reverse Transcriptase kit (Invitrogen, Karlsruhe, Germany) followed by a DNase digestion step (Fermentas, St Leon Rot, Germany).2.10. Quantification of microbial and tissue gene expressions by real-time qPCR
Specific primers targeting different bacterial genera were used to characterize the fecal microbiota by real-time qPCR.22 Briefly, real-time qPCR experiments were performed with a Light Cycler 2.0 PCR sequence detection system using a Fast Start DNA Master SYBR Green kit (Roche Diagnostics, Indianapolis, IN). All PCR tests were carried out in duplicate with a final volume of 20 μL containing 1 μL of each fecal microbiota DNA preparation and 200 nM of each primer (Table 2). The PCR primers for the different strains used were obtained from the Bioresource Collection and Research Center (BCRC) (Bifidobacterium bifidum, BCRC 11845, Lactobacillus reuteri, BCRC 14691, Clostridium difficile, BCRC 17678, Pseudomonas aeruginosa, BCRC 10261). The data presented here were the mean values of duplicate qPCR analyses.| Target | Sequence |
|---|---|
| β-Actin | Forward: 5′-TGACAGGTGCAGAAGGAGA-3′ |
| Reverse: 5′-TAGAGCCACCAATCCACACA-3′ | |
| PPAR-γ | Forward: 5′-AACCGGAACAAATGCCAGTA-3′ |
| Reverse: 5′-TGGCAGCAGTG-GAAGAATCG-3′ | |
| SREBP-1c | Forward: 5′-TAGAGCATATCCCCCAGGTG-3′ |
| Reverse: 5′-GGTACGGGCCACAAGAAGTA-3′ | |
| TNF-α | Forward: 5′-CTGTCTACTGAACTTCGGGGTGAT-3′ |
| Reverse: 5′-CATCAGTTCTATGGCCCAGACC-3′ | |
| IL-6 | Forward: 5′-GACTGATGTTGTTGACAGCCACTGC-3′ |
| Reverse: 5′-TAGCCACTCCTTCTGTGACTCTAACT-3′ | |
| Lactobacillus | Forward: 5′-GAGGCAGCAGTAGGGAATCTTC-3′ |
| Reverse: 5′-GGCCAGTTACTACCTCTATCCTTCTTC-3′ | |
| Bifidobacterium | Forward: 5′-CGCGTCYGGTGTGAAAG-3′ |
| Reverse: 5′-CCCCACATCCAGCATCCA-3′ | |
| Clostridium | Forward: 5′-GACGCCGCGTGAAGGA-3′ |
| Reverse: 5′-AGCCCCAGCCTTTCACATC-3′ | |
| Pseudomonas | Forward: 5′-CAAAACTACTGAGCTAGAGTACG-3′ |
| Reverse: 5′-TAAGATCTCAAGGATCCCAACGGCT-3′ |
The PCR primers for adipose and liver tissue genes were designed using Primer3 software (Table 2). Real-time qPCR experiments were performed with a Light Cycler 2.0 PCR sequence detection system using a Fast Start DNA Master SYBR Green kit (Roche Diagnostics). All PCR tests were carried out in duplicate with a final volume of 20 μL containing 1 μL of each adipose or hepatic tissue cDNA preparation and 200 nM of each primer (Table 2). The comparative CT-method was used to determine the amount of the target gene, normalized to an endogenous reference gene (β-actin) and relative to a calibrator (2−ΔΔCT).
2.11. Immunohistochemistry
Adipose tissues collected from the rats were fixed and stained with anti-F4/80 antibodies (Nichirei, Tokyo, Japan) to detect macrophage infiltrations. Sections were incubated with an anti-F4/80 antibody (dilution, 1:100) overnight at 4 °C and a secondary antibody of Histofine Simple Stain Max PO (rat) for 30 min. Images were captured using a Zeiss Stallion Dual Detector Imaging System with the Intelligent Imaging Innovations Software (Carl Zeiss Microimaging Inc., NY, USA).
2.12. Liver steatosis evaluation and histological analysis
Approximately 50 mg of each liver tissue was homogenized in PBS. The levels of total cholesterol and TG levels were analysed by a colorimetric assay (Thermo Fisher Scientific Inc., Middletown, USA).19 Additionally, oil red O staining was performed using freshly isolated rat hepatic tissues fixed in 10% neutral buffered formalin at 4 °C for two days, transferred to a 20% sucrose solution for two days, and then frozen in Tissue-Tek. Frozen tissues were sectioned at 8 μm on a Microm cryostat set to −19 °C and then air dried. The liver sections were rehydrated and stained with oil red O from 5% stock solution in isopropanol diluted 3:2 with distilled water for 20 min. Mayer's hematoxylin was used for counterstaining.23 For hematoxylin and eosin staining, the liver tissues isolated from rats were fixed in 10% formalin and embedded in paraffin. Three micrometer thick sections were obtained, and deparaffinized in xylene. The sections were stained with hematoxylin–eosin. The results of staining were viewed and photographed with an Olympus microscope (Olympus Corporation, Tokyo, Japan).
2.13. Statistical analysis
All experimental data were presented as the mean ± standard error of the mean (SEM). Data were analyzed using a one-way analysis of variance using SPSS 17.0 statistical software (SPSS Inc., Chicago, IL, USA). A value of P < 0.05 was considered to be statistically significant.3. Results
3.1. Effects of Lactobacillus reuteri GMNL-263 on body weights and various tissue weights in HFD rats
At the beginning and the end of the experimental periods the body weights of rats in all six groups were recorded. The initial body weights of rats showed no significant difference among groups. After a 12 week experimental period, the average body weight of rats in the HFD group was significantly increased compared to the control group (Table 3). However, the body weight and weight gain were reduced in heat-killed or live Lr263 groups. In addition, liver and adipose tissue weights, which were significantly increased in the HFD group, were significantly decreased after heat-killed Lr263 and live Lr263 treatments compared to the HFD group (Table 3). The body weights as well as liver and adipose tissue weights among the control group, the control + HK Lr263 group, and the control + live Lr263 group showed no significant difference.| Control | Control + HK Lr263 | Control + live Lr263 | HFD | HFD + HK Lr263 | HFD + live Lr263 | |
|---|---|---|---|---|---|---|
| Initial body weight (g) | 190.0 ± 8.60 | 188.5 ± 9.90 | 189.5 ± 7.10 | 188.6 ± 6.50 | 191.5 ± 5.60 | 191.3 ± 2.50 |
| Final body weight (g) | 462.5 ± 20.85 | 420.2 ± 34.56 | 433.2 ± 28.42 | 542.7 ± 34.93# | 482.2 ± 23.91* | 490.6 ± 31.50* |
| Weight gain (g) | 272.2 ± 18.05 | 232.7 ± 10.71 | 241.7 ± 9.41 | 354.0 ± 29.66# | 291.8 ± 26.18* | 299.8 ± 31.10* |
| Adipose tissue/body weight ratio (%) | 3.7 ± 0.61 | 2.8 ± 0.55 | 2.9 ± 0.85 | 6.7 ± 0.94# | 4.7 ± 0.42* | 5.6 ± 0.72* |
| Liver/body weight ratio (%) | 2.6 ± 0.24 | 2.6 ± 0.15 | 2.5 ± 0.32 | 2.9 ± 0.11# | 2.5 ± 0.11* | 2.3 ± 0.10* |
3.2. Effects of Lactobacillus reuteri GMNL-263 on various biochemical markers
The results from Table 4 revealed that liver toxicity biomarkers, namely alanine transaminase (ALT) and aspartate transaminase (AST), were significantly elevated in the HFD group as compared to the control group. However, serum ALT and AST were reduced in the heat-killed or live Lr263 groups. Regarding serum lipid profile assays, the HFD group showed a significant increase in serum triglyceride, total cholesterol and LDL/HDL levels compared to the normal group. Administration of heat-killed or live Lr263 significantly reduced serum triglyceride, total cholesterol and LDL/HDL ratio levels. These results suggested that both heat-killed and live Lr263 have lipid-lowering effects. In order to investigate whether Lr263 influenced insulin sensitivity, circulating insulin and glucose concentrations were monitored for all the groups under fasting conditions. We found higher serum insulin levels and blood glucose concentrations in the HFD group as compared to the control group. However, the elevated serum insulin and glucose levels in the HFD group were reduced after the administration of heat-killed or live Lr263. Additionally, the biochemical markers among the control group, control + HK Lr263 group, and control + live Lr263 group were not significantly different.| Control | Control + HK Lr263 | Control + live Lr263 | HFD | HFD + HK Lr263 | HFD + live Lr263 | |
|---|---|---|---|---|---|---|
| AST conc. (U dl−1) | 113.98 ± 19.19 | 146.37 ± 22.13 | 131.37 ± 19.88 | 185.16 ± 5.40# | 115.37 ± 16.26* | 127.7 ± 14.05* |
| ALT conc. (U dl−1) | 51.13 ± 9.57 | 53.25 ± 2.13 | 55.47 ± 3.26 | 103.37 ± 58.68# | 48.67 ± 8.34* | 56.55 ± 11.53* |
| Total cholesterol conc. (mg dl−1) | 59.22 ± 1.90 | 54.77 ± 11.81 | 53.98 ± 8.44 | 96.27 ± 19.05# | 47.63 ± 3.31* | 52.43 ± 11.20* |
| Triglyceride (mg dl−1) | 52.17 ± 16.02 | 34.98 ± 6.22 | 36.98 ± 4.92 | 96.27 ± 19.05# | 39.03 ± 9.73* | 45.88 ± 8.69* |
| LDL/HDL ratio (%) | 14.12 ± 2.66 | 15.74 ± 3.44 | 16.22 ± 4.15 | 21.60 ± 4.86# | 12.54 ± 2.21* | 13.14 ± 2.03* |
| Insulin conc. (ng ml−1) | 1.06 ± 0.25 | 0.90 ± 0.36 | 1.14 ± 0.26 | 3.26 ± 0.60# | 1.14 ± 0.31* | 1.38 ± 0.25* |
| Glucose conc. (mg dl−1) | 79.83 ± 4.31 | 81.83 ± 4.88 | 87.74 ± 5.17 | 89.83 ± 6.68 | 79.8 ± 6.18* | 80.75 ± 4.50* |
3.3. Effects of Lactobacillus reuteri GMNL-263 on glucose tolerance
To assess the impact of Lr263 on blood glucose homeostasis, an oral glucose tolerance test (OGTT) at week 12 was performed. The results showed that the AUCglucose value in the HFD group was higher than that of the control group (Fig. 1). Furthermore, the AUCglucose values shown in Fig. 1 clearly reflected that administration of heat-killed or live Lr263 downregulated the blood glucose concentrations increased by HFD treatment. However, no significant differences in AUCglucose values among the control group, the control + HK Lr263 group, and the control + live Lr263 group were observed. | ||
| Fig. 1 Effect of Lactobacillus reuteri GMNL-263 on the oral glucose tolerance test (OGTT) in HFD rats treated for 12 weeks. The test was performed at 12 weeks. The blood samples were collected from the tail veins at 0, 15, 30, 60, 90, and 120 min after glucose administration (2 g per kg of body weight). The blood glucose concentrations were determined by using a glucometer. AUCglucose represented the area under the curve for glucose. Data were expressed as the mean ± SEM (n = 6). Control: control diet group; control + HK Lr263: heat-killed Lactobacillus reuteri GMNL-263 and control diet group; control + live Lr263: live Lactobacillus reuteri GMNL-263 and control diet group; HFD: high-fat diet group; HFD + HK Lr263: heat-killed Lactobacillus reuteri GMNL-263 and high-fat diet group; HFD + live Lr263: live Lactobacillus reuteri GMNL-263 and high-fat diet group. #P < 0.05 compared with control group; *P < 0.05 compared with HFD group. | ||
3.4. Effects of Lactobacillus reuteri GMNL-263 on fecal microbial populations and intestinal barrier
In general, Bifidobacteria and Lactobacilli have been regarded as beneficial microflora species,24,25 whereas Clostridia and Pseudomonas would be harmful as a consequence of their metabolic activities.26,27 In our present study, HFD treatment resulted in an increase in Clostridium and Pseudomonas copy numbers, which was ameliorated after heat-killed or live Lr263 treatments (Fig. 2A). On the other hand, the decreased Bifidobacterium and Lactobacillus copy numbers observed in the HFD group were significantly increased after treatments with heat-killed or live Lr263 (Fig. 2B). Furthermore, the copy numbers of pathogenic and probiotic bacteria among the control group, the control + HK Lr263 group, and the control + live Lr263 group were not significantly different. The gut barrier function was evaluated through the measurement of the transepithelial electrical resistance (TEER) in the rat intestine. Compared to the control rats, the TEER in HFD rats was markedly decreased. However, administration of heat-killed or live Lr263 significantly reversed the decreased TEER in HFD rats (Fig. 2C). | ||
| Fig. 2 Effect of Lactobacillus reuteri GMNL-263 on intestinal microbiota profiles and gut permeability in HFD rats treated for 12 weeks. Real-time quantitative RT-PCR analysis of (A) Clostridium and Pseudomonas and (B) Bifidobacterium and Lactobacillus copy numbers of HFD rats. (C) Effect of Lr263 on transepithelial resistance (TEER) in HFD rats. Data were expressed as the mean ± SEM (n = 6). Control: control diet group; control + HK Lr263: heat-killed Lactobacillus reuteri GMNL-263 and control diet group; control + live Lr263: live Lactobacillus reuteri GMNL-263 and control diet group; HFD: high-fat diet group; HFD + HK Lr263: heat-killed Lactobacillus reuteri GMNL-263 and high-fat diet group; HFD + live Lr263: live Lactobacillus reuteri GMNL-263 and high-fat diet group. #P < 0.05 compared with control group; *P < 0.05 compared with HFD group. | ||
3.5. Effects of Lactobacillus reuteri GMNL-263 on inflammation
The plasma monocyte chemoattractant protein-1 (MCP-1) level in the HFD group was markedly increased when compared to the control group. However, the elevation of MCP-1 in the HFD group was downregulated by administration of HK or live Lr263 (Fig. 3A). Previous studies indicated that infiltrated macrophages were strongly involved in adipocyte inflammation;5,28 therefore, we investigated the effect of Lr263 on macrophage infiltration in adipose tissue by immunohistochemical analysis with an anti-F4/80 antibody. As shown in Fig. 3B, the number of F4/80-positive cells was increased significantly in HFD (Fig. 3B-d) fed rats compared to the control group (Fig. 3B-a), but was significantly reduced by HK or live Lr263 treatments (Fig. 3B-e and B-f, respectively). No significant differences in macrophages infiltration in adipose tissue among the control group, the control + HK Lr263 group, and the control + live Lr263 group (Fig. 3B-a, B-b, and B-c, respectively) were noticed. These results revealed that amelioration of adipocyte inflammation by HK or live Lr263 may be attributed to decreased macrophage infiltration. To investigate the potential mechanisms by which HK or live Lr263 improve the metabolic disorder in HFD fed rats, we analyzed the gene expressions related to inflammation in adipose and hepatic tissues. Two important insulin resistance-related inflammatory cytokines, namely TNF-α and IL-6, produced by adipose and hepatic tissue were evaluated by real-time PCR. As shown in Fig. 3C, the levels of TNF-α and IL-6 in adipose and hepatic tissues were significantly increased in the HFD group as compared to the control group, and this enhancing effect was markedly abrogated by HK or live Lr263 treatments. Furthermore, there was no significant difference in these inflammatory parameters among the control group, the control + HK Lr263 group, and the control + live Lr263 group.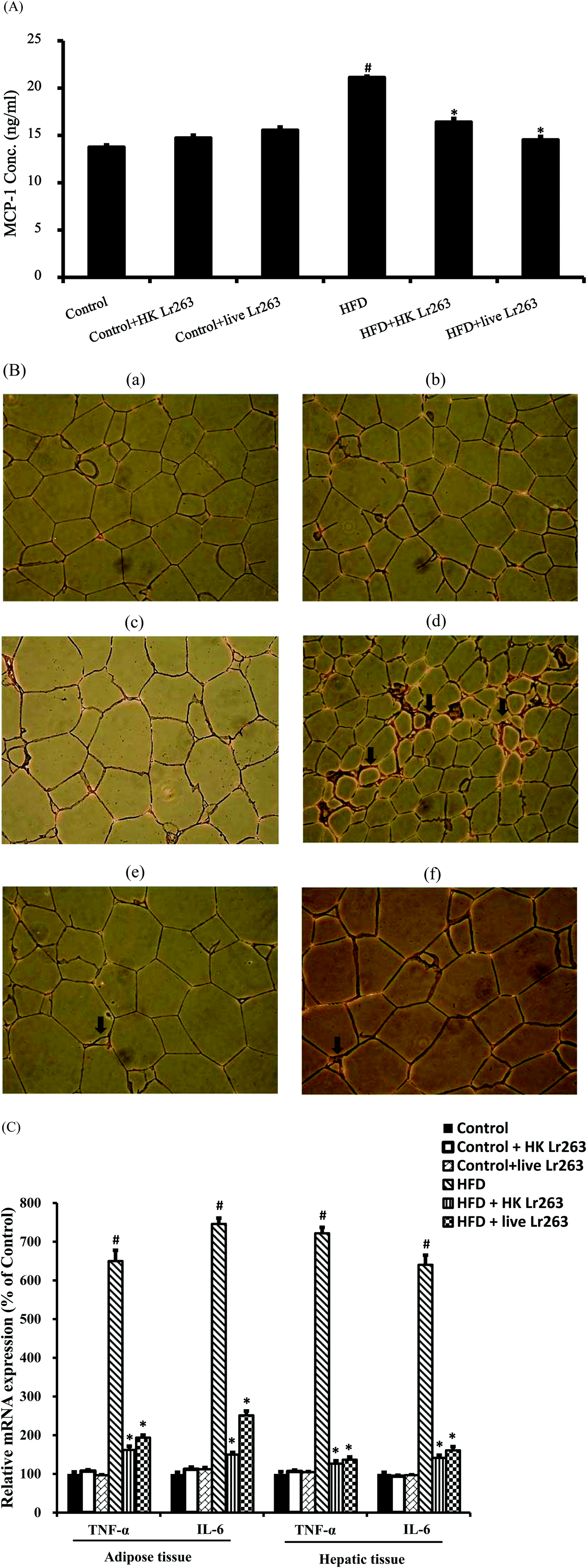 | ||
| Fig. 3 Lactobacillus reuteri GMNL-263 attenuates the state of inflammation in high-fat diet rats. (A) Serum levels of MCP-1; (B) at the end of the experimental period (12 weeks), the adipose tissue obtained from sacrificed rats was subjected to F4/80 immunostaining for evaluation of macrophage infiltration in adipose tissue. The histological sections of rat adipose tissue were observed at a magnification of 200×. (B-a) Control: control diet group; (B-b) heat killed Lactobacillus reuteri GMNL-263 and control diet group; (B-c) live Lactobacillus reuteri GMNL-263 and control diet group; (B-d) high-fat diet group; (B-e) heat killed Lactobacillus reuteri GMNL-263 and high-fat diet group; (B-f) live Lactobacillus reuteri GMNL-263 and high-fat diet group. (C) Pro-inflammation cytokine gene expressions (TNF-α and IL-6) in adipose and hepatic tissues. Data were expressed as the mean ± SEM (n = 6). #P < 0.05 compared with control group; *P < 0.05 compared with HFD group. | ||
3.6. Effects of Lactobacillus reuteri GMNL-263 on high-fat diet-induced insulin resistance
Assessment of insulin resistance using the HOMA index has been the most frequently employed technique in both clinical practice and epidemiological studies. In the present study, the HOMA index was significantly higher in the HFD group compared to the control group, but was significantly reduced by HK or live Lr263 treatments (Fig. 4A). There was no significant difference in the HOMA index among the control group, the control + HK Lr263 group, and the control + live Lr263 group. We next examined the insulin resistance associated gene expressions in the adipose and hepatic tissues. The results showed that the gene expressions of peroxisome proliferator-activated receptor-γ (PPAR-γ) and insulin receptor substrate-1 (IRS-1), decreased by HFD treatment, were significantly upregulated after HK or live Lr263 treatments in both adipose and hepatic tissues (Fig. 4B). No significant differences in the expressions of PPAR-γ and IRS-1 mRNA among the control group, the control + HK Lr263 group, and the control + live Lr263 group were observed.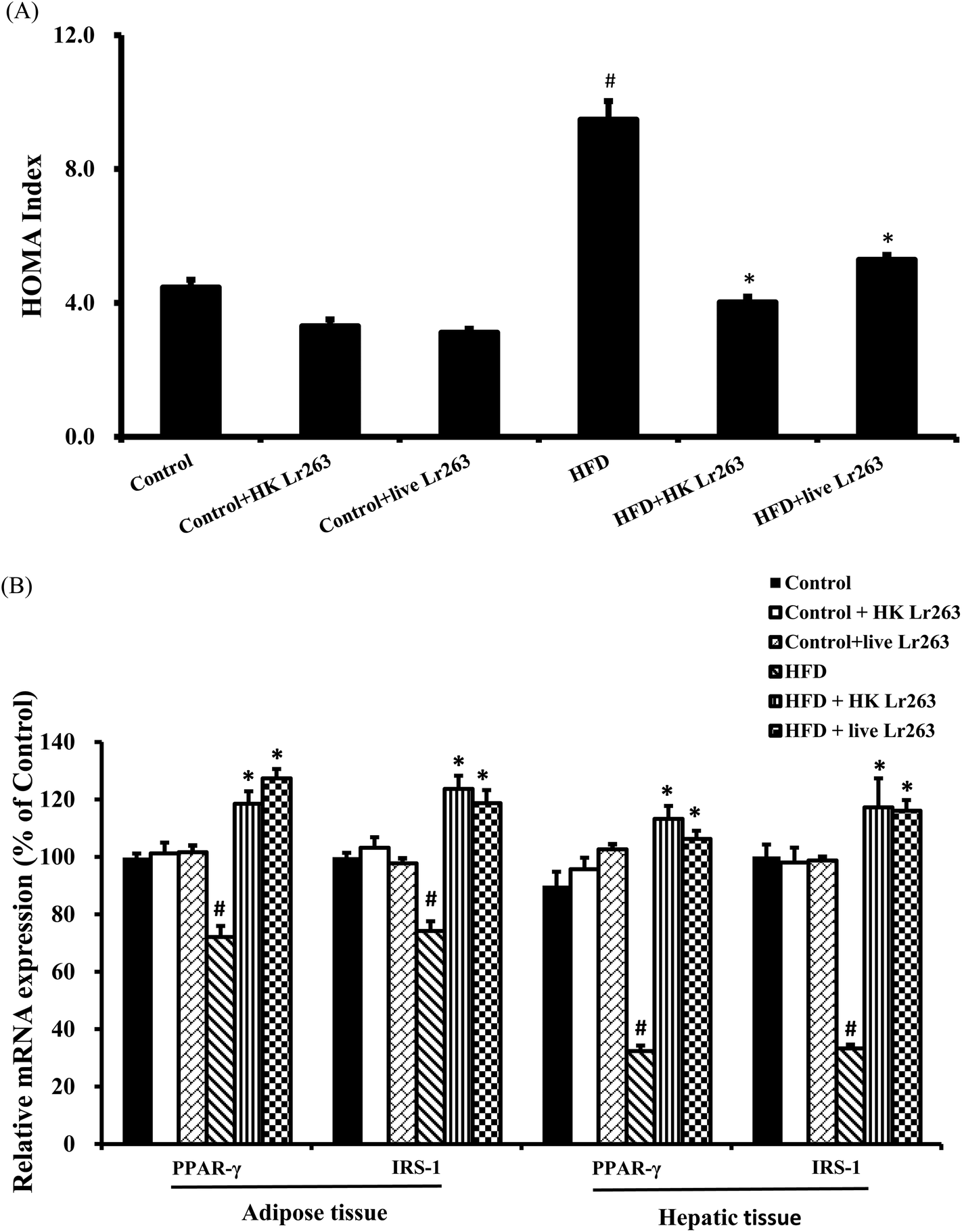 | ||
| Fig. 4 Effect of Lactobacillus reuteri GMNL-263 on the homeostasis model assessment (HOMA) index and expression of insulin resistance related genes. (A) HOMA index; (B) insulin sensitivity regulation related gene expressions (PPAR-γ and IRS-1) in adipose and hepatic tissues. Data were expressed as the mean ± SEM (n = 6). Control: control diet group; control + HK Lr263: heat killed Lactobacillus reuteri GMNL-263 and control diet group; control + live Lr263: live Lactobacillus reuteri GMNL-263 and control diet group; HFD: high-fat diet group; HFD + HK Lr263: heat killed Lactobacillus reuteri GMNL-263 and high-fat diet group; HFD + live Lr263: live Lactobacillus reuteri GMNL-263 and high-fat diet group. #P < 0.05 compared with control group; *P < 0.05 compared with HFD group. | ||
3.7. Effects of Lactobacillus reuteri GMNL-263 on liver lipid accumulation
Our present findings demonstrated that feeding a HFD caused a significant increase in hepatic TG and cholesterol levels (Fig. 5A). Administration of HK or live Lr263 reversed the increased levels of hepatic TG and cholesterol (Fig. 5A). Additionally, the expressions of lipogenic-related genes including fatty acid synthase (FAS), sterol regulatory element-binding transcription factor 1c (SREBP-1c), and carbohydrate-responsive element-binding protein (ChREBP) were markedly increased in the HFD group as compared to the control group. However, following the treatments with HK or live Lr263, the increased expressions of these lipogenic-related genes were significantly decreased (Fig. 5B). There was no significant difference in hepatic lipid profiles and lipogenic-related gene expressions among the control group, the control + HK Lr263 group, and the control + live Lr263 group.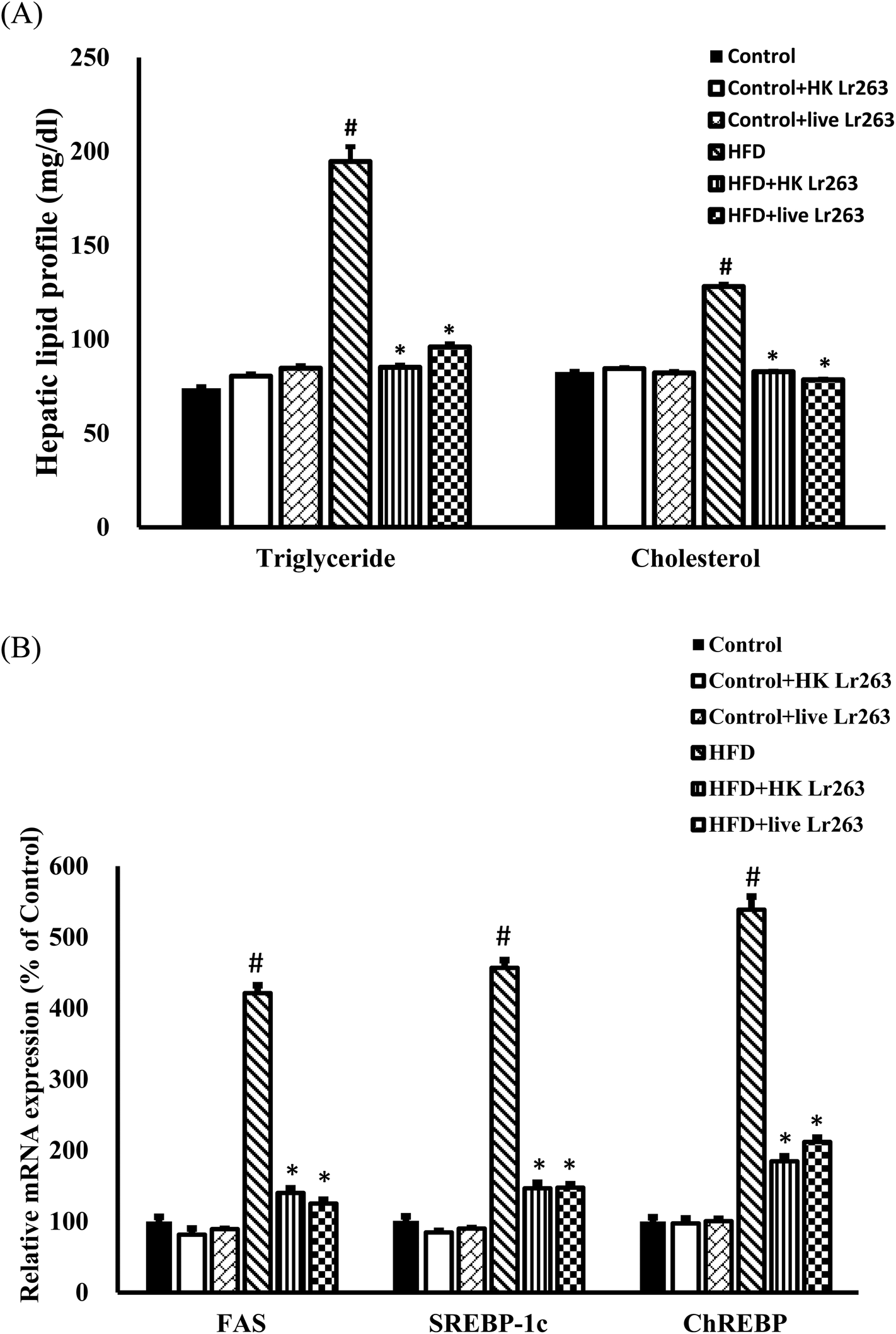 | ||
| Fig. 5 Effect of Lactobacillus reuteri GMNL-263 on hepatic lipid profiles and expression of hepatic lipogenic related genes in high-fat diet rats treated for 12 weeks. (A) Hepatic lipid profile. (B) Real-time quantitative RT-PCR analysis of lipogenic related genes in the liver (FAS, SREBP-1c and ChREBP). Data were expressed as the mean ± SEM (n = 6). Control: control diet group; control + HK Lr263: heat killed Lactobacillus reuteri GMNL-263 and control diet group; control + live Lr263: live Lactobacillus reuteri GMNL-263 and control diet group; HFD: high-fat diet group; HFD + HK Lr263: heat killed Lactobacillus reuteri GMNL-263 and high-fat diet group; HFD + live Lr263: live Lactobacillus reuteri GMNL-263 and high-fat diet group. #P < 0.05 compared with control group; *P < 0.05 compared with HFD group. | ||
3.8. Effects of Lactobacillus reuteri GMNL-263 on hepatic histopathology
Representative histological photomicrographs of liver H&E staining (Fig. 6, left column) as well as oil red O staining (Fig. 6, right column) are shown in Fig. 6. Compared to the control group (Fig. 6A, left), high-fat diet treatment (Fig. 6D, left) caused prominent diffuse macrovesicular steatosis in the liver. However, administration of HK or live Lr263 (Fig. 6E, left and F, left, respectively) significantly reversed the formation of hepatic steatosis induced by high-fat diet treatment. In addition, high-fat diet feeding (Fig. 6D, right) induced obvious fat accumulation in liver, demonstrated by oil red O staining, when compared to the control group (Fig. 6A, right). However, administration of HK or live Lr263 significantly reduced hepatic lipid droplet accumulation (Fig. 6E, right and F, right, respectively) in the rats. No significant differences in hepatic steatosis and fat accumulation between the control group and the control + HK Lr263 group (Fig. 6B, left and right) or between the control group and the control + Live Lr263 group (Fig. 6C, left and right) were noticed.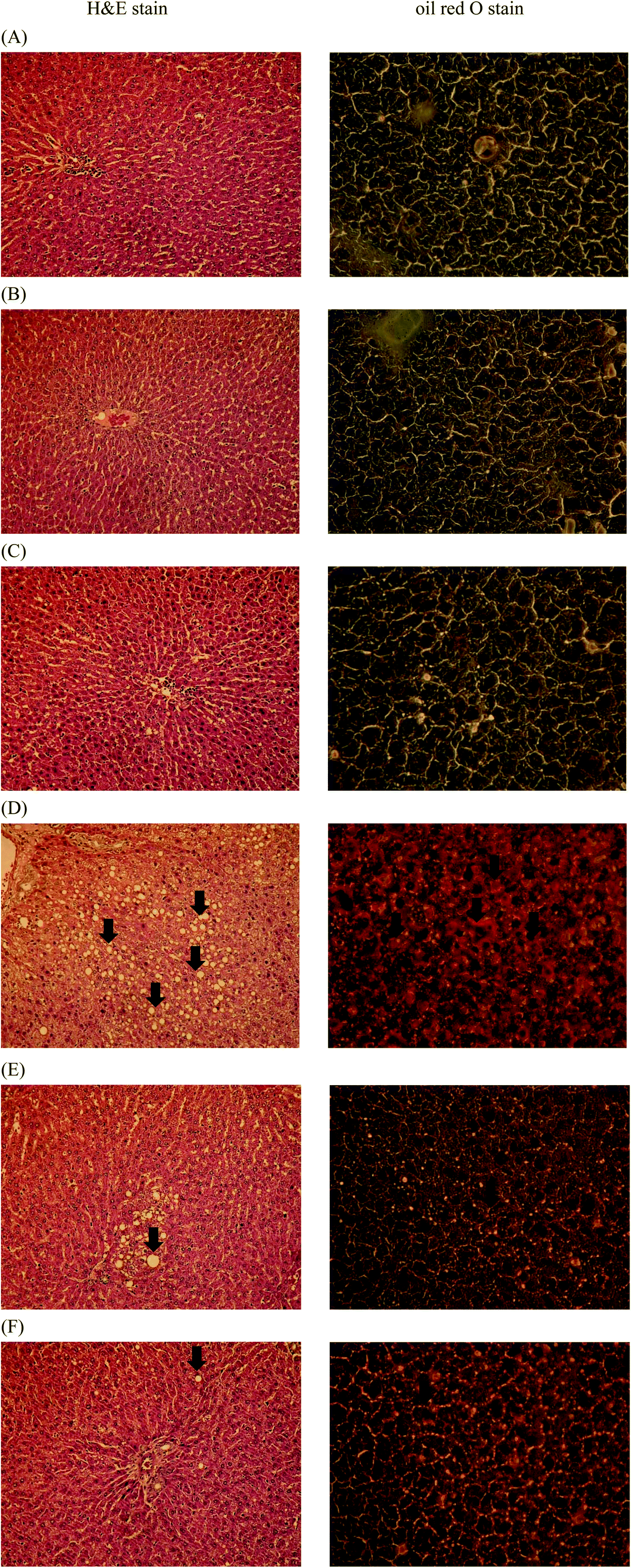 | ||
| Fig. 6 Effects of Lactobacillus reuteri GMNL-263 on hepatic steatosis in treated rats. At the end of the experimental period (12 weeks), the livers obtained from sacrificed rats were subjected to hematoxylin and eosin (H&E) staining (left column) and oil red O staining (right column) for the evaluation of hepatic steatosis. The histological sections of rat liver were observed at a magnification of 200×. (A) Control diet group; (B) heat killed Lactobacillus reuteri GMNL-263 and control diet group; (C) live Lactobacillus reuteri GMNL-263 and control diet group; (D) high-fat diet group; (E) heat killed Lactobacillus reuteri GMNL-263 and high-fat diet group; (F) live Lactobacillus reuteri GMNL-263 and high-fat diet group. The results shown here were from one representative experiment of six different samples with similar results. | ||
4. Discussion
In our current study, feeding a HFD resulted in increased body weight gain, induced glucose intolerance, and promoted fat accumulation in the liver. However, administration of HK or live Lr263 was able to prevent obesity, insulin resistance and hepatosteatosis in HFD rats. Furthermore, our findings demonstrated that feeding HK or live Lr263 could ameliorate the metabolic consequences of the high-fat diet-induced obesity in rats by suppressing the inflammatory response.A previous study demonstrated that feeding a high-fat diet influenced the type and amounts of gut microbes. These alterations in guts have shown a close association with impaired gut barrier function, endotoxemia, and increased adipocytes hyperplasia in visceral white adipose tissue (WAT).29 Furthermore, growing evidence demonstrated that probiotics administration significantly improved the integrity of the intestinal barrier30,31 which reduced intestinal endotoxin leakage and consequently resulted in suppression of inflammatory response.32 Our present studies demonstrated that the proportions of Bifidobacterium spp. and Lactobacillus spp. (generally considered to be typical probiotic bacteria) were reduced, whereas Clostridium spp. and Pseudomonas spp. (generally considered to be typical pathogenic bacteria) were increased in HFD rats. In addition, the level of TEER in the HFD group was lower than that of the control group. This result was in agreement with a previous study suggesting that HFD treatment resulted in metabolic changes that impaired gut barrier function as reflected by a decreased TEER in the proximal colon.33 However, in our current results, HK or live Lr263 consumption not only significantly increased the number of Bifidobacteria and Lactobacilli, and in contrast, decreased the number of Clostridia and Pseudomonas in the feces but also decreased the value of TEER when compared to those of the HFD rats.
One of the important characteristics of obesity is massive macrophage infiltration in adipose tissue. Numerous studies have reported that corresponding with chronic inflammation of adipose tissue was an increase in macrophage infiltration.5,28,34 Indeed, several studies have shown that abrogation of macrophage infiltrations in adipose tissue could be a potential intervention attenuating insulin resistance and other obesity complications.6,24,28 Recent studies have shown that MCP-1, belonging to the chemotactic cytokines family, played a critical role in macrophage activation and recruitment.35 In our study, we found that in comparison with the control group, the enhanced serum levels of MCP-1 in the HFD group were significantly decreased by HK or live Lr263 treatments. Furthermore, the results from immunohistochemical staining in adipose tissue demonstrated that the increased macrophage infiltrations in the HFD group were dramatically decreased by HK or live Lr263 administration. It was demonstrated that hyperglycemia resulted in the generation of reactive oxygen species (ROS), ultimately leading to increased oxidative stress in a variety of tissues. Oxidative stress is supposed to stimulate the production of a variety of pro-inflammatory cytokines, which may ultimately lead to insulin resistance.35–37 Both TNF-α and IL-6, two main pro-inflammatory cytokines released by adipose and other tissues, can inhibit insulin signaling, and TNF-α may have a crucial role in the development of insulin resistance in obesity-diabetes.38 As shown in our study, the expressions of TNF-α and IL-6 mRNA in adipose and hepatic tissues in the HFD group were higher than those of the control group. Feeding HK or live Lr263 abrogated the TNF-α and IL-6 gene expressions in the HFD group. These results implied that reducing macrophage infiltrations in adipose tissue and downregulated pro-inflammatory cytokine expression in both adipose and hepatic tissues might be one potential mechanism by which HK or live Lr263 improved insulin resistance in HFD rats.
It was shown that IRS-1 played an important role in the metabolic actions of insulin in skeletal muscle and adipose tissue.39 In obese and type 2 diabetic patients, IRS-1 expression was markedly reduced in adipocytes, resulting in decreased IRS-1-associated PI 3-kinase activity, and was presumably responsible for the failure of insulin in suppressing lipolysis as well as reducing insulin-stimulated glucose transport.40 In addition, PPAR-γ has been strongly implicated in the regulation of systemic insulin sensitivity.41 There was some evidence indicating that TNF-α, IL-1β, and IL-6 impaired adipogenesis and lipid accumulation in 3T3-L1 preadipocytes.42 More specifically, IL-6 treatment reduced PPAR-γ and IRS-1 expressions; while TNF-α treatment increased secretion of IL-6 and MCP-1 from 3T3-L1 preadipocytes.43 Our study demonstrated that the expressions of IRS-1 and PPAR-γ mRNA in both adipose and hepatic tissues of HFD rats were decreased. However, HK or live Lr263 administration restored the IRS-1 and PPAR-γ mRNA expressions reduced by high fat treatment. Our data implied that HK or live Lr263 exerted their effects on decreasing inflammation and improving insulin resistance by altering intestinal microbiota and restoring the integrity of guts in HFD rats.
The major pathogenesis of metabolic syndrome is the development of insulin resistance caused by accumulation of visceral fat, which promotes the elevation of blood pressure, dyslipidemia, and dysregulation of glucose metabolism.44 Among the target tissues of insulin, liver is the principal regulator of glucose and lipid metabolism by controlling hepatic glucose production, glycogen storage, and lipogenesis.44 As shown in our study, administration of HK or live Lr263 dramatically decreased the serum levels of important components of metabolic syndrome, including serum glucose, insulin, LDL/HDL ratio, TG, and cholesterol, enhanced by high fat treatment. In addition to serum levels, the increased hepatic levels of TG and cholesterol treated with HFD were also found to be suppressed by oral administration of HK or live Lr263. Our data implicated that administration of HK or live Lr263 could improve insulin resistance via downregulation of both serum and hepatic lipid concentrations. To elucidate the possible mechanisms by which HK or live Lr263 suppressed hepatic lipid accumulation in HFD rats, we assessed liver mRNA levels of genes involved in lipogenesis. SREBP-1c and ChREBP were two important transcription factors reported to be involved in the transcriptional activation of genes encoding rate-limiting enzymes in the lipogenesis, and were also associated with increased de novo lipogenesis in NAFLD.45 Upon insulin stimulation, SREBP-1c regulates hepatic fatty acid and TG biosynthesis by upregulating the expression of key genes, such as FAS.46 Furthermore, administration of probiotics has been reported to lower the hepatic TG and cholesterol content in rats with high-fat diet-induced obesity.47,48 Our current results revealed that high fat treatment markedly induced the expressions of the lipogenic genes SREBP-1c, ChREBP, and FAS in the rat liver. Administration of HK or live Lr263 counteracted the increase in hepatic lipogenic genes in HFD rats. These results suggested that HK or live Lr263 consumption not only suppressed hepatic lipid accumulation, but also subsequently improved insulin resistance in HFD rats.
Insulin resistance plays a crucial role in hepatic steatosis,49 which is closely associated with obesity and often accompanied by marked abdominal adiposity.50 Fat accumulation in the liver and insulin resistance cause as well as potentiate each other, creating a vicious cycle of metabolic dysfunction resulting in the development of hepatic steatosis.51 The results from our histological analysis demonstrated that a prominent hepatic steatosis was observed in HFD rats, whereas administration of HK or live Lr263 significantly ameliorated fat accumulation in the liver as compared to the HFD group. These findings indicated that hyperlipidemia and insulin resistance caused by high fat treatment were improved by administration of HK or live Lr263, which may ameliorate the development of hepatic steatosis in HFD rats.
Previous studies indicated that the therapeutic effects of live probiotics might be related to a variety of direct and indirect mechanisms, including modulation of local microbiota, epithelial barrier function, and the immune system. Recently, however, some non-viable probiotics have been shown to exhibit beneficial effects on human health similarly to live probiotics.52 Based on our present studies, we proposed two possible mechanisms by which HK Lr263 improved metabolic syndrome. The first was that consumption of HK Lr263 might suppress dietary fat absorption in the intestine. Previous studies suggested that the decreased fat absorption with probiotic was probably due to its binding of intestinal fat.53 An in vitro study evaluating probiotic cholesterol assimilation showed that Lactobacillus reuteri NCIMB 701089 assimilated over 67% of the cholesterol. Moreover, the cholesterol assimilation ability of a probiotic was independent of whether they were alive or in a probiotic bacterial culture.54 In addition, one study showed that the levels of TG and cholesterol were significantly decreased in the liver and significantly increased in the feces after HK Lr263 treatment.55 These results suggested that HK Lr263 exerted a lipid-elimination effect in the gut of hamsters.55 The second possible mechanism by which administration of HK Lr263 ameliorated metabolic syndrome was attenuation of systemic inflammation. Substantial evidence indicated that metabolic syndrome was closely linked to low grade chronic systemic inflammation.56,57 Previous studies showed that both live and HK Lactobacillus GG had an anti-inflammatory effect in rats suffering from experimental arthritis.58 Clearly, the anti-inflammatory effect of Lactobacillus GG was independent of the viability of the micro-organism.58 Furthermore, a study reported that HK Lactobacillus pentosus SPT84 was more effective than its live counterpart in the production of IL-12 and IFN-γ, two important anti-inflammatory cytokines produced by Th1 cells.59 These results may provide explanations for the similar effects on improving obesity-related metabolic syndromes exerted by HK Lr263 and live Lr263.
One aspect of probiotic research that has been often questioned is whether the entire effect is due to cell wall components or whether there are further advantages provided by live, metabolically active probiotic bacteria.60 Nevertheless, recent studies showed that bacterial non-protein cell-wall components, such as lipoteichoic acid and polysaccharides, played key roles not only in the adherence of Lactobacilli to the intestinal epithelium but also in the properties of anti-inflammation of the host.61,62 Other components of probiotic Lactobacilli may also engage in cross-talk with the host immune system by means of their surface layer (S-layer), a monomolecular crystalline envelope produced by the self-assembly of protein or glycoprotein subunits on the outer cell surface.63 For instance, the S-layer protein from L. helveticus MIMLh5, a bacterium demonstrated to interact with host cells and modulate immune responses,64 markedly altered cytokine production through the inhibition of NF-κB activation in human intestinal epithelial cells.65 Besides, a considerable amount of published data indicates that the use of non-viable probiotics or their components can influence the host's immune system and improve the onset of allergy, influenza virus infection and NAFLD.55,66,67 Therefore, it was reasonable to speculate that the protective effects of live and HK Lr263 on obesity-induced metabolic syndrome were achieved by suppressing dietary fat absorption in intestines and reduced the inflammatory tone of the intestine by the cell wall components of Lr263, such as lipoteichoic acid and lipopolysaccharides.
In summary, our results demonstrated that HK Lr263 exerted similar beneficial effects on improving insulin resistance, glucose intolerance, and fatty liver in high-fat diet treated rats as live Lr263 did. Therefore, we suggested that HK Lr263 will be expected to have commercial potential in ameliorating obesity-induced metabolic abnormalities.
Funding statement
This work was supported by the National Science Council Taiwan, which was renamed the Ministry of Science and Technology in 2014 (grant number NSC 101-2622-B-037-004-CC3). The funders had no role in the study design, data collection and analysis, decision to publish, or the preparation of the manuscript.Authors’ contribution
Ching-Shuang Wu and Yi-Hsing Chen were responsible for the study design and manuscript preparation; Feng-Ching Hsieh, Tsui-Yin Huang, Kuan-Wei Chen and Ai-Hui Fang performed animal experiments and analyzed the data; Chee-Yin Chai and Wan-Tzu Chen contributed to the oil red O staining of liver tissues; Ching-Shuang Wu, Cheng-Che E. Lan and Feng-Ching Hsieh contributed to the manuscript writing, data processing, interpretation, and analyses. All authors have read and approved the final manuscript.Abbreviations
| NAFLD | Non-alcoholic fatty liver disease |
| CHD | Coronary heart disease |
| ATM | Adipose tissue macrophage |
| TNF-α | Tumor necrosis factor-α |
| IL-6 | Interleukin-6 |
| IL-1β | Interleukin-1β |
| Lr263 | Lactobacillus reuteri GMNL-263 |
| HFD | High-fat diet |
| MRS | Man Rogosa Sharpe |
| IACUC | Institutional Animal Care and Use Committee |
| HK Lr263 | Heat-killed Lr263 |
| TG | Triglyceride |
| LDL | Low density lipoprotein |
| AST | Aspartate aminotransferase |
| ALT | Alanine aminotransferase |
| HOMA | Homeostasis model assessment |
| FPI | Fasting plasma insulin |
| FPG | Fasting plasma glucose |
| OGTT | Oral glucose tolerance test |
| AUCglucose | Total glucose areas under the curve |
| TEER | Transepithelial electrical resistance |
| PBS | Phosphate buffered saline |
| BCRC | Bioresource Collection and Research Center |
| SD | Standard deviations |
| WAT | White adipose tissue |
| LPS | Lipopolysaccharide |
| TLR-4 | Toll-like receptor-4 |
| ROS | Reactive oxygen species |
| PPAR-γ | Peroxisome proliferator-activated receptor-γ |
| IRS-1 | Insulin receptor substrate-1 |
| MCP-1 | Monocyte chemoattractant protein-1 |
| SREBP-1c | Sterol regulatory element-binding transcription factor 1c |
| ChREBP | Carbohydrate-responsive element-binding protein |
| FAS | Fatty acid synthase |
References
- C. Nishida, R. Uauy, S. Kumanyika and P. Shetty, Public Health Nutr., 2004, 7, 245–250 Search PubMed.
- B. M. Nathan and A. Moran, Curr. Opin. Endocrinol., Diabetes Obes., 2008, 15, 21–29 CrossRef PubMed.
- N. Chu, D. Wang, S. Shieh and E. Rimm, Int. J. Obes. Relat. Metab. Disord., 2000, 24, 1265–1271 CrossRef CAS PubMed.
- H. K. Gill and G. Y. Wu, World J. Gastroenterol., 2006, 12, 345–353 CrossRef CAS PubMed.
- S. P. Weisberg, D. McCann, M. Desai, M. Rosenbaum, R. L. Leibel and A. W. Ferrante Jr., J. Clin. Invest., 2003, 112, 1796–1808 CAS.
- R. Cancello, C. Henegar, N. Viguerie, S. Taleb, C. Poitou, C. Rouault, M. Coupaye, V. Pelloux, D. Hugol and J. L. Bouillot, Diabetes, 2005, 54, 2277–2286 CrossRef CAS PubMed.
- H. Morinaga, S. Talukdar, E. J. Bae and J. M. Olefsky, Diabetes, 2012, 61, 346–354 CrossRef PubMed.
- C. Lagathu, L. Yvan-Charvet, J. P. Bastard, M. Maachi, A. Quignard-Boulange, J. Capeau and M. Caron, Diabetologia, 2006, 49, 2162–2173 CrossRef CAS PubMed.
- P. A. Kern, S. Ranganathan, C. Li, L. Wood and G. Ranganathan, Am. J. Physiol., 2001, 280, E745–E751 CAS.
- P. J. Turnbaugh, R. E. Ley, M. A. Mahowald, V. Magrini, E. R. Mardis and J. I. Gordon, Nature, 2006, 444, 1027–1131 CrossRef PubMed.
- R. E. Ley, P. J. Turnbaugh, S. Klein and J. I. Gordon, Nature, 2006, 444, 1022–1023 CrossRef CAS PubMed.
- R. E. Ley, F. Bäckhed, P. Turnbaugh, C. A. Lozupone, R. D. Knight and J. I. Gordon, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 11070–11075 CrossRef CAS PubMed.
- P. D. Cani and N. M. Delzenne, Curr. Pharm. Des., 2009, 15, 1546–1558 CrossRef CAS PubMed.
- R. Y. Xu, Y. P. Wan, Q. Y. Fang, W. Lu and W. Cai, J. Clin. Biochem. Nutr., 2012, 50, 72–77 CrossRef CAS PubMed.
- B. Goldin and S. Gorbach, Clin. Infect. Dis., 2008, 46, S96–S100 CrossRef PubMed.
- G. MacGregor, A. Smith, B. Thakker and J. Kinsella, Postgrad. Med. J., 2002, 78, 366–367 CrossRef PubMed.
- L. Chuang, K. G. Wu, C. Pai, P. S. Hsieh, J. J. Tsai, J. H. Yen and M. Y. Lin, J. Agric. Food Chem., 2007, 55, 11080–11086 CrossRef CAS PubMed.
- C. A. Adams, Nutr. Res. Rev., 2010, 23, 37–46 CrossRef CAS PubMed.
- F. C. Hsieh, C. L. Lee, C. Y. Chai, W. T. Chen, Y. C. Lu and C. S. Wu, Nutr. Metab., 2013, 10, 35–48 CAS.
- T. M. Wallace, J. C. Levy and D. R. Matthews, Diabetes Care, 2004, 27, 1487–1495 CrossRef PubMed.
- K. Atisook, S. Carlson and J. L. Madara, Am. J. Physiol., 1990, 258, C77–C85 CAS.
- J. M. Delroisse, A. L. Boulvin, I. Parmentier, R. D. Dauphin, M. Vandenbol and D. Portetelle, Microbiol. Res., 2008, 163, 663–670 CrossRef CAS PubMed.
- M. N. VanSaun, I. K. Lee, M. K. Washington, L. Matrisian and D. L. Gorden, Am. J. Pathol., 2009, 175, 355–364 CrossRef PubMed.
- P. D. Cani, N. M. Delzenne, J. Amar and R. Burcelin, Pathol. Biol., 2008, 56, 305–309 CrossRef CAS PubMed.
- P. Heczko, M. Strus and P. Kochan, J. Physiol. Pharmacol., 2006, 57, 5–12 Search PubMed.
- J. Leung, B. Burke, D. Ford, G. Garvin, C. Korn, C. Sulis and N. Bhadelia, Emerging Infect. Dis., 2013, 19, 1791–1798 CrossRef PubMed.
- Y. Qiao, J. Sun, Z. Xie, Y. Shi and G. Le, J. Clin. Biochem. Nutr., 2014, 55, 120–128 CrossRef CAS PubMed.
- H. Xu, G. T. Barnes, Q. Yang, G. Tan, D. Yang, C. J. Chou, J. Sole, A. Nichols, J. S. Ross and L. A. Tartaglia, J. Clin. Invest., 2003, 112, 1821–1830 CAS.
- M. Serino, E. Luche, S. Gres, A. Baylac, M. Bergé, C. Cenac, A. Waget, P. Klopp, J. Iacovoni and C. Klopp, Gut, 2012, 61, 543–553 CrossRef CAS PubMed.
- P. D. Cani, R. Bibiloni, C. Knauf, A. Waget, A. M. Neyrinck, N. M. Delzenne and R. Burcelin, Diabetes, 2008, 57, 1470–1481 CrossRef CAS PubMed.
- P. D. Cani, A. M. Neyrinck, F. Fava, C. Knauf, R. G. Burcelin, K. M. Tuohy, G. Gibson and N. M. Delzenne, Diabetologia, 2007, 50, 2374–2383 CrossRef CAS PubMed.
- P. D. Cani, S. Possemiers, T. Van de Wiele, Y. Guiot, A. Everard, O. Rottier, L. Geurts, D. Naslain, A. Neyrinck and D. M. Lambert, Gut, 2009, 58, 1091–1103 CrossRef CAS PubMed.
- D. Adawi, S. Ahrné and G. Molin, Int. J. Food Microbiol., 2001, 70, 213–220 CrossRef CAS PubMed.
- D. Adawi, F. B. Kasravi, G. Molin and B. Jeppsson, Hepatology, 1997, 25, 642–647 CrossRef CAS PubMed.
- F. Bäckhed, J. K. Manchester, C. F. Semenkovich and J. I. Gordon, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 979–984 CrossRef PubMed.
- P. D. Cani, J. Amar, M. A. Iglesias, M. Poggi, C. Knauf, D. Bastelica, A. M. Neyrinck, F. Fava, K. M. Tuohy and C. Chabo, Diabetes, 2007, 56, 1761–1772 CrossRef CAS PubMed.
- K. E. Wellen and G. S. Hotamisligil, J. Clin. Invest., 2003, 112, 1785–1788 CrossRef CAS PubMed.
- D. Huang, J. Wang, P. Kivisakk, B. J. Rollins and R. M. Ransohoff, J. Exp. Med., 2001, 193, 713–726 CrossRef CAS PubMed.
- J. Brüning, J. Winnay, B. Cheatham and C. R. Kahn, Mol. Cell Biol., 1997, 17, 1513–1521 CrossRef.
- C. M. Rondinone, L. M. Wang, P. Lonnroth, C. Wesslau, J. H. Pierce and U. Smith, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 4171–4175 CrossRef CAS.
- N. Zeghari, H. Vidal, M. Younsi, O. Ziegler, P. Drouin and M. Donner, Am. J. Physiol., 2000, 279, E736–E743 CAS.
- B. Gustafson and U. Smith, J. Biol. Chem., 2006, 281, 9507–9516 CrossRef CAS PubMed.
- S. Chung, K. LaPoint, K. Martinez, A. Kennedy, M. Boysen Sandberg and M. K. McIntosh, Endocrinology, 2006, 147, 5340–5351 CrossRef CAS PubMed.
- M. A. Cornier, D. Dabelea, T. L. Hernandez, R. C. Lindstrom, A. J. Steig, N. R. Stob, R. E. Van Pelt, H. Wang and R. H. Eckel, Endocr. Rev., 2008, 29, 777–822 CrossRef CAS PubMed.
- C. Postic and J. Girard, J. Clin. Invest., 2008, 118, 829–838 CrossRef CAS PubMed.
- M. Kohjima, M. Enjoji, N. Higuchi, M. Kato, K. Kotoh, T. Yoshimoto, T. Fujino, M. Yada, R. Yada and N. Harada, Int. J. Mol. Med., 2007, 20, 351–358 CAS.
- N. Xie, Y. Cui, Y. N. Yin, X. Zhao, J. W. Yang, Z. G. Wang, N. Fu, Y. Tang, X. H. Wang and X. W. Liu, BMC Complementary Altern. Med., 2011, 11, 53–63 CrossRef CAS PubMed.
- S. R. Yoo, Y. J. Kim, D. Y. Park, U. J. Jung, S. M. Jeon, Y. T. Ahn, C. S. Huh, R. McGregor and M. S. Choi, Obesity, 2013, 21, 2571–2578 CrossRef CAS PubMed.
- C. P. Day and O. F. James, Gastroenterology, 1998, 114, 842–845 CrossRef CAS.
- N. Sabir, Y. Sermez, S. Kazil and M. Zencir, Eur. J. Ultrasound, 2001, 14, 121–128 CrossRef CAS PubMed.
- J. Shang, L. L. Chen, F. X. Xiao, H. Sun, H. C. Ding and H. Xiao, Acta Pharmacol. Sin., 2008, 29, 698–706 CrossRef CAS PubMed.
- N. Li, W. M. Russell, M. Douglas-Escobar, N. Hauser, M. Lopez and J. Neu, Pediatr. Res., 2009, 66, 203–207 CrossRef CAS PubMed.
- S. A. Joyce, J. MacSharry, P. G. Casey, M. Kinsella, E. F. Murphy, F. Shanahan, C. Hill and C. G. Gahan, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 7421–7426 CrossRef CAS PubMed.
- C. Tomaro-Duchesneau, M. L. Jones, D. Shah, P. Jain, S. Saha and S. Prakash, BioMed. Res. Int., 2014, 2014, 380316–380324 Search PubMed.
- W. J. Ting, W. W. Kuo, D. J. Y. Hsieh, Y. L. Yeh, C. H. Day, Y. H. Chen, R. J. Chen, V. V. Padma, Y. H. Chen and C. Y. Huang, Int. J. Mol. Sci., 2015, 16, 25881–25896 CrossRef PubMed.
- B. B. Kahn and J. S. Flier, J. Clin. Invest., 2000, 106, 473–481 CrossRef CAS PubMed.
- K. E. Wellen and G. S. Hotamisligil, J. Clin. Invest., 2005, 115, 1111–1119 CAS.
- E. Baharav, F. Mor, M. Halpern and A. Weinberger, J. Nutr., 2004, 134, 1964–1969 CAS.
- T. Izumo, M. Ida, T. Maekawa, Y. Furukawa, Y. Kitagawa and Y. Kiso, J. Health Sci., 2011, 57, 304–310 CrossRef CAS.
- S. Ostad, A. Salarian, M. Ghahramani, M. Fazeli, N. Samadi and H. Jamalifar, Folia Microbiol., 2009, 54, 157–160 CrossRef CAS PubMed.
- M. P. Vélez, T. L. Verhoeven, C. Draing, S. Von Aulock, M. Pfitzenmaier, A. Geyer, I. Lambrichts, C. Grangette, B. Pot and J. Vanderleyden, Appl. Environ. Microbiol., 2007, 73, 3595–3604 CrossRef PubMed.
- C. Grangette, S. Nutten, E. Palumbo, S. Morath, C. Hermann, J. Dewulf, B. Pot, T. Hartung, P. Hols and A. Mercenier, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10321–10326 CrossRef CAS PubMed.
- S. R. Konstantinov, H. Smidt, W. M. De Vos, S. C. Bruijns, S. K. Singh, F. Valence, D. Molle, S. Lortal, E. Altermann and T. R. Klaenhammer, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 19474–19479 CrossRef CAS PubMed.
- S. Guglielmetti, V. Taverniti, M. Minuzzo, S. Arioli, I. Zanoni, M. Stuknyte, F. Granucci, M. Karp and D. Mora, Infect. Immun., 2010, 78, 4734–4743 CrossRef CAS PubMed.
- V. Taverniti, M. Stuknyte, M. Minuzzo, S. Arioli, I. De Noni, C. Scabiosi, Z. M. Cordova, I. Junttila, S. Hämäläinen and H. Turpeinen, Appl. Environ. Microbiol., 2013, 79, 1221–1231 CrossRef CAS PubMed.
- Y. Liu, T. Fu, W. Peng, Y. Chen, Y. Cao, C. Chen, W. Hung and Y. Tsai, Benefic. Microbes, 2015, 6, 697–705 CrossRef CAS PubMed.
- M. Kawase, F. He, A. Kubota, K. Yoda, K. Miyazawa and M. Hiramatsu, FEMS Immunol. Med. Microbiol., 2012, 64, 280–288 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this paper. |
| This journal is © The Royal Society of Chemistry 2016 |