
Impact of gastric pH profiles on the proteolytic digestion of mixed βlg-Xanthan biopolymer gels
B. L.
Dekkers†
,
E.
Kolodziejczyk
,
S.
Acquistapace
,
J.
Engmann
and
T. J.
Wooster
*
Nestec S.A., Nestlé Research Centre, Vers-chez-les-Blanc, CH 1000 Switzerland. E-mail: timothyjames.wooster@rdls.nestle.com
First published on 18th November 2015
Abstract
The understanding of how foods are digested and metabolised is essential to enable the design/selection of foods as part of a balanced diet. Essential to this endeavour is the development of appropriate biorelevant in vitro digestion tools. In this work, the influence of gastric pH profile on the in vitro digestion of mixtures of β-lactoglobulin (βlg) and xanthan gum prior to and after heat induced gelation was investigated. A conventional highly acidic (pH 1.9) gastric pH profile was compared to two dynamic gastric pH profiles (initial pH of 6.0 vs. 5.2 and H+ secretion rates of 60 vs. 36 mmol h−1) designed to mimic the changes in gastric pH observed during clinical trials with high protein meals. In moving away from the pH 1.9 model, to a pH profile reflecting in vivo conditions, the initial rate and degree of protein digestion halved during the first 45 minutes. After 90 minutes of gastric digestion, all three pH profiles caused similar extents of protein digestion. Given that 50% gastric emptying times of (test) meals are in range of 30–90 min, it would seem highly relevant to use a dynamic pH gastric model rather than a pH 1.9 (USP) or pH 3 model (INFOGEST) in assessing the impact of food structuring approaches on protein digestion. The impact that heat induced gelation had on the degree of gel digestion by pepsin was also investigated. Surprisingly, it was found that heat induced gelation of βlg-xanthan mixtures at 70–90 °C for 20 minutes lead to a considerable decrease in the rate of proteolysis, which contrasts many studies of dispersed aggregates and gels of βlg alone whose heating accelerates pepsin activity due to unfolding. In the present case, the formation of a dense protein network created a fine pore structure which restricted pepsin access into the gel thereby slowing proteolysis. This work not only has implications for the in vitro assessment of protein digestion, but also highlights how protein digestion might be slowed, learnings that might have an influence on the design of foods as part of a satisfying balanced diet.
1. Introduction
The imbalance between energy consumption through diet and energy expenditure through physical activity is cited as the main cause of the increase in non-communicable diseases, such as obesity, diabetes and cardiovascular disease.1 An increased consumption of highly appealing foods, rich in calories but low in satiation is thought to be a key cause of this imbalance. Therefore there has been a considerable increase in the research into the psychological, sensorial, and biochemical mechanisms that drive food consumption. A particularly important area, is how the digestion and physical transformation of foods during consumption impacts short term (satiety – i.e. size of meal consumed) and long term (satiation – i.e. frequency of snacking) food intake.2–8 The physical accommodation of foods within the stomach along with regulation of gastrointestinal motility act to suppress feelings of hunger via neuro-hormonal pathways involving Ghrelin, CCK, PYY and GLP-1.3,5,6 The magnitude of these effects is a function of both the site and duration of small intestinal exposure to nutrient digestion products, and is therefore largely affected by the speed of food digestion.4,7,8 The implications that different rates/extents of digestion have on human health are increasingly being recognized and understood,9,10 hence there has been consistent interest in understanding how to control macronutrient digestion.11–13 Since proteins are widely considered to be the most potent at triggering of CCK, PYY and GLP-1 mediated satiety, there is strong interest in understanding how to measure and control the digestion of proteins.6,14There has been extensive research into regulation of the kinetics of protein digestion using in vitro and to a lesser extent in vivo techniques.15–23 This research has focused on how a proteins native conformation,17 thermal denaturation, interfacial adsorption15,16 and formation of cross linked gels impact the kinetics of digestion. For example, the native globular conformation of bovine β-lactoglobulin is much more resistant to pepsin digestion compared to the open/rheomorphic structure of caseins because its compact structure limits pepsin access.17 Similarly, the unfolding of proteins upon adsorption to emulsion interfaces has been shown to facilitate their gastric and intestinal proteolysis.15,16,24 The interaction of proteins with the gastric environment can also have a considerable impact on the kinetics of protein digestion.18–20 The concept of fast and slow protein digestion was developed based on the different aggregation states of whey proteins and casein micelles within the stomach.18,19 Whilst whey proteins are soluble under gastric conditions and hence undergo fast digestion due to moderately fast rates of gastric emptying, micellar caseins become insoluble, forming a curd in the stomach that results in slow protein release in the intestine.20 The deliberate modification of a proteins supramolecular structure via heat/chemical aggregation and/or gelation has also been shown to alter proteolysis kinetics.21–23 Whilst heating individual whey proteins to 90 °C leads to an acceleration of their peptic digestion in vitro,21 heat induced aggregation of whey proteins at mildly acidic pH to form microgels was found to slow digestion in vivo.23 Coagulation of the microgels within the stomach lead to a moderate delay in the peak of amino acid absorption.23 Complete gelation of whey proteins (to form gel pieces) via heating has also been found to slow the peptic digestion of proteins.22
It is clear from the above research that the proteolysis and absorption of peptides can be significantly altered depending on protein conformation and aggregate structure. It is also apparent that a key feature of these differences is the coagulation of protein structures and/or the action of pepsin within the mildly acidic stomach. Much of the work on modifying the kinetics of protein digestion has focused on using in vitro models to develop a mechanistic understanding of the factors that control protein digestion. To achieve greater potential clinical relevance, the conditions used in the in vitro models have progressed considerably in their complexity in the past two decades to mimic the processes occurring in vivo.25–28 However, the specific biochemical and the physical conditions that best mimic the physiological conditions have been under discussion for several years.27,28 A key omission of most of the in vitro studies on protein digestion is consideration of the effect that gastric pH has on protein digestion. The digestion of proteins within the stomach is facilitated by pepsin, whose activity is highly pH dependent having a maximum at pH 2.29 Classical in vitro models such as those based on USP Pharmacopeia typically use the pH of the fasted state (i.e. a pH of 1.9) during simulated gastric digestion.30 A recent “standardised” static model proposed by INFOGEST suggests using a pH of 3 in order to represent the average pH the meal sees over the two hours of gastric digestion.27 However neither of these types of models reflect the dynamic changes in gastric pH that occur in vivo during the consumption of protein rich meals.31–34
The consumption of a protein rich meal causes a rapid increase in gastric pH from fasted pH of 1.9 to a pH of 4.5–6.7 within 15 minutes.31,32,34–36 Clinical studies showed that a constant secretion of HCl (0.16 M at a rate of 36 mmol h−1) by parietal cells in the stomach causes a steady decrease in gastric pH as digestion progresses (Fig. 1). Moreover, a meal exits the stomach progressively during digestion and it is the extent of digestion of the initial boluses leaving the stomach that has the greatest influence on neuro-hormonal regulation of gastric emptying and hence the total time a meal spends within the stomach at a certain pH. Several dynamic in vitro models of gastric digestion (e.g. “TIM 1” and dynamic gastric model “DGM” models) have been created to mimic this constant change in gastric pH upon meal consumption.28,37–42 The TIM series of models (TIM 1 and recent tinyTIM) were created by the team of Minekus et al. at TNO and are a highly relevant mimic of the dynamics of gastric secretions and peristalsis in a series of interconnected compartments.28,37,38 The DGM of the team at IFR and Nottingham uses knowledge from MRI studies to mimic antral grinding and gastric emptying in addition to dynamically mimicking gastric secretions (i.e. pH).41,42 A limitation of both of these highly accurate in vitro digestion models is that their complexity limits sample throughput and requires considerable amounts of enzymes for each run. To date, the number of studies published with these two models is dwarfed by the number of studies with simple static in vitro models. A simple in vitro model with dynamic gastric pH profile has been proposed by Lesmes et al.,39 however this model has a fixed pH gradient, and is far too acidic compared to clinical measurements of gastric pHs during the consumption of protein rich meals.31,32,35 Guo et al. propose a dynamic in vitro model that mimics in parallel the rate of gastric secretions, gastric emptying, and the effects of antral grinding.40 It is clear that there are a couple of excellent complex dynamic in vitro models that mimic in parallel many processes of the stomach.40 However a gap is the existence of a simple in vitro model with dynamic representation of gastric secretions (pH).
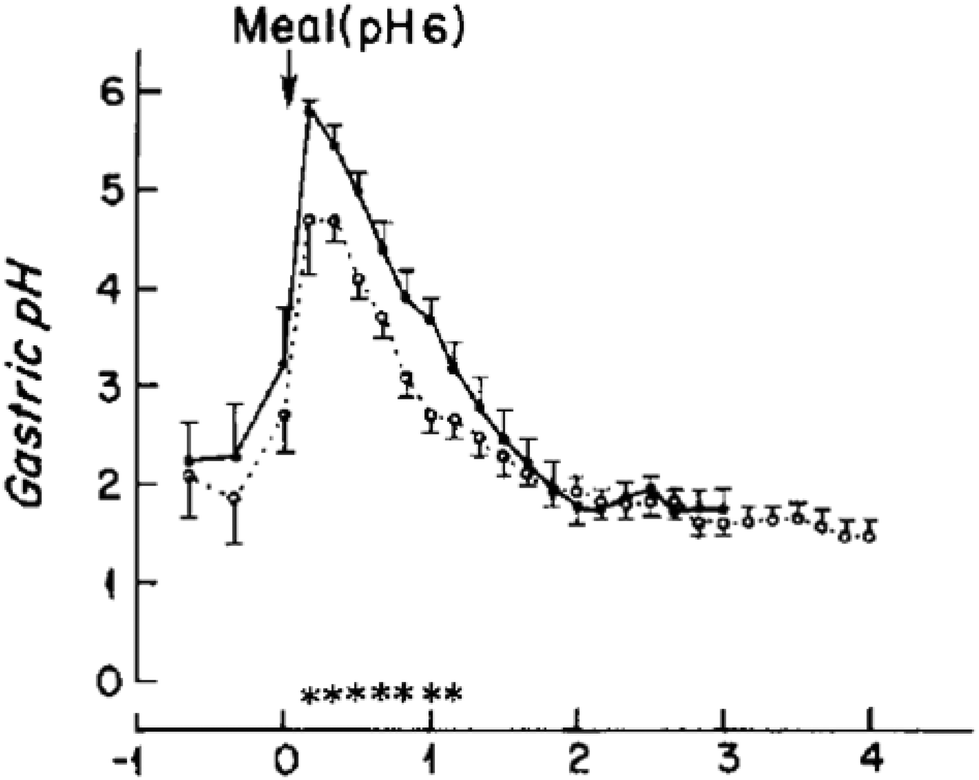 | ||
| Fig. 1 Change in gastric pH of six healthy male (39.7 ± 7.1 years) after the consumption of a standard evening meal (90 g steak, 25 g white bread, 8 g butter, and 60 g vanilla ice cream topped with 35 g chocolate syrup) either as; ● – a “solid meal” where it has been consumed after cooking or ○ – a “homogenised” meal where it has been puréed in a blender. Reproduced with permission from Malagelada et al.34 | ||
Given the high pH dependence of pepsin activity, and the significant deviations in gastric pH seen in vivo, the present study sought to develop a dynamic gastric pH in vitro model that replicates the pH profile in vivo. There has been extensive study of the digestion of single protein/biopolymer gels and comparatively little on mixed protein – biopolymer gels.13,18,19,21,22,43,44 Hence, the impact of different gastric pH in vitro models have on protein digestion was assessed using a model mixed biopolymer gel consisting of βlg and xanthan gum. This system was chosen due to the formation of particulate/fractal gels induced by the onset of thermodynamic incompatibility between βlg and xanthan, which affects protein gel structure, and will likely affect digestion.45 To investigate the impact of different gastric pH models on protein digestion, the gelation temperature was chosen as a variable parameter. It is expected that a higher gelation temperature induces more protein aggregation, making the gels less digestible.46 The viscoelastic properties of the gel and confocal scanning laser microscopy (CSLM) were used to assess the effects of gelation temperature on digestibility.
2. Experimental section
2.1. Materials
The protein gel was made of β-lactoglobulin (Biopro, batch 48317, Davisco) and xanthan (Ketrol Sf, N1D1595K, CP Kelco). Sodium azide was added to the gel to inhibit microbial growth. For the digestion pepsin (porcine, 800–2500 U mg−1, P7000, actual activities used 674 UI mg−1 and 561 U mg−1) was used as obtained from Sigma Aldrich. For the OPA assay, sodium tetraborate decahydrate (<99.5%, S9640), sodium dodecyl sulfate (>98.5%, L4509), mercapto-ethanol (>99%, M6250), o-phthaldialdehyde (>97%, PI378) were obtained from Sigma Aldrich and ethanol (absolute for analysis) from Merck. A sodium acetate buffer was made from sodium acetate (ensure, >99.0%, A0346768) and acetic acid (ensure, 100% anhydrous for analysis) from Merck. For dissolution, double-distilled water was used.2.2. Methods
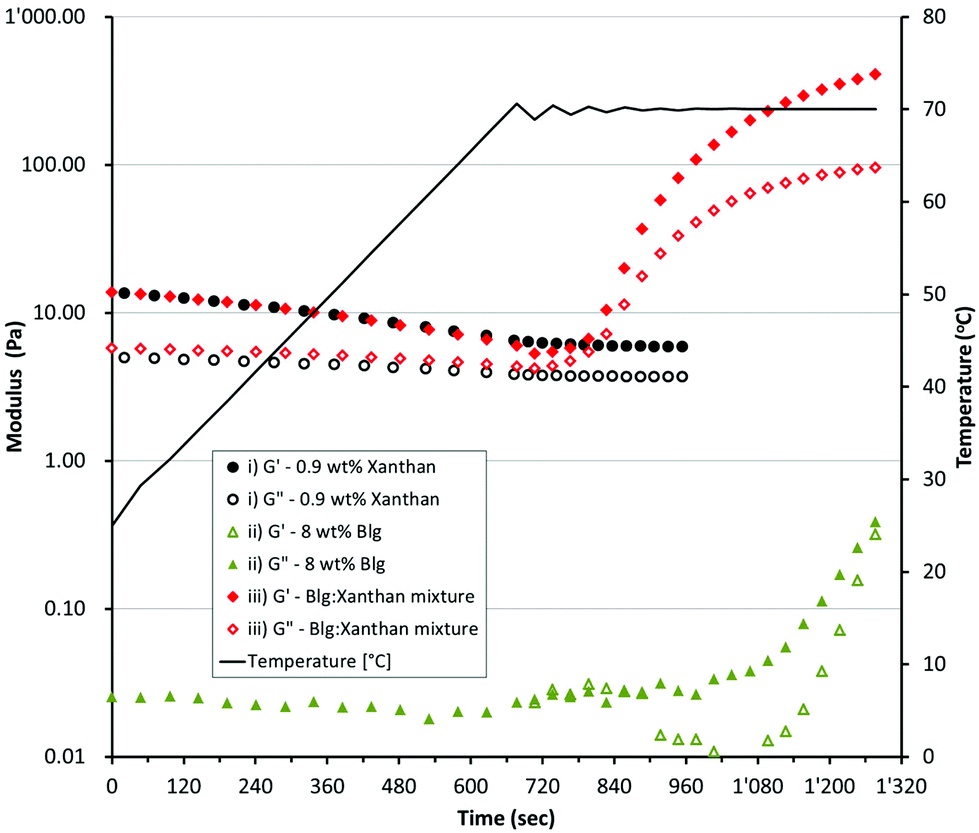 | ||
| Fig. 2 Storage modulus G′ (full symbols) and Loss modulus G′′ (empty symbols) as a function of heating time for a βlg-xanthan mixture (8 wt% βlg, 0.9 wt% xanthan, 100 mM NaCl, pH 8.2, 70 °C 20 min) undergoing gelation. | ||
Frequency sweep measurements were performed using a Rheometer model Physica MCR501 (Anton Paar) operating in oscillatory mode. A frequency sweep test was done for both the liquid βlg-xanthan mixture and for the different gels. For the liquid material a bob and cup geometry was used and for the gels a parallel plate geometry with serrated plates and a diameter of 25 mm was used. The storage modulus (G′) and loss modulus (G′′) were recorded during a frequency sweep test (10−1–101 Hz) performed with a strain of 0.5% at 25 °C. To minimize slip a normal force of 0.1 N was used. Several normal forces were tested (0.05 to 1 N), and a normal force of 0.1 N was the most optimal condition for performing a frequency sweep test on these gels when considering slip and forces applied to the gel. The mechanical spectra of the gels were recorded. The entire test was replicated three times.
The rate of peptide release during digestion of the protein gel was measured with the o-phthaldialdehyde (OPA) spectrophotometric assay. In the presence of reduced sulfhydryl groups, NH2 groups of primary amines in amino acids, peptide and proteins, can react with o-phthaldialdehyde to form a fluorescent molecule. At several time points (t = 0, 5, 15, 30, 60 and 90 min) 40 μL was taken out and 2 mL of OPA reagent was added. The absorbance was measured at 340 nm after incubation of 2 min at 25 °C with a UV-Vis spectrophotometer (Evolution 300, Thermo). The OPA reagent consists of 80 mg of o-phthaldialdehyde, 50 mL of 0.1 M sodium tetra borate buffer (pH 9.7–10), 5 mL of 20 wt% sodium dodecyl sulphate, 0.2 mL 2-mercaptoethanol (2 M) and the total volume was made up to 100 mL with double distilled water. The OPA solution was prepared fresh daily. To determine the peptide release a calibration curve was made with lysine 0.01–0.1 g mL−1 (Sigma, >98%). All in vitro digestion experiments were performed in triplicate.
3. Results and discussion
The main question of this study is to understand if the pH profile of the gastric phase of digestion has an impact on the kinetics/extent of protein digestion. The main hypothesis is that the microstructure of a protein gel is the main factor that controls the rate of proteolysis, a number of model mixed βlg-xanthan were created via thermal gelation. In the following sections, the microstructure of the mixed protein polysaccharide gels is characterised, followed by details of how dynamic gastric pH profiles were developed and then the impact of gastric pH profile on the kinetics of mixed protein polysaccharide gel digestion. Finally the mechanisms governing the digestion of mixed protein polysaccharide gels as they relate gel microstructure and gastric pH are discussed, highlighting why the implications of this study have an important impact on how in vitro models for digestion are designed.3.1. Structure of mixed βlg-xanthan gels
Mixed βlg-xanthan gels were formed by heating a solution containing 8 wt% βlg and 0.9 wt% xanthan at pre-determined temperatures for 20 minutes. Fig. 2 shows how the rheology of individual solutions of 8 wt% βlg, 0.9 wt% xanthan and their mixture changes with heating. The 8 wt% βlg solution is a liquid having a complex viscosity (η*) of 3.5 ± 0.35 mPa s. Heating the 8 wt% βlg up to 70 °C had no effect on solution viscosity upon initial heating to 70 °C (G′ – 0.023 Pa, G′′ – 0.027 Pa). However, after 6 minutes of heating at 70 °C there was a gradual increase in both the loss and storage modulus. Heating for a further 2 minutes increased both the loss/storage moduli tenfold to 0.38 and 0.32 Pa respectively, and the solution begins to exhibit gel like behaviour (tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ increases from 15° to 40° over the last 20 second of heating at 70 °C). The changes in rheology of the 8 wt% βlg solution can be understood from the denaturation, and thermal gelation of βlg molecules.47 In solution at a pH of 8.2 βlg has undergone the Tanford transition, the dimer has dissociated and cys121 is exposed due to a conformational change of the EF loop.48 These changes expose the previously buried free cysteine (cys121) and increase its reactivity due to partial dissociation of the thiol (pKa 9.35). Heating above 65 °C increases thiol reactivity, and the tertiary and secondary structures of βlg begins to unfold facilitating formation of extensive intra- and inter-molecular disulphide exchange reactions.49
δ increases from 15° to 40° over the last 20 second of heating at 70 °C). The changes in rheology of the 8 wt% βlg solution can be understood from the denaturation, and thermal gelation of βlg molecules.47 In solution at a pH of 8.2 βlg has undergone the Tanford transition, the dimer has dissociated and cys121 is exposed due to a conformational change of the EF loop.48 These changes expose the previously buried free cysteine (cys121) and increase its reactivity due to partial dissociation of the thiol (pKa 9.35). Heating above 65 °C increases thiol reactivity, and the tertiary and secondary structures of βlg begins to unfold facilitating formation of extensive intra- and inter-molecular disulphide exchange reactions.49
At room temperature the 0.9 wt% xanthan solution exhibits weak gel like behaviour, G′ > G′′ but the overall magnitude of G′ is low (13.6 Pa). Heating the xanthan solution causes a halving of G′ (down to 6.6 Pa at 60 °C), and a slight decrease in G′′ (5.1 to 4 Pa). The gel-like behaviour of xanthan arises from the formation of a network of xanthan molecules, which is stabilised by a combination of intermolecular interactions between ordered and disordered xanthan segments via polymer entanglement.50,51 Heating the xanthan solution causes an ordered-disordered transition, which disrupts the network of xanthan helices causing a reduction in solution viscosity.51–53 The solutions containing a mixture of βlg (8 wt%) and xanthan (0.9 wt%) had a rheological profile that was a combination of the rheology of the individual solutions (Fig. 2iii). At temperatures below 55 °C the rheology of the mixture was very similar to the xanthan solution, exhibiting the same decrease in G′ and G′′ with temperature. At temperatures above 60 °C the rheology of the mixture shows similar behaviour to the βlg solution, undergoing a rapid increase in both G′ and G′′ indicating aggregation and then complete gelation of the βlg phase. Confocal micrographs indicate that incorporation of xanthan into solutions of βlg causes a significant change in the microstructure of the resulting gels (Fig. 3). Gels created from only βlg form a dense network with a fine microstructure which is difficult to discern (Fig. 3A). Gels created from mixtures of βlg and xanthan result in a phase separated structure, which is caused by the increase in thermodynamic incompatibility (between xanthan and βlg) as βlg unfolds and begins to aggregate (Fig. 3B–D).
 | ||
| Fig. 3 Confocal microscopy images of (A) βlg gel (8 wt%, 100 mM NaCl, pH 8.2, 70 °C) (B)–(D) mixed βlg-xanthan gels (8 wt% βlg, 0.9 wt% xanthan, 100 mM NaCl, pH 8.2) prepared by heating for 20 minutes at; (B) 70 °C (C) 80 °C and (D) 90 °C. | ||
3.2. Creating a dynamic gastric pH in vitro model and effect of gastric pH on protein gel digestion
Our principle question in this study was to understand if the pH profile of the gastric phase had an impact on the kinetics/extent of protein digestion. Fig. 4 presents the three different gastric pH profiles that were used to understand the impact that different gastric pH models have on the in vitro digestion of protein gels. The reference profile is based on using the fasted gastric pH (1.9) as the starting pH, a common approach in many in vitro studies (e.g. USP 24 simulated gastric fluid).30Fig. 4i shows that peptide release due to digestion, results in an increase of the pH over time, increasing the pH to 3 after 30 minutes and 3.7 after 90 minutes. The other two profiles are dynamic gastric pH profiles based on clinical observations of; meal stimulated increase in post prandial pH, a gastric acid molarity of 0.16 M,54,55 and average secretion rates of gastric juice found in vivo (Table 1). Dynamic profile A was designed to reflect the more extreme values of meal induced increase in gastric pH and acid secretion rates (60 mmol h−1 H+). Dynamic profile A undergoes a rapid increase in gastric pH to 6.0 after 2 minutes, followed by a steady decrease in pH to 4.8 after 30 minutes, 3.5 after 60 minutes and 2.5 after 90 minutes. Dynamic profile B was designed to follow the lower end of meal specific increase in gastric pH and acid secretion rate (36 mmol h−1 H+). Dynamic profile B undergoes a rapid increase, to pH 5.2 after 2 minutes, followed by a steady decrease in pH to 4.7 after 30 minutes, 4.2 after 60 minutes and 3.6 after 90 minutes.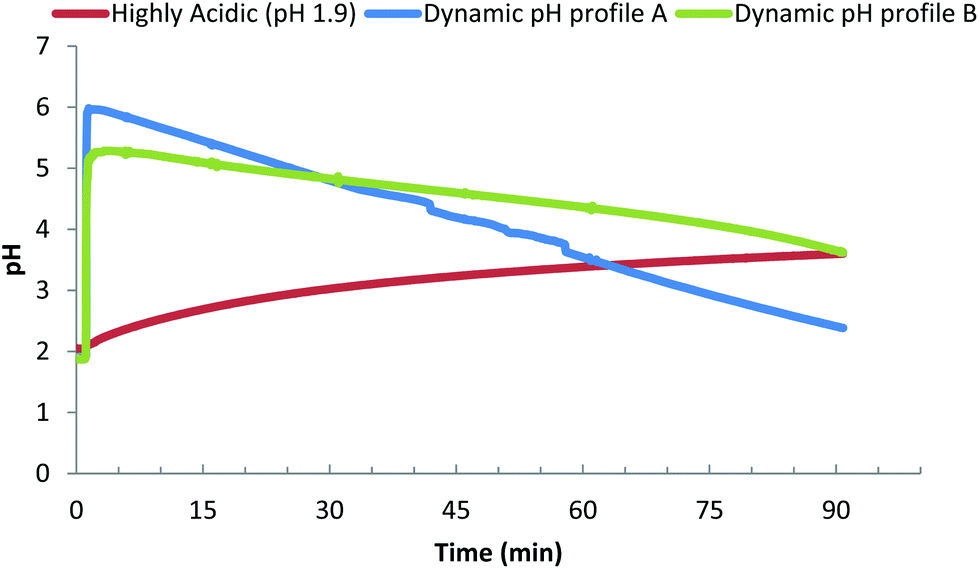 | ||
| Fig. 4 Example pH profiles obtained from protein gel pieces during in vitro gastric digestion. The pH profiles were, (i) highly acidic (pH 1.9) – initial pH 1.9 with no active control via acid addition, (ii) dynamic pH profile A – meal stimulated increase to pH 6 followed by acid addition at 60 mmol h−1 H+ and (iii) dynamic pH profile B – meal stimulated increase to pH 6 followed by acid addition at 36 mmol h−1 H+. | ||
| Meal stimulated gastric acid max pH secretion (mmol h−1) | pH | References |
|---|---|---|
| 60 (40–71) | 4.5 | (Gardner, Ciociola et al. 2002)36 |
| 41 (38–53) | (Gardner, Sloan et al. 2003)32 | |
| 13 | 6.4 | (Kalantzi, Goumas et al. 2006)35 |
| 12 | (Sauter, Curcic et al. 2012)76 | |
| 6.0 | (Malagelada, Go et al. 1979)34 | |
| 6.7 | (Dressman, Berardi et al. 1990)31 | |
| 36 | 5.9 | Average |
The effect that these three different gastric pH profiles had on the digestion of a mixed βlg-xanthan gel (8 wt% βlg, 0.9 wt% xanthan, 100 mM NaCl, pH 8.2, 70 °C, 20 min) was assessed by measuring peptide release (NH2 end group) due to digestion, Fig. 5. Digestion of the βlg-xanthan gel using the “static” pH 1.9 model results in an initial peptide hydrolysis rate of 0.23 μmol NH2 min−1 for the first 45 minutes, releasing 14.3 μmol of NH2 over the 90 minutes of gastric digestion. Digestion of the βlg-xanthan gel by “Dynamic profile A” was much slower during the initial phase, having an initial peptide hydrolysis rate of 0.1 μmol NH2 min−1 during the first 45 minutes. However, after 90 minutes of digestion by “Dynamic profile A” the βlg-xanthan gel created 14.9 μmol of NH2 which was slightly, but not significantly, higher than the “static” pH 1.9 profile. Digestion of the βlg-xanthan gel by “Dynamic profile B” resulted in intermediate peptide hydrolysis rate of 0.185 μmol NH2 min−1 for the first 45 minutes. The hydrolysis of the βlg-xanthan gel by “Dynamic profile B” between 45 and 90 minutes was not significantly different from “Dynamic profile A” and released 13.5 μmol NH2 after 90 minutes.
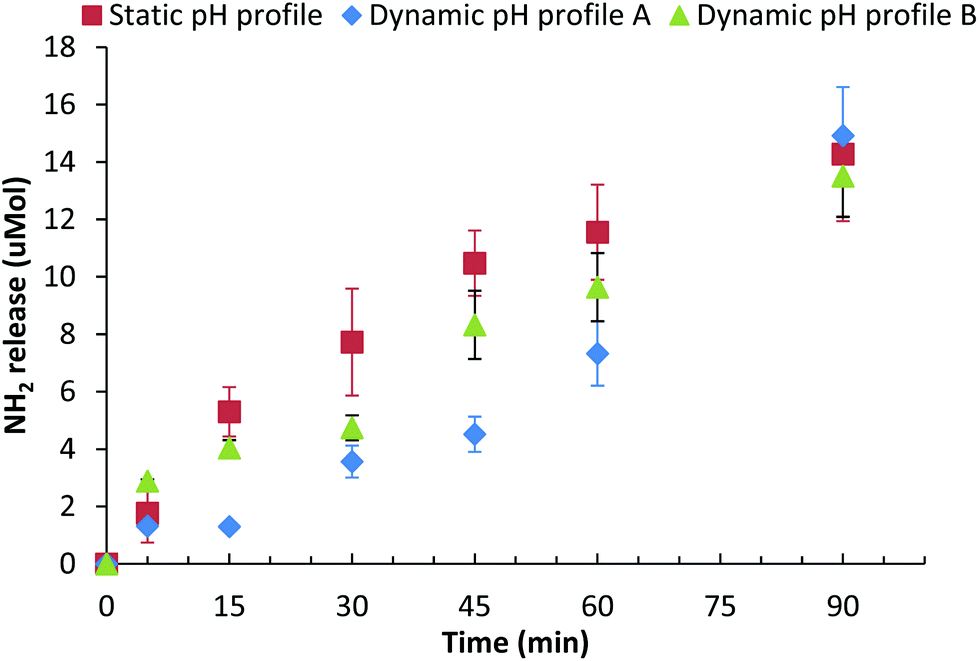 | ||
| Fig. 5 Amount of protein digestion (μmol NH2 exposed) from phase separated βlg-xanthan gels (8 wt% βlg, 0.9 wt% xanthan, 100 mM NaCl, pH 8.2, 70 °C 20 min) over time during in vitro gastric digestion; (i) highly acidic (pH 1.9) – initial pH 1.9 with no active control via acid addition, (ii) dynamic pH profile A (initial pH 6.0, 60 mmol h−1 H+) and (iii) dynamic pH profile B (initial pH 5.2, 36 mmol h−1 H+). | ||
The results of Fig. 5 highlight that the pH profile used during gastric digestion can have a considerable impact on the degree of digestion of a protein based gel. In moving away from the pH 1.9 model, to a pH profile reflective in vivo conditions, the initial rate and degree of protein digestion halved during the first 45 minutes. After 90 minutes of gastric digestion all three pH profiles caused similar extents of gel digestion. The differences in extent of digestion vs. time for the three pH profiles can be explained from the pH dependence of pepsin activity. Pepsin has maximal hydrolytic activity between pH 1.5 and 2.5, at pH's between 3 and 4.5 activity is approximately 70% of maximal, above pH 4.5 pepsin activity drops dramatically, to 40% at pH 5 and <5% above pH 5.29 The significant differences between the “static pH 1.9” gastric model and “dynamic pH profile A” are due to considerably lower pepsin activity during the initial phase (first 30 min) of gastric digestion when the pH was above 5. Given that 50% gastric emptying times of (test) meals are in range of 30–90 min,50,56–59 it would seem highly relevant to use a dynamic pH gastric model rather than a static pH 1.9 or pH 3 model, both of which would likely over-estimate the degree of proteolysis of the boluses entering the intestinal compartment during the initial phase of digestion.
3.3. Impact of gelation temperature on gel digestion
The different gastric pH in vitro models were also used to examine the digestion of; a solution of native βlg-xanthan and mixed protein βlg-xanthan gels prepared at different temperatures, e.g. 70, 80 and 90 °C. In Fig. 6A and B, peptide release is shown for different protein gels for a ‘static’ pH model. Relatively high concentrations of peptides were released from the native βlg-xanthan solution during the gastric step compared to that released from the gels prepared at different temperatures. NH2 release from the native βlg-xanthan solution was rapid during the first 30 minutes, resulting in 114 μmol of peptides. A final amount of 133.6 μmol NH2 was released after 90 minutes. This amount of NH2 release corresponds to an average degree of hydrolysis of 6.7%, which reflects the observation of numerous studies that βlg is resistant to proteolysis by pepsin.17,21,43,44,60 Heat induced gelation of the βlg-xanthan mixture lead to a considerable decrease in the rate and extent of peptide release, which systematically decreased with increasing gelation temperature. βlg-xanthan gels prepared at 70 °C released 14.3 μmol NH2 after 90 minutes, which was considerably higher compared to gels prepared at 80 and 90 °C, being 9.6 and 8.7 μmol respectively. Both the pH profile (not shown) and hydrolysis rates during static gastric pH digestion indicate that gelation of the βlg-xanthan mixture at higher temperatures resulted in gel microstructures that were less susceptible to breakdown by pepsin.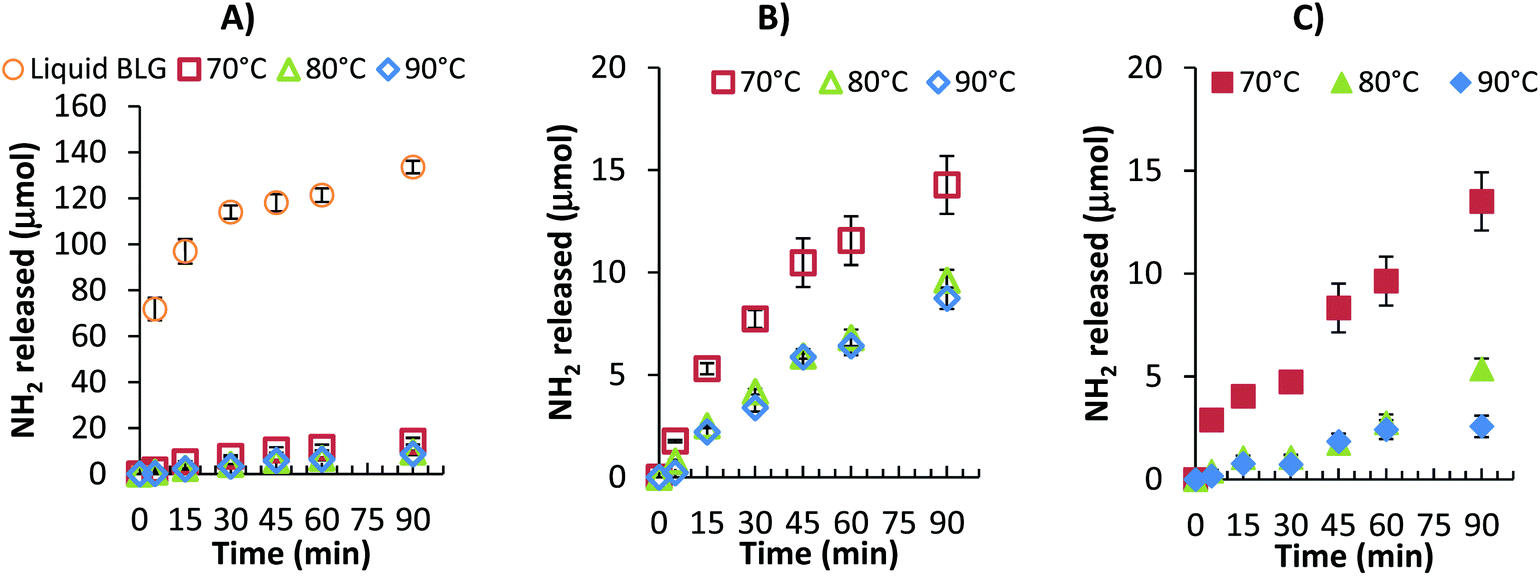 | ||
| Fig. 6 Effect of gelation temperature on the amount of protein digestion (μmol NH2 exposed) from phase separated βlg-xanthan gels (8 wt% βlg, 0.9 wt% xanthan, 100 mM NaCl, pH 8.2, 70 °C 20 min) over time during in vitro gastric digestion; (A) & (B) static pH profile (initial pH 1.9) and (C) dynamic pH profile B (initial pH 5.2, 36 mmol h−1 H+). | ||
To understand if the pH profile of the gastric phase of digestion had an impact on the kinetics/extent of protein digestion samples gelled at the three different temperatures were also tested under dynamic pH conditions (dynamic pH profile B). Fig. 6C shows the peptide release for samples that were prepared with a gelation temperature of 70, 80 and 90 °C with dynamic pH profile B. A similar trend in hydrolysis was found; the hydrolysis rate was lowered when proteins were gelled at higher temperatures. Again a statistical difference was found for gelation temperature 70 and 80 °C, but not for gels prepared at 80 and 90 °C. A comparison between the static and dynamic gastric pH models led us believe that the effect of pH on hydrolysis rate has a stronger effect when gels were prepared at higher temperatures. Gels that were prepared at 70 °C released only 5% less peptide when digested under dynamic pH conditions compared to a static pH, while gels that were prepared at 80 °C resulted in a 45% difference and for gels prepared at 90 °C this difference was even higher i.e. 71%. These large differences in the extent of gel proteolysis between different models of gastric pH could lead to a significant over estimation of the extent of digestion. This is particularly important given that food is sequentially emptied from the stomach, and the pH of the gastric milieu is quite elevated just after meal consumption.
It is interesting to note that heat induced gelation of the βlg-xanthan mixture led to a considerable decrease in the rate and extent of proteolysis catalysed by pepsin. It is well documented that βlg is resistant to digestion to pepsin, with between 5 to 35% of “native” βlg undergoing digestion depending on the pH and enzyme to substrate ratio.21,43,60,61 βlg is resistant to digestion by pepsin because most of the hydrophobic amino acids, which are the main potential sites for pepsin, are buried within the hydrophobic core limiting their accessibility.60,62 There has been extensive research that has shown that heating of βlg and/or heat induced aggregation of βlg enhances its susceptibility to pepsin digestion.17,21,47,63 Heating near or above its denaturation temperature causes denaturation of βlg, which exposes the aromatic peptic cleavage sites, facilitating digestion by pepsin.17,60 It appears that aggregation of βlg to form dimers/trimers, dispersed aggregates or even complete gels does little to limit the heat induced enhancement of the action of pepsin once the aromatic residues have been exposed.21,43,44 It is surprising then, that in the present study the opposite was observed, that heat induced gelation of the βlg-xanthan mixture lead to a considerable reduction in pepsin activity, despite heating to temperatures that ensure denaturation of βlg. A possible explanation for the considerable reduction in βlg proteolysis upon heat induced gelation of βlg-xanthan mixtures is the fact that the current gels were created at pH 8.2 compared to the neutral pHs used in other studies.17,21,43,44,60 Using βlg solutions with an alkaline pH (i.e. 8.2) induces considerable structural change to βlg prior to heating that have and impact on the extent of gel cross linking. In solution, at room temperature and at a pH of ≥7; (i) βlg undergoes the Tanford transition where a local conformational change of the EF loop exposes Cys121,48 (ii) the βlg dimer dissociates into two monomers64 and (iii) there is increased dissociation of the cysteine thiol group (pKa – 9.35).65 Combined these changes considerably increase the reactivity of the free thiol group of Cys121 and likely result in more extensive di-sulfide cross-linking in the current gels prepared at pH 8.2 compared to aggregates/gels prepared at more neutral pHs.
Limitations of the model used in the present work are the simplistic ionic composition of the gastric juice (100 mM NaCl), and the lack of an oral phase. Clinical studies of adult (fasted) gastric juice have found that it has an osmolality of 191 ± 36 mOsm and largely consists of monovalent ions [Na+ – 68 ± 29 mM, K+ – 13.4 ± mM, Cl− ± mM], with small amounts of divalent ions [Ca2+ – 0.6 ± 0.2, Mg2+ – 0.6 ± 0.2 mM].66 Since the overall monovalent ion content is similar to in vivo levels, and the fact that pepsin does not rely Ca2+ or Mg2+ for its activity, the simplistic composition of the gastric juice is unlikely to have an effect on the outcomes of this study. Of greater potential impact is the lack of an oral phase/trituration during gastric digestion. The gels created in this study could be imagined to either be swallowed directly (i.e. as a capsule/nutraceutical) or chewed during the course of eating. Jalabert-Malbos et al. found that after mastication gelled proteins (cooked egg white and Emmental cheese) have an average particle size (D50) in the range of 2.29 to 2.4 mm.67 Whilst Marciani et al. have shown that antral grinding forces of the stomach are a maximum of 0.53 to 0.78 N and can fracture gels with storage moduli of up to 10000 to 45000 Pa into millimeter sized pieces.68,69 Since surface erosion is the main degradation mechanism of the protein gels in the current study, trituration by mastication/antral grinding could possibly led to a 2.6 times increase in digestion rate due to the 2.6 times increase in surface area. Overall, the key strength of this study is that it provides a simple in vitro approach to mimic digestion with biorelevant rates of gastric acid secretion. Comparison of these biorelevant gastric pH profiles with static pH 1.9 gastric profiles clearly demonstrated an overestimation of the extent of proteolysis.
3.4. Mechanisms by which protein gels impact digestion
We propose that βlg-xanthan gel microstructure is the main factor that controls the rate of proteolysis, via the formation of a dense particulate protein network that restricts the mobility of pepsin. Gel crosslinking and porosity are key aspects of this effect. These properties can be evaluated from the viscoelastic properties of the gel (storage and loss moduli, G′ and G′′ respectively), which were measured using small-amplitude (γ = 0.5%) oscillatory rheology as a function of frequency, Fig. 7. For both the unheated mixture and the heat-treated gels, the storage modulus (G′) was higher than the loss modulus (G′′) indicating the formation of a gel. Only a slight dependence of G′ on oscillation frequency (ω) was found for both the unheated mixture and the gels, which indicates that the gels are formed above the critical (percolation) regime. The difference between G′ and G′′ was much more pronounced for the heat-treated systems, where both moduli are also much higher than for the native solution. The strength of mixed βlg-xanthan gels increased with gelation temperature, the storage modulus increased by a factor of two when the gelation temperature was increased from 70 to 80 °C and increased by a further 50% when it increased from 80 to 90 °C. Higher gelation temperatures result in greater denaturation/unfolding of βlg and more reactivity of cysteine residues, hence a more compact network structure (i.e. higher fractal dimension, smaller floc size and smaller pore size) is expected, which is consistent with the higher storage moduli observed at the same protein concentration. The confocal micrographs in Fig. 3 however, did not show a clear trend between heating temperature and floc size.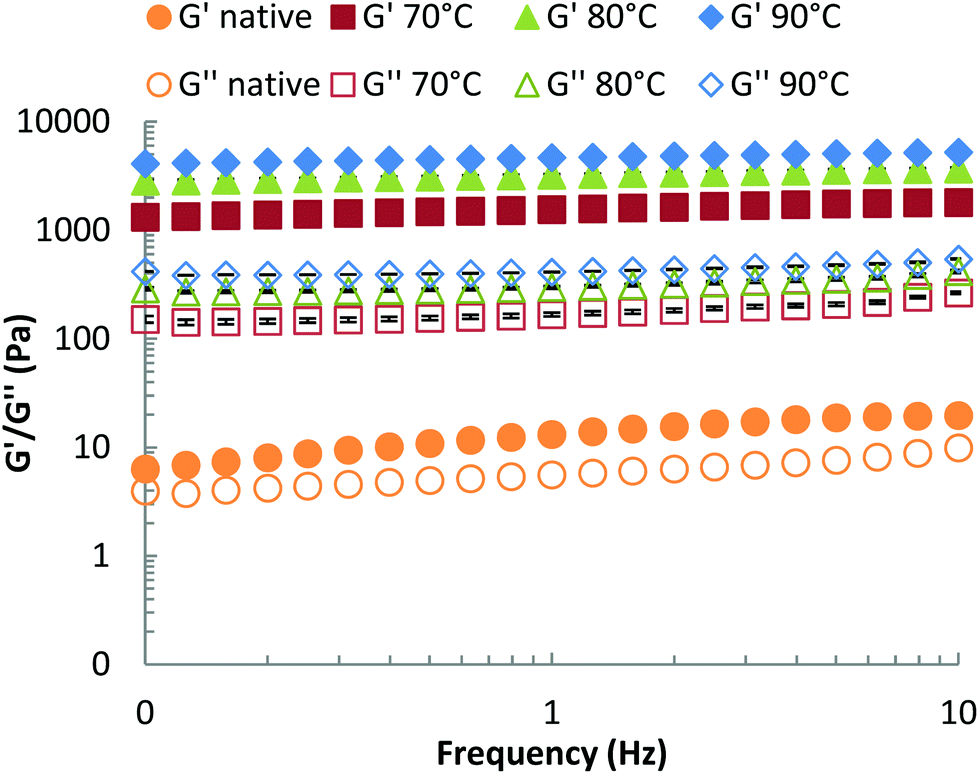 | ||
| Fig. 7 Storage G′ (full symbols) and loss modulus G′′ (open symbols) as a function of frequency for native and gelled mixtures of βlg-xanthan (8 wt% βlg, 0.9 wt% xanthan, 100 mM NaCl, pH 8.2) heated at different temperatures (70, 80 & 90 °C). | ||
A challenge is, how to relate the viscoelastic properties of the mixed βlg-xanthan particulate gels to the rate of their digestion? The rate of proteolysis of simple polymeric gelatine gels has been found to rely strongly on the porosity of the gel.70 In classical cross-linked polymer gels the porosity (average mesh size) can be related to the storage modulus using scaling theory based on; (a) affine deformation of the cross-linked polymer strands with the macroscopic deformation, and (b) an entropic restoring force in the deformed polymer strands linearly proportional with the temperature of the system.71,72 Such models are not directly applicable to particulate gels because of the different mechanisms of restoring forces in the particulate network strands (bending and deformation of junctions between particle aggregates). Scaling theory for the elastic properties of such particulate gels shares some features with theories for polymeric gels,73,74 but the temperature dependence and the relation to structures on different length scales is a different one. Analogous to polymers, the theory allows to relate a characteristic “floc size” ξ in the particle gel (equivalent to a “blob size” in the case of polymers) to the storage modulus G′. The modulus scales with a fractional power of this floc size,
| G′ ∝ ξ−(1+2ε+δ) | (1) |
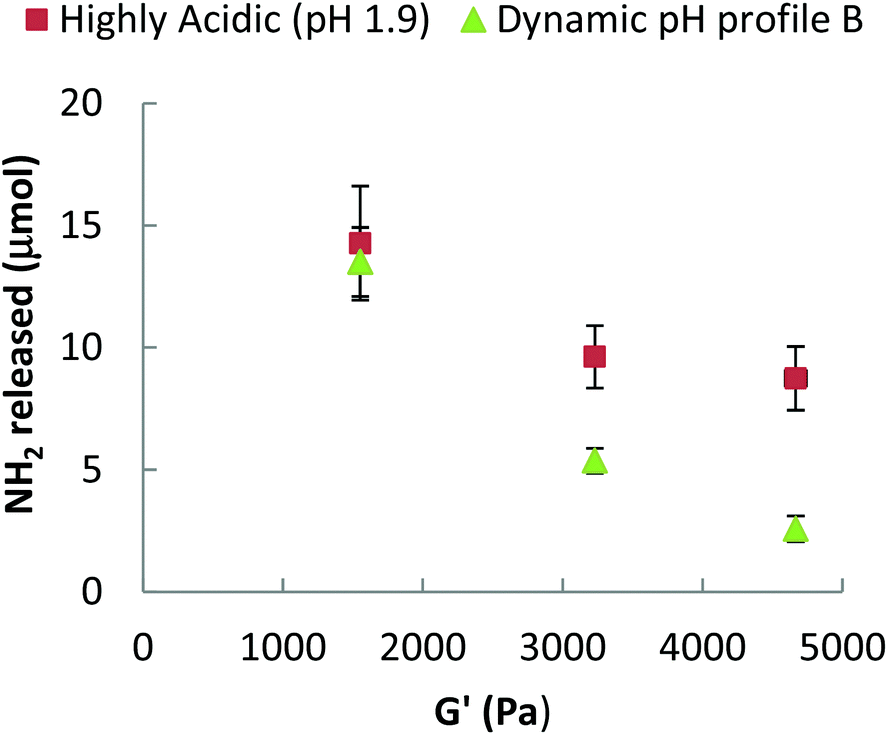 | ||
| Fig. 8 Storage G′ as a function of NH2 release for mixed βlg-xanthan (8 wt% βlg, 0.9 wt% xanthan, 100 mM NaCl, pH 8.2) heated at different temperatures (70, 80 & 90 °C) for 20 minutes and undergoing gastric digestion in the presence of pepsin following either; ■ a highly acidic (pH 1.9) or ▲ dynamic gastric pH profile B. G′ was measured at γ 0.5% and 1 = Hz. | ||
4. Summary and conclusions
The main aim of this study was to understand if the pH profile of the gastric phase of digestion had an impact on the kinetics/extent of digestion of protein gels. The premise was that in vitro models based on a highly acidic fasting gastric pH (1.9 or 3) could potentially have a bias because they do not represent the dynamic changes in gastric pH that are seen in vivo. In moving away from the pH 1.9 model, to a pH profile reflective of in vivo conditions, the initial rate and degree of protein digestion halved during the first 45 minutes. After 90 minutes of gastric digestion all three pH profiles caused similar extents of gel digestion. The significant differences between the “pH 1.9” gastric model and “dynamic pH profile A” are due to considerably lower pepsin activity during the initial phase (first 30 min) of gastric digestion when the pH was above 5. Given that 50% gastric emptying times of (test) meals are in range of 30–90 min, it would seem highly relevant to use a dynamic pH gastric model rather than a pH 1.9 or an averaged pH 3 model in assessing the impact of food structuring approaches on protein digestion. What is unknown is the effect of switching to a dynamic pH model might have on lipid digestion, which involves gastric lipase which has a different pH optima.How a protein gels structure impacts gastric digestion was also examined by preparing gels at different temperatures (70, 80 and 90 °C). The formation of a dense phase separated mixed biopolymer network lead to a considerable reduction in digestibility by pepsin. This finding was surprising as it is opposite to studies of simple βlg aggregates and gels which normally find that heat aggregation enhances proteolysis by pepsin. The reason why the βlg-xanthan gels resist digestion by pepsin possibly arises from the microstructure of the gels. Confocal microscopy describes a structure with phase separated regions of xanthan dispersed within a continuous βlg network. Rheological measurements demonstrated that the heating the βlg-xanthan mixture results in a dense elastic network. The systematic relationship between the extent of digestion, and the gel storage modulus highlight that the gel stiffness/density (i.e. porosity) is a major factor contributing to the ability of the gel to resist digestion by pepsin. The combination of these results indicates that the microstructure of phase separated βlg-xanthan limits digestion by restricting the ability of pepsin to migrate into the gel and access the exposed aromatic peptide bonds. Given the previous research on heat set βlg gels this is a rather novel finding and points to a new direction for using microstructural elements to control digestion.
Acknowledgements
The authors would like to thank Prof. Karen Schroën Wageningen University for excellent discussions during the internship of B. Dekkers. We would also like to thank CPKelco for generous donation of the xanthan gum used in this work.References
- J. WHO and F. E. Consultation, WHO technical report series, 2003, pp. 1–60.
- J. Bowen, M. Noakes and P. M. Clifton, J. Clin. Endocrinol. Metab., 2006, 91, 2913–2919 CrossRef CAS PubMed.
- D. Matzinger, L. Degen, J. Drewe, J. Meuli, R. Duebendorfer, N. Ruckstuhl, M. D'Amato, L. Rovati and C. Beglinger, Gut, 2000, 46, 688–693 CrossRef CAS PubMed.
- A. N. Pilichiewicz, T. J. Little, I. M. Brennan, J. H. Meyer, J. M. Wishart, B. Otto, M. Horowitz and C. Feinle-Bisset, Am. J. Physiol.: Regul., Integr. Comp. Physiol., 2006, 290, R668–R677 CrossRef CAS PubMed.
- C. Feinle, M. Christen, D. Grundy, H. Faas, O. Meier, B. Otto and M. Fried, Neurogastroenterol. Motil., 2002, 14, 205–213 CrossRef CAS PubMed.
- R. D. Mattes, Can. J. Diet. Pract. Res., 2007, 68 Search PubMed.
- T. J. Little, A. Russo, J. H. Meyer, M. Horowitz, D. R. Smyth, M. Bellon, J. M. Wishart, K. L. Jones and C. Feinle-Bisset, Gastroenterology, 2007, 133, 1124–1131 CrossRef CAS PubMed.
- L. Marciani, M. Wickham, G. Singh, D. Bush, B. Pick, E. Cox, A. Fillery-Travis, R. Faulks, C. Marsden, P. A. Gowland and R. C. Spiller, Gastroenterology, 2006, 130, A227–A227 Search PubMed.
- L. Sjostrom, A. Rissanen, T. Andersen, M. Boldrin, A. Golay, H. P. F. Koppeschaar and M. Krempf, Lancet, 1998, 352, 167–173 CrossRef CAS.
- R. V. Seimon, T. Wooster, B. Otto, M. Golding, L. Day, T. J. Little, M. Horowitz, P. M. Clifton and C. Feinle-Bisset, Am. J. Clin. Nutr., 2009, 89, 1729–1736 CrossRef CAS PubMed.
- Y. Li, M. Hu, Y.-M. Du, H. Xiao and D. J. McClements, Food Hydrocolloids, 2010, 25, 122–130 CrossRef.
- T. J. Wooster, L. Day, M. Xu, M. Golding, S. Oiseth, J. Keogh and P. Clifton, Food Hydrocolloids, 2014, 36, 102–114 CrossRef CAS.
- Q. Guo, A. Ye, M. Lad, D. Dalgleish and H. Singh, Soft Matter, 2014, 10, 1214–1223 RSC.
- M. Foltz, P. Ansems, J. Schwarz, M. C. Tasker, A. Lourbakos and C. C. Gerhardt, J. Agric. Food Chem., 2008, 56, 837–843 CrossRef CAS PubMed.
- A. Macierzanka, A. I. Sancho, E. N. C. Mills, N. M. Rigby and A. R. Mackie, Soft Matter, 2009, 5, 538–550 RSC.
- A. Sarkar, K. K. T. Goh and H. Singh, Food Hydrocolloids, 2010, 24, 534–541 CrossRef CAS.
- M. R. Guo, P. F. Fox, A. Flynn and P. S. Kindstedt, J. Dairy Sci., 1995, 78, 2336–2344 CrossRef CAS PubMed.
- W. L. Hall, D. J. Millward, S. J. Long and L. M. Morgan, Br. J. Nutr., 2003, 89, 239–248 CrossRef CAS PubMed.
- Y. Boirie, M. Dangin, P. Gachon, M. P. Vasson, J. L. Maubois and B. Beaufrère, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 14930–14935 CrossRef CAS.
- S. Mahe, N. Roos, R. Benamouzig, L. Davin, C. Luengo, L. Gagnon, N. Gausserges, J. Rautureau and D. Tome, Am. J. Clin. Nutr., 1996, 63, 546–552 CAS.
- M. R. Peram, S. M. Loveday, A. Ye and H. Singh, J. Dairy Sci., 2013, 96, 63–74 CrossRef CAS PubMed.
- Q. Luo, R. M. Boom and A. E. M. Janssen, LWT–Food Sci. Technol., 2015, 63, 161–168 CrossRef CAS.
- E. Pouteau, L. Bovetto, G. Schlup-Ollivier, D. Grathwohl, H. Macharia, M. Beaumont and K. Macé, presented in part at the EPSEN Barcelona, 2012.
- J. l. Zhai, L. Day, M.-I. Aguilar and T. J. Wooster, Curr. Opin. Colloid Interface Sci., 2013, 18, 257–271 CrossRef CAS.
- A. Guerra, L. Etienne-Mesmin, V. Livrelli, S. Denis, S. Blanquet-Diot and M. Alric, Trends Biotechnol., 2012, 30, 591–600 CrossRef CAS PubMed.
- S. B. Doherty, V. L. Gee, R. P. Ross, C. Stanton, G. F. Fitzgerald and A. Brodkorb, Food Hydrocolloids, 2011, 25, 1604–1617 CrossRef CAS.
- M. Minekus, M. Alminger, P. Alvito, S. Ballance, T. Bohn, C. Bourlieu, F. Carriere, R. Boutrou, M. Corredig, D. Dupont, C. Dufour, L. Egger, M. Golding, S. Karakaya, B. Kirkhus, S. Le Feunteun, U. Lesmes, A. Macierzanka, A. Mackie, S. Marze, D. J. McClements, O. Menard, I. Recio, C. N. Santos, R. P. Singh, G. E. Vegarud, M. S. Wickham, W. Weitschies and A. Brodkorb, Food Funct., 2014, 5, 1113–1124 CAS.
- M. Minekus, P. Marteau and R. Havenaar, Altern. Lab. Anim., 1995, 197–209 Search PubMed.
- D. W. Piper and B. H. Fenton, Gut, 1965, 6, 506–508 CrossRef CAS PubMed.
- Anon, U S Pharmacopeia, U S Pharmacopeial Convention 2000, 24 rev edn, 1941 Search PubMed.
- J. B. Dressman, R. R. Berardi, L. C. Dermentzoglou, T. L. Russell, S. P. Schmaltz, J. L. Barnett and K. M. Jarvenpaa, Pharm. Res., 1990, 7, 756–761 CrossRef CAS.
- J. Gardner, S. Sloan, P. Miner and M. Robinson, Aliment. Pharmacol. Ther., 2003, 17, 945–953 CrossRef CAS PubMed.
- L. Kalantzi, E. Persson, B. Polentarutti, B. Abrahamsson, K. Goumas, J. B. Dressman and C. Reppas, Pharm. Res., 2006, 23, 1373–1381 CrossRef CAS PubMed.
- J.-R. Malagelada, V. L. Go and W. Summerskill, Dig. Dis. Sci., 1979, 24, 101–110 CrossRef CAS PubMed.
- L. Kalantzi, K. Goumas, V. Kalioras, B. Abrahamsson, J. Dressman and C. Reppas, Pharm. Res., 2006, 23, 165–176 CrossRef CAS PubMed.
- J. D. Gardner, A. A. Ciociola and M. Robinson, J. Appl. Physiol., 2002, 92, 427–434 CrossRef CAS.
- M. Minekus, M. Smeets-Peeters, A. Bernalier, S. Marol-Bonnin, R. Havenaar, P. Marteau, M. Alric, G. Fonty and J. H. J. Huis in't Veld, Appl. Microbiol. Biotechnol., 1999, 53, 108–114 CrossRef CAS PubMed.
- S. Blanquet, E. Zeijdner, E. Beyssac, J.-P. Meunier, S. Denis, R. Havenaar and M. Alric, Pharm. Res., 2004, 21, 585–591 CrossRef CAS.
- C. Shani-Levi, S. Levi-Tal and U. Lesmes, Food Hydrocolloids, 2013, 32, 349–357 CrossRef CAS.
- Q. Guo, A. Ye, M. Lad, M. Ferrua, D. Dalgleish and H. Singh, Food Funct., 2015, 6, 756–764 CAS.
- A. Mercuri, A. Lo Curto, M. S. J. Wickham, D. Q. M. Craig and S. A. Barker, J. Pharm. Pharmacol., 2008, 60, 4 CrossRef.
- M. Wickham, R. Faulks and C. Mills, Mol. Nutr. Food Res., 2009, 53, 952–958 CAS.
- A. Macierzanka, F. Bottger, L. Lansonneur, R. Groizard, A.-S. Jean, N. M. Rigby, K. Cross, N. Wellner and A. R. Mackie, Food Chem., 2012, 134, 2156–2163 CrossRef CAS PubMed.
- T. K. Singh, S. K. Øiseth, L. Lundin and L. Day, Food Funct., 2014, 5, 2686–2698 CAS.
- S. L. Turgeon, M. Beaulieu, C. Schmitt and C. Sanchez, Curr. Opin. Colloid Interface Sci., 2003, 8, 401–414 CrossRef CAS.
- A. Mackie and A. Macierzanka, Curr. Opin. Colloid Interface Sci., 2010, 15, 102–108 CrossRef CAS.
- T. Considine, H. A. Patel, S. G. Anema, H. Singh and L. K. Creamer, Innovative Food Sci. Emerging Technol., 2007, 8, 1–23 CrossRef CAS.
- C. Tanford, L. G. Bunville and Y. Nozaki, J. Am. Chem. Soc., 1959, 81, 4032–4036 CrossRef CAS.
- M. A. Hoffmann and P. J. van Mil, J. Agric. Food Chem., 1997, 45, 2942–2948 CrossRef CAS.
- O. Goetze, A. Steingoetter, D. Menne, I. R. van der Voort, M. A. Kwiatek, P. Boesiger, D. Weishaupt, M. Thumshirn, M. Fried and W. Schwizer, Am. J. Physiol.: Gastrointest. Liver Physiol., 2007, 292, G11–G17 CrossRef CAS PubMed.
- E. R. Morris, D. A. Rees, G. Young, M. D. Walkinshaw and A. Darke, J. Mol. Biol., 1977, 110, 1–16 CrossRef CAS PubMed.
- S. B. Ross-Murphy, V. J. Morris and E. R. Morris, Faraday Symp. Chem. Soc., 1983, 115–129, 10.1039/fs9831800115.
- M. M. Kool, PhD, Wageningnen, 2014.
- J. G. Forte and L. Zhu, Annu. Rev. Physiol., 2010, 72, 273–296 CrossRef CAS PubMed.
- A. Allen and A. Garner, Gut, 1980, 21, 249–262 CrossRef CAS PubMed.
- C. L. Hoad, P. Rayment, R. C. Spiller, L. Marciani, B. D. Alonso, C. Traynor, D. J. Mela, H. P. F. Peters and P. A. Gowland, J. Nutr., 2004, 134, 2293–2300 CAS.
- L. Marciani, N. S. Coleman, S. P. Dunlop, G. Singh, C. A. Marsden, G. K. Holmes, R. C. Spiller and P. A. Gowland, J. Magn. Reson. Imaging, 2005, 22, 634–638 CrossRef PubMed.
- L. Marciani, M. S. J. Wickham, D. Bush, R. Faulks, J. Wright, A. J. Fillery-Travis, R. C. Spiller and P. A. Gowland, Br. J. Nutr., 2006, 95, 331–339 CrossRef CAS PubMed.
- W. Schwizer, A. Steingoetter and M. Fox, Scand. J. Gastroenterol., 2006, 41, 1245–1260 CrossRef PubMed.
- I. M. Reddy, N. K. D. Kella and J. E. Kinsella, J. Agric. Food Chem., 1988, 36, 737–741 CrossRef CAS.
- M. Dalgalarrondo, E. Dufour, J.-M. Chobert, C. Bertrand-Harb and T. Haertle, Int. Dairy J., 1995, 5, 1–14 CrossRef CAS.
- G. Miranda and J. P. Pelissier, J. Dairy Res., 1983, 50, 27–36 CrossRef CAS PubMed.
- M. Zeece, T. Huppertz and A. Kelly, Innovative Food Sci. Emerging Technol., 2008, 9, 62–69 CrossRef CAS.
- D. Renard, J. Lefebvre, M. C. A. Griffin and W. G. Griffin, Int. J. Biol. Macromol., 1998, 22, 41–49 CrossRef CAS PubMed.
- N. K. D. Kella and J. E. Kinsella, Int. J. Pept. Protein Res., 1988, 32, 396–405 CrossRef CAS PubMed.
- A. Lindahl, A. L. Ungell, L. Knutson and H. Lennernas, 2nd European Congress of Pharmaceutical Sciences, Plenum Publ Corp, Berlin, Germany, 1994, pp. 497–502 Search PubMed.
- M.-L. Jalabert-Malbos, A. Mishellany-Dutour, A. Woda and M.-A. Peyron, Food Qual. Preference, 2007, 18, 803–812 CrossRef.
- L. Marciani, P. A. Gowland, A. Fillery-Travis, P. Manoj, J. Wright, A. Smith, P. Young, R. Moore and R. C. Spiller, Am. J. Physiol., 2001, 280, G844–G849 CAS.
- K. C. Labropoulos, D. E. Niesz, S. C. Danforth and P. G. Kevrekidis, Carbohydr. Polym., 2002, 50, 407–415 CrossRef CAS.
- M.-C. Klak, J. Picard, S. Giraudier and V. Larreta-Garde, Soft Matter, 2012, 8, 4750–4755 RSC.
- P. G. De Gennes, Scaling Concepts in Polymer Physics, Cornell Univ. Press, 1979 Search PubMed.
- M. Rubinstein and R. H. Colby, Polymer Physics, Oxford University Press, Oxford, UK, 2003 Search PubMed.
- W. H. Shih, W. Y. Shih, S. I. Kim, J. Liu and I. A. Aksay, Phys. Rev. A, 1990, 42, 4772–4779 CrossRef CAS.
- M. Mellema, J. H. J. van Opheusden and T. van Vliet, J. Rheol., 2002, 46, 11–29 CrossRef CAS.
- S. Ghosh, S. Dolai, T. Patra and J. Dey, J. Phys. Chem. B, 2015, 119, 12632–12643 CrossRef CAS PubMed.
- M. Sauter, J. Curcic, D. Menne, O. Goetze, M. Fried, W. Schwizer and A. Steingoetter, Neurogastroenterol. Motil. 24, 2012, 7, 632–628 CrossRef PubMed.
Footnote |
| † Current address: Wageningen UR, Agrotechnology and Food Sciences, Food Process Engineering, Netherlands. |
| This journal is © The Royal Society of Chemistry 2016 |