 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Linear assembly of patchy and non-patchy nanoparticles
Rachelle M.
Choueiri†
a,
Elizabeth
Galati†
a,
Anna
Klinkova
a,
Héloïse
Thérien-Aubin
a and
Eugenia
Kumacheva
*abc
aDepartment of Chemistry, University of Toronto, 80 Saint George Street, Toronto, Ontario M5S 3H6, Canada. E-mail: ekumache@chem.utoronto.ca
bDepartment of Chemical Engineering and Applied Chemistry, University of Toronto, 200 College Street, Toronto, Ontario M5S 3E5, Canada
cThe Institute of Biomaterials and Biomedical Engineering, University of Toronto, 4 Taddle Creek Road, Toronto, Ontario M5S 3G9, Canada
First published on 19th April 2016
Abstract
Linear assemblies of nanoparticles show promising applications due to their collective electronic, optical and magnetic properties. Rational design and controllable organization of nanoparticles in one-dimensional structures can strongly benefit from the marked similarity between conventional step-growth polymerization reactions and directional step-wise assembly of nanoparticles in linear chains. Here we show different aspects of the “polymerization” approach to the solution-based self-assembly of polymer-functionalized metal nanoparticles with different chemical compositions, shapes and dimensions. The self-assembly was triggered by inducing solvophobic attraction between polymer ligands, due to the change in solvent quality. We show that both anisotropic (patchy) nanoparticles and nanoparticles uniformly capped with polymer molecules can self-assemble in linear chains. We explore the control of chain length, morphology, and composition, discuss the ability to form isotropic and hierarchical structures and show the properties and potential applications of linear assemblies of plasmonic nanoparticles.
Introduction
Linear, one-dimensional assemblies of metal nanoparticles (NPs) have promising applications in the sensing of chemical and biological species and in the fabrication of waveguides and antennas.1 The collective optical properties of NP assemblies depend on their aggregation number, the distance between the NPs in the chain, NP collinearity and chain rigidity, to name just a few important characteristics.2,3 For example, the structure and composition of NP chains influence the spatial distribution of charge polarization, as well as the splitting and position of the plasmon resonance peak. Specific design rules have to be applied to the fabrication of such assemblies, in order to achieve their best performance.One of the promising approaches to the rational design of self-assembled NP structures relies on a remarkable resemblance between linear polymer molecules and one-dimensional NP assemblies, in which NP building blocks act as repeat units linked with physical or chemical bonds. For example, for the chain of rod-like NPs shown schematically in Fig. 1, it is possible to define the degree of polymerization, x (equal to 3), the bond length and the bond angle.
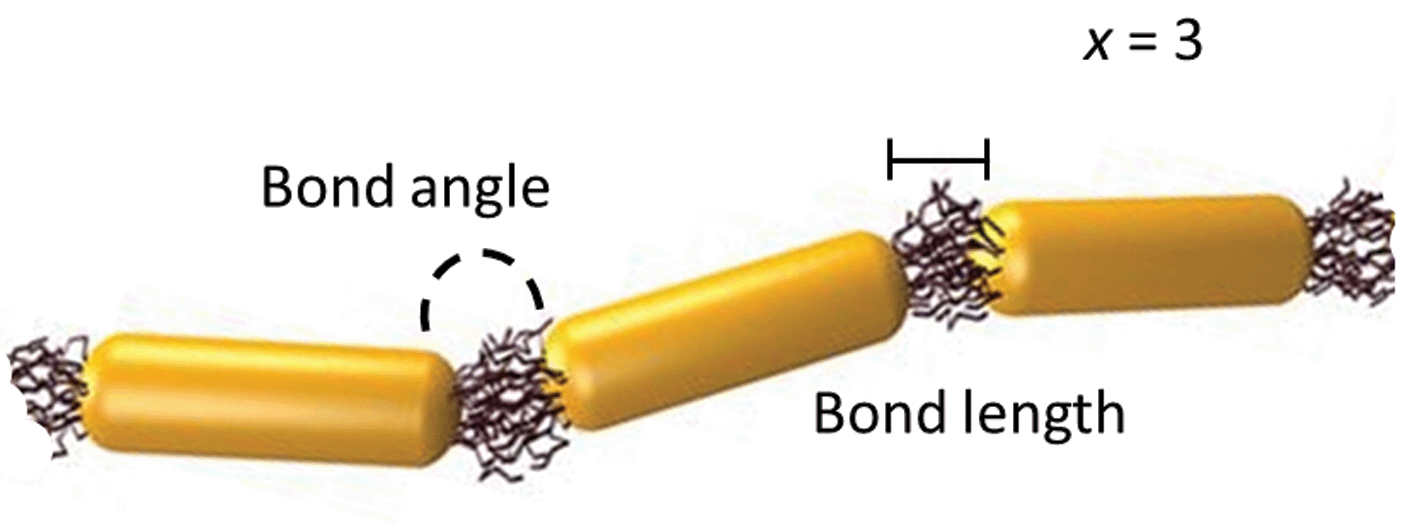 | ||
| Fig. 1 A chain of three polymer-functionalized nanorod repeat units forming bonds with a particular angle and length. | ||
Furthermore, building on this analogy, it would be possible to make the correlation between the mean aggregation number of the NP chains and their number average or weight average degree of polymerization, as well as polydispersity index, PDI4 as
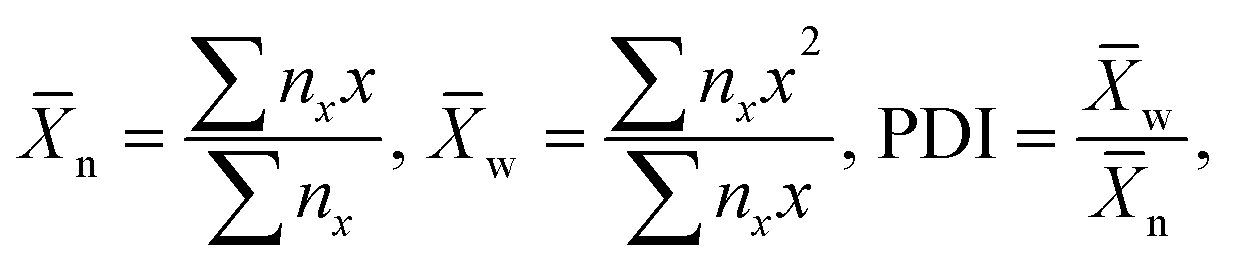 | (1) |
The analogy between molecular polymerization reactions and linear self-assembly of NPs can benefit both areas of research. Direct visualization of nanostructures formed in different stages of self-assembly and analysis of their shape, composition and size distribution offers the capability to examine the effect of reactivity of nanomers and explore the role of side reactions on nanopolymer structure and composition. Such analysis would enable validation of theoretical models developed for synthetic polymer chemistry. Furthermore, nanoscience-specific techniques, e.g., the measurement of the red-shift of surface plasmon resonance of metal NPs or small-angle X-ray scattering can be used for the characterization of colloidal polymerization. On the other hand, strategies developed in synthetic polymer chemistry, e.g., the copolymerization of comonomers with distinct properties, the synthesis of specific isomers, and control of polymer molecular weight by using “chain stoppers”, offer a platform for the self-assembly of nanopolymers with well-defined structure and properties.
Here we discuss different aspects of one-dimensional self-assembly of polymer-functionalized metal NPs with different compositions, shapes and dimensions. We show that both patchy and non-patchy NPs coated with polymer ligands can assemble in nanopolymer chains, with a striking similarity between molecular step-growth polymerization and nanoparticle assembly. We also show an example of a higher-level organization of NP assemblies in complex, hierarchical structures. Finally, we demonstrate optical properties of linear assemblies of plasmonic NPs, such as their surface enhanced Raman scattering (SERS), as well as new properties of the chains of face-to-face assembled metal nanocubes, due to the formation of a Fabry–Perot resonator structure.5
Experimental
Gold, silver and palladium NPs were synthesized using metal precursor salts as described elsewhere.6–12 The syntheses were carried out in water, with cetyltrimethylammonium bromide (CTAB) used as a stabilizer. To exchange the CTAB ligands with polymer molecules, a concentrated NP solution in water was added to the solution of thiol-terminated polystyrene (PS) or thiol-terminated poly(styrene-co-isoprene) in tetrahydrofuran. The resulting solution was sonicated for 30 min and equilibrated for at least 24 h. The synthesis of two-component heterostructured iron oxide–gold NPs is reported elsewhere.13 For these NPs, after the replacement of CTAB ligands with PS molecules on the surface of the gold constituent, an additional ligand exchange step was carried out to replace oleic acid with dopamine-terminated polyethylene glycol on the iron oxide surface.After completion of the ligand exchange procedure, the NPs were transferred into dimethylformamide (DMF), a good solvent for both CTAB and PS. Linear NP self-assembly was triggered by reducing the quality of solvent for the polymer ligands. A solvent mixture containing DMF and water was added to the NP solution to reach a pre-determined concentration of water, Cw, in the solution. When thiol-terminated poly(styrene-co-isoprene) was used as a ligand, after a particular self-assembly time, the NP solution in the DMF–water mixture in the presence of azobisisobutyronitrile (AIBN) was subjected to 5–25-min-long ultra-violet irradiation using a UV-A lamp (Hönle, UVAPrint 40C, λ = 365 nm, intensity 30 mW cm−2), to achieve permanent crosslinking of the self-assembled NP chains.
The evolution of the self-assembled structures was followed by monitoring the shift in the extinction peak of the NPs using a Varian Cary 5000 spectrometer. Raman scattering measurements were carried out using a Raman spectrometer (DeltaNu Advantage Raman Series with a 785 nm laser line). Characterization of NP dimensions and electrokinetic potential were carried on a Zetasizer Nano instrument (Malvern) using a 633 nm HeNe laser. Imaging of the self-assembled structures was carried out by transmission electron microscopy (TEM) and scanning electron microscopy (SEM) using a Hitachi H-7000 transmission electron microscope and a Hitachi HD-2000 scanning transmission electron microscope, respectively. A droplet of the NP solution was deposited on the carbon-coated grid, and after ∼60 s the solvent was removed with a tissue.
Results and discussion
Linear assembly of anisotropic (patchy) nanoparticles
End-functionalization of gold spherocylindrical and arrow-headed nanorods (both denoted as NRs) with PS ligands was achieved by fine-tuning the concentration of PS in the solution in tetrahydrofuran in the ligand exchange procedure, so that CTAB was replaced only at the NR edges due to its weaker attachment to the 111 crystalline plane at the NR ends.14 The end-attachment of PS endowed patchy NRs with two reactive sites (Fig. 2A). To induce linear NR self-assembly, the reactivity of the patches was activated by introducing water to the NR solution in DMF, thereby reducing solvent quality for the hydrophobic PS molecules. To reduce the free surface energy of the system, the PS ligands screened unfavorable interactions with the poor solvent by PS–PS association, thus forming a physical bond between the NR ends. This process resulted in the formation of linear NR chains shown in Fig. 2B. The self-assembly resembled molecular polymerization, with individual NRs acting as monomers (or nanomers) forming a colloidal analogue of a molecular polymer (nanopolymer). Similar to molecular polymers, we observed the formation of other nanopolymer structures such as branched H-polymers (Fig. 2C) and networks (Fig. 2D). | ||
| Fig. 2 (A) Schematic (top) and TEM image (bottom) of a gold NR end-tethered with PS ligands. Scale bar is 100 nm. (B) SEM image of a linear chain of gold NRs end-tethered with PS ligands. Scale bar is 500 nm. (C) Dark-field TEM image of PS-end-tethered gold arrow-headed NRs self-assembled into a branched polymer (H-polymer). Scale bar 200 nm. SEM image of the self-assembled network of gold NRs end-terminated with PS molecules. Scale bar is 500 nm. In (B–D) the self-assembly occurred in the DMF–water solution at Cw = 15 vol%. | ||
The evolution of the nanopolymers in the course of self-assembly was characterized by statistical analysis of the self-assembled structures. By analogy with molecular polymerization, the average number of NRs in the chains was defined as the number average degree of polymerization, ![[X with combining macron]](https://www.rsc.org/images/entities/i_char_0058_0304.gif) n.
n.
The self-assembly kinetics and statistics of the distribution of the degree of polymerization of the nanopolymer followed closely those of a conventional molecular step-growth polymerization.15 The value of n of the nanopolymer increased linearly with the self-assembly time t as
| n = 2[M]0kpt + 1, | (2) |
n ∼ t was characteristic of reaction-controlled step growth polymerization, in which the reactivity of monomer functional groups does not depend on the polymer’s degree of polymerization (Flory's assumption).4 The rate of assembly characterized as dn/dt linearly increased with nanomer concentration (Fig. 3A). Furthermore, the variation in polydispersity index, PDI, of the nanopolymers was similar to the change in PDI of step-growth polymers, which increase as4,16 PDI = 2 − 1/n and approach 2 at high conversion (Fig. 3B).
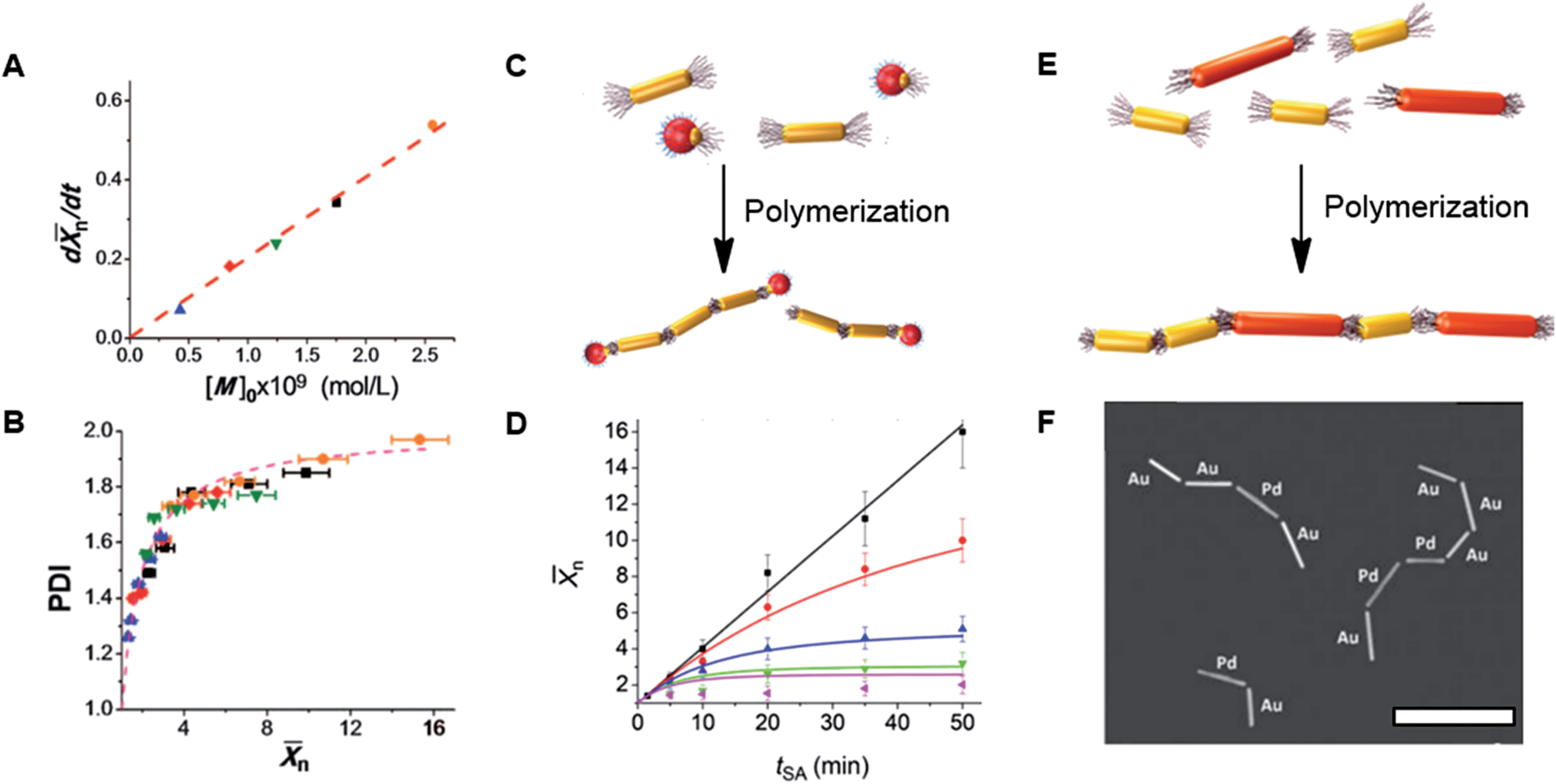 | ||
| Fig. 3 (A and B) Polymerization of NRs at initial NR concentration, [M]0, of 0.42 × 10−9 (solid blue triangles), 0.84 × 10−9 (solid red diamonds), 1.24 × 10−9 (solid green inverted triangles), 1.76 × 10−9 (solid black squares), and 2.56 × 10−9 (solid orange circles) (mol L−1). (A) Variation in the rate of NR polymerization, dn/dt, plotted as a function of NR concentration. (B) Variation in polydispersity index, PDI, in the course of NR polymerization. The dashed line shows the theoretically expected variation PDI = (2 − 1/n). (A) and (B) are adapted from ref. 15. (C) Schematic of NR polymerization in the presence of colloidal chain stoppers. The yellow and red colours correspond to the gold and iron oxide constituents of the colloidal chain stoppers. (D) Variation in n of the NR chains with self-assembly time at different chain stopper concentrations. (C) and (D) are adapted from ref. 17. (E) Schematic of copolymerization of distinct nanomers shown with different colours; (F) dark field TEM image of chains of copolymerized palladium and gold NRs. Scale bar is 500 nm. (F) is adapted from ref. 20. | ||
By analogy with molecular polymers, we estimated the mean contour and persistence lengths of the nanopolymers formed by the NRs, which at varying solvent quality were up to 800 and 250 nm, respectively. We note that these characteristics should be treated with caution, since they were determined by analyzing NR images and may be affected by drying NR solutions.
We further explored the similarity between the self-assembly of patchy NRs and polymer synthesis by using the strategies used in molecular step-growth polymerization, namely, control of polymer molecular weight by using monofunctional chain stoppers and copolymerization of chemically distinct nanomers.
Control of nanopolymer chain length
To control the value ofn in conventional step-growth polymerization, the stoichiometry between bifunctional monomers is broken by adding a small amount of monofunctional comonomers which “zip” growing polymer chains.4 We explored this approach for nanopolymers by introducing in the solution of self-assembling bifunctional patchy NRs a small quantity of “monofunctional” nanospheres (NSs), as illustrated in Fig. 3C.17 The colloidal analogues of monofunctional chain stoppers (CSs) were heterostructured NSs, which contained two constituents: a reactive gold constituent capped with PS ligands and an iron oxide constituent capped with dopamine-tethered polyethylene glycol. The ratio of the dimensions of the gold and iron oxide components of the NSs was carefully tuned to ensure size complementarity between the end of the gold NR nanomer and the reactive PS-capped gold domain of the CS (polyethylene glycol end-tethered to the non-reactive iron oxide domain acted as a stabilizer). A barrier preventing further NR or NS attachment to the “zipped” chain end was provided by a sufficiently large size of the iron oxide domain.
The heterostructured CSs were mixed with the NRs in DMF in the ratio 0 ≤ [CS]/[NR] ≤ 1.55, where [CS] and [NR] were the molar concentrations of the CSs and the NRs, respectively. When the quality of the solvent for the PS ligands was reduced by adding water to DMF to achieve Cw = 15 vol%, the two-patch NRs assembled in linear chains, while the single-patch patch NSs attached to chain end(s) and in this manner, “zipped” them to suppress further NR self-assembly. The processes of self-assembly and chain zipping, as well as side reactions of CSs were visualized at different stages of the self-assembly process by using electron microscopy. Imaging of NPs was complemented by the analysis of the shift in surface plasmon resonance of the NRs. In this manner, we were able to ascertain the prevalence of side reactions of the monofunctional CSs such as their dimerization or binding to junctions between adjacent NRs in the chains. Based on the kinetic and statistical analysis of chain growth, we developed a model of the kinetics of the colloidal polymerization in the presence of a particular concentration of CSs. In particular, via statistical analysis of the TEM images we accounted quantitatively for the contribution of side reactions in the course of NR chain growth. The efficiency, E, of the CS attachment to the end of the chain (the desired “zipping” step) enabled the prediction of n of the nanopolymer as
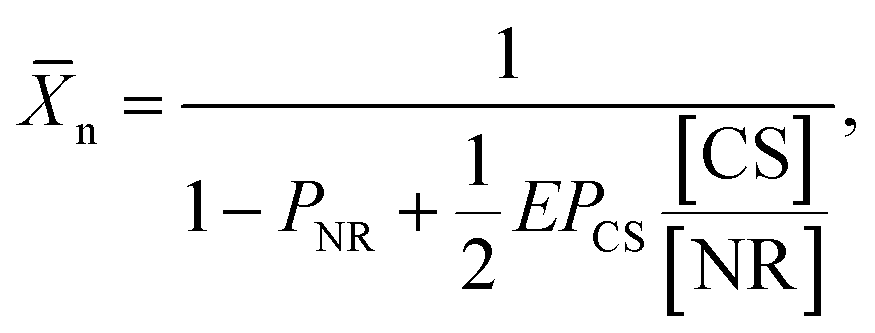 | (3) |
The ratio [CS]/[NR] affected the length of the NR chains: for a particular self-assembly time, the value of n of the nanopolymer reduced with increasing concentration of monofunctional CSs (Fig. 3D). The co-assembly of CSs with the NR chains also resulted in a deviation from the linear dependence of nvs. t, which was characteristic of the NR self-assembly in the absence of CSs.
By varying the ratio [CS]/[NR] at the beginning of the self-assembly process, a particular value of n of the nanopolymer could be achieved and effectively controlled by adjusting the initial concentration of colloidal CSs, in striking similarity with molecular step-growth polymerization.
Step-growth copolymerization
Future applications of self-assembled nanostructures require NP ensembles with a high degree of complexity, which may be achieved by heteroassembly of NPs with different dimensions, shapes, and chemical compositions. For example, new optical modes, such as Fano resonance are expected for heterostructures of NPs.18,19 Since linear self-assembly of NPs is analogous to a polymerization reaction, we explored the step-growth polymerization-based approach for the assembly of nanocopolymers,20 in which distinct NP repeat units were assembled in linear chains, as shown in the scheme in Fig. 3E. Experimentally, this approach was explored for the self-assembly of PS end-tethered gold and palladium NR co-nanomers, and for the self-assembly of gold NRs with difference lengths in the DMF–water mixture. A study of the co-assembly of gold NRs with different lengths revealed that the polymerization rate reduced with increasing NR length, due to the combined effect of reduced NR mobility and increased excluded volume. The copolymerization of gold NRs with different lengths was examined to determine the composition and microstructure of the resulting copolymer chains. When the NRs with different length were co-assembled, the rate of copolymerization was related to the rate of the homopolymerization as| kcopol = k11f2 + k22(1 − f)2 + 2k12f(1 − f) | (4) |
The structure of the nanocopolymer was found to be random. The microhomogeneity factor (H) of the nanocopolymer was determined by analyzing the distribution of dyads of two NP types in the nanocopolymer as
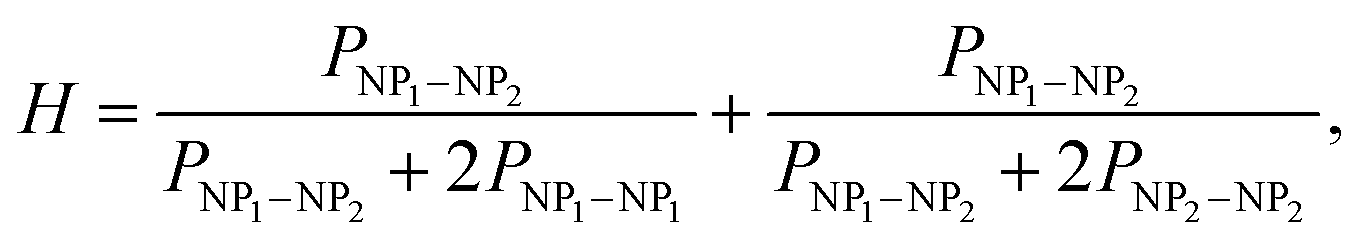 | (5) |
Following the same strategy, we co-assembled (copolymerized) gold and palladium NRs with lengths of ∼180 and ∼270 nm, respectively, that were introduced in the solution at concentrations of 43 and 8.6 nM, respectively. The coassembly yielded a random copolymer (shown in Fig. 3F) with a microhomogeneity factor H = 1.13.
Photocrosslinking of nanopolymers
Photocrosslinking of the polymer ligands bridging NRs in the nanopolymer was used to gain control over the properties of the self-assembled chains.21 The NRs end-terminated with poly(styrene-co-isoprene) (PS-co-PI) were dispersed in DMF containing an initiator azobisisobutyronitrile (AIBN). The addition of water to this mixed solution led to a reduction of the quality of the solvent for both PS-co-PI22 and AIBN, which resulted in two effects: the self-assembly of the NRs in linear chains and the partition of the hydrophobic initiator in the PS-co-PI ligands bridging the NRs (Fig. 4A). After a particular self-assembly time (determining the value ofn), the crosslinking reaction was triggered by illuminating the NR chains with ultra-violet (UV) light. The UV-irradiation of the AIBN molecules created free radicals which initiated the crosslinking of the 3,4-isoprene units in adjacent PS-co-PI molecules (Fig. 4B).
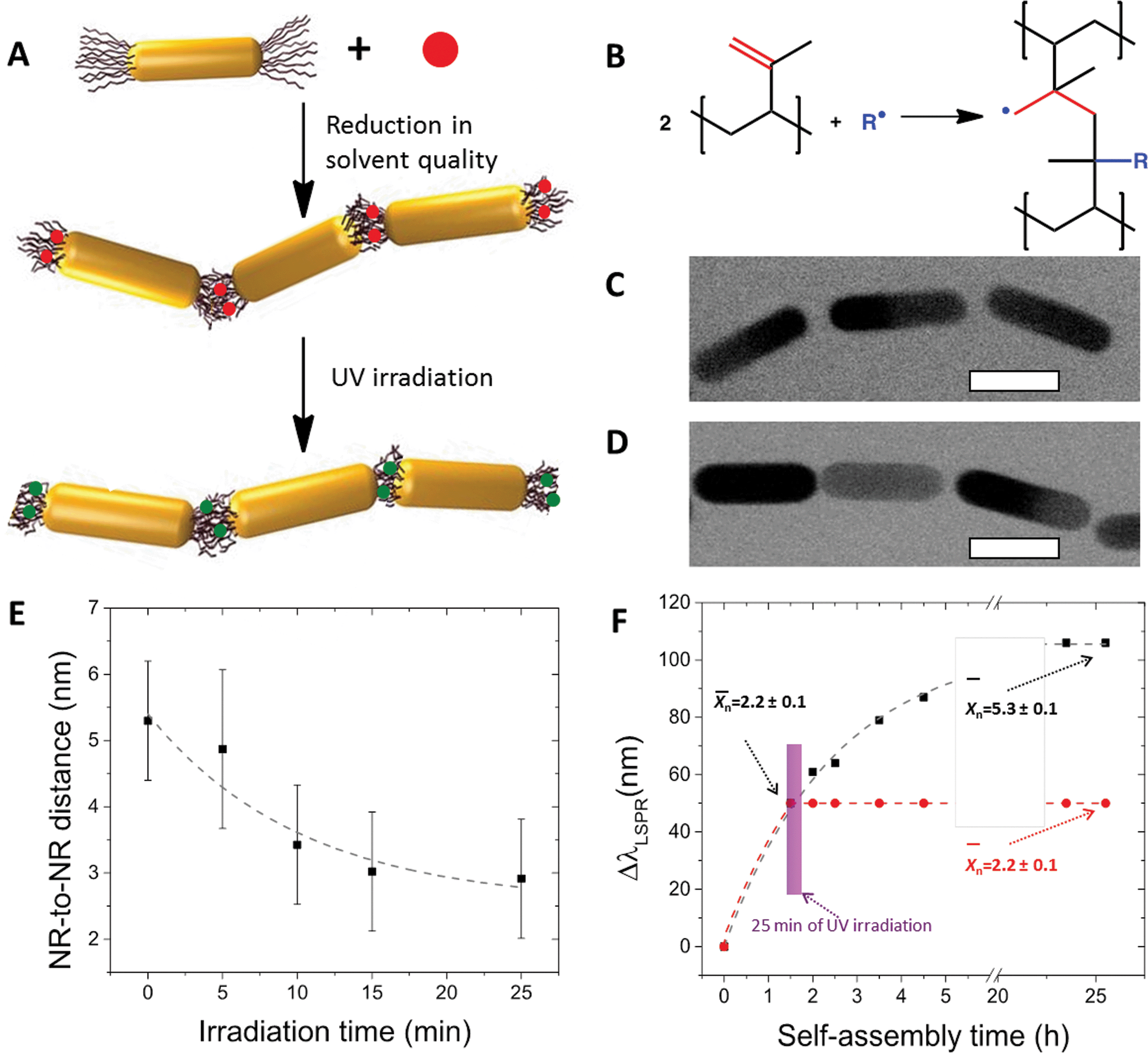 | ||
| Fig. 4 Photocrosslinking of self-assembled NR chains. (A) Schematic of chain crosslinking. Top: The NRs end-functionalized with poly(styrene-co-isoprene) ligands are mixed with a photoinitiator, AIBN (red dot), in the solution in DMF; middle: NR self-assembly is triggered by adding of water to the NR solution in DMF. The hydrophobic AIBN molecules partition in the copolymer between adjacent NRs in the chains; bottom: chain photocrosslinking is initiated by the irradiation at 365 nm to crosslink the 3,4-isoprene units in the copolymer. (B) Chemical reaction taking place in the copolymer during photocrosslinking. A fragment of the NR chain before (C) and after (D) photocrosslinking. The scale bars are 25 nm. (E) Effect of the photocrosslinking time on the average distance between two adjacent NRs in the chain. (F) Effect of photocrosslinking on the NR self-assembly, presented as a temporal shift of the surface plasmon resonance with time. After 1.5 h assembly (at n = 2.2), the NRs were irradiated for 25 min at 365 nm. The NR chain growth was frozen at n = 2.2 (red circles), while without irradiation the self-assembly continued and the value of n increased. (A, C, D, E and F) are reproduced from ref. 21. | ||
One of the important outcomes of the NR chain photocrosslinking was a decrease in the volume of the copolymer ligands bridging NR ends in the chain. This effect resulted in a reduction of the gap separating two adjacent NRs (Fig. 4C–E). Approximately 55% reduction in the inter-rod distance was achieved after 25 min of UV-irradiation, in comparison with the non-crosslinked nanopolymer. This effect has important implications for potential applications of NR ensembles in surface-enhanced Raman scattering (SERS)-based sensing, since the enhancement of the SERS signal increases with a reducing distance between plasmonic NPs.21,23 Furthermore, the crosslinking of the copolymer also resulted in the quenching of the growth of the nanopolymer. This effect was caused by the crosslinking of the terminal copolymer ligands at the NR chain ends, thus suppressing the interpenetration of the copolymer molecules and preventing further addition of nanomers to the chain. This effect is explicitly shown in Fig. 4F. After the value of n of 2.2 was reached (corresponding to a particular shift in the surface plasmon resonance of the NR chains) and the polymer was crosslinked, no further increase in n occurred within 25 h, nor variation in the NR chain optical properties, while without irradiation the NR chains continued to grow reaching a value of n of 5.3 after 24 h.
Finally, the photocrosslinking of the copolymer ligands led to an increase of collinearity of adjacent NRs in the chains. Crosslinked NR chains had a higher fraction of bond angles exceeding 120°, compared to the non-crosslinked NR chains. A higher rigidity of the photocrosslinked NR chains was attributed to a reduced conformational entropy due to the reduction in the volume of the polymer ligands connecting the NRs in the chains.
Linear assembly of isotropic (non-patchy) nanoparticles
We found that surface or shape anisotropy was not required for directional linear self-assembly of polymer-functionalized NPs, in comparison with linear assembly of bifunctional “patchy” NPs.24 In a poor solvent, NPs uniformly coated with PS ligands also assembled in nanopolymers. This effect was attributed to directional, anisotropic forces governing linear NP assembly and was examined for gold NSs and silver and palladium nanocubes (NCs), all coated with PS ligands.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1), we induced NS assembly in linear and globular structures (Fig. 5B–D).25 The effect of solvent quality and polymer molecular weight on the resulting structure of NS assemblies is summarized in a phase-like diagram (Fig. 5G), which shows globular (G), chain (C), a mixed (G + C) and individual NS (I) regimes. The formation of globules was favored for the NSs capped with high molecular weight PS ligands at low water content, whereas for higher Cw values (dependent on polymer molecular weight), linear NS assemblies were the dominant species. At Cw ≥ 15 vol%, regardless of the polymer molecular weight, the NSs assembled only in a linear manner.
000 g mol−1), we induced NS assembly in linear and globular structures (Fig. 5B–D).25 The effect of solvent quality and polymer molecular weight on the resulting structure of NS assemblies is summarized in a phase-like diagram (Fig. 5G), which shows globular (G), chain (C), a mixed (G + C) and individual NS (I) regimes. The formation of globules was favored for the NSs capped with high molecular weight PS ligands at low water content, whereas for higher Cw values (dependent on polymer molecular weight), linear NS assemblies were the dominant species. At Cw ≥ 15 vol%, regardless of the polymer molecular weight, the NSs assembled only in a linear manner.
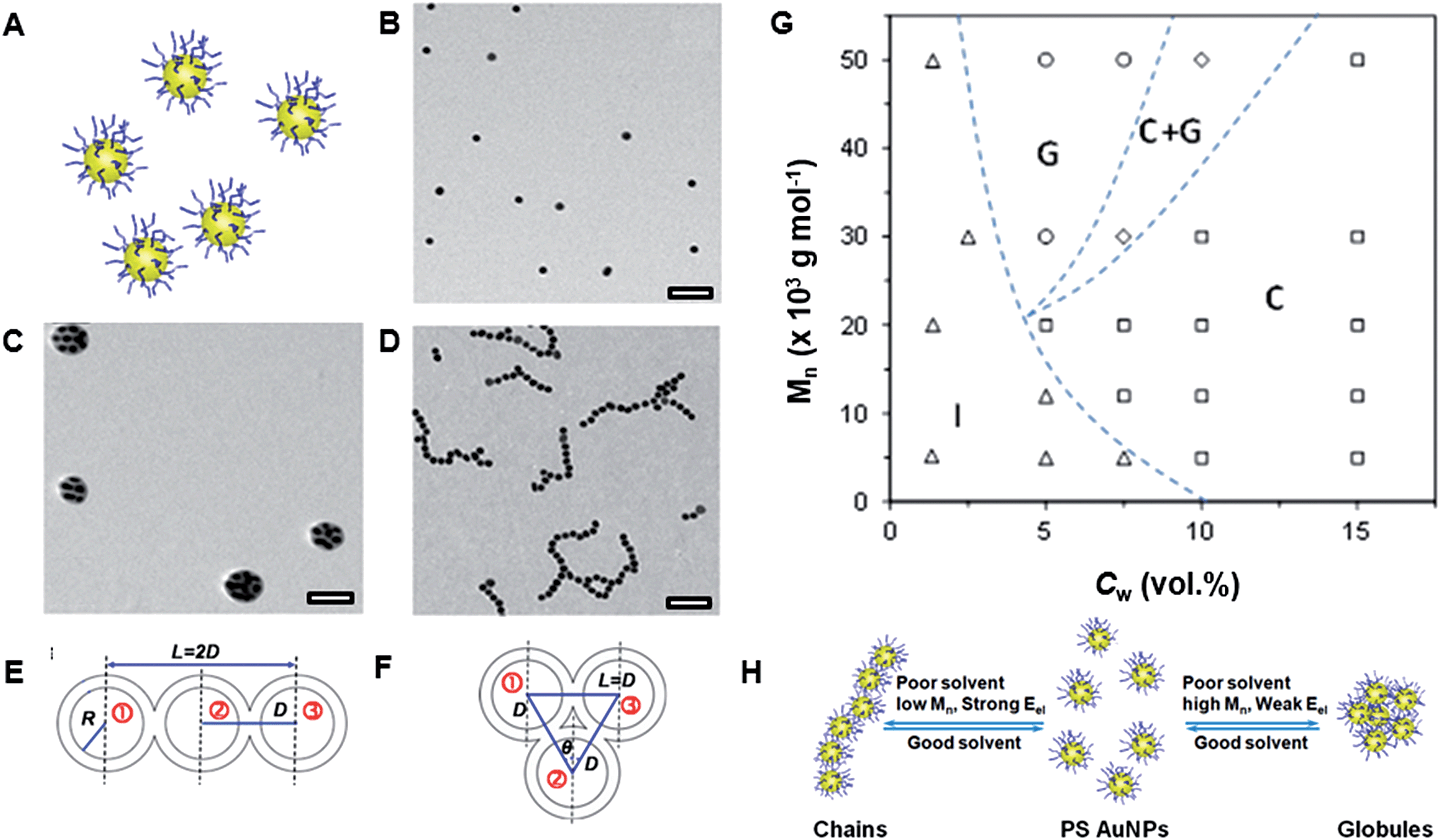 | ||
| Fig. 5 (A) Schematic of individual gold NSs functionalized with PS ligands. (B) TEM image of individual PS-50k-functionalized gold NSs deposited from DMF. (C) TEM image of globules formed by gold NSs functionalized with PS-50k after 1 h assembly in the DMF–water solution at Cw = 5%. (D) TEM image of chains of gold NSs functionalized with PS-5k after 1 h assembly in the DMF–water solution at Cw = 15%. (G) Phase diagram of self-assembly of gold NSs. Schematics of three assembled gold NSs with 180° (E) and 60° (F) the center of the second NS with the centers of adjacent 1st and 3rd NSs (H) schematic of the factors determining NS self-assembly in linear and globular structures. The grafting density of PS-50k on the NS surface was ∼0.08 chains per nm2 and the mean square end-to-end distance for PS-50k was ∼220 nm2. Adapted from ref. 25. | ||
The schematics of two limiting configurations formed by three adjacent NSs in the chains and globules are shown in Fig. 5E and H, respectively. The angle θ between the lines connecting the center of the 2nd NS with the centers of adjacent 1st and 3rd NSs was 60° and 180° for the globular and linear assembly, and thus determined the type of structure formed.
The formation of a particular structure (globular vs. linear) was governed by the interplay of forces leading to the minimum total free energy of the system,
| ΔEt = ΔEel + ΔEh, | (6) |
The value of ΔEh was calculated as
| ΔEh = ΔEγ × ΔA, | (7) |
The value of ΔEel for two associating NSs was calculated as26
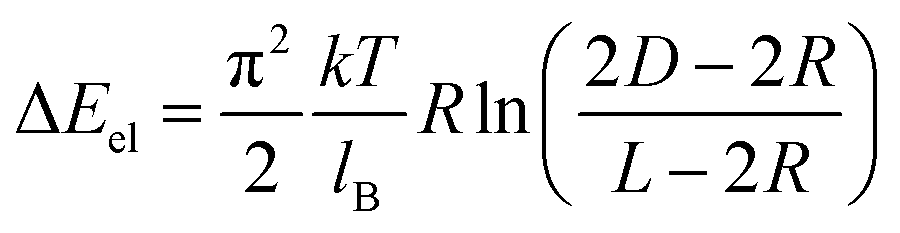 | (8) |
The variation in ΔEt was evaluated for the assembly of NSs stabilized with PS molecules with a molecular weight of 50000 g mol−1 at Cw of 5 and 15 vol%, at which the NPs assembled into the globules and the chains, respectively. Indeed, in agreement with experimental results, for Cw = 5 vol%, due to the stronger contribution of the surface energy, the globular NS assembly (θ = 60°) had a minimum in ΔEt and was thus favored. At Cw = 15 vol%, the minimum value of ΔEt was achieved at θ = 180° (corresponding to the NS chains), due to the stronger and larger contribution of electrostatic repulsion.
We found that reduction in electrostatic repulsion achieved by addition of salts or tetrahydrofuran (a solvent with a lower dielectric constant than DMF) to the NS solution in the DMF/water mixture, resulted in a chain-to-globule transition. In addition, variation in the molecular weight of the polymer ligands could be used to control the formation of a particular structure. The utilization of longer polymer ligands resulted in a greater screening of the NS charge and the propensity of NSs to form globular structures increased even at high solvent polarity. Fig. 5H summarizes the role of different factors on the ability to form linear nanopolymers or globular structures from non-patchy NSs.
For PS-coated NCs, it was expected that due to the NC geometry, their face-to-face assembly is preferred, due to the maximum reduction in solvent-exposed PS surface area. For this binding modality, the formation of rigid chains was expected. We found, however, that PS ligands with a high molecular weight screened the NC geometry, thus yielding fewer face-to-face NC contacts. Consequently, chains with a larger number of kinks were formed, resembling assembly of PS-coated NSs. Fig. 6 shows the chains formed by PS-capped silver NSs (Fig. 6A) and the chains of NCs at a reducing ratio of the PS length-to-NC size (top-to-bottom, Fig. 6B–D). When the ratio of polymer length-to-NC size reduced, the fraction of face-to-face NC contacts increased, the NC chains became more rigid and exhibited a smaller number of kinks (Fig. 6E and F). Quantification of the number of face-to-face contacts was carried out using a MATLAB code that analysed the TEM images of the self-assembled chains and measured the angles formed by two or three adjacent NPs in the chains (Fig. 6E and F, insets). The statistical analysis of the angles formed by two or three adjacent NCs confirmed quantitatively the trend of increased NC collinearity with increasing ratio of NC size-to-polymer length.
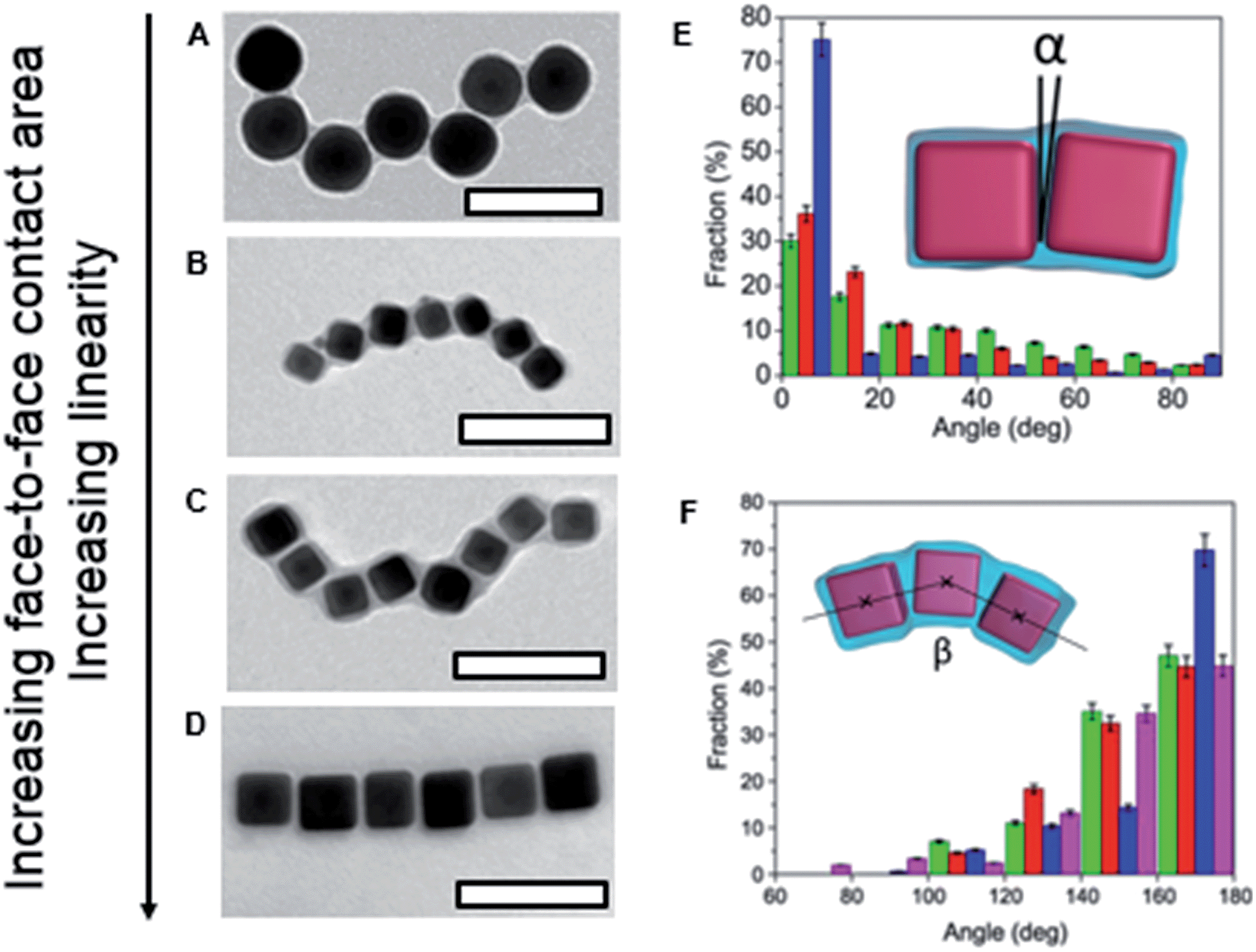 | ||
| Fig. 6 (A) Chain of silver NSs; (B, C and D) representative TEM images of silver NCs assembled in nanopolymer structures. Scale bars are 100 nm. (E) Fraction of α angles for 25 (green), 35 (red) and 45 nm (blue) silver NCs. (F) Fraction of β angles for 25 (green), 35 (red) and 45 nm (blue) silver NCs and silver NSs (pink). Adapted from ref. 5. | ||
000 g mol−1 (PS-5k and PS-50k, respectively).
The morphology of the self-assembled structures in the DMF/water mixture at Cw = 20 vol% depended on the length of the polymer ligands (controlled by its molecular weight), in qualitative agreement with the results obtained for assembly of gold NSs and silver NCs. Fig. 7 shows that for the same self-assembly time and solvent quality, the structures formed by palladium NCs functionalized with PS-5k exhibited a stronger trend to form linear structures than those formed by the NCs capped with PS-50k. The latter had a larger aggregation number due to the stronger solvophobic attraction between the longer polymer ligands. In addition, the assemblies formed by PS-50k-capped palladium NCs had fewer face-to-face contacts between adjacent NCs. The latter effect stemmed from the screening of the cubic shape of the NCs with longer polymer ligands. In contrast, palladium NCs capped with PS-5k showed the propensity to form chain-like and open network assemblies.
 | ||
| Fig. 7 TEM images of nanostructures formed by palladium NCs capped with (A) PS-5k and (B) PS-50k in the DMF/water mixture at Cw = 20 vol%. Scale bars are 200 nm. Inset in (A) shows an enlarged fragment of the NC chain. Scale bar is 10 nm. | ||
Hierarchical assembly of nanoparticles
An extension of the anisotropic assembly of isotropically PS functionalized gold NSs was the self-assembly of networks of globules composed of these NSs. Given a sufficiently long incubation time of 24–48 h in the DMF–water mixture, the globules of with an average diameter of approximately 85 nm (Fig. 8A) assembled into chains and networks (Fig. 8B). We note that in the chains, the globules did not merge to form larger entities, which could be rationalized by the enthalpic penalty involved in the breaking of physical bonds to maximize PS–PS interactions. In addition, although the solvent was not sufficiently polar to cause anisotropic assembly of individual NSs, linear and network diffusion-controlled assembly of the globules was favored due to the larger overall charge of the NS globules, in comparison with individual NSs.27 | ||
| Fig. 8 (A) Individual globules formed by gold NSs isotropically functionalized with PS-50k in the DMF–water solution at Cw = 7.5% after 1 h self-assembly. Networks of globules formed from the globules after 24 h assembly (B) and after increasing Cw to 15 vol% at the self-assembly time of 2 h. | ||
Furthermore, reducing solvent quality by increasing Cw from 5 to 15 vol% resulted in qualitatively similar self-assembly of chains and networks of globules at a reduced time of 2 h (Fig. 8C), due to the more efficient bonding of the NS globules.
Plasmonic properties of nanopolymers
Nanopolymers display directional coupling of optical, electronic, and magnetic properties as a result of interactions between the surface plasmons, excitons, or magnetic moments, respectively, of individual NPs. The formation of a periodic array of hot spots in the gaps between the NPs assembled in chains is important for SERS applications of plasmonic nanopolymers, as they provide significant signal enhancement of up to 1015 orders of magnitude.28 Thus, the use of hot spots enables the observation of single-molecule SERS and sets the limit for the spatial resolution in scanning probe microscopy with high chemical specificity (tip-enhanced Raman spectroscopy).29Dynamic linear self-assembly of metal NPs provides the capability to explore the role of order in NP assemblies on their SERS properties. While a direct qualitative correlation exists between extinction and SERS of individual metal NPs, it deteriorates dramatically in strongly coupled systems with a large distribution of spatially localized resonances. At the same time, there is a direct correlation between SERS enhancement and the dynamic evolution of electric field enhancement in the hot spots, as assessed by solving numerically Maxwell's equations by iteration over time.23
We assembled nanopolymers from gold NRs end-tethered with PS ligands and found that for the nanopolymers formed by gold NRs exhibited a non-linear increase with increasing n. Fig. 9B shows the variation in the normalized intensities of the Raman peak of a reporter molecule (oxazine) uptaken by the chains of gold NRs and localized in the inter-NR gaps filled with PS (Fig. 9A). The collinearity of the NR chains had a prodigious effect on the electric field, and therefore resulted in SERS enhancement: for bent chains, there was a significant reduction in extinction and SERS enhancement, compared to the co-linear NR assemblies.
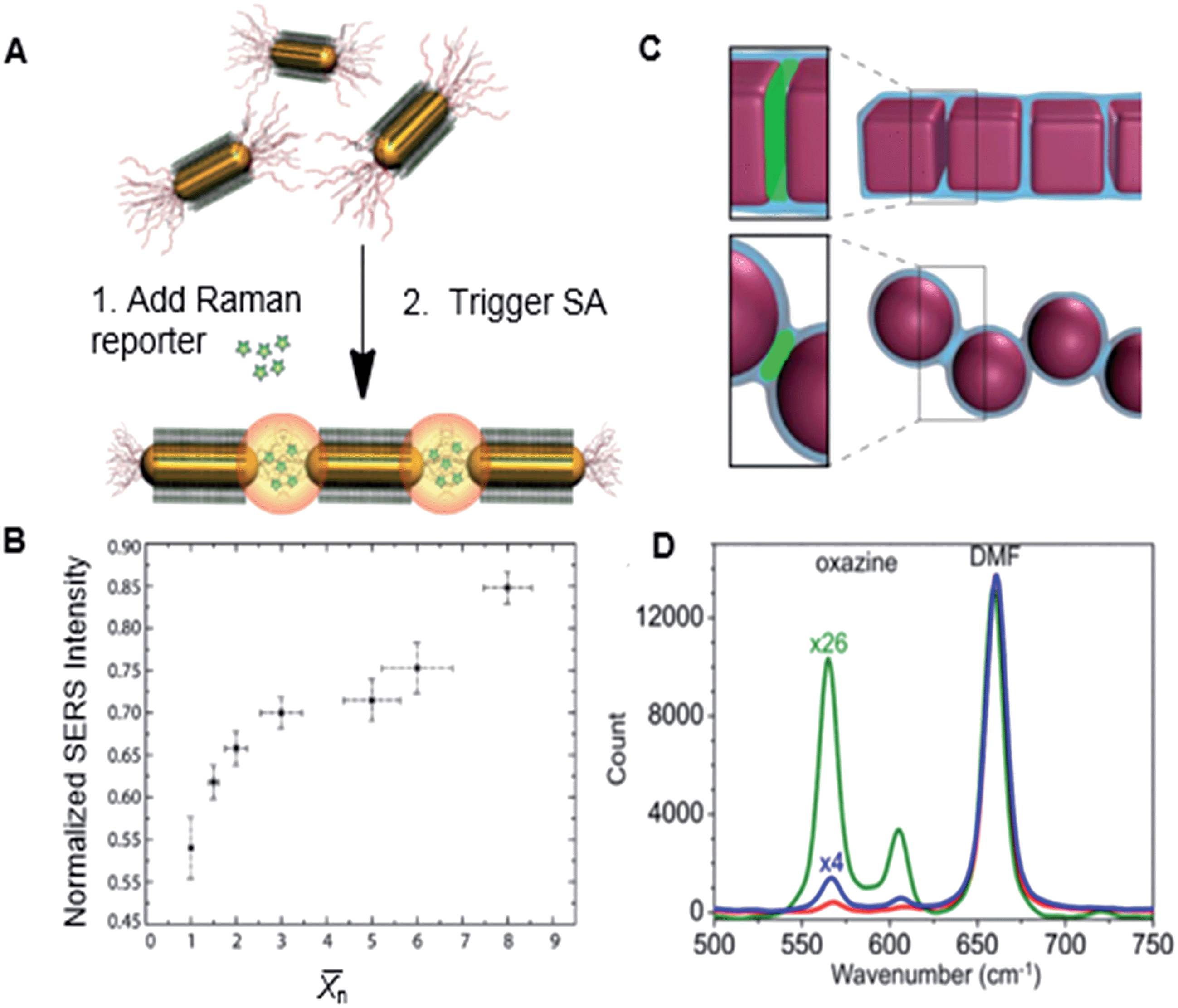 | ||
| Fig. 9 (A) Schematic representation of the Raman reporter partitioning in the gaps between linearly self-assembled gold NRs. (B) Normalized SERS intensity with respect to n of gold NR chains. Adapted from ref. 22. (C) Illustration of the hot spots localized between adjacent NCs and adjacent NSs in the nanopolymers. (D) SERS profiles of oxazine solution in DMF (red trace), and oxazine partitioned in the PS junctions binding adjacent NSs (blue trace) in the chains and NCs (green traces) in the nanopolymer. Adapted from ref. 5. | ||
Furthermore, for plasmonic nanopolymers formed by face-to-face assembled silver NCs (shown in Fig. 6), we found that the geometry of hot spots played an important role in associated SERS enhancement factors (Fig. 9C). In comparison with chains of silver NSs, nanopolymers formed by face-to-face bonded NCs showed a higher SERS intensity (Fig. 9D). This effect was attributed to the larger volume of hot spots and a nearly uniform electric field in the gaps between collinear NCs, as well as new coupling modes in the plasmonic polymer formed by the NCs. A pair of face-to-face assembled NCs formed a Fabry–Perot resonator30 with a resonance wavelength determined by the NC dimensions and the size of the inter-NC gap. When Fabry–Perot resonance corresponded to the excitation wavelength, strong enhancement in electric field and SERS was observed.5 Thus, the optical properties of plasmonic polymers can be programmed by using NP building blocks with specific shapes in sub-wavelength sensing and imaging enhancement.
Conclusions
We have shown that linear nanopolymer chains can be assembled from nanomers with surface and shape anisotropy. This process follows the kinetics and statistics of molecular step-growth polymerization. Following this analogy, we used the strategies of molecular polymerization such as the utilization of chain stoppers and copolymerization to precisely tune the morphology of the nanopolymers. Photocrosslinking of the polymer ligands binding NPs in the nanopolymers enabled control over interparticle distance, chain rigidity and the degree of polymerization of the nanopolymer. We have also demonstrated that surface and shape anisotropy of NPs is not a requirement for assembling one-dimensional NP structures, since anisotropy may arise from the competition of nanoscale forces. Isotropically functionalized metal NSs and NCs self-assembled into chains when polymer ligand and self-assembly medium characteristics were judiciously selected. We also demonstrate that the generated self-assembled structures may form hierarchical self-assemblies, including chain networks and globular networks. Lastly, the effect of self-assembled structure morphology with respect to SERS enhancement was explored yielding important insight into the effects of hot spot geometry and nanopolymer length on plasmonic performance. These findings pave the way for the systematic design of self-assembled nanostructures in the quest for their applications or in the exploration of new self-assembly parameter spaces.Acknowledgements
We thank the Connaught Foundation and the Natural Sciences and Engineering Research Council of Canada (Discovery and Canada Research Chair programs) for financial support of this work. R. M. C. acknowledges Natural Sciences and Engineering Research Council of Canada for a PGS-D scholarship. A. K. acknowledges the Connaught fund. The authors are grateful to Ilya Gourevich for help with imaging experiments.Notes and references
- A. Klinkova, R. M. Choueiri and E. Kumacheva, Chem. Soc. Rev., 2014, 43, 3976–3991 RSC.
- P. K. Jain, K. S. Lee, I. H. El-Sayed and M. A. El-Sayed, J. Phys. Chem. B, 2006, 110, 7238–7248 CrossRef CAS PubMed.
- L. S. Slaughter, B. A. Willingham, W.-S. Chang, M. H. Chester, N. Ogden and S. Link, Nano Lett., 2012, 12, 3967–3972 CrossRef CAS PubMed.
- G. Odian, Principles of Polymerization, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2004 Search PubMed.
- A. Klinkova, H. Thérien-Aubin, A. Ahmed, D. Nykypanchuk, R. M. Choueiri, B. Gagnon, A. Muntyanu, O. Gang, G. C. Walker and E. Kumacheva, Nano Lett., 2014, 14, 6314–6321 CrossRef CAS PubMed.
- B. Nikoobakht and M. A. El-Sayed, Chem. Mater., 2003, 15, 1957–1962 CrossRef CAS.
- B. D. Busbee, S. O. Obare and C. J. Murphy, Adv. Mater., 2003, 15, 414–416 CrossRef CAS.
- Y. Xiang, X. Wu, D. Liu, L. Feng, K. Zhang, W. Chu, W. Zhou and S. Xie, J. Phys. Chem. C, 2008, 112, 3203–3208 CAS.
- Y.-H. Chen, H.-H. Hung and M. H. Huang, J. Am. Chem. Soc., 2009, 131, 9114–9121 CrossRef CAS PubMed.
- N. R. Jana, L. Gearheart and C. J. Murphy, Langmuir, 2001, 17, 6782–6786 CrossRef CAS.
- F. Lu, Y. Tian, M. Liu, D. Su, H. Zhang, A. O. Govorov and O. Gang, Nano Lett., 2013, 13, 3145–3151 CrossRef CAS PubMed.
- W. Niu, Z.-Y. Li, L. Shi, X. Liu, H. Li, S. Han, J. Chen and G. Xu, Cryst. Growth Des., 2008, 8, 4440–4444 CAS.
- H. Yu, C. Min, P. M. Rice, S. X. Wang, R. L. White and S. Sun, Nano Lett., 2005, 5, 379–382 CrossRef CAS PubMed.
- Z. Nie, D. Fava, E. Kumacheva, S. Zou, G. C. Walker and M. Rubinstein, Nat. Mater., 2007, 6, 609–614 CrossRef CAS PubMed.
- K. Liu, Z. Nie, N. Zhao, W. Li, M. Rubinstein and E. Kumacheva, Science, 2010, 329, 197–200 CrossRef CAS PubMed.
- W. H. Carothers, Trans. Faraday Soc., 1936, 32, 39–49 RSC.
- A. Klinkova, H. Thérien-Aubin, R. M. Choueiri, M. Rubinstein and E. Kumacheva, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 18775–18779 CrossRef CAS PubMed.
- L. Hu, Y. Huang, L. Fang, G. Chen, H. Wei and Y. Fang, Sci. Rep., 2015, 5, 16069 CrossRef CAS PubMed.
- K. C. Woo, L. Shao, H. Chen, Y. Liang, J. Wang and H.-Q. Lin, ACS Nano, 2011, 5, 5976–5986 CrossRef CAS PubMed.
- K. Liu, A. Lukach, K. Sugikawa, L. Chung, J. Vickery, H. Therien-Aubin, B. Yang, M. Rubinstein and E. Kumacheva, Angew. Chem., Int. Ed., 2014, 53, 2648–2653 CrossRef CAS PubMed.
- A. Lukach, K. Liu, H. Therien-Aubin and E. Kumacheva, J. Am. Chem. Soc., 2012, 134, 18853–18859 CrossRef CAS PubMed.
- A. Stewart, A. Lee, S. Ip, E. Kumacheva and G. A. Walker, ACS Nano, 2014, 8, 5462–5467 CrossRef CAS PubMed.
- A. Lee, G. F. S. Andrade, A. Ahmed, M. L. Souza, N. Coombs, E. Tumarkin, K. Liu, R. Gordon, A. G. Brolo and E. Kumacheva, J. Am. Chem. Soc., 2011, 133, 7563–7570 CrossRef CAS PubMed.
- G. A. DeVries, et al. , Science, 2007, 315, 358–361 CrossRef CAS PubMed.
- R. Choueiri, A. Klinkova, H. Thérien-Aubin, M. Rubinstein and E. Kumacheva, J. Am. Chem. Soc., 2013, 135, 10262–10265 CrossRef CAS PubMed.
- E. B. Zhulina, A. B. Boulakh and O. V. Borisov, Z. Phys. Chem., 2012, 226, 625–643 CrossRef CAS.
- M. Yang, G. Chen, Y. Zhao, G. Silber, Y. Wang, S. Xing, Y. Han and H. Chen, Phys. Chem. Chem. Phys., 2010, 12, 11850–11860 RSC.
- D. J. Anderson and M. Moskovits, J. Phys. Chem. B, 2006, 110, 13722–13727 CrossRef CAS PubMed.
- R. C. Maher, in Raman Spectroscopy for Nanomaterials Characterization, Springer Berlin Heidelberg, Berlin, Heidelberg, 2012, pp. 215–260 Search PubMed.
- S. A. Maier, Plasmonics Fundamentals and Applications, Springer, New York, 2007 Search PubMed.
Footnote |
| † R. M. Choueiri and E. Galati contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |