 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Anisotropic lanthanide-based nano-clusters for imaging applications†
Xiaoping
Yang
a,
Shiqing
Wang
a,
Tyler L.
King
b,
Christopher J.
Kerr
 c,
Clement
Blanchet
c,
Dmitri
Svergun
c,
Robert
Pal
d,
Andrew
Beeby
d,
Jamuna
Vadivelu
e,
Katherine A.
Brown
*cf,
Richard A.
Jones
*c,
Lijie
Zhang
a and
Shaoming
Huang
*a
c,
Clement
Blanchet
c,
Dmitri
Svergun
c,
Robert
Pal
d,
Andrew
Beeby
d,
Jamuna
Vadivelu
e,
Katherine A.
Brown
*cf,
Richard A.
Jones
*c,
Lijie
Zhang
a and
Shaoming
Huang
*a
aCollege of Chemistry and Materials Engineering, Wenzhou University, Zhejiang Key Laboratory of Carbon Materials, Wenzhou 325035, China. E-mail: smhuang@wzu.edu.cn; Tel: +86 577 88373064
bThe University of Texas at Austin, Department of Chemistry, Austin, Texas 78712, USA. E-mail: rajones@cm.utexas.edu; Tel: +1 512 4711706
cEuropean Molecular Biology Laboratory, Hamburg Unit, EMBL c/o DESY, Hamburg, 22607, Germany
dDepartment of Chemistry, University of Durham, South Road, Durham, DH1 3LE, UK
eDepartment of Medical Microbiology, University of Malaya, Kuala Lumpur 50603, Malaysia
fCavendish Laboratory, Department of Physics, University of Cambridge, Cambridge CB3 0HE, UK. E-mail: kb518@cam.ac.uk
First published on 31st March 2016
Abstract
We have developed a new class of lanthanide nano-clusters that self-assemble using flexible Schiff base ligands. Cd–Ln and Ni–Ln clusters, [Ln8Cd24(L1)12(OAc)39Cl7(OH)2] (Ln = Nd, Eu), [Eu8Cd24(L1)12(OAc)44], [Ln8Cd24(L2)12(OAc)44] (Ln = Nd, Yb, Sm) and [Nd2Ni4(L3)2(acac)6(NO3)2(OH)2], were constructed using different types of flexible Schiff base ligands. These molecular nano-clusters exhibit anisotropic architectures that differ considerably depending upon the presence of Cd (nano-drum) or Ni (square-like nano-cluster). Structural characterization of the self-assembled particles has been undertaken using crystallography, transmission electron microscopy and small-angle X-ray scattering. Comparison of the metric dimensions of the nano-drums shows a consistency of size using these techniques, suggesting that these molecules may share similar structural features in both solid and solution states. Photophysical properties were studied by excitation of the ligand-centered absorption bands in the solid state and in solution, and using confocal microscopy of microspheres loaded with the compounds. The emissive properties of these compounds vary depending upon the combination of lanthanide and Cd or Ni present in these clusters. The results provide new insights into the construction of novel high-nuclearity nano-clusters and offer a promising foundation for the development of new functional nanomaterials.
Introduction
Lanthanide nanomaterials are being increasingly investigated for applications in bio-imaging and molecular recognition,1 due in part to their advantageous photophysical properties, which include a very large pseudo-Stokes shift between the excitation and emission wavelengths, the absence of photo-bleaching, long lived excited states and narrow emission bands.1,2 Since emissions via 4f–4f transitions are parity forbidden,3 Ln3+ ions usually have a relatively long photoluminescence lifetime of the excited state. This can allow for gated and time-resolved photoluminescence detection and thus can eliminate interference from short-lived autofluorescence.High-nuclearity heterometallic nano-clusters composed of d-block transition metals and lanthanide ions (Ln) have received extensive attention due not only to their aesthetically stunning molecular structures but also their versatile applications in optoelectronics, magnetism, and as probes in biological systems.4 Most trivalent lanthanide ions exhibit long-lived (in the microsecond-to-millisecond range) and line-like emission bands at characteristic wavelengths, e.g. Tb3+ (green), Eu3+ (red) and Yb3+, Nd3+, and Er3+ (near infra-red, NIR). These characteristics allow time-gated detection. More specifically, the NIR emitters have some potential applications in bioassays and luminescent probes because of the transparency of biological tissues to electromagnetic radiation in the range of 0.8–1.3 μm.5 In high-nuclearity d–f clusters, light-absorbing d-block metal chromophores (i.e. PtII,6a,b RuII,6c,d ZnII,6e,f and CdII6g,h) can act as sensitizers for luminescence from Ln(III) centers following d → f energy-transfer. Many reports have so far been focused on high-nuclearity 3d–4f clusters, such as Cu/Ln,7a Mn/Ln7b,c and Ni/Ln7d,e cluster systems, in order to study their magnetic properties as single-molecule magnets. In contrast, high-nuclearity 4d–f systems with luminescence properties have received much less attention.
Compartmental Schiff bases with two dissimilar metal-binding sites, one being specific for the d metal ion and another for the f metal ion, have been employed to synthesize d–f heteronuclear clusters.8 Recent studies in our laboratories have focused on the construction of luminescent d–f clusters with flexible Schiff base ligands.9 Flexible ligands may provide more possibilities for the construction of unique frameworks because of their freedom of conformation. In our previous studies, the use of Schiff base ligands which have flexible carbon–carbon backbones containing from 2 to 4 methylene (CH2) units, resulted in a variety of binuclear,10a trinuclear,10b tetranuclear,10c and hexanuclear10d,e 3d–4f complexes (3d = Zn, Cu and Ni). Recently, we discovered a series of 24-metal Cd–Ln complexes [Ln6Cd18(L1)9Cl8(OAc)28] from reactions of a flexible Schiff base ligand, N,N′-bis(5-bromo-3-methoxysalicylidene)hexane-1,6-diamine (H2L1), which has a 6 carbon backbone (Scheme 1), with Cd(OAc)2·4H2O and LnCl3·6H2O.9b The structures of these metal clusters are often influenced by a variety of factors such as the ligand structure, the nature of the counter anions, and the pH value of the surrounding environment. As part of our continuing studies focused on the construction of luminescent polynuclear lanthanide-based frameworks, we report here seven d–f clusters with H2L1, H2L2 and long-chain Schiff base ligand N,N′-bis(5-bromo-3-methoxysalicylidene) (N′′-(3-amion-propyl)-propane)-1,6-diamine (H2L3), which has a flexible (CH2)3NH(CH2)3 backbone (Scheme 1b). They are [Ln8Cd24(L1)12(OAc)39Cl7(OH)2] (Ln = Nd (1) and Eu (2)), [Nd2Ni4(L3)2(acac)6(NO3)2(OH)2] (3), [Ln8Cd24(L2)12(OAc)48] (Ln = Eu (4) and Sm (5)) and [Ln8Cd24(L2)12(OAc)44Cl4] (Ln = Nd (6) and Yb (7)). 1 and 2 were synthesized from the reactions of H2L1 with CdCl2·2H2O and Ln(OAc)3·4H2O in the presence of NaOH. Interestingly, differing from our previously reported Cd–Ln complexes [Ln6Cd18(L1)9Cl8(OAc)28], 1 and 2 exhibit 32-metal anisotropic nano-drum-like architectures. Hydroxide (OH−) anions are found in the structures of 1 and 2, indicating that the basic environment favors the formation of these complexes. The structures appear to be ligand and anion dependent. Compared with H2L1 and H2L2, H2L3 features a backbone NH group. Thus, 3 shows a 6-metal square-like structure. The flexible Schiff-base ligands H2L1, H2L2 and H2L3 exhibit “stretched” coordination modes with metal ions (Scheme 1b), resulting in the large sizes of 1–7. For example, the sizes of 1 and 2 are approximately 19 × 20 × 20 Å, which are much larger than most other d–f Schiff base polynuclear complexes thus far reported. To further explore the self-assembly properties and imaging of these molecules, we also report preliminary small angle X-ray scattering measurements and confocal imaging of Cd–Ln complexes containing the H2L2 Schiff base ligand.
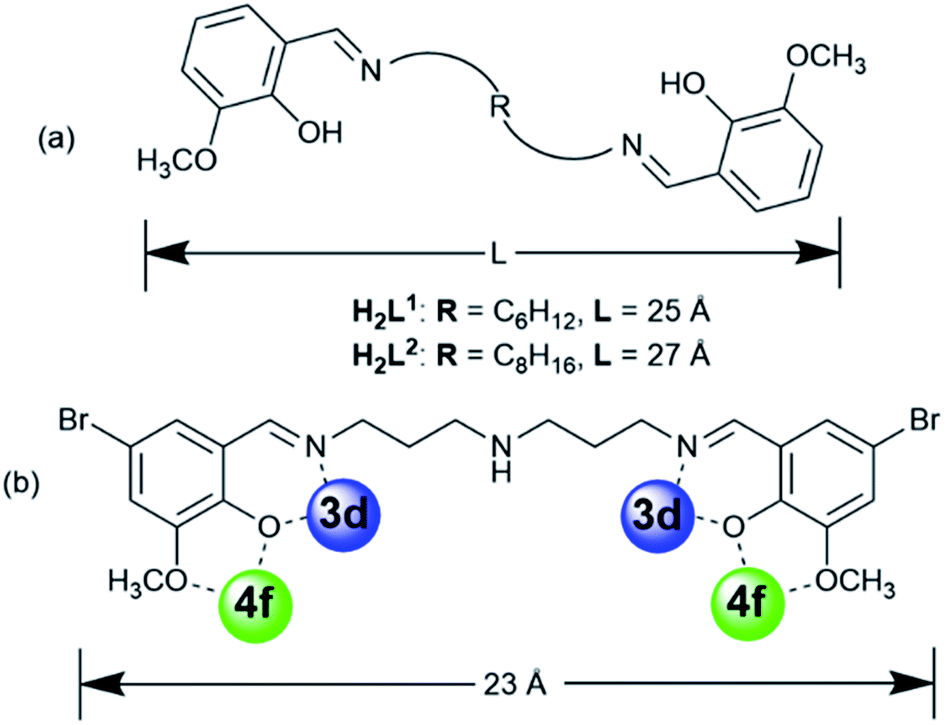 | ||
| Scheme 1 (a) Flexible Schiff base ligands; (b) the “stretching” coordination mode of H2L3. | ||
Experimental section
General information
All reactions were performed under dry oxygen-free dinitrogen atmospheres using standard Schlenk techniques. The Schiff-base ligands H2L1,2,3 were prepared according to well-established procedures.11 Physical measurements: NMR: VARIAN UNITY-plus 600 spectrometer (1H, 600 MHz) at 298 K; IR: Nicolet IR 200 FTIR spectrometer. Elemental analyses (C, H, N) were carried out on a EA1112 elemental analyser. Melting points were obtained in sealed glass capillaries under dinitrogen and are uncorrected. Conductivity measurements were carried out with a DDS-11 conductivity bridge for 10−3 M solutions in CH3CN. Transmission electron microscopy (TEM) images were recorded on a JEOL JEM-1200EX transmission electron microscope. Scanning electron microscopy (SEM) images were recorded on a Nova NanoSEM 200 scanning electron microscope. Absorption spectra were obtained on a UV-3600 spectrophotometer, and excitation and emission spectra on a QuantaMaster PTI fluorimeter.Syntheses
X-ray analysis
Data were collected using a Smart APEX CCD diffractometer with graphite monochromated Mo-Kα radiation (λ = 0.71073 Å) at 223 K. The data set was corrected for absorption based on multiple scans and reduced using standard methods. Data reduction was performed using DENZO-SMN.12 The structures were solved by direct methods and refined anisotropically using full-matrix least-squares methods with the SHELX 97 program package.12 Coordinates of the non-hydrogen atoms were refined anisotropically, while hydrogen atoms were included in the calculation isotropically but not refined. Neutral atom scattering factors were taken from Cromer and Waber.14For the crystal structures of 1 and 2, some uncoordinated solvent molecules such as CH3OH, C2H5OH, C2H5OC2H5 and H2O molecules were found to be badly disordered. Attempts to model the disorder were unsatisfactory. The contributions to the scattering factors due to these solvent molecules were removed by use of the utility SQUEEZE15a in PLATON98.15b,c PLATON98 was used as incorporated in WinGX.15d Crystallographic data for 1–3 (CCDC reference numbers 1450317–1450319) are presented in Table S1 and selected bond lengths are given in Tables S2–S4.†
1: C342H428Cd24Cl7N24Nd8O128, orthorhombic, space group Pna2(1), a = 54.734(11), b = 27.619(12), c = 35.243(18) Å, α = 90°, β = 90°, γ = 90°, V = 53![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 277(18) Å3, Z = 4, Dc = 1.374 g cm−3, μ(Mo-Kα) = 1.798 mm−1, F(000) = 21692, T = 223 K. R1 = 0.1018, wR2 = 0.2608 for 90178 independent reflections with a goodness-of-fit of 1.038.
277(18) Å3, Z = 4, Dc = 1.374 g cm−3, μ(Mo-Kα) = 1.798 mm−1, F(000) = 21692, T = 223 K. R1 = 0.1018, wR2 = 0.2608 for 90178 independent reflections with a goodness-of-fit of 1.038.
2: C342H428Cd24Cl7N24Eu8O128, orthorhombic, space group Pna2(1), a = 54.106(11), b = 27.127(5), c = 34.876(7) Å, α = 90°, β = 90°, γ = 90°, V = 51189(18) Å3, Z = 4, Dc = 1.438 g cm−3, μ(Mo-Kα) = 2.040 mm−1, F(000) = 21788, T = 223 K. R1 = 0.1035, wR2 = 0.2090 for 86518 independent reflections with a goodness-of-fit of 0.922.
3: C56H50Br4N8Nd2Ni4O52, monoclinic, space group P21/c, a = 15.462(3), b = 19.156(4), c = 19.871(4) Å, α = 90°, β = 107.63°(3), γ = 90°, V = 5609.2(19) Å3, Z = 2, Dc = 1.481 g cm−3, μ(Mo-Kα) = 3.073 mm−1, F(000) = 2452, T = 223 K. R1 = 0.0781, wR2 = 0.2092 for 9806 independent reflections with a goodness-of-fit of 1.061.
Small-angle X-ray scattering (SAXS)
SAXS data for [Eu8Cd24(L2)12(OAc)48] (4) were collected on the SAXS beamline P12 at the PETRA III storage ring (Deutsches Elektronen-Synchrotron, Hamburg).13 Using a PILATUS 2M pixel detector at a sample-detector distance of 3.0 m and at an energy of 9.7 keV (λ = 1.28 Å), the range of momentum transfer 0.01 < s < 0.45 Å−1 was covered (s = 4πsinθ/λ, where 2θ is the scattering angle). Solute concentrations of 1.8 and 3.7 mg mL−1 were measured in MeOH/MeCN (50:50) at 20 °C. Samples were loaded manually into the observation capillary. Primary data processing steps were performed using the automated data pipeline SASFLOW.16
Polystyrene bead loading
Luminex MicroPlex microspheres (6 μm cross-linked polystyrene beads with surface carboxyl groups) were prepared for loading by drying 12.5 × 106 beads under high vacuum for 24 hours. The dried beads were resuspended in a chloroform/MeOH solution (1 mL, 50:50) containing 5 mg of sample. This suspension was rotated slowly for 2 days. The suspension was then centrifuged, the supernatant discarded and the beads washed with MeOH (3 × 1 mL) followed by drying under high vacuum (24 h).
Confocal microscopy of polystyrene beads
Images were recorded using a Leica SP5 II confocal microscope equipped with a HCX PL APO 63x/1.40 NA LambdaBlue Oil immersion objective. Data were collected using 5× digital magnification at a 400 Hz/line scan speed (4 line average, bidirectional scanning) at 355 nm (3rd harmonic NdYAG laser) with 3 mW laser power. In order to achieve excitation with maximal probe emission, the microscope was equipped with a triple channel imaging detector, comprising two conventional PMT systems and a HyD hybrid avalanche photodiode detector. The latter part of the detection system, when operated in the BrightRed mode, is capable of improving imaging sensitivity by 25%, reducing signal to noise by a factor of 5. Frame size was determined at 2048 × 2048 pixels, with a 0.6 airy disc unit determining the applied pinhole diameter rendering one voxel to correspond to 24.02 × 24.02 nm (frame size 49.16 × 49.16 μm) with a section thickness of 380 nm. This particular LSCM is equipped with a novel structural illumination module called PhMoNa (achievable resolution 62 × 62 × 280 nm).17 To detect sensitized Ln emission, a corresponding detection window of 400–800 nm was used to record images using only the above detailed 355 nm laser line.Results and discussion
In the presence of NaOH (0.01 mol L−1), reactions of H2L1 with CdCl2·2H2O and Ln(OAc)3·4H2O in refluxing methanol/ethanol produced yellow solutions from which 1 and 2 were isolated as pale yellow crystalline solids. 1 and 2 are isomorphous and have 32-metal drum-like structures. Two views of the crystal structure of 1 are shown in Fig. 1a. The top view is essentially a side-on view while the lower one is looking down into the top of the drum. The ends of the drum are created by two rings of 16 metals (4 Nd(III) and 12 Cd(II)) coordinated to half of the N, O binding groups of the 12 Schiff base ligands. The sides of the drum are formed by the –(CH2)6– linkers of the Schiff base ligands. Regarding the two Nd4Cd12 rings, one includes four Cl−, one OH− and nineteen OAc− anions and the other has three Cl−, one OH− and twenty OAc− anions to balance the charge of the cluster. In 1, the Cd–O and Cd–N bond lengths range from 2.163 Å to 2.646 Å and 2.210 Å to 2.406 Å, respectively. The Nd–O and Nd–N bond lengths range from 2.208 Å to 2.652 Å and 2.527 Å to 2.650 Å, respectively. In 1 and 2, each Ln3+ ion and its closest two Cd2+ ions are linked by phenolic oxygen atoms of the L1 ligand and OAc− anions.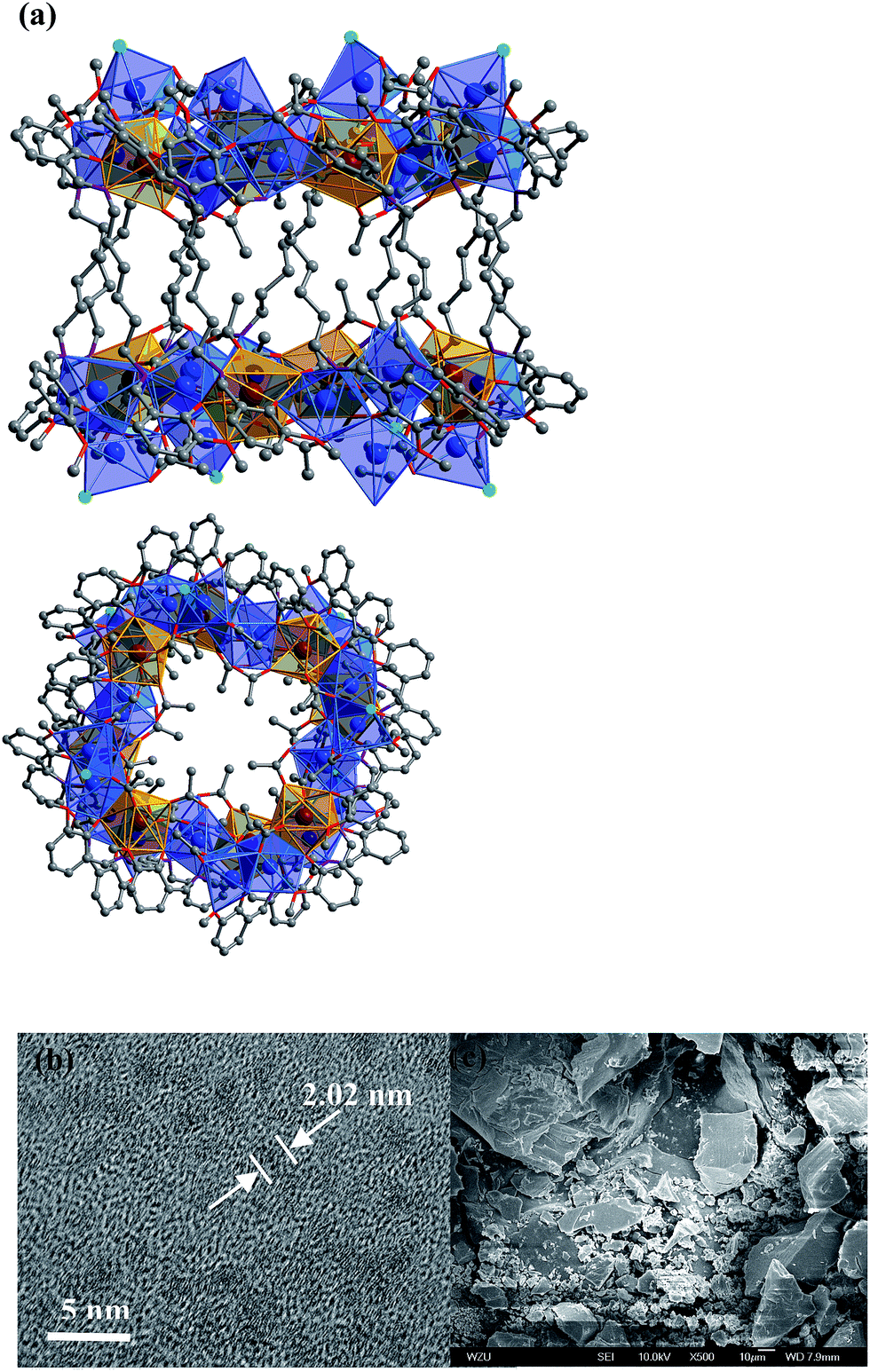 | ||
| Fig. 1 (a) Two views of the nano-drum-like structure of 1: viewed along the b-axis (top) and the ac-axis (lower) (Nd3+: brown; Cd2+: blue; Cl−: green); (b and c) TEM and SEM images of 1. | ||
The Cd(II) ion tends to have relatively high coordination numbers (6–8) and exhibits variable coordination geometries, somewhat similar to Ln(III). This geometry may favor the formation of high-nuclearity Cd–Ln clusters 1 and 2. Compared with Cd(II), the Ni(II) ion tends to have lower coordination numbers (4–6) and exhibits some common coordination geometries such as square-planar, tetragonal pyramid and octahedral. Thus, the reaction of H2L3 with Ni(acac)2·2H2O and Nd(NO3)3·6H2O under the same experimental conditions as above produced green solutions from which 3 was isolated as a green crystalline solid. Two views of the square-like structure of 3 are shown in Fig. 2. The top view is looking down into the top of the rectangle while the lower view is essentially a side-on view. The X-ray structure of 3 reveals a centrosymmetric core with two equivalent NdNi2(acac)3(NO3)(OH) moieties linked by two L2 ligands. In each NdNi2(acac)3(NO3)(OH) moiety, the Nd3+ ion is 8-coordinate, surrounded by eight oxygen atoms from two L2 ligands, one acac−, one NO3− and one OH− ion. Both Ni2+ ions have bi-pyramidal geometries. They are bridged by one acac− and one OH− ion with a separation of 3.237 Å. The Nd3+ ion and two Ni2+ ions are linked by phenolic oxygen atoms of the L2 ligand and OH− anions. The average distance between the Nd3+ and Ni2+ ions is 3.413 Å.
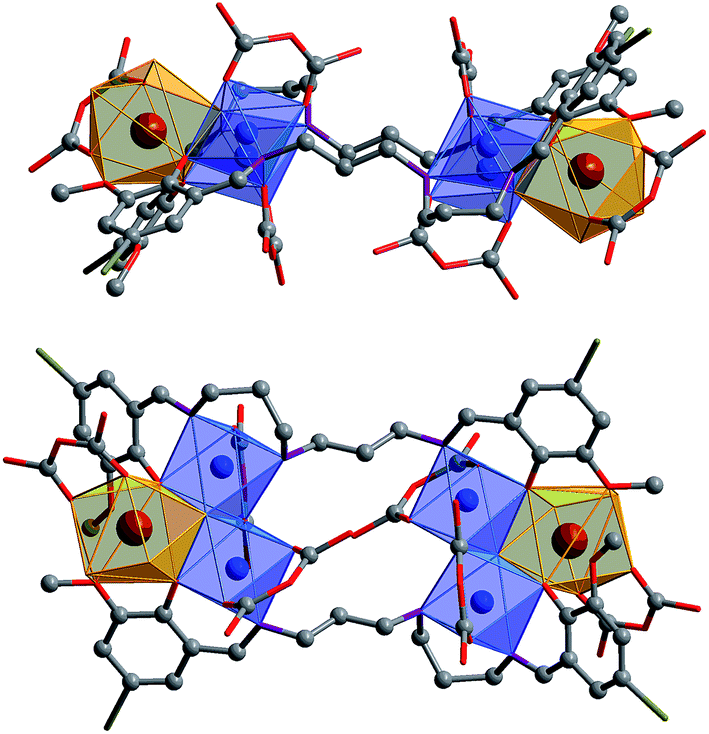 | ||
| Fig. 2 Two views of the square-like structure of 3: viewed along the ac-axis (top) and the b-axis (lower) (Nd3+: brown; Ni2+: blue). | ||
It is noticeable that the Schiff base ligands exhibit a “stretched” coordination mode with metal ions in 1–3 (Scheme 1b). This may be due to the fact that, compared with those Schiff base ligands with shorter carbon-carbon backbones, there is less of a chelating or templating effect.1,2 Thus the heights of the drum-like and square-like structures are mainly decided by the lengths of the Schiff base ligands, resulting in the formation of nanoscale clusters. The molecular dimensions of 1 and 2 are bigger than those of 3 (i.e. 19 × 20 × 20 Å for 1vs. 8 × 12 × 18 Å for 3). It is also possible to obtain images of these Cd–Ln molecular nanoparticles using transmission electron microscopy (TEM). Dilute solutions of 1 in CH3CN were contacted with a Cu grid and the solvent was carefully evaporated under vacuum. The TEM images obtained (Fig. 1b) show uniform nanoparticles with diameters measuring approximately 2.02 nm which corresponds well with the diameter of the 16-metal ring end of the drum found in the crystal structure, indicating that the nano-cluster retains its unique molecular structure in solution. In Fig. 1c a panoramic scanning electron microscopy (SEM) image shows the crystalline nature of 1.
The nature of 1 in solution has been probed using molar conductivity and 1H NMR studies. Molar conductivity studies of 1 in CH3CN also confirm that this molecular nanoparticle is neutral in solution, in accordance with the solid state structure. The 1H NMR spectrum of 1 in CDCl3 contains multiple broad peaks ranging from −17 to +18 ppm, due to the paramagnetic Nd3+ ion. The 1H NMR spectrum of 1 remains unchanged for several weeks, indicating that it is stable in solution (Fig. S1†).
The nature of 4 in solution has been probed using small-angle X-ray scattering (SAXS). SAXS data were obtained at 1.8 and 3.7 mg mL−1, as described above, and merged using the ALMERGE18 software as shown in Fig. 3a. Bragg peaks in the scattering data appear to be arising from undissolved material still in a crystalline state. Evaluation of the particle distance distribution function P(r) was undertaken using GNOM19 and shows a bi-modal distribution, with a maximum particle dimension of approximately 2.7 nm, consistent with the crystal structure of this nano-drum.20 However, further studies are required to establish conditions that are purely monodisperse, without the presence of undissolved crystalline material and/or nano-cluster substructures.
 | ||
| Fig. 3 SAXS analysis of compound 4. (a) SAXS curves of data collected at 1.8 and 3.7 mg mL−1 are shown with the merged curve that was used as an input to derive (b) the particle distance function, P(r), plot. | ||
The photophysical properties of 1–3 were studied in both solution and the solid state. The d–f clusters' absorption bands in the UV-vis region are all red-shifted compared to those of the ligands alone (H2L1,2) (Fig. 4). For the free ligands, H2L1,2, excitations produce broad emission bands at λmax = 515 nm and 509 nm, respectively (Fig. S2†). The emission and excitation spectra of 1–3 in CH3CN are shown in Fig. 5, 6 and S3.† Upon excitation of the ligand-centered absorption bands, 1 and 3 show the NIR luminescence of Nd3+ (4F3/2 → 4Ij/2 transitions, j = 9, 11 and 13). As shown in Fig. 6, the emissions at 873 nm can be assigned to the 4F3/2 → 4I9/2 transition, 1063 nm to the 4F3/2 → 4I11/2 transition and 1350 nm to the 4F3/2 → 4I13/2 transition of Nd3+. The free Schiff base ligands H2L1,2 and Nd(OAc)3·4H2O do not exhibit NIR luminescence in CH3CN under similar conditions. Upon excitation of the ligand centered absorption bands, 2 shows typical visible emission bands of the Eu3+ ion (5D0 → 7Fj transitions, j = 0, 1, 2, 3 and 4). As shown in Fig. 5, the appearance of the symmetry-forbidden emission 5D0 → 7F0 at 579 nm indicates that the Eu3+ ions in 2 occupy sites with low symmetry and have no inversion center.21
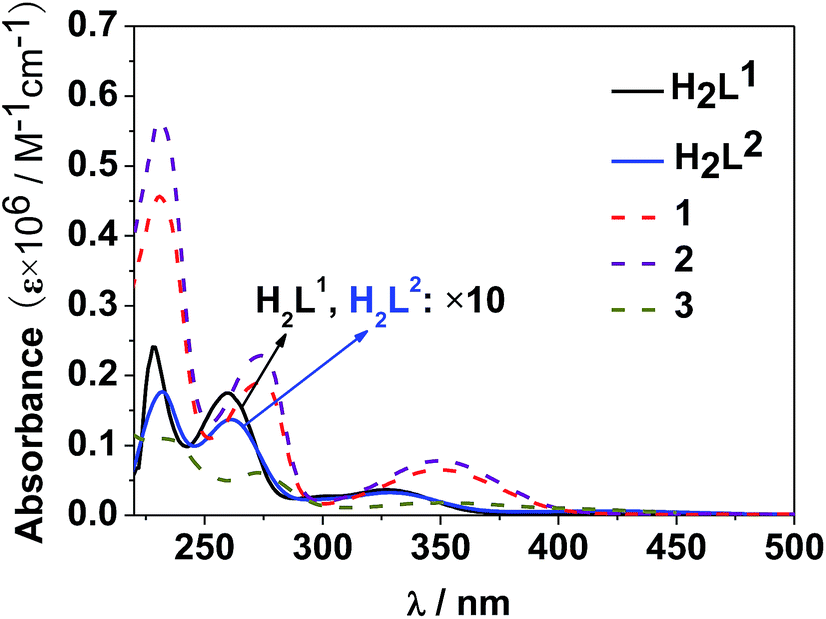 | ||
| Fig. 4 UV-vis spectra of the free ligands H2L1,2 and clusters 1–3 in CH3CN (C = 10−8 to 10−7 M). | ||
 | ||
| Fig. 5 The visible emission spectrum of 2 in CH3CN. | ||
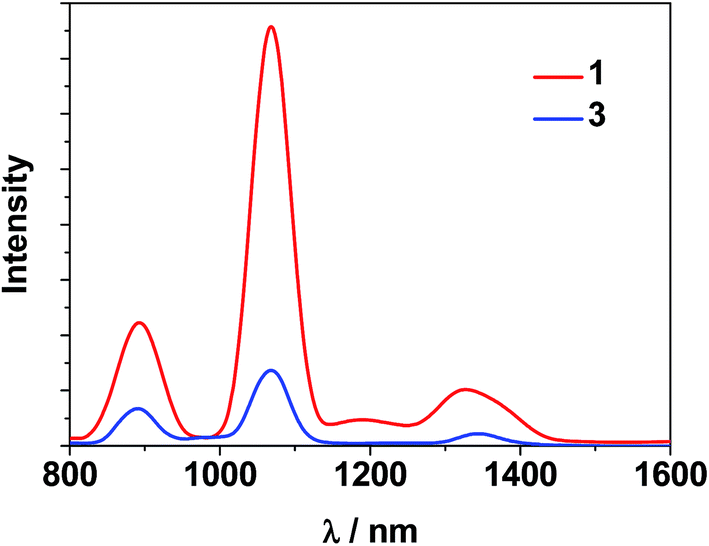 | ||
| Fig. 6 NIR emission spectra of clusters 1 and 3 with the same absorbance value at λex = 355 nm in CH3CN. | ||
This is further confirmed by the intensity ratio of 3.9 for I(5D0 → 7F2)/I(5D0 → 7F1), which is a good measure of the nature and symmetry of the first coordination sphere of the Eu3+ ion.22 The overall quantum yield of 2 in CH3CN was determined as 0.083 relative to [Ru(bipy)3]Cl2 in water (bipy = 2,2′-bipyridine; Φem = 0.028)23 and corrected for the refractive index of the solvent. 1 and 2 show two excitation bands at approximately 280 nm and 350 nm, while 3 has one at approximately 360 nm, in agreement with their absorption spectra, confirming that the energy transfers from the ligand centers to Ln3+ ions occur (Scheme 2). For each Cd–Ln or Ni–Nd nano-cluster the luminescence spectrum in the solid state is similar to that in solution.
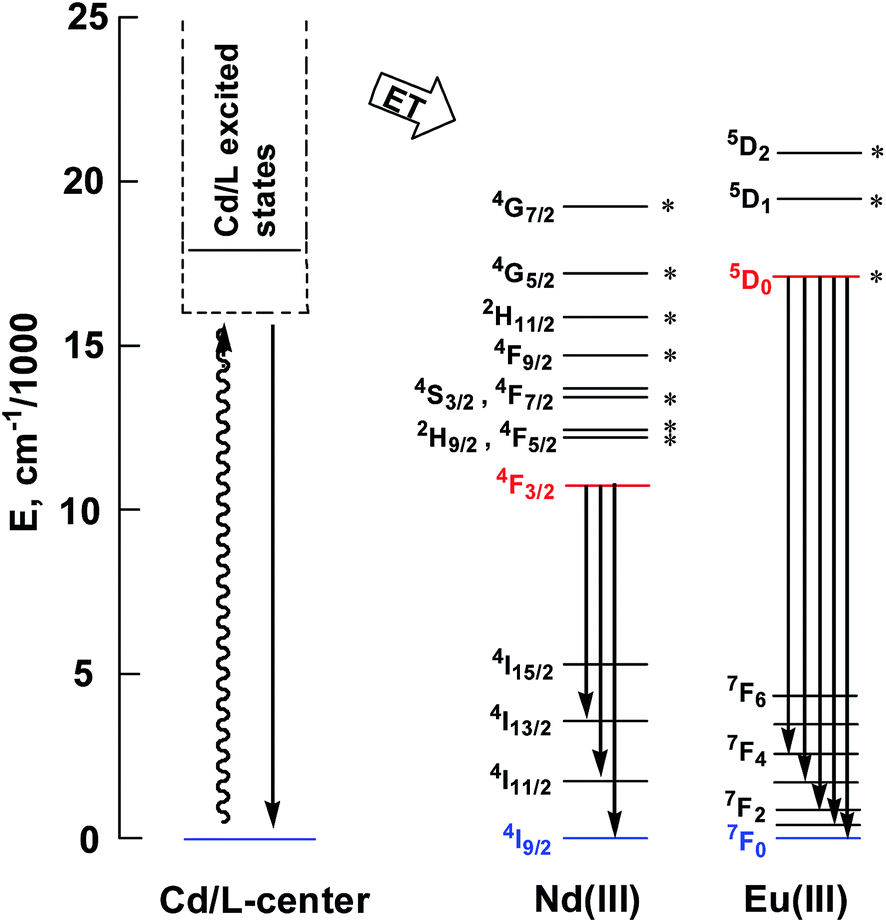 | ||
| Scheme 2 Relevant energy levels in 1–3. Those marked with * can act as energy acceptors by either Förster or Dexter mechanisms (the former requiring |ΔJ| = 2, 4, or 6 at the lanthanide, and the latter requiring |ΔJ| = 0 or 1 with the exception of |ΔJ| = |ΔJ′| = 0, which is forbidden).27 | ||
For the Nd(III) nano-clusters 1 and 3, we were naturally interested in the influence of their structures and d metals on their photophysical properties. The relative emission intensities of 1 and 3 were determined under the same experimental conditions. With the same absorbance value at 355 nm, the relative emission intensity at 1068 nm was estimated to be 5.4 for 1:3 in CH3CN (Fig. 6), indicating that with the central metal ions encapsulated by chromophoric Cd/L1 components (energy transfer donors), 1 has superior luminescence properties compared to 3. It is also noticeable that, compared to the “enclosed” nano-drum-like structure in 1 the “open” square-like structure of 3 results in metal centers which are less well shielded from outside solvents which may quench lanthanide luminescence.24 In addition, the d-block metal ions introduced into the lanthanide-based clusters may play different roles in the luminescence properties of the Ln3+ ions. For the Cd2+ ion, its saturated d10 electronic configuration prevents the quenching of lanthanide luminescence through a d–d transition (f → d energy transfer), while the Ni2+ ion may quench the luminescence via f → d energy transfer.25 The emission lifetimes (τ) of 1 and 3 are 1.9 μs and 0.5 μs, respectively. The intrinsic quantum yields (ΦLn) of Nd3+ emission in 1 and 3 are calculated as 0.76% and 0.20%, respectively, using ΦLn = τ/τ0 (τ0 = 250 μs, the natural lifetime of Nd3+).26
To explore the imaging utility of Cd–Ln nano-clusters on an imaging platform used for biological materials, cross-linked polystyrene beads (6–7 μm) containing complexes 5, 6 and 7 were visualized using confocal microscopy (Fig. 7). Images were collected to show total emission (400–800 nm). All experimental parameters are identical and bead emissions shown are a true representation of brightness on a color scale of black to red to white to blue, where blue indicates saturation. As shown in Fig. 7, beads loaded with 5 are considerably brighter, suggesting that the Nd–Cd nano-cluster may have enhanced emission properties with the H2L2 Schiff base ligand, compared to the Sm–Cd and Yb–Cd nano-clusters. Further analysis of the relative emission intensities between the bead sets can be undertaken to improve the confidence of this observation.
 | ||
| Fig. 7 Confocal images of polystyrene beads loaded with Ln–Cd nano-clusters (a) 5, Nd; (b) 6, Sm, and (c) 7, Yb. | ||
Conclusions
Nanostructured materials are increasingly being developed as imaging agents for both fundamental research imaging and diagnostic imaging. Here, we have presented data characterizing a new class of self-assembling lanthanide nano-clusters. These compounds have structures and imaging properties making them suitable candidates for cell and deep tissue imaging agents. Unlike classical nanoparticles, these materials are self-assembling homogeneous particles with asymmetric structures. The Cd–Ln clusters adopt nano-drum-like structures. The Ni–Ln cluster 3 has a square-like architecture. TEM and SAXS data are consistent with these molecular compounds having solution structures similar to those found in the solid state. The different architectures give rise to different imaging properties. With the Ln(III) centers enclosed within its nano-drum-like structure, Cd–Nd cluster 1 exhibits better NIR luminescence properties than Ni–Nd cluster 3. The photophysical properties of the nano-drum-like clusters demonstrate their potential as lanthanide emitters, and their utility as bio-imaging agents in confocal microscopy platforms. Future studies will focus on understanding how their molecular structures influence their stability, photophysical properties and ability to be delivered to cells and tissues in nano-formulations.Acknowledgements
The work was supported by the Welch Foundation (F-816) (RAJ), NSFC (51025207) (SMH), the Ministry of High Education (MOHE), Malaysia under High Impact Research (HIR) – MOHE project (UM.C/625/1/HIR/MoE/CHAN/13/6 H-50001-00-A000034) (KAB), NIH/NIAID 1U01AI078008-3, the Centre for Blast Injury Study at Imperial College London (KAB), the Royal Society (University Research Fellowship to RP), and the EU infrastructure grant BioStruct-X (contract number: 283570) (DIS).Notes and references
- (a) Y. Sun, W. Feng, P.-Y. Yang, C.-H. Huang and F.-Y. Li, Chem. Soc. Rev., 2015, 44, 1509–1525 RSC; (b) L. Prodi, E. Rampazzo, F. Rastrelli, A. Speghini and N. Zaccheroni, Chem. Soc. Rev., 2015, 44, 4922–4952 RSC; (c) S. Ranjan, M. K. Jayakumar and Y. Zhang, Nanomedicine, 2015, 10, 1477–1491 CrossRef CAS PubMed; (d) Q. Liu, W. Feng and F.-Y. Li, Coord. Chem. Rev., 2014, 273/274, 100–110 CrossRef; (e) A. Foucault-Collet, C. M. Shade, I. Nazarenko, S. Petoud and S. V. Eliseeva, Angew. Chem., Int. Ed., 2014, 53, 2927–2930 CrossRef CAS PubMed.
- (a) E. G. Moore, A. P. S. Samuel and K. N. Raymond, Acc. Chem. Res., 2009, 42, 542–552 CrossRef CAS PubMed; (b) J.-C. G. Bünzli, Chem. Rev., 2010, 110, 2729–2755 CrossRef PubMed; (c) C. P. Montgomery, B. S. Murray, E. J. New, R. Pal and D. Parker, Acc. Chem. Res., 2009, 42, 925–937 CrossRef CAS PubMed; (d) A. Thibon and V. C. Pierre, Anal. Bioanal. Chem., 2009, 394, 107–120 CrossRef CAS PubMed.
- T. Nishioka, K. Fukui and K. Matsumoto, Handbook on the Physics and Chemistry of Rare Earth, ed. K. A. Gschneidner, J.-C. G. Bünzli and V. K. Pecharsky, Elsevier Science B. V., Amsterdam, 2007, p. 37 Search PubMed.
- (a) J.-B. Peng, Q.-C. Zhang, X.-J. Kong, Y.-Z. Zheng, Y.-P. Ren, L.-S. Long, R.-B. Huang, L.-S. Zheng and Z. Zheng, J. Am. Chem. Soc., 2012, 134, 3314–3317 CrossRef CAS PubMed; (b) B. Wang, Z. Zang, H. Wang, W. Dou, X. Tang, W. Liu, Y. Shao, J. Ma, Y. Li and J. Zhou, Angew. Chem., Int. Ed., 2013, 52, 3756–3759 CrossRef CAS PubMed; (c) A. Müller, Nature, 2007, 447, 1034–1035 CrossRef; (d) A. Müller, P. Kögerler and A. W. M. Dress, Coord. Chem. Rev., 2001, 222, 193–218 CrossRef; (e) E. G. Mednikov, M. C. Jewell and L. F. Dahl, J. Am. Chem. Soc., 2007, 129, 11619–11630 CrossRef CAS PubMed.
- (a) J.-C. G. Bünzli, Metal Ions in Biological Systems, ed. M. Dekker, New-York, 2004, p. 42 Search PubMed; (b) I. Hemmila and S. Webb, Drug Discovery Today, 1997, 2, 373–380 CrossRef CAS; (c) J. W. Stouwdam, G. A. Hebbink, J. Huskens and F. C. J. M. Van Veggel, Chem. Mater., 2003, 15, 4604–4616 CrossRef CAS.
- (a) X.-L. Li, L.-X. Shi, L.-Y. Zhang, H. M. Wen and Z. N. Chen, Inorg. Chem., 2007, 46, 10892–10900 CrossRef CAS PubMed; (b) H.-B. Xu, L.-X. Shi, E. Ma, L.-Y. Zhang, Q.-H. Wei and Z.-N. Chen, Chem. Commun., 2006, 1601–1603 RSC; (c) H.-M. Wen, Y. Yang, X.-S. Zhou, J.-Y. Liu, D.-B. Zhang, Z.-B. Chen, J.-Y. Wang, Z.-N. Chen and Z.-Q. Tian, Chem. Sci., 2013, 4, 2471–2477 RSC; (d) S. G. Baca, H. Adams, C. S. Grange, A. P. Smith, I. Sazanovich and M. D. Ward, Inorg. Chem., 2007, 46, 9779–9789 CrossRef CAS PubMed; (e) M. Yin, X. Lei, M. Li, L. Yuan and J. Sun, J. Phys. Chem. Solids, 2006, 67, 1372–1378 CrossRef CAS; (f) H.-B. Xu, H.-M. Wen, Z.-H. Chen, J. Li, L.-X. Shi and Z.-N. Chen, J. Chem. Soc., Dalton Trans., 2010, 39, 1948–1953 RSC; (g) Y.-X. Chi, S.-Y. Niu, J. Jin, R. Wang and Y. Li, J. Chem. Soc., Dalton Trans., 2009, 47, 7653–7659 RSC; (h) Y.-X. Chi, S.-Y. Niu, Z.-L. Wang and J. Jin, Eur. J. Inorg. Chem., 2008, 2336–2343 CrossRef CAS.
- (a) S.-C. Xiang, S.-M. Hu, T.-L. Sheng, R.-B. Fu, X.-T. Wu and X.-D. Zhang, J. Am. Chem. Soc., 2007, 129, 15144–15146 CrossRef CAS PubMed; (b) C. M. Zaleski, E. C. Depperman, J. W. Kampf, M. L. Kirk and V. L. Pecoraro, Angew. Chem., Int. Ed., 2004, 43, 3912–3914 CrossRef CAS PubMed; (c) V. M. Mereacre, A. M. Ako, R. Clerac, W. Wernsdorfer, G. Filoti, J. Bartolome, C. E. Anson and A. K. Powell, J. Am. Chem. Soc., 2007, 129, 9248–9249 CrossRef CAS PubMed; (d) X.-J. Kong, Y.-P. Ren, L.-S. Long, Z. Zheng, R.-B. Huang and L.-S. Zheng, J. Am. Chem. Soc., 2007, 129, 7016–7017 CrossRef CAS PubMed; (e) J.-B. Peng, Q.-C. Zhang, X.-J. Kong, Y.-Z. Zheng, Y.-P. Ren, L.-S. Long, R.-B. Huang, L.-S. Zheng and Z.-P. Zheng, J. Am. Chem. Soc., 2012, 134, 3314–3317 CrossRef CAS PubMed.
- (a) M. Sakamoto, K. Manseki and H. Okawa, Coord. Chem. Rev., 2001, 219, 379–414 CrossRef; (b) R. E. P. Winpenny, Chem. Soc. Rev., 1998, 27, 447–452 RSC; (c) C. Camp, V. Guidal, B. Biswas, J. Pecaut, L. Dubois and M. Mazzanti, Chem. Sci., 2012, 3, 2433–2448 RSC; (d) M. Andruh, Chem. Commun., 2011, 47, 3025–3042 RSC; (e) X. Feng, W. Zhou, Y. Li, H. Ke, J. Tang, R. Clerac, Y. Wang, Z. Su and E. Wang, Inorg. Chem., 2012, 51, 2722–2724 CrossRef CAS PubMed; (f) T. Yamaguchi, J.-P. Costes, Y. Kishima, M. Kojima, Y. Sunatsuki, N. Brefuel, J.-P. Tuchagues, L. Vendier and W. Wernsdorfer, Inorg. Chem., 2010, 49, 9125–9135 CrossRef CAS PubMed.
- (a) X.-P. Yang, R. A. Jones and S.-M. Huang, Coord. Chem. Rev., 2014, 273, 63–75 CrossRef; (b) X.-P. Yang, D. Schipper, R. A. Jones, L. A. Lytwak, B. J. Holliday and S.-M. Huang, J. Am. Chem. Soc., 2013, 135, 8468–8471 CrossRef CAS PubMed.
- (a) W.-K. Lo, W.-K. Wong, W.-Y. Wong, J. Guo, K.-T. Yeung, Y.-K. Cheng, X.-P. Yang and R. A. Jones, Inorg. Chem., 2006, 9315–9325 CrossRef CAS PubMed; (b) X.-P. Yang, S. Desmond, A. Liao, J. M. Stanley, R. A. Jones and B. J. Holliday, Polyhedron, 2013, 52, 165–168 CrossRef CAS; (c) X.-P. Yang, R. A. Jones, V. Lynch, M. M. Oye and A. L. Holmes, J. Chem. Soc., Dalton Trans., 2005, 5, 849–851 RSC; (d) X.-P. Yang, R. A. Jones, W.-K. Wong, V. Lynch, M. M. Oye and A. L. Holmes, Chem. Commun., 2006, 1836–1838 RSC; (e) X.-P. Yang, C. Chan, D. Lam, D. Schipper, J. M. Stanley, X. Chen, R. A. Jones, B. J. Holliday, W.-K. Wong, S.-C. Chen and Q. Chen, J. Chem. Soc., Dalton Trans., 2012, 41, 11449–11453 RSC.
- F. Lam, J.-X. Xu and K.-S. Chan, J. Org. Chem., 1996, 61, 8414–8418 CrossRef CAS.
- Z. Otwinowski and W. Minor, Methods in Enzymology, 276: Macromolecular Crystallography, Part a, ed. C. W. J. Carter, M. I. Simon and R. M. Sweet, Academic Press, 1997, pp. 307–326, DENZO-SMN Search PubMed.
- G. H. Sheldrick, SHELX 97, a Software Package for the Solution and Refinement of X-ray Data, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
- D. T. Cromer and J. T. Waber, International Tables for X-Ray Crystallography, Kynoch Press, Birmingham, 1974, vol. 4, Table 2.2A Search PubMed.
- (a) P. van der Sluis and A. L. Spec, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 194–201 CrossRef; (b) A. L. Spec, Platon, A Multipurpose Crystallographic Tool, Utrecht, the Netherlands, 1998 Search PubMed; (c) A. L. Spec, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef; (d) L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef CAS.
- D. Franke, A. G. Kikhney and D. I. Svergun, Nucl. Instrum. Methods Phys. Res., Sect. A, 2012, 689, 52–59 CrossRef CAS.
- R. Pal, Faraday Discuss., 2015, 177, 507–515 RSC.
- M. V. Petoukhov, D. Franke, A. V. Shkumatov, G. Tria, A. G. Kikhney, M. Gajda, C. Gorba, H. D. Mertens, P. V. Konarev and D. I. Svergun, J. Appl. Crystallogr., 2012, 45(2), 342–350 CAS.
- D. I. Svergun, J. Appl. Crystallogr., 1992, 25, 495–503 CrossRef.
- K. A. Brown, X. Yang, D. Schipper, J. W. Hall, L. J. DePue, A. J. Gnanam, J. F. Arambula, J. N. Jones, J. Swaminathan, Y. Dieye, J. Vadivelu, D. J. Chandler, E. M. Marcotte, J. L. Sessler, L. I. R. Ehrlich and R. A. Jones, Dalton Trans., 2015, 44, 2667–2675 RSC.
- (a) D. H. Metcalf, R. G. Ghirardelli and R. A. Palmer, Inorg. Chem., 1985, 24, 634–636 CrossRef CAS; (b) M. Albin, R. R. Whittle and W. D. Horrocks Jr, Inorg. Chem., 1985, 24, 4591–4594 CrossRef CAS.
- (a) Q. H. Xu, L. S. Li, X. S. Liu and R. R. Xu, Chem. Mater., 2002, 14, 549–555 CrossRef CAS; (b) J.-C. G. Bünzli and C. Piguet, Chem. Soc. Rev., 2005, 34, 1048–1077 RSC; (c) Y. B. Katsimirski and N. K. Davidenko, Coord. Chem. Rev., 1979, 27, 223–273 CrossRef.
- K. Nakamaru, Bull. Chem. Soc. Jpn., 1982, 5, 2697–2705 CrossRef.
- (a) F. S. Richardson, Chem. Rev., 1982, 82, 541–552 CrossRef CAS; (b) S. Yanagida, Y. Hasegawa, K. Murakoshi, Y. Wada, N. Nakashima and T. Yamanaka, Coord. Chem. Rev., 1998, 171, 461–480 CrossRef CAS.
- (a) V. Wen, Y. Zhao, L. Wang, M. Zhang and D. Gao, J. Rare Earths, 2007, 25, 679–683 CrossRef; (b) E. Wolcan, L. Villata, A. L. Capparelli and M. R. Feliz, Photochem. Photobiol. Sci., 2004, 3, 322–328 RSC; (c) B. C. Barja, A. Remorino, M. J. Roberti and P. F. Aramendia, J. Argent. Chem. Soc., 2005, 93, 81–88 CAS; (d) Q.-Y. Chen, C.-J. Li and Q.-H. Luo, Synth. React. Inorg. Met.-Org. Chem., 2003, 33(9), 1701–1715 CrossRef CAS.
- (a) S. B. Meshkova, Z. M. Topilova, D. V. Bolshoy, S. V. Beltyukova, M. P. Tsvirko, V. Y. Venchikov and V. Ya, Acta Phys. Pol., A, 1999, 95, 983–990 CrossRef CAS; (b) S. I. Klink, L. Grave, D. N. Reinhoudt and F. C. J. M. v. Veggel, J. Phys. Chem. A, 2000, 104, 5457–5468 CrossRef CAS.
- (a) D. L. Dexter, J. Chem. Phys., 1953, 21, 836–850 CrossRef CAS; (b) G. F. de Sá, O. L. Malta, C. de Mello Donegá, A. M. Simas, R. L. Longo, P. A. Santa-Cruz and E. F. da Silva Jr, Coord. Chem. Rev., 2000, 196, 165–195 CrossRef; (c) J.-C. G. Bünzli and C. Piguet, Chem. Soc. Rev., 2005, 34, 1048–1077 RSC; (d) N. M. Shavaleev, G. Accorsi, D. Virgili, D. R. Bell, T. Lazarides, G. Calogero, N. Armaroli and M. D. Ward, Inorg. Chem., 2005, 44, 61–72 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization details for clusters 1–3. CCDC 1450317–1450319. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6fd00018e |
| This journal is © The Royal Society of Chemistry 2016 |