 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect oxygen removal technique for recycling titanium using molten MgCl2 salt
Toru H.
Okabe
*a,
Yuki
Hamanaka†
b and
Yu-ki
Taninouchi
c
aInternational Research Center for Sustainable Materials, Institute of Industrial Science, The University of Tokyo, 4-6-1 Komaba, Meguro-ku, Tokyo 153-8505, Japan. E-mail: okabe@iis.u-tokyo.ac.jp
bDepartment of Materials Engineering, Graduate School of Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo, 113-8656, Japan
cInstitute of Industrial Science, The University of Tokyo, 4-6-1 Komaba, Meguro-ku, Tokyo 153-8505, Japan
First published on 20th January 2016
Abstract
Deoxidation of Ti, or direct removal of O dissolved in metallic Ti, is known to be extremely difficult when Mg is used as the deoxidizing agent. This difficulty arises because the chemical potential of O2, pO2, under Mg/MgO equilibrium is high (approximately 10−41 atm at 1200 K) and is equivalent to that of Ti containing ∼2 mass% O at 1200 K. Therefore, when deoxidizing Ti to the commercial level of high-grade pure Ti (below 0.05 mass% O) using an Mg reductant at 1200 K, the activity of the reaction product MgO (aMgO) must be decreased to below ∼0.025, which is difficult in practice. In this study, the removal of O in Ti in molten MgCl2 salt using an electrochemical technique was examined at ∼1173 K with the objective of obtaining Ti containing less than 0.05 mass% O. Ti samples and graphite electrodes immersed in molten MgCl2 served as the cathode and anode, respectively. A constant voltage was applied between the electrodes using an external DC source. Molten MgCl2 was employed to produce the deoxidizing agent Mg and to facilitate deoxidation of Ti by decreasing the activity of the reaction product MgO. By applying a voltage of approximately 3.1 V between the electrodes, the chemical potential of Mg in the molten MgCl2 was increased at the surface of the Ti cathode, and the Ti samples were deoxidized. The resulting O species, mainly formed O2− dissolved in the molten MgCl2, was removed from the molten salt by reacting with the C anode to form CO (or CO2) gas. Ti wires containing 0.12 mass% O were deoxidized to less than 0.02 mass% O. In some cases, the O concentration in the Ti samples was reduced to the level of 0.01 mass%, which cannot be accomplished using the conventional Kroll process. The possible application of this deoxidation technique to practical industrial recycling processes is discussed.
Introduction
In recent years, the recycling of Ti and Ti alloys has become an important issue.1,2 The demand for Ti and Ti alloys in the aerospace industry is increasing, and large amounts of Ti scraps are generated from the fabrication of Ti and Ti alloy products. Fig. 1 shows the material flow of Ti in the US.1 The volume of scrap generated is greater than the volume of fabricated Ti products.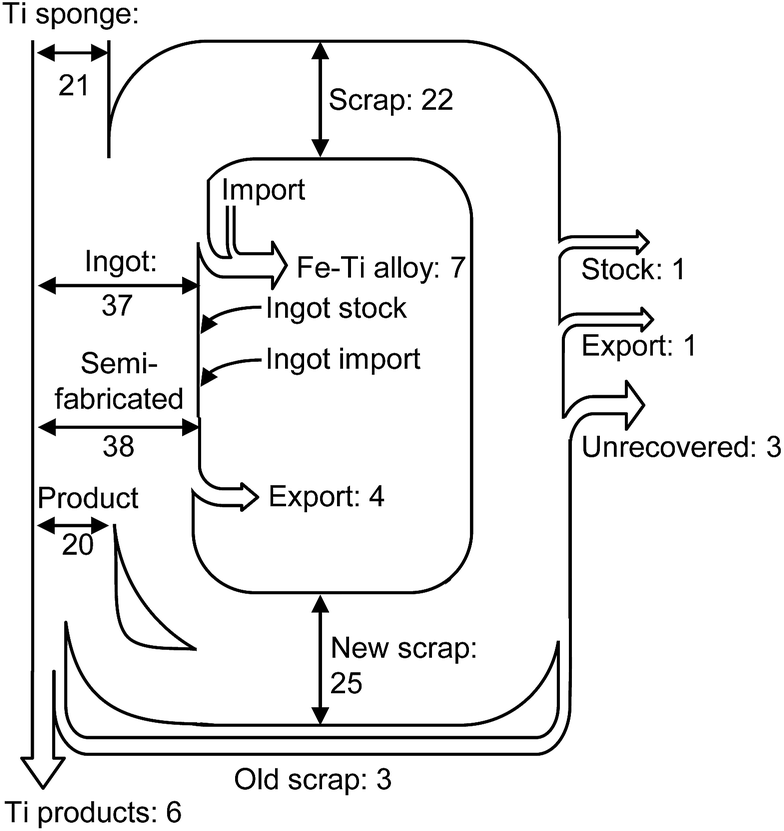 | ||
| Fig. 1 Estimated material flow of titanium scrap in the U.S. in 2004.1 Units in the figure are 103 ton. Values are rounded to the nearest thousand metric tons of contained titanium. | ||
Commercial Ti and its alloys contain oxygen at approximately 0.1 mass% (1000 mass ppm) as a major impurity. Oxygen contamination of the product must be avoided, because the presence of O deteriorates the mechanical properties of the metals. Table 1 shows the ASTM grades and acceptable impurity concentrations of Ti and its alloys.3 In producing commercially pure Ti of ASTM Gr. 1, low-O-containing Ti feed such as high-purity Ti sponge (0.03–0.1 mass% O) produced by the Kroll process4 is used. Using large amounts of scrap metal to produce low-O Ti is hindered by the high O level in Ti scrap, which typically exceeds 0.1 mass%. Titanium and its alloys containing more than 0.4 mass% O cannot be recycled into titanium alloys, and these metals are mainly used in cascade recycling as additive elements for steel, aluminum, etc.
| Specification | Concentration of element i, Ci (mass%) | |||||||
|---|---|---|---|---|---|---|---|---|
| C | H | O | N | Fe | Al | V | ||
| Pure Ti | ASTM Gr. 1 | ≤0.08 | ≤0.015 | ≤0.18 | ≤0.03 | ≤0.2 | — | — |
| ASTM Gr. 2 | ≤0.08 | ≤0.015 | ≤0.25 | ≤0.03 | ≤0.3 | — | — | |
| ASTM Gr. 3 | ≤0.08 | ≤0.015 | ≤0.35 | ≤0.05 | ≤0.3 | — | — | |
| ASTM Gr. 4 | ≤0.08 | ≤0.015 | ≤0.40 | ≤0.05 | ≤0.5 | — | — | |
| Ti–Al–V alloy | ASTM Gr. 5 | ≤0.08 | ≤0.015 | ≤0.20 | ≤0.05 | ≤0.40 | 5.5–6.75 | 3.5–4.5 |
| ASTM Gr. 23 | ≤0.08 | ≤0.0125 | ≤0.13 | ≤0.03 | ≤0.25 | 5.5–6.5 | 3.5–4.5 | |
O removal directly from Ti and its alloys to levels below 0.1 mass% (1000 mass ppm) is very difficult because O dissolves in Ti to form an interstitial solid solution, and because Ti has a strong affinity with O.5,6Table 2 lists some effective methods for direct removal of O from Ti and from TiAl.2,6–48 For example, the deoxidation of solid Ti by reaction with chemically active Ca dissolved in CaCl2 in the temperature range of 1273 to 1473 K has been examined. Okabe et al. successfully produced Ti containing less than 50 ppm by mass of O with a high residual resistivity ratio (RRR > 120).18,19 Ca metal is one of the most effective deoxidation agents because it has an extremely strong affinity for O and because it has very low solubility in solid Ti. In the Ca-halide flux deoxidation process shown in Table 2, the CaCl2 flux contains the deoxidation agent Ca and facilitates the reaction by diluting the reaction product CaO, thus decreasing the activity of the by-product. However, the purity of flux and the initial O content of the Ti are subject to limits, since the ultimate limit of deoxidation depends on the amounts of both O present as an impurity in the CaCl2 flux and CaO produced during deoxidation.
| Method | Reference | Advantages (deox. limit) | Disadvantages |
|---|---|---|---|
| Deoxidation (Deox.) | |||
| Deox. by M/MOx eq. (DOSS process) |
Ono and Miyazaki,6
Fisher,7–10 Okabe et al.,11–14 Oh et al.,15,16 Roh et al.17 |
Ti powder can be deoxidized. (∼500 ppm O) | Low capability of deox. |
| Calcium halide flux deox. | Okabe et al.18–20 | High capability of deox. (∼50 ppm O) | Not suitable for removing large amounts of O. Leaching of molten salt bath is needed |
| Electrochemical deox. |
Okabe et al.,21,22
Nakamura et al.,23 Hirota et al.,24 Taninouchi et al.25 |
Ultra-high capability of deox. CaCl2 bath (<10 ppm O), MgCl2 bath (<100 ppm O) | Long processing time |
| FFC process |
Chen et al.,26–28
Fray,29 Mohandas and Fray,30 Tripathy et al.31 |
Production of low-oxygen titanium directly from oxides | Long processing time |
| OS process |
Ono and Suzuki,32
Suzuki et al.33–37 |
Production of low-oxygen titanium directly from oxides | Long processing time |
| Deox. during EB remelting with Al | Yahata et al.38 | Short processing time (∼100 ppm O) | Excess Al is contained in Ti |
| VIM with CaAl2 | Rotmann et al.2 | Short processing time (∼1500 ppm O) | Capability of deox. is insufficient |
| PESR with Ca and CaF2 flux | Reitz et al.39 | Short processing time (∼250 ppm O) | Subsequent VAR is required to remove residual Ca. Applicability to deoxidation of pure Ti is uncertain |
| Electric arc remelting under H2–Ar atmosphere | Su et al.40 | Short processing time (∼300 ppm O) | Capability of deox. is insufficient. Applicability to deoxidation of pure Ti is uncertain |
| Deox. of Ti alloy by HDH |
Oh et al.,16
Roh et al.17 |
Simple process | Capability of deox. is insufficient |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||
| Others | |||
| Electrorefining in molten salt |
Nettle et al.,41
Suchkov et al.,42 Takeuchi et al.,43,44 Hashimoto et al.,45–47 Miyazaki et al.48 |
Almost all impurities can be removed | Contamination from molten salt. Difficulty of morphology control of Ti deposition |
An effective technique for the preparation of O-free Ti by electrochemical deoxidation using CaCl2 molten salt has been developed.21 Removal of O2− (present as CaO in the flux) and production of Ca deoxidant using electrochemical techniques improved the deoxidation limit of the Ca-halide flux deoxidation process. Under certain conditions, Ti with 0.001 mass% (10 mass ppm) O has been successfully produced by the electrochemical deoxidation technique.
Hashimoto et al. proposed a method for the deoxidation of Ti during electrowinning using molten fluoride salt baths,45–47 but contamination of Ti by O or C is unavoidable using this method. In 2001, Chen et al. developed a novel process, called the FFC process, for producing Ti directly from TiO2 immersed in CaCl2 molten salt using electrochemical methods, and they succeeded in obtaining low-O Ti using this process.26–28 Ono and Suzuki developed a new Ti reduction process that utilizes electrochemically produced Ca reductant in molten CaCl2 salt.32 To obtain low-O Ti, a highly reducing atmosphere is required. For this reason, molten CaCl2 with high metallic Ca activity is employed as the reaction medium. Other recent developments in deoxidation techniques for Ti and its alloys are also listed in Table 2.
Deoxidation of Ti using Mg as the deoxidizing agent is believed to be almost impossible. This is because the deoxidation limit of a Ti–O solid solution by Mg under MgO saturation is about 2 mass% (20000 mass ppm) at 1200 K; reducing the deoxidation limit to the level of 100 mass ppm is difficult from a practical perspective. However, if deoxidation of Ti using a MgCl2 medium becomes feasible, the reduction and electrolysis facilities of the conventional Kroll process4 could be utilized. Therefore, developing an effective deoxidation process for Ti and its alloys using molten MgCl2 is expected to significantly enhance the recovery rate for Ti scraps and the production of low-O-containing Ti.
Principle
Because the principle of electrochemical deoxidation of Ti has been reported in detail previously,5,21 the deoxidation process is only briefly outlined here. A solid Ti–O solution is deoxidized by Mg in the following reaction:| O (in Ti) + Mg (in flux) = MgO (in flux) | (1) |
The equilibrium O concentration in Ti depends thermodynamically on the temperature T according to the following equation,
| [mass% O] = (aMgO/aMg)(1/fO)exp(ΔG°/RT) | (2) |
Fig. 2 shows the binary phase diagram for the Ti–O system.49 The equilibrium O concentration of Ti under Mg/MgO equilibrium at 1273 K is plotted as an open circle. For reference, the equilibrium O concentration of Ti under Ca/CaO equilibrium at 1273 K is plotted as a filled circle (about 0.05 mass% O). The equilibrium O concentration under Mg/MgO equilibrium is approximately 2.6 mass% (7.4 mol%) at 1273 K. This shows that it is thermodynamically difficult to remove O directly from Ti to levels below 1 mass% by Mg deoxidant when the activity of MgO (aMgO) is high.
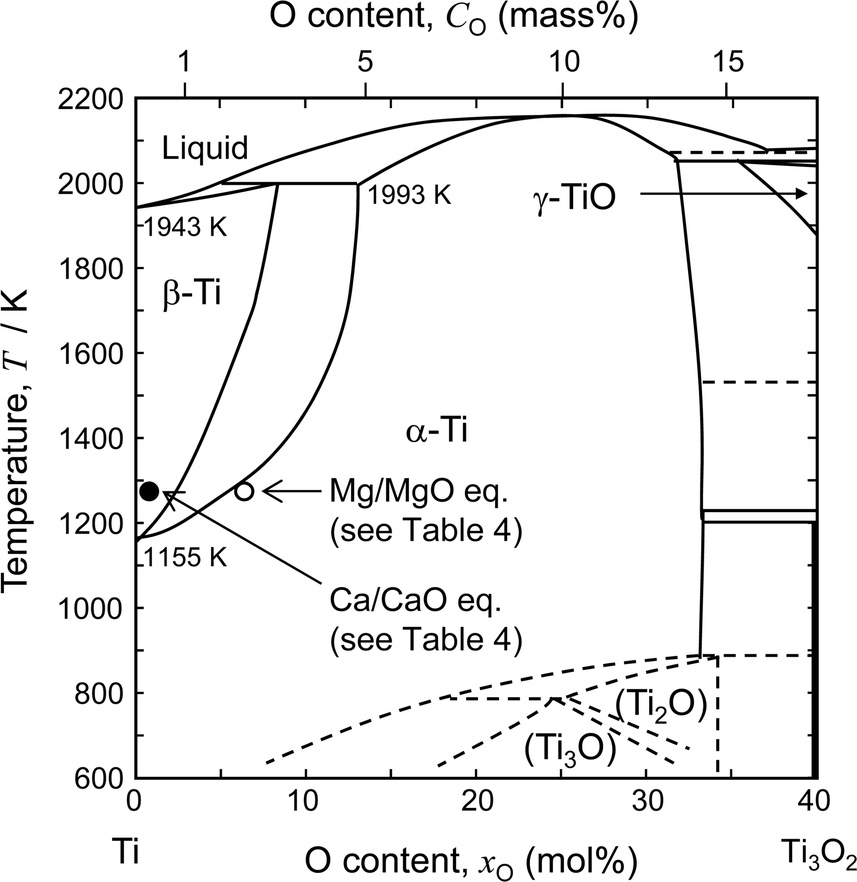 | ||
| Fig. 2 Phase diagram of the Ti–O binary system.49 Compositions of Ti–O solution under Ca/CaO and Mg/MgO equilibrium at 1273 K are plotted in the figure. | ||
The O potentials of β-Ti–O solid solution at temperatures between 1173 and 1473 K have been determined,11 and the feasibility of preparing low-O Ti by decreasing the activity of the by-product CaO (aCaO) has been examined using various Ca-halide fluxes in the presence of Ca metal (Ca-halide flux deox. shown in Table 2).18,19 Several factors have been considered in selecting a flux for deoxidation. CaCl2 has been found to be the most desirable flux at temperatures of ∼1300 K because the deoxidation product CaO dissolves easily in CaCl2 flux, which decreases its activity and thus lowers the limit of deoxidation. By submerging Ti wires and small pieces in Ca-saturated CaCl2, Ti samples has been deoxidized to a level of 50 mass ppm (see also Table 3).5,19
| Methoda | Oxygen (mass ppm) | Nitrogenc (mass ppm) | |||||
|---|---|---|---|---|---|---|---|
| Init. | After deox. | Init. | After deox. | Note and references (Exp. no.) | |||
| a “M-MOx”: deoxidation by metal/metal oxide, “Ca-CaCl2”: calcium-halide flux deoxidation, “Electrochem.”: electrochemical deoxidation. b Salt in parentheses shows molten salt used for electrochemical deoxidation. c Values in parenthesis include uncertainties. d Residual resistivity ratio, ρ298/ρ4.2, determined by measurement at 298 and 4.2 K. e Vickers micro hardness (kg f mm−2), measured using 500 g load at room temperature. f “N.A.”: not analyzed or not reported. g Ti wire (ϕ1.0 mm) was used as sample. Oxygen and nitrogen contents were analyzed by KOBELCO Research Institute, Inc. h Ti rod (ϕ3.0 mm) was used as sample. Oxygen and nitrogen contents were analyzed by KOBELCO Research Institute, Inc. | |||||||
| Ca-CaO | 430 | → | 460 | 20 | → | 340 | 1273 K, 86 ks, ref. 11 |
| 1430 | → | 470 | 100 | → | 470 | Eqn (3) | |
| Y-Y2O3 | 200 | → | 120 | N.A.f | 1231 K, 260 ks, ref. 12 | ||
| 1100 | → | 150, 170 | N.A.f | (#2U) | |||
| Y-Y2O3 | 210 | → | 90 | 10 | → | (30) | 1213 K, 551 ks, ref. 14 |
| 950 | → | 100, 220 | 20 | → | (50) | (#U) | |
| 670 | → | 80, 100 | 30 | → | (60) | ||
| Y-Y2O3 | 210 | → | 40, 80 | 10 | → | (50) | 1173 K, 518 ks, ref. 14 |
| 950 | → | 160, 160 | 20 | → | (120) | (#T) | |
| 670 | → | 30, 240 | 30 | → | (80) | ||
| Ca-Y-Y2O3 | 210 | → | 130 | 10 | → | (40) | 1173 K, 604 ks, ref. 14 |
| 950 | → | 60, 90 | 20 | → | (50) | (#W) | |
| 670 | → | 60, 90 | 30 | → | (120) | ||
| Ca-CaCl2 | 130 | → | 16 | 10 | → | 41 | 1273 K, 90 ks, ref. 19 |
| 130 | → | 17 | 10 | → | 34 | (#17B1) | |
| RRR4.2d = 98, Hve = 85 | |||||||
| Ca-CaCl2 | 130 | → | 35 | 10 | → | 49 | 1373 K, 40 ks, ref. 19 |
| 130 | → | 22 | 10 | → | 45 | (#19B1) | |
| RRR4.2d = 95, Hve = 92 | |||||||
| Ca-CaCl2 | 150 | → | 62 | 10 | → | 86 | 1273 K, 86 ks, ref. 19 |
| (#3C) | |||||||
| RRR4.2d = 122, Hve = 82 | |||||||
| Electrochem. (CaCl2)b | 900 | → | <10 | 40 | → | 20 | 1223 K, 22 ks, ref. 21 |
| 1400 | → | <10 | 20 | → | 20 | (#49, contaminated by carbon) | |
| Electrochem. (CaCl2)b | 110 | → | 10 | <10 | → | <10 | 1223 K, 27 ks, ref. 23 |
| 900 | → | 20 | 30 | → | 50 | (#69, carbon: 20 → 30, 50 → 80 ppm) | |
| RRR4.2d = 155, Hve = 64 | |||||||
| Electrochem. (CaCl2)b | 210 | → | 40, 50 | 10 | → | 10 | 1189 K, 36 ks, ref. 24 |
| (#12) | |||||||
| (O in Gd: 2100 → 10, 50 ppm, O in Tb: 2600 → 20, 20 ppm, O in Dy: 2300 → 20, 30 ppm O in Er: 2800 → 40, 40 ppm, O in Y: 11200 → 340, 380 ppm) | |||||||
| Electrochem. (MgCl2)b | 110 | → | 100 | <10 | → | 40 | 1173 K, 86 ks, ref. 23 |
| 900 | → | 60 | 30 | → | 70 | (#75, carbon: 20 → 50, 50 → 120 ppm) | |
| 840 | → | 90 | 20 | → | 440 | 1173 K, 86 ks, ref. 25 (D13_150121g) | |
| 1200 | → | 270 | 40 | → | 260 | 1173 K, 86 ks, ref. 25 (D15_150123h) | |
In the Ca-halide flux deoxidation method, the ultimate limit of deoxidation depends on the final O concentration in the flux, which means that the purity of the flux and the initial O content of the Ti will rule the deoxidation limit. To decrease the deoxidation limit set by aCaO, an electrochemical method was developed (electrochemical deox. shown in Table 2).21,22 The modified Ca-halide flux deoxidation method is characterized by both the production of Ca from the CaCl2 flux and the effective removal of O2− (present as CaO) dissolved in the flux by electrochemical means. In this method, as expressed in eqn (3), a Ti cathode is deoxidized by Ca, which forms electrochemically on the surface of the cathode according to eqn (4). O2− species in the flux are carried continuously to the C anode, and O in the flux is removed from the system as CO (or CO2) gas by the anodic reaction expressed in eqn (5).
| O (in Ti) + Ca (in flux) = Ca2+ (in flux) + O2− (in flux) | (3) |
| Ca2+ (in flux) + 2e− = Ca (on Ti cathode: in flux) | (4) |
| O2− (in flux) + C (C anode) = CO (gas) + 2e− | (5) |
Unlike the Ca-halide flux deoxidation, the electrochemical method does not require the addition of metallic Ca as a deoxidant, because the activity of Ca near the cathode can be increased by controlling the applied voltage between the Ti cathode and the C anode. In some cases, Ca is precipitated on the cathode using an applied voltage exceeding the theoretical decomposition voltage of CaCl2 (3.25 V at 1223 K). Furthermore, CaCl2 flux can be purified through pre-electrolysis. This electrochemical method therefore has the advantage that contamination-free Ca deoxidant and CaCl2 flux can be used in the deoxidation process. By applying the electrochemical technique to molten CaCl2, Ti containing O below 10 mass ppm has been successfully produced under certain conditions.5,21
In this study, the feasibility of the electrochemical deoxidation technique using MgCl2 as flux was examined. A schematic of the electrochemical deoxidation of Ti in molten MgCl2 is shown in Fig. 3.
| O (in Ti) + Mg (in flux) = Mg2+ (in flux) + O2− (in flux) | (6) |
 | ||
| Fig. 3 Schematic illustration of electrochemical deoxidation in MgCl2 molten salt. Electrolysis is achieved using titanium as the cathode and carbon as the anode. At the cathode, a deoxidant of magnesium is produced by electrolysis, and the deoxidation product of magnesium oxide is dissolved in molten salt. At the anode, an oxide anion generated by the cathode is removed as carbon monoxide or dioxide gas. The anode reaction occurs in parallel with chlorine evolution. During this process, the activity of MgO can be kept at a low level on the cathode surface. | ||
Based on the thermodynamic calculation shown in Fig. 4,5,11,50 when aMgO is decreased to the level of 10−2 in the presence of metallic Mg at 1200 K, the chemical potential of O2 (pO2) in the system is decreased to ∼10−45 atm, and Ti containing 0.018 mass% (180 mass ppm) O can be obtained. In other words, when Ti is deoxidized down to commercial O levels for high-grade Ti (0.05 mass% O, or 500 mass ppm) using Mg as reductant, aMgO at 1200 K must be decreased to the level of 0.025.
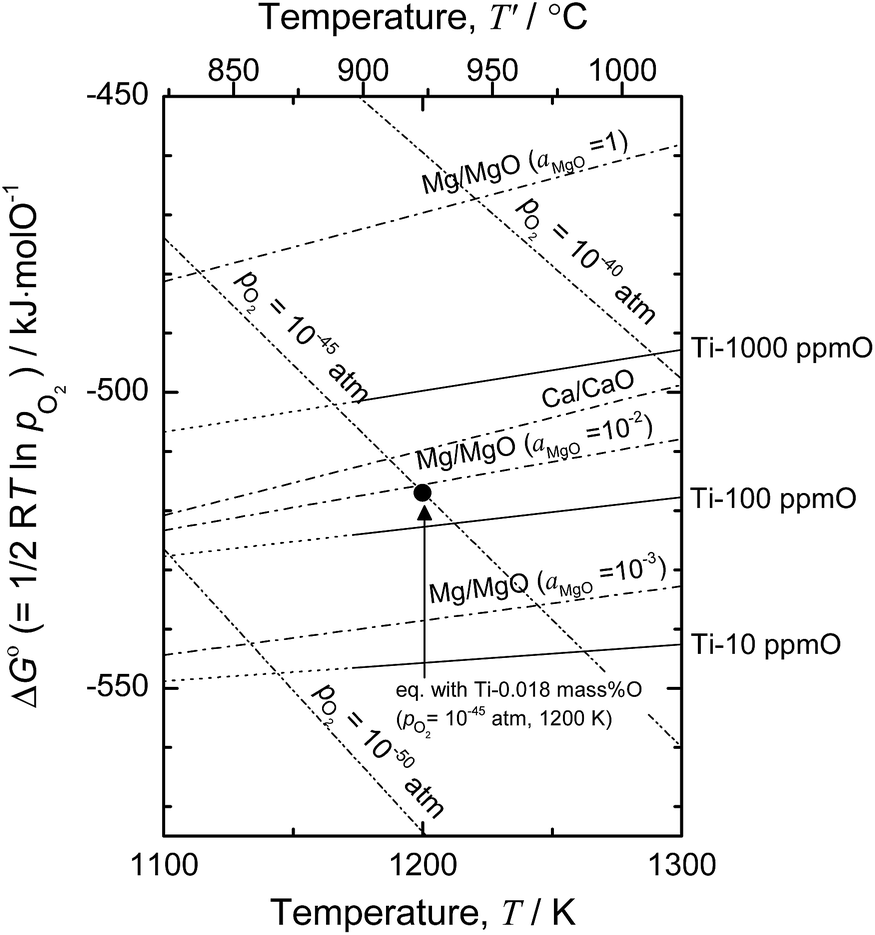 | ||
| Fig. 4 Ellingham diagram of selected oxides50 and solid solutions of Ti–O.5,11 Oxygen chemical potential for Ti-0.018 mass% O (pO2 = 10−45 atm) at 1200 K is plotted for reference. | ||
Experimental procedure
Fig. 5 shows a flowchart of the experimental procedure for the electrochemical deoxidation of Ti in MgCl2. A schematic of the experimental apparatus used for the electrochemical deoxidation of Ti in MgCl2 is shown in Fig. 6. Ti in the form of small pieces and wires with different O concentrations and configurations was used as the starting material. The flux was reagent-grade anhydrous MgCl2 (>97.0%) in flake form, dried at 523 K for more than 43 ks (12 h).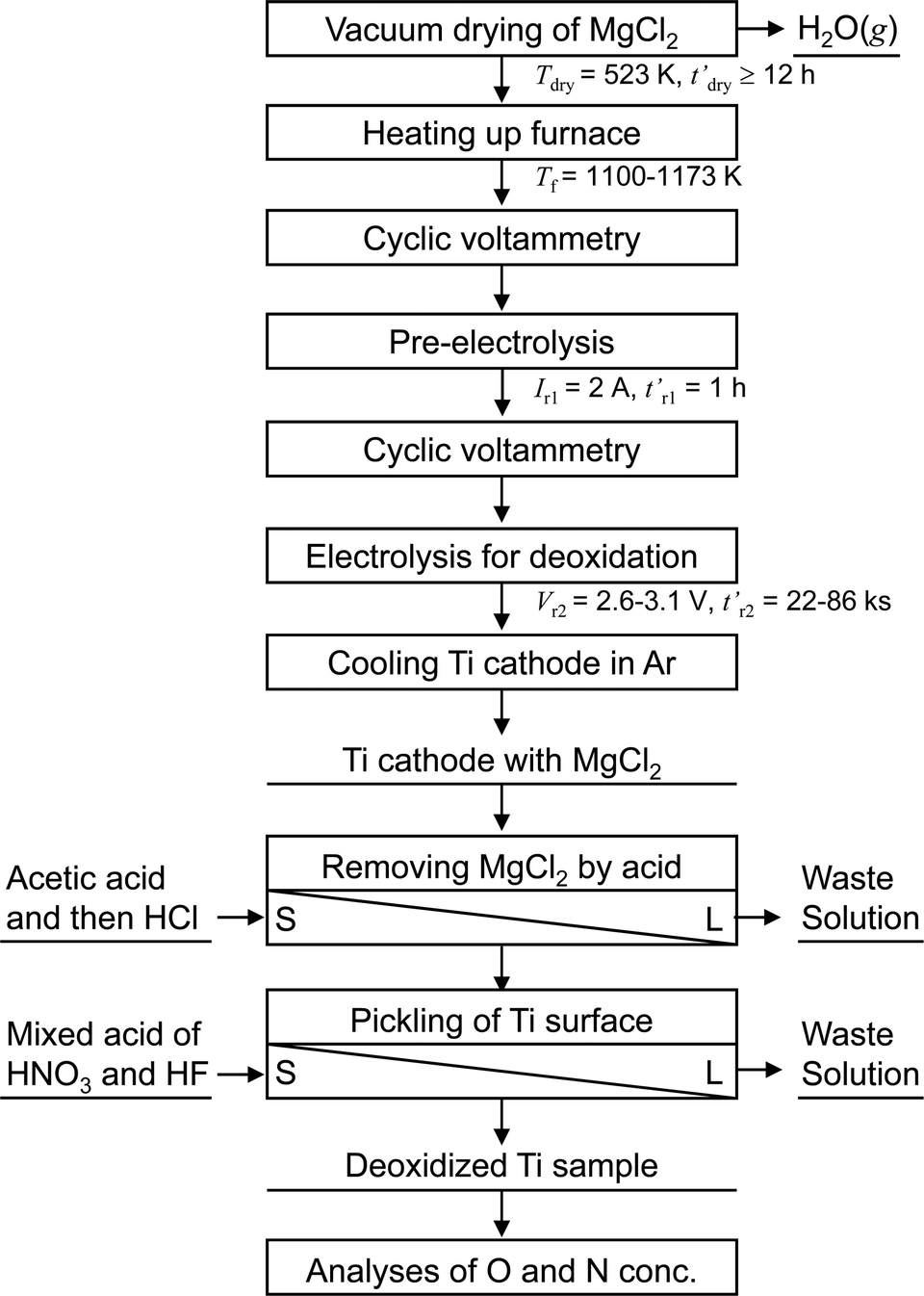 | ||
| Fig. 5 Flowchart of experimental procedure for electrochemical deoxidation of titanium in molten MgCl2. | ||
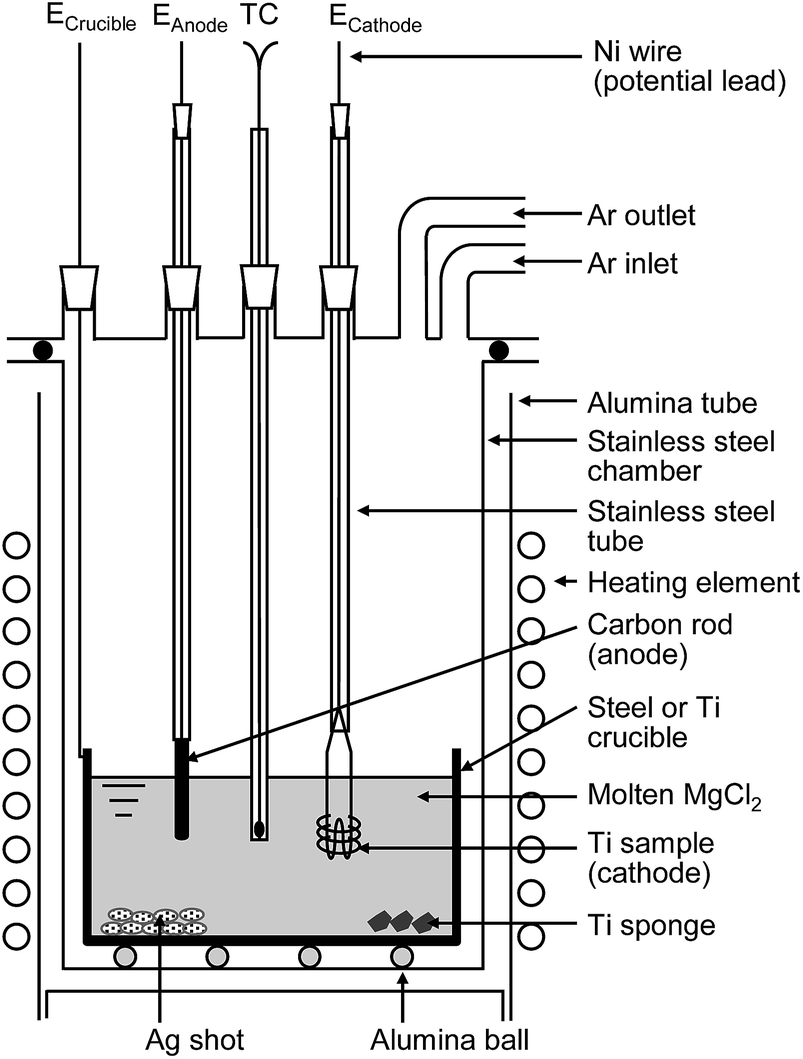 | ||
| Fig. 6 Schematic illustration of experimental apparatus for electrochemical deoxidation of titanium in MgCl2. | ||
The dehydrated MgCl2 flux was contained in a Ti or mild steel crucible (89 mm in diameter, 200 mm in height) and set in a gas-tight stainless steel chamber, as shown in Fig. 6. The Ti cathode consisted of several strands of Ti wire ∼50 mm in length and 1 mm in diameter. The total electrode surface area was approximately 1000 mm2. In the same experiment, a Ti rod (approximately 30 mm in length, 3 mm in diameter) was used as the cathode. High-purity (99.9%) graphite was used as the C anode (approximately 35 mm in length, 3 or 6 mm in diameter). These electrodes, as well as thermocouple tubing, were inserted into the reaction tube through a gas-tight water-cooled stainless steel head.
After the assembled cell was evacuated at ∼673 K to ensure a completely gas-tight and moisture-free system, Ar was introduced to the reaction tube. The furnace was then heated to a constant temperature between 1100 and 1173 K. After the MgCl2 was melted, the graphite anode was inserted into the molten salt, and pre-electrolysis was conducted. In the pre-electrolysis, a 2 A current was passed for ∼3.6 ks (1 h) between the Ti crucible (or Ni electrode immersed in molten salt) and the C anode to eliminate residual metal and gaseous impurities in the molten salt. Approximately 50 g of silver was placed at the bottom of the crucible to absorb the magnesium metal deposited during electrolysis. The temperature of the molten salt was measured directly using a chromel–alumel thermocouple protected by a stainless steel sheath. After pre-electrolysis, the Ti cathode containing the Ti samples was inserted into the molten MgCl2, and deoxidation experiments were performed by applying voltages between 0 and 3.1 V between the electrodes, which were separated by a distance of ∼30 mm. In most cases, the deoxidation of the Ti cathode occurred with the application of voltages exceeding 2.6 V at 1100–1173 K for longer than 22 ks (6 h).
Before and after the pre-electrolysis of MgCl2 and the deoxidation experiments, the electrochemical properties of the flux were measured by cyclic voltammetry (CV). An electrochemical interface (Solartron SI1287) was used for the CV measurements.
After the deoxidation experiment, the electrodes were removed from the molten MgCl2, and the cell was cooled in a stream of Ar gas. In some cases, only the Ti cathode was replaced after cooling for the next experimental run. The fused salt that adhered to the surface of the Ti electrode was removed by leaching with (1 + 1) acetic acid, after which the Ti samples of the electrode were carefully cleaned in (1 + 10) aqueous HCl solution, followed by water, alcohol, and acetone, and then dried.
The Ti samples obtained were subjected to O and N analyses using LECO analyzers. Prior to the analyses, the samples were surface-etched with a 1:4:10 mixture of HF–HNO3–H2O. To increase the accuracy of the O and N analyses, 1.1 g of nickel capsule containing 8.8 ± 1.4 μg O and 0.2 ± 0.2 μg N was used as an extraction bath for each 0.05–0.1 g Ti sample. The instrument for the O analysis was calibrated using Ti standard samples containing 470 ± 30 and 980 ± 40 mass ppm O.
Results and discussion
Fig. 7(a) shows the CV of the MgCl2 salt at 1173 K using a Ni quasi-reference before pre-electrolysis. The deposition of Mg occurs at approximately −0.7 V with respect to the Ni quasi-reference, and gas evolution occurs at approximately +1.6 V. The oxidation wave between 1 and 1.6 V probably results from oxygen impurities in the molten salt, and the gas evolution peak at ∼1.6 V corresponds to the evolution of Cl2 gas.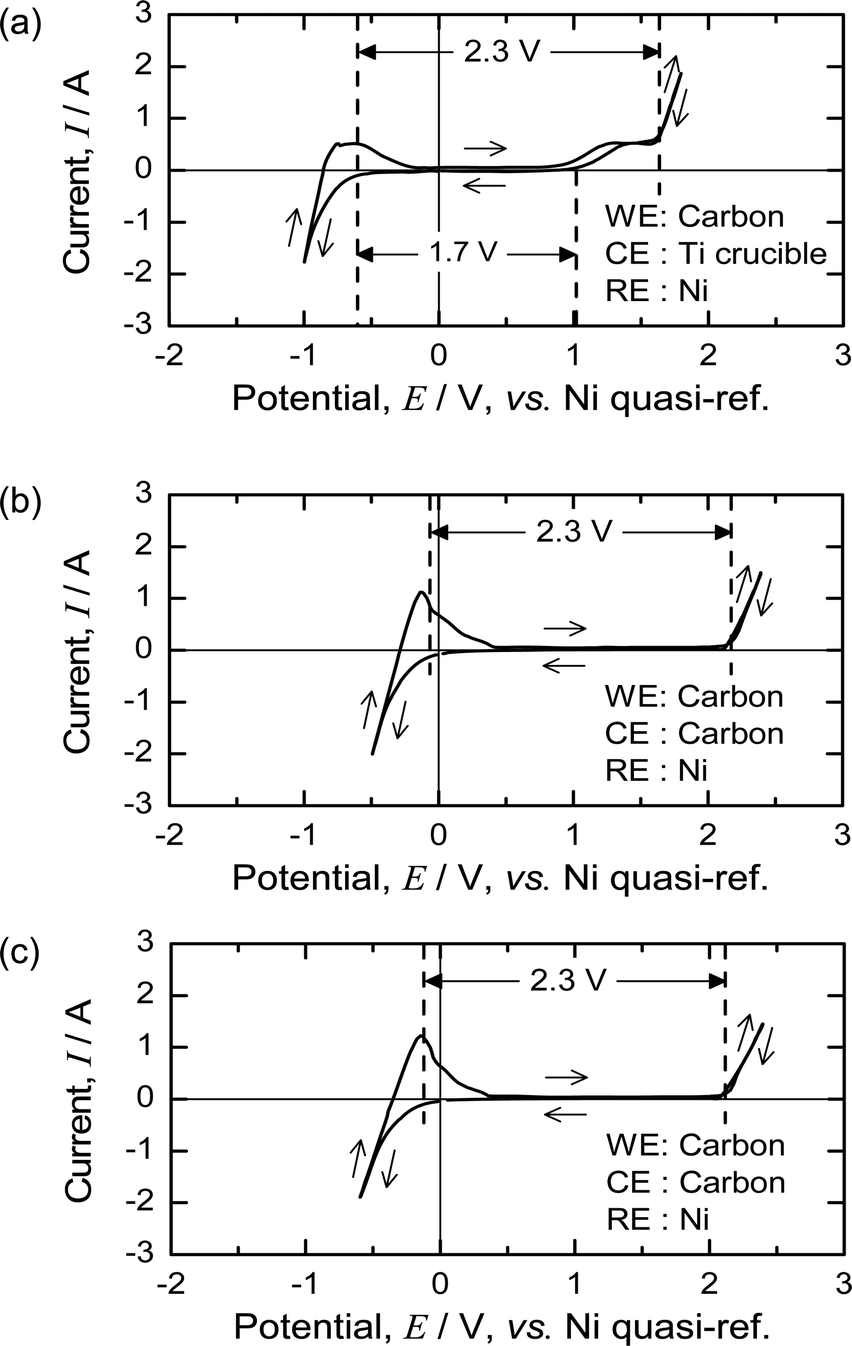 | ||
| Fig. 7 Cyclic voltammograms in MgCl2 molten salt at 1173 K. Surface area of electrode: 2 cm2 (C rod, Ni rod). Surface area of electrode: 260 cm2 (Ti crucible). Scan rate: 100 mV s−1. (a) Before pre-electrolysis. (b) After pre-electrolysis. (c) After electrolysis for Ti deoxidation. | ||
During pre-electrolysis using a Ni electrode as a cathode, Mg metal was electrochemically deposited on the Ni electrode. After pre-electrolysis, the Mg deposition and gas evolution peaks shifted to 0 and 2.3 V, respectively. Fig. 7(b) and (c) show the CV of the molten MgCl2 before and after the deoxidation experiments. As the theoretical decomposition voltage of MgCl2 at 1173 K is 2.3 V,50 the reference electrode worked correctly, and the Ni quasi-reference electrode became a reference for Mg/Mg2+ after pre-electrolysis.
The results shown in Fig. 7 indicate that the electrochemical properties of MgCl2 are unchanged before and after the deoxidation experiments. After pre-electrolysis, only the Mg deposition wave and Cl2 evolution are observed, and therefore, the reaction system in the molten salt can be regarded as contamination-free MgCl2.
By applying a voltage of ∼3.1 V between the electrodes, the chemical potential of metallic Mg (aMg) in MgCl2 was increased on the surface of the Ti cathode to a value approaching 1, and the Ti samples were deoxidized by the electrochemically produced Mg. The resulting O species, present mainly as O2− dissolved in the molten MgCl2, reacted at the C anode to form CO (or CO2) gas, which was removed from the molten salt system. For example, after electrochemical deoxidation at 1173 K for 86 ks, Ti wires 1.0 mm in diameter and containing 0.084 mass% (840 mass ppm) O were deoxidized to 0.009 mass% (90 mass ppm) (Exp. no. D13_150121 shown in Table 3).25 When a Ti rod of 3.0 mm diameter containing 0.12 mass% (1200 mass ppm) O was deoxidized at 1173 K for 86 ks, Ti with 0.027 mass% (270 mass ppm) O was obtained (Exp. no. D15_150123 shown in Table 3).25 The N concentration in the sample was increased from ∼10–40 to 40–440 mass ppm after deoxidation.
In Table 3, some representative analytical results for the O and N levels in Ti before and after deoxidation are listed, as well as results from previous studies.11,12,14,19,21,23–25 The analytical values below 0.01 mass% (100 mass ppm) listed in the table include some uncertainty because gas analysis of Ti samples below 0.01 mass% (100 mass ppm) is difficult. However, notably, Ti with an oxygen level of about 0.01 mass% (100 mass ppm) was obtained by using Mg as the deoxidant when using the electrochemical technique.
Table 4 summarizes the deoxidation limits for the Mg/MgO, Ca/CaO, and Y/Y2O3 systems. The activity of the metal oxides for obtaining Ti with 100 mass ppm O when using the respective metal deoxidants was calculated using the available thermodynamic data11,50 and are listed in the table. In this study, Ti wires containing 0.12 mass% (1200 mass ppm) O were deoxidized to less than 0.02 mass% (200 mass ppm) O. In some cases, the O concentration in the Ti samples was reduced to below 0.01 mass% (100 mass ppm) O, which cannot be accomplished using the current Kroll process. These experimental results show that the activity of MgO, aMgO, was decreased to below 0.01.
| System | Equilibrium O conc. in Ti at 1273 K, CO,eq (mass ppm) | Required aMOx to obtain Ti with 1000 ppm O at 1273 K, aMOx | Required aMOx to obtain Ti with 100 ppm O at 1273 K, aMOx |
|---|---|---|---|
| Mg/MgO | 26000 |
0.039 | 0.0039 |
| Ca/CaO | 500 | 2.0 | 0.20 |
| Y/YO1.5 | 180 | 13 | 0.41 |
When electrolysis in CaCl2 is utilized to remove O directly from Ti, the deoxidation limit can be easily decreased to below 0.01 mass% (100 mass ppm). However, after deoxidation, CaCl2 flux attached on the surface of Ti products must be removed by dissolving in aqueous solution, which causes O pickup. It is impractical to remove CaCl2 by evaporation, because the vapor pressure of CaCl2 is low even at elevated temperatures. Fig. 8 shows the temperature dependence of the vapor pressures of CaCl2 and MgCl2.50 The vapor pressure of MgCl2 is two or three orders of magnitude higher than that of CaCl2. The high vapor pressure of MgCl2 might enhance operational challenges during electrolysis.51 However, MgCl2 can be removed directly from titanium products by vacuum treatment; a technique for MgCl2 removal via vacuum distillation is already established in the Kroll process. This MgCl2 removal technique can easily be applied to the deoxidation process investigated in this study.
 | ||
| Fig. 8 Vapor pressures of metal chlorides as functions of reciprocal temperature.50 | ||
Fig. 9 shows the new recycling process for Ti scrap using electrochemical deoxidation in molten MgCl2 followed by the removal of MgCl2 by vacuum distillation. The reduction chamber used in the conventional Kroll process can be utilized in the electrochemical deoxidation process, as schematically shown in Fig. 10. As Ti production is increased, scrap treatment becomes increasingly important. In the future, the direct O removal technique proposed in this study may be applied to recycling Ti scrap.
 | ||
| Fig. 9 Recycling process proposed in this study. Oxygen dissolved in Ti scrap is directly removed by molten salt electrolysis in MgCl2. | ||
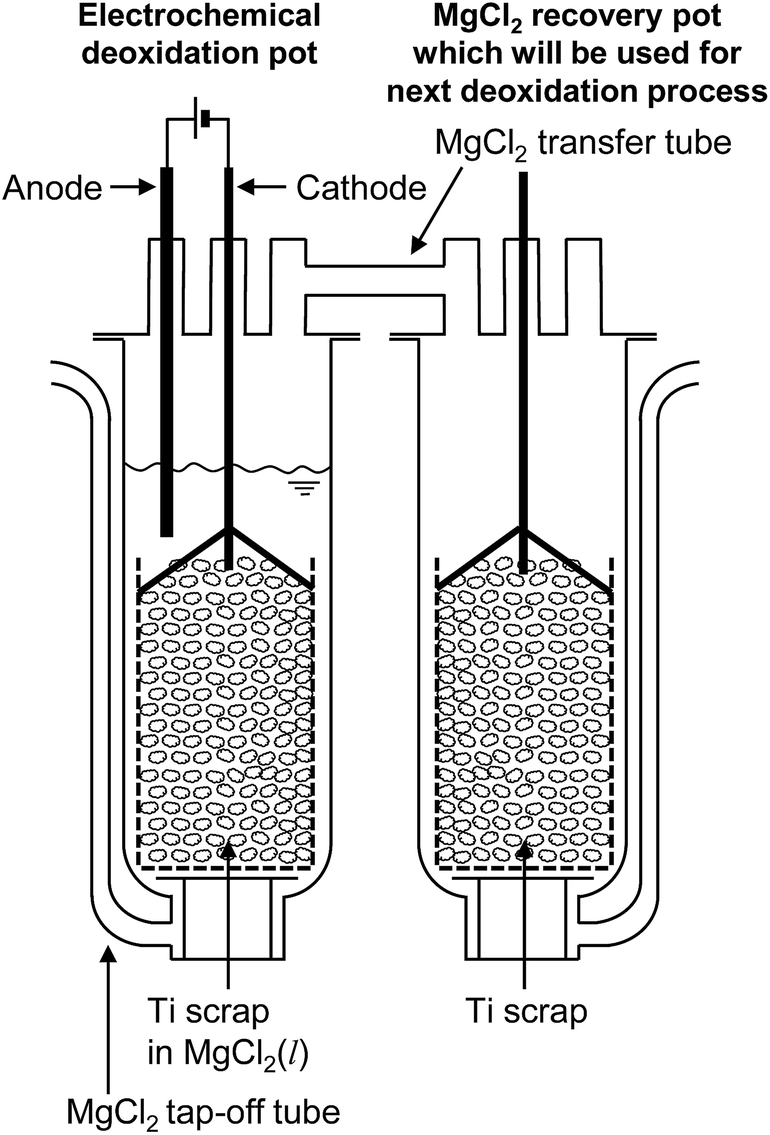 | ||
| Fig. 10 Industrial application of the recycling process based on electrolysis in MgCl2. Process is feasible in existing Ti smelters based on the Kroll process. | ||
Conclusions
The removal of O from Ti using an electrochemical technique in molten MgCl2 was examined at temperatures of ∼1173 K. The activity of Mg was increased near the Ti cathode surface and the activity of MgO was decreased by applying voltages between the Ti cathode and C anode immersed in molten MgCl2. These activity changes facilitated the deoxidation of a Ti cathode consisting of the Ti samples. A Ti sample containing approximately 1000 mass ppm O were deoxidized to less than 200 mass ppm by applying 3.1 V between the Ti and C electrodes for 86 ks. In some cases, the O concentration in Ti was decreased to below 100 mass ppm, which cannot be accomplished using the conventional Kroll process. The application of this deoxidation technique to industrial recycling processes may become practical in the future.Acknowledgements
The authors are grateful to Professor Masafumi Maeda and Professor Kazuki Morita of The University of Tokyo and to Dr Tetsushi Deura, and Messrs. Daisuke Matsuwaka and Fumiaki Kudo of Kobe Steel, Ltd. for their sample analysis. The authors thank Dr Katsuhiro Nose and Mr Hisao Kimura for their valuable suggestions and technical assistance. This research was partly funded by a Grant-in-Aid for the Next Generation of World-Leading Researchers (NEXT Program) for the Research Project for Development of Environmentally Sound Recycling Technology of Rare Metals (NEXT Program #GR019), and a Grant-in-Aid for Scientific Research (S) (KAKENHI Grant #26220910) by JSPS.References
- T. G. Goonan, Titanium Recycling in the United States in 2004, Flow studies for recycling metal commodities in the United States, ed. S. F. Sibley, U. S. Geological Survey Circular, 1196, 2010, ch. Y, pp. Y1–Y16 Search PubMed.
- B. Rotmann, C. Lochbichler and B. Friedrich, Challenges in Titanium Recycling-Do We Need a New Specification for Secondary Alloys?, Proceedings of EMC 2011, 2011 Search PubMed.
- American Society for Testing and Materials, Standard Specification for Titanium and Titanium Alloy Strip, Sheet, and Plate, B265–06b, ASTM International, West Conshohocken, PA, 2006 Search PubMed.
- W. Kroll, The Production of Ductile Titanium, Trans. Electrochem. Soc., 1940, 78, 35–47 CrossRef.
- T. H. Okabe, K. T. Jacob and Y. Waseda, Removal of Oxygen in Reactive Metals, in Purification Process and Characterization of Ultra High Purity Metals, ed. Y. Waseda and M. Isshiki, Springer, Berlin, 2001, pp. 3−37 Search PubMed.
- K. Ono and S. Miyazaki, Study on the Limit of Deoxidation of Titanium and the Reduction of Titanium Dioxide by Saturated Calcium Vapors, J. Jpn. Inst. Met., 1985, 49(10), 871–875 CAS , (in Japanese).
- R. L. Fisher, Deoxidation of Titanium and Similar Metals Using a Deoxidant in a Molten Metal Carrier, US Pat. No. 4923531A, 1990, (UK Patent No. GB 2224749A, 1989).
- R. L. Fisher, Deoxidation of a Refractory Metal, US Pat. No. 5022935, 1991.
- R. L. Fisher and S. R. Seagle, Removal of Oxide Layers from Titanium Castings Using an Alkaline Earth Deoxidizing Agent, US Patent No. 5211775 A, 1993.
- R. L. Fisher and S. R. Seagle, DOSS, An Industrial Process for Removing Oxygen From Titanium Turnings Scrap, Titanium Science and Technology, ed. F. H. Froes and I. Caplan, The Minerals, Metals & Materials Society (Proceedings of the 7th World Conference on Titanium, 1992), 1993, vol. 3, pp. 2265−2272 Search PubMed.
- T. H. Okabe, R. O. Suzuki, T. Oishi and K. Ono, Thermodynamic Properties of Dilute Titanium-Oxygen Solid Solution in Beta Phase, Mater. Trans. JIM, 1991, 32(5), 485–488 CrossRef CAS.
- T. H. Okabe, T. Deura, T. Oishi, K. Ono and D. R. Sadoway, Thermodynamic Properties of Oxygen in Yttrium Solid Solution, Metall. Trans. B, 1996, 27B, 841–847 CrossRef CAS.
- T. H. Okabe, K. Hirota, Y. Waseda and K. T. Jacob, Thermodynamic Properties of Oxygen in Ln–O (Ln = La, Pr, Nd) Solid Solutions and Their Deoxidation by Molten Salt Electrolysis, J. MMIJ, 1998, 114(11), 813–818 CrossRef CAS.
- T. H. Okabe, K. Hirota, E. Kasai, F. Saito, Y. Waseda and K. T. Jacob, Thermodynamic Properties of Oxygen in RE–O (RE = Gd, Tb, Dy, Er) Solid Solutions, J. Alloys Compd., 1998, 279, 184–191 CrossRef CAS.
- J.-M. Oh, B.-K. Lee, C.-Y. Suh, S.-W. Cho and J.-W. Lim, Preparation Method of Ti Powder with Oxygen Concentration of < 1000 ppm Using Ca, Powder Metall., 2012, 55(5), 402–404 CrossRef CAS.
- J.-M. Oh, K.-M. Roh, B.-K. Lee, C.-Y. Suh, W. Kim, H. Kwon and J.-W. Lim, Preparation of Low Oxygen Content Alloy Powder from Ti Binary Alloy Scrap by Hydrogen-Dehydrogenation and Deoxidation Process, J. Alloys Compd., 2014, 593, 61–66 CrossRef CAS.
- K.-M. Roh, C.-Y. Suh, J.-M. Oh, W. Kim, H. Kwon and J.-W. Lim, Comparison of Deoxidation Capability for Preparation of Low Oxygen Content Powder from TiNi Alloy Scraps, Powder Technol., 2014, 253, 266–269 CrossRef CAS.
- T. H. Okabe, R. O. Suzuki, T. Oishi and K. Ono, Preparation of Extra Low Oxygen Titanium by Calcium-Halide Flux Deoxidation, J. Iron Steel Inst. Jpn., 1991, 77(1), 93–99 CAS , (in Japanese).
- T. H. Okabe, T. Oishi and K. Ono, Preparation and Characterization of Extra-Low-Oxygen Titanium, J. Alloys Compd., 1992, 184, 43–56 CrossRef CAS.
- T. H. Okabe, T. Oishi and K. Ono, Deoxidation of Titanium Aluminide by Ca–Al Alloy under Controlled Aluminum Activity, Metall. Trans. B, 1992, 23B, 583–590 CrossRef CAS.
- T. H. Okabe, M. Nakamura, T. Oishi and K. Ono, Electrochemical Deoxidation of Titanium, Metall. Trans. B, 1993, 24B, 449–455 CrossRef CAS.
- T. H. Okabe, T. Deura, T. Oishi, K. Ono and D. R. Sadoway, Electrochemical Deoxidation of Yttrium–Oxygen Solid Solution, J. Alloys Compd., 1996, 237, 150–154 CrossRef CAS.
- M. Nakamura, T. H. Okabe, T. Oishi and K. Ono, Electrochemical Deoxidation of Titanium, Proc. Int. Symp. Molten Salt Chem. Technol., 1993, 529–540 CAS.
- K. Hirota, T. H. Okabe, F. Saito, Y. Waseda and K. T. Jacob, Electrochemical Deoxidation of RE–O (RE = Gd, Tb, Dy, Er) Solid Solution, J. Alloys Compd., 1999, 282, 101–108 CrossRef CAS.
- Y. Taninouchi, Y. Hamanaka and T. H. Okabe, submitted.
- G. Z. Chen, D. J. Fray and T. W. Farthing, Direct Electrochemical Reduction of Titanium Dioxide to Titanium in Molten Calcium Chloride, Nature, 2000, 407(6802), 361–364 Search PubMed.
- G. Z. Chen, D. J. Fray and T. W. Farthing, Cathodic Deoxidation of the Alpha Case on Titanium and Alloys in Molten Calcium Chloride, Metall. Trans. B, 2001, 32B, 1041–1052 CrossRef CAS.
- G. Z. Chen, D. J. Fray and T. W. Farthing, Removal of Oxygen from Metal Oxides and Solid Solutions by Electrolysis in a Fused Salt, US Pat. No. 2004/0159559 A1, 2004.
- D. J. Fray, Emerging Molten Salt Technologies for Metals Production, JOM, 2001, 53(10), 26–31 CrossRef CAS.
- K. S. Mohandas and D. J. Fray, FFC Cambridge Process and Removal of Oxygen from Metal-Oxygen Systems by Molten Salt Electrolysis: an Overview, Trans. Indian Inst. Met., 2004, 57(6), 579–592 CAS.
- P. K. Tripathy, M. Gauthier and D. J. Fray, Electrochemical Deoxidation of Titanium Foam in Molten Calcium Chloride, Metall. Trans. B, 2007, 38B, 893–900 CrossRef CAS.
- K. Ono and R. O. Suzuki, A New Concept for Producing Ti Sponge: Calciothermic Reduction, JOM, 2002, 54(2), 59–61 CrossRef CAS.
- R. O. Suzuki and S. Inoue, Calciothermic Reduction of Titanium Oxide in Molten CaCl2, Metall. Trans. B, 2003, 34B(3), 277–285 CrossRef CAS.
- R. O. Suzuki, K. Ono and K. Teranuma, Calciothermic Reduction of Titanium Oxide and In-situ Electrolysis in Molten CaCl2, Metall. Trans. B, 2003, 34B(3), 287–295 CrossRef CAS.
- R. O. Suzuki and S. Fukui, Reduction of TiO2 in Molten CaCl2 by Ca Deposited during CaO Electrolysis, Mater. Trans., 2004, 45(5), 1665–1671 CrossRef CAS.
- R. O. Suzuki, Calciothermic Reduction of TiO2 and In-situ Electrolysis of CaO in Molten CaCl2, J. Phys. Chem. Solids, 2005, 66(2), 461–465 CrossRef CAS.
- R. O. Suzuki, Direct Reduction Process for Titanium Oxide in Molten Salt, JOM, 2007, 59(1), 68–71 CrossRef CAS.
- T. Yahata, T. Ikeda and M. Maeda, Deoxidation of Molten Titanium by Electron-Beam Remelting Technique, Metall. Trans. B, 1993, 24B, 599–604 CrossRef CAS.
- J. Reitz, C. Lochbichler and B. Friedrich, Recycling of Gamma Titanium Aluminide Scrap from Investment Casting Operations, Intermetallics, 2011, 19, 762–768 CrossRef CAS.
- Y. Su, L. Wang, L. Luo, X. Jiang, J. Guo and H. Fu, Deoxidation of Titanium Alloy Using Hydrogen, Int. J. Hydrogen Energy, 2009, 34, 8958–8963 CrossRef CAS.
- J. R. Nettle, D. H. Baxer Jr and F. S. Wartman, Electrorefining Titanium Metal, United States Bureau of Mines, Report of Investigations 5315, Washington, D.C., U.S., 1957 Search PubMed.
- A. B. Suchkov, Z. A. Tubyshki, Z. I. Sokolova and N. V. Zhukova, Electrolytic Refining of Titanium Alloy Scrap Using Solid Intermediate Electrodes, Russ. Metall., 1969, 6, 52 Search PubMed.
- S. Takeuchi and O. Watanabe, Studies on the Method for the Electro-Refining of Titanium from the Molten Salts (1) Effect of the Various Molten Salt Baths on the Electro-Refining, J. Jpn. Inst. Met., 1964, 28(10), 633–638 CAS , (in Japanese).
- S. Takeuchi and O. Watanabe, The Investigation on the Electrorefining of Titanium from the Molten Salts (II) Crystal Shapes and Growth of Titanium from the Various Molten Salt in the Electro-refining, J. Jpn. Inst. Met., 1964, 28(11), 728–734 CAS , (in Japanese).
- Y. Hashimoto, K. Uriya and R. Kono, Electro-winning of Titanium Metal from Its Oxide by Fused Salt Electrolysis at Temperatures above the Melting Point of the Metal, Denki Kagaku, 1971, 39(6), 516–522 CAS , (in Japanese).
- Y. Hashimoto, Influences of Fluoride Salt Baths on Fused-Salt Electrodeposition of Titanium Metal from TiO2, Denki Kagaku, 1971, 39(12), 938–943 CAS , (in Japanese).
- Y. Hashimoto, Electro-Winning of Titanium from TiO2 or CaTiO3 in CaF2-MgF2 Molten Salt Bath, Denki Kagaku, 1972, 40(1), 39–44 CAS , (in Japanese).
- H. Miyazaki, Y. Yamakoshi and Y. Shindo, Electrorefining of Titanium in Molten Salts, Mater. Jpn., 1994, 33(1), 51–54 CrossRef CAS , (in Japanese).
- T. B. Massalski, Binary Alloy Phase Diagrams, ASM international, Materials Park, OH, United States of America, 2nd edn, 1990 Search PubMed.
- I. Barin, Thermochemical Data of Pure Substances, VCH Verlagsgesellschaft mbH, Weinheim, Germany, 3rd edn, 1995 Search PubMed.
- W. Li, Y. Yuan, X. Jin, H. Chen and G. Z. Chen, Environmental and Energy Gains from Using Molten Magnesium-sodium-potassium Chlorides for Electro-metallisation of Refractory Metal Oxides, Prog. Nat. Sci., 2015, 25(6), 650–653 CrossRef.
Footnote |
| † Currently at Mitsubishi Materials Corporation. |
| This journal is © The Royal Society of Chemistry 2016 |