 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective hydrogenation of halogenated arenes using porous manganese oxide (OMS-2) and platinum supported OMS-2 catalysts†
Iain J.
McManus
,
Helen
Daly
,
Haresh G.
Manyar
,
S. F. Rebecca
Taylor
,
Jillian M.
Thompson
* and
Christopher
Hardacre
*
CenTACat, School of Chemistry and Chemical Engineering, Queen's University, Belfast, David Keir Building, Stranmillis Road, Belfast, BT9 5AG, UK. E-mail: jillian.thompson@qub.ac.uk; c.hardacre@qub.ac.uk
First published on 21st December 2015
Abstract
Porous manganese oxide (OMS-2) and platinum supported on OMS-2 catalysts have been shown to facilitate the hydrogenation of the nitro group in chloronitrobenzene to give chloroaniline with no dehalogenation. Complete conversion was obtained within 2 h at 25 °C and, although the rate of reaction increased with increasing temperature up to 100 °C, the selectivity to chloroaniline remained at 99.0%. Use of Pd/OMS-2 or Pt/Al2O3 resulted in significant dechlorination even at 25 °C and 2 bar hydrogen pressure giving a selectivity to chloroaniline of 34.5% and 77.8%, respectively, at complete conversion. This demonstrates the potential of using platinum group metal free catalysts for the selective hydrogenation of halogenated aromatics. Two pathways were observed for the analogous nitrobenzene hydrogenation depending on the catalyst used. The hydrogenation of nitrobenzene was found to follow a direct pathway to aniline and nitrosobenzene over Pd/OMS-2 in contrast to the OMS and Pt/OMS-2 catalysts which resulted in formation of nitrosobenzene, azoxybenzene and azobenzene/hydrazobenzene intermediates before complete conversion to aniline. These results indicate that for Pt/OMS-2 the hydrogenation proceeds predominantly over the support with the metal acting to dissociate hydrogen. In the case of Pd/OMS-2 both the hydrogenation and hydrogen adsorption occur on the metal sites.
Introduction
Halogenated anilines are used in the manufacture of polymers, pharmaceuticals, dyes and agrochemicals1,2 and are most easily prepared by the hydrogenation of the corresponding halonitrobenzene. High selectivity for the hydrogenation reaction over any dehalogenation is required to optimise yield as well as minimise poisoning of the metal catalyst by strong adsorption of the halogen.High selectivities to haloanilines have been observed over a range of heterogeneous catalysts including ruthenium,3 nickel,4 platinum5 and a range of transition metal sulphides.6 In many cases the source of hydrogen is from a stoichiometric reducing agent such as NaBH4;3,4,6 however, the use of hydrogen gas is preferred to minimise the waste formed. It is generally accepted that platinum is the preferred metal for hydrogenation, giving minimum amounts of dehalogenation with palladium enhancing the dehalogenation reaction leading to increased side products. This is highlighted by the observation that palladium is the catalyst of choice in dehalogenation for pollution control,7,8 although it should be noted that homogeneous catalysts based on iron,9 nickel10,11 and rhodium12 are also reported to be highly active for dehalogenation reactions. Despite the propensity of palladium to dehalogenate aromatics, a recent study has highlighted the importance of the size of palladium nanoparticles for the selective hydrogenation of halonitrobenzenes over Pd/C. Using hydrogen gas, high conversion and greater than 99% selectivity to chloroaniline were obtained in the absence of a solvent on a 2000 L pilot plant scale over 30 runs.7
Dehalogenation can, therefore, occur to varying degrees over different catalysts; however, the overall selectivity of the hydrogenation reaction depends on a number of other factors such as the halogen involved. For example, iodo-substituted aromatics are more easily dehalogenated than the corresponding bromo and chloro molecules and the fluoro molecules are the most stable.13 This has been attributed to a combination of the C–X bond strength, with C–F > C–Cl > C–Br > C–I, as well as the adsorption strength of the different substrates on the catalyst.14,15 In addition, severe reaction conditions such as high temperatures or pressures can cause dehalogenation and complete hydrogenation of the aromatic ring. For example, Wang et al. showed that for the gas phase hydrogenation of para-chloronitrobenzene, the product selectivity was sensitive to the H2 to substrate ratio with a range of products from para-chloroaniline to cyclohexane being formed.16
The present study reports on the hydrogenation of halogenated and non-halogenated nitrobenzene over a series of manganese oxide supported catalysts. Despite many reports examining nitrobenzene hydrogenation, the mechanism by which the reaction occurs is still not well understood. The first reported mechanism for this reaction was by Haber in 1898 and involved hydrogenation of nitrobenzene (1) to nitrosobenzene (2) which could then further hydrogenate to form phenyl hydroxylamine (3) or react with either phenyl hydroxylamine or aniline to form azoxybenzene (4) or azobenzene (5), respectively.17 The azoxybenzene could also be further hydrogenated to azobenzene and on to hydroazobenzene (6) and aniline (7). Meanwhile the phenyl hydroxylamine is further hydrogenated to aniline. This process is illustrated in Scheme 1. However, Gelder et al. used kinetic isotope effects and the relative rates of reaction between nitrobenzene and nitrosobenzene to show that the nitrosobenzene species could not be the main intermediate in the reaction mechanism over Pd/C.18 They proposed that a PhNOH intermediate was formed which is hydrogenated to phenylhydroxylamine. In this case, the hydrogen involved results in a kinetic isotope effect however, in the nitrosobenzene reaction, the intermediate is dimerised to give azoxybenzene where no hydrogen is involved and no kinetic isotope effect is observed. A similar idea was proposed by Richner et al. who used infra-red spectroscopic analysis of the species adsorbed on the catalyst as well as in the liquid phase to propose the direct hydrogenation mechanism through nitrosobenzene and phenylhydroxylamine over Au/TiO2. Therein, the hydrogenation of nitrosobenzene was so fast that it was not observed either in the liquid phase or on the catalyst surface;19 however, adsorbed phenylhydroxylamine was proposed as an adsorbed species. Hydrogenation of either nitrosobenzene or phenyl hydroxylamine formed azoxybenzene and with nitrosobenzene poisoning the catalyst the subsequent reaction pathway depended on the route taken to form azoxybenzene. Currently, as the reaction mechanism is still unclear, it cannot be said whether this reaction occurs via different routes over different metals, supports or under different experimental conditions.
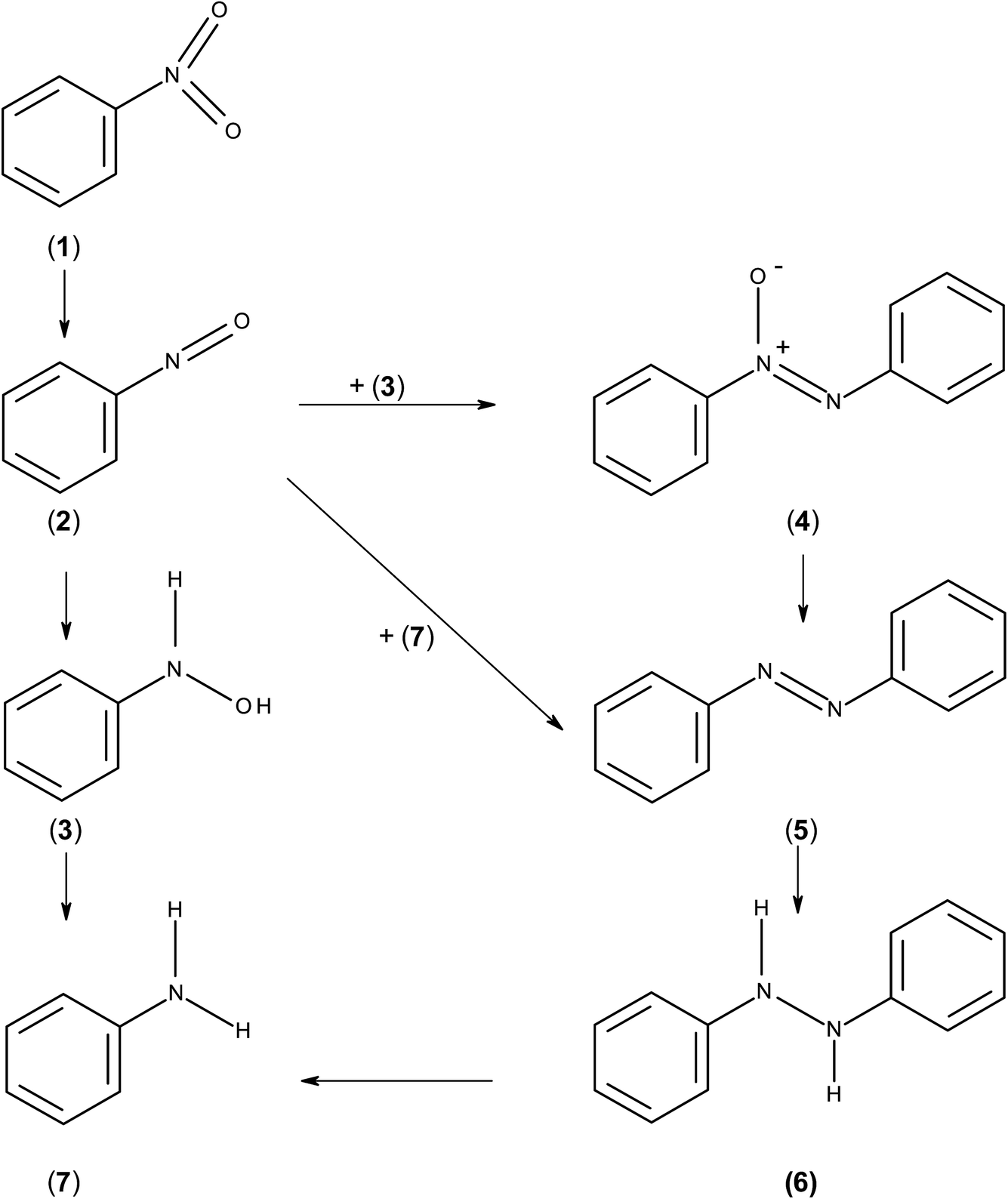 | ||
| Scheme 1 Mechanism of hydrogenation of nitrobenzene as proposed by Haber. | ||
Octahedral molecular sieves (OMS) are porous materials based on linked MnO6 octahedra which utilise the redox behaviour of manganese for use in a variety of applications including catalysis, adsorption20 and in batteries.21 These materials can be prepared with varying pore size22 and OMS-2 with a pore diameter of 4.6 Å has been used extensively as a catalyst23 predominantly for oxidation reactions in the liquid24 and gas phase.25 Although most reports are concerned with oxidation catalysis, OMS-2 and metals supported on OMS-2 have also been used in a number of liquid phase carbonyl hydrogenation reactions including acetophenone,26 cinnamaldehyde and ketoisophorone.27 A recent report on the formation of azobenzene from nitrobenzene over a 15% Ag–OMS-2 catalyst with aniline as a minor product28 shows that the hydrogenation of a nitro group is also possible with this catalyst.
Herein, we report the hydrogenation of chloronitrobenzene over OMS-2, Pt/OMS-2 and Pd/OMS-2. A study into the hydrogenation of nitrobenzene over these catalysts has provided insight into the mechanism of the reaction over the OMS-2 catalysts.
Experimental
Materials
Nitrobenzene (99%), nitrosobenzene (97%), azoxybenzene, azobenzene (98%), hydrazobenzene, phenylhydroxylamine (97%), 4-chloronitrobenzene (99%), 4-chloroaniline (98%), 4-chlorobenzaldehyde (97%), 4-chlorobenzylalcohol (99%), 4-iodonitrobenzene (98%), 4-iodoaniline (98%), 4-iodotoluene (99%), methanol (99.9%) and 1-butanol (99.7%) were all obtained from Sigma Aldrich. Potassium permanganate (≥99%) was obtained from Fluka as was maleic acid (≥98%). Metal precursors used were platinum nitrate (assay 15.14%) supplied by Johnson Matthey and palladium(II)nitrate hydrate procured from Sigma Aldrich. Research grade hydrogen was supplied by BOC. All chemicals were used as supplied.Catalyst preparation
OMS-2 was synthesized via a sol–gel method following the procedure reported by Genuino et al.25 A 5 wt% Pt/OMS-2 catalyst was prepared by incipient wetness impregnation using aqueous platinum nitrate as the metal precursor. After impregnation, the material was dried at 120 °C for 12 h followed by calcination at 450 °C in air for 4 h. The 5 wt% Pd/OMS-2 catalyst was prepared by wet impregnation using palladium nitrate in methanol as the metal precursor at room temperature under agitation for 24 h. The catalyst was dried by removing methanol in vacuo and then the material was calcined in air at 450 °C for 4 h. All catalysts were ground and sieved to a less than 45 μm particle size for use in liquid phase hydrogenation reactions. 5 wt% Pt/Al2O3 (code 125) was obtained from Johnson Matthey and used as received.Catalyst characterization
X-ray diffraction measurements were performed using Cu Kα radiation (1.5405 Å) on a PANalytical X'PERT PRO MPD diffractometer in reflection geometry using a NaI scintillation counter, a curved graphite crystal monochromator and a nickel filter. The scattered intensities were collected from 5° to 90° (2θ) by scanning at 0.017° (2θ) steps with a counting time of 0.5 s at each step.The surface area, pore volume and average pore diameter were measured by N2 adsorption–desorption isotherms at 77 K using Micromeritics ASAP 2010. The pore size was calculated in the adsorption branch of the isotherms using the Barrett–Joyner–Halenda (BJH) method and the surface area was calculated using the Brunauer–Emmett–Teller (BET) method.
The metal content of samples of OMS-2 and metal supported on OMS-2 was determined by ICP-OES using a PE Optima spectrometer.
The transmission electron microscopy (TEM) studies were performed on a JEOL 2010-FEG instrument, at 100 kV. Platinum particle size distributions were obtained by counting up to 100 particles.
XPS spectra were obtained using a Kratos AXIS Ultra DLD XPS spectrometer with monochromated Al Kα X-rays and a hemispherical analyser with a pass energy of 160 eV. The powdered samples were mounted on conducting copper tape and the binding energies were normalized to the C 1s signal from adventitious carbon at 284.6 eV. Background subtraction was performed using a Shirley background and CasaXPS.29
Typical reaction sequence
All reactions were performed in a Hazard Evaluation Laboratory (HEL) autoclave reactor incorporating a gas inducing impeller, baffles and online hydrogen consumption monitoring. The reactor was heated by an internal heating coil and the temperature was monitored by a thermocouple. Hydrogen pressure was maintained throughout the reaction by addition through a mass flow controller.The reactor was charged with a catalyst (0.03 g) and methanol (30 cm3) and the reactor was sealed and purged with hydrogen before heating to 60 °C while stirring at 800 rpm. Upon reaching 60 °C the catalyst was reduced by stirring at 1000 rpm for 1 h under 1 bar H2. The substrate (0.002 mol), 1-butanol (0.4 cm3) as the internal standard and fresh solvent (30 cm3) were then added to the reactor which was sealed, purged with hydrogen and heated to the reaction temperature, with stirring at 800 rpm. When the required temperature was reached the stirring was stopped and the reactor pressurised to the required pressure for the reaction which was started by commencing the stirring at 1400 rpm. Reaction samples (2 cm3) were removed over time, passed through a syringe filter to remove the catalyst and analyzed by GC using a Perkin-Elmer Clarus 500 GC equipped with a FID and an Agilent HP-5 column. For the chloronitrobenzene and halotoluene reactions the products were identified by GC/MS analysis and those for the nitrobenzene reactions were compared with known standards. It was not possible to separate the pure samples of azobenzene and hydrazobenzene using this method on the GC.
Results and discussion
Catalyst characterization
N2 adsorption–desorption isotherms of OMS-2, Pt/OMS-2 and Pd/OMS-2 catalysts showed a type IV behaviour with a hysteresis loop of type H3, which is characteristic of aggregates of particles with rod-like morphology with tunnel or slit type pores.30 The BET surface area for OMS-2 was 59.0 m2 g−1 and on addition of metal, the surface area of the catalyst decreased to 43.2 m2 g−1 for Pt/OMS-2 and 43.9 m2 g−1 for Pd/OMS-2. The surface textural properties of the catalysts used are shown in Table S1.†The XRD patterns obtained for Pt/OMS-2 and Pd/OMS-2 only show peaks assigned to the OMS-2 support (Fig. S1†). The diffractograms show that the material was a pure cryptomelane phase with no impurities of other manganese oxide forms. No peaks due to Pd or Pt were observed by XRD suggesting Pd and Pt particles of less than 4 nm in diameter were present. This is consistent with the TEM characterisation of the Pt/OMS-2 catalyst (Fig. S2†) which shows uniformly distributed spherical Pt nanoparticles with a mean diameter of ∼ 2 nm and the OMS-2 molecular sieves having a rod like morphology.
Hydrogenation of chloronitrobenzene and halogenated reagents
It is desirable for the halogen atom to be retained in the product following hydrogenation giving a high selectivity to halogenated products as well as minimising catalyst poisoning from strong adsorption of halogens on the active metal sites. Fig. 1 shows the selectivity to halogenated products for the hydrogenation of chloronitrobenzene over OMS-2, Pt/OMS-2 and Pd/OMS-2. The reaction over OMS-2 formed chloroaniline as the major product with selectivities of >96% over the temperature range of 25–100 °C. Similarly, high selectivities to halogenated products were observed over Pt/OMS-2 while for a commercial 5% Pt/Al2O3 catalyst, the selectivity to halogenated products was reduced to 89% for a reaction at 25 °C and further decreased with increasing temperature. At elevated reaction temperatures over OMS-2 and Pt/OMS-2, no change in selectivity was observed with selectivities to halogenated products of 99% observed at 100 °C over both catalysts.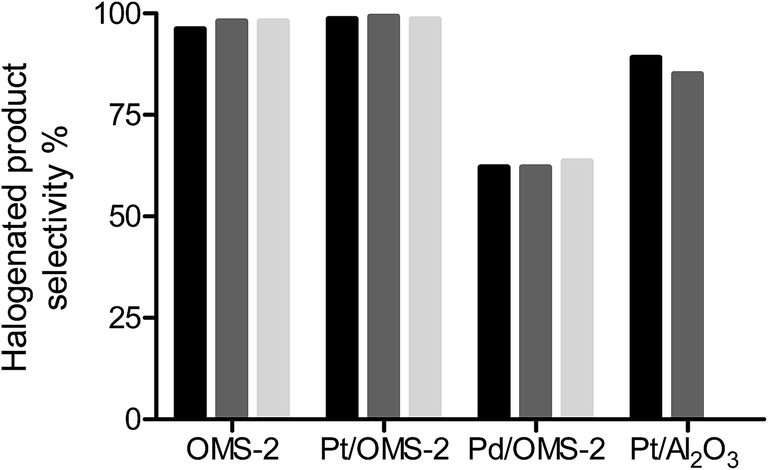 | ||
| Fig. 1 % selectivity to halogenated products for the hydrogenation of chloronitrobenzene over OMS-2, Pt/OMS-2, Pd/OMS-2 and Pt/Al2O3 at 2 bar H2 and reaction temperatures of 25 °C (black), 50 °C (mid grey) and 100 °C (light grey). | ||
In contrast, significant dehalogenation of chloronitrobenzene was observed over Pd/OMS-2 where 62% selectivity to all halogenated products was observed again with little variation on increasing temperature. At low chloronitrobenzene conversions (<20%) the selectivity was the highest to nitrobenzene (38.6%) and aniline (37.8%) with a selectivity to chloroaniline of only 8.5%. Therefore, the initial reaction occurring on Pd/OMS-2 is rapid dehalogenation and subsequent hydrogenation of nitrobenzene to aniline. As the reaction proceeded to complete conversion of chloronitrobenzene, the selectivity to chloroaniline increased to 30.3% while the selectivity to aniline decreased to 30.4% as a result of reduction of the aromatic ring. The increase in chloroaniline selectivity suggests that the catalyst is either modified by the presence of chlorine or the strong adsorption of the products on the surface which alters the adsorption geometry of chloronitrobenzene. Alternatively, this adsorption could result in a poisoning of the metal which forces the hydrogenation to occur on the OMS-2 support leading to lower hydro-dehalogenation occurring. The further hydrogenation of aniline through to cyclohexane has been previously reported for hydrogenation of chloronitrobenzene over Au and Pd supported on alumina catalysts16 and selectivity to these products is observed to increase as aniline reacts further over Pd/OMS-2.
To determine the extent to which the halogen is stable under hydrogenation conditions, a range of substrates were tested under different reaction conditions. It is known that iodo-based substrates are less stable to dehalogenation than the analogous chloro molecules, therefore, iodonitrobenzene was tested over OMS-2 under the same reaction conditions as chloronitrobenzene. Here the selectivity to halogenated products decreased to 62% as expected from the relative carbon–halogen bond strengths. Fig. 2 shows that, at low conversions, there was an initially rapid dehalogenation to produce up to 77% selectivity to nitrobenzene. However, as the conversion increased there was no further dehalogenation and the selective hydrogenation of iodonitrobenzene occurred to give a selectivity of 38.5% iodoaniline. It is proposed that iodide formed from dehalogenation adsorbed strongly on the sites responsible for dehalogenation thereby poisoning them to further reaction or modifies the adsorption geometry for the substrate promoting nitro group reduction vs. hydro-dehalogenation. In contrast, Fig. 3 shows that the extent of dehalogenation of iodotoluene was again high over the palladium catalyst whilst, even with this easily dehalogenated substrate at elevated temperatures of 100 °C, there was negligible reaction over either OMS-2 or Pt/OMS-2 with only 3.4% conversion observed after 3 h.
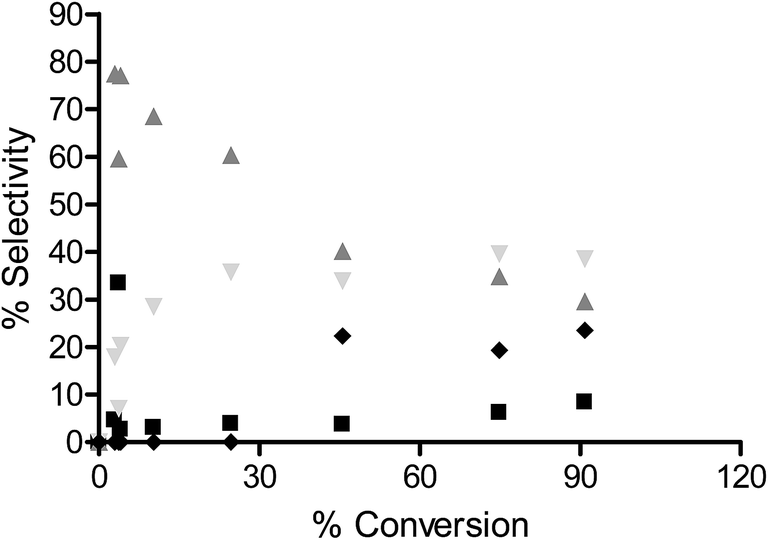 | ||
Fig. 2 % selectivity as a function of % conversion for hydrogenation of iodo-nitrobenzene (0.002 mol) to nitrobenzene over OMS-2 at 2 bar H2 and 25 °C in 60 ml methanol; nitrobenzene ( ), aniline ( ), aniline ( ), iodoaniline ( ), iodoaniline ( ) and coupling products ( ) and coupling products ( ). ). | ||
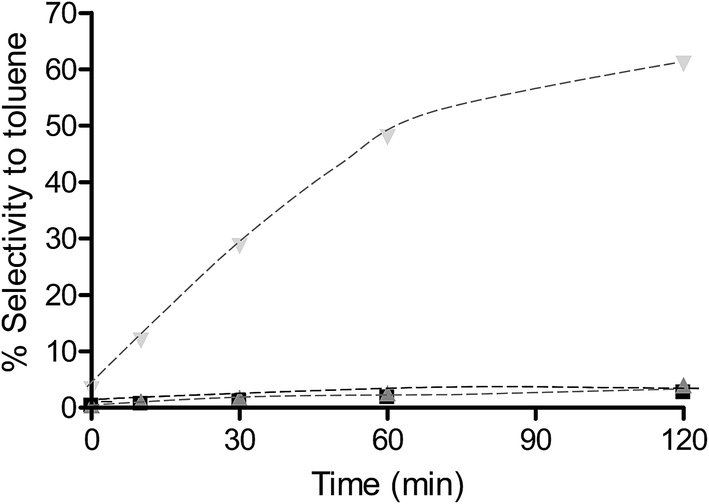 | ||
Fig. 3 % selectivity to toluene for reaction of iodotoluene over OMS-2 ( ), Pt/OMS-2 ( ), Pt/OMS-2 ( ) and Pd/OMS-2 ( ) and Pd/OMS-2 ( ). Reactions were carried out using 0.1 g catalyst and 0.002 mol iodotoluene at 2 bar H2 and 100 °C in 60 cm3 methanol. ). Reactions were carried out using 0.1 g catalyst and 0.002 mol iodotoluene at 2 bar H2 and 100 °C in 60 cm3 methanol. | ||
To exemplify the system further, hydrogenation of a halogenated carbonyl compound was tested using chlorobenzaldehyde. The results given in Fig. S3† show that for OMS-2 and Pt/OMS-2 there was less than 1% dehalogenation with the major product being chlorobenzyl alcohol. However, when the reaction was carried out over Pd/OMS-2, a rapid reaction occurred to form chlorobenzyl alcohol (58.9% selectivity to chlorobenzyl alcohol at 10% conversion of chlorobenzaldehyde) with the major products at the complete conversion of chlorobenzaldehyde found to be dechlorinated benzaldehyde (50.0%) and toluene (38.9%). In the case of Pt/Al2O3, the reaction profile was initially very different to the reaction over any of the OMS-2 supported catalysts. In the case of the alumina supported catalyst, half of the substrate formed an acetal with the methanol solvent before the first sample could be taken. However, there was no dehalogenation with the acetal and chlorobenzaldehyde with both undergoing hydrogenation to a chlorobenzyl alcohol and a small amount of di-chlorobenzyl ether (3%), which was probably formed through the reaction between chlorobenzaldehyde and chlorobenzyl alcohol.
In general, the selectivity observed over OMS-2 and Pt/OMS-2 is the same for all reagents although the rate of reaction is typically faster over the platinum containing catalyst. The rate of reaction is much faster over Pd/OMS-2 with, in general, significantly different selectivities being observed compared with that of OMS-2 and Pt/OMS-2 which is mainly associated with dehalogenation reactions. The similarity in the selectivity for OMS-2 and Pt/OMS-2 indicates that the reaction over the Pt containing catalyst may be occurring over the support rather than the metal. The faster rate of reaction is found due to the more efficient dissociation of hydrogen over the metal which can then spillover onto the support, as reported previously for the hydrogenation of unsaturated aldehydes and ketones.30 A comparison of the rates of reaction and significant differences in selectivity of the two platinum catalysts supported on OMS-2 and alumina is also consistent with this proposal. In the case of Pd/OMS-2, the chloronitrobenzene is converted significantly faster than over Pt/OMS-2 or OMS-2 with a different selectivity implying that the reaction occurs over the metal rather than the support. The higher rate of dehalogenation is consistent with the reported ability of palladium to facilitate this reaction.
This study shows the applicability of OMS-2 either on its own or as a support for platinum for the selective hydrogenation of halogenated aromatic compounds such as chloronitrobenzene and chlorobenzaldehyde. This is in contrast to other supports such as alumina where it has been found that little hydrogenation occurs in the absence of a metal.31 Although the presence of platinum allows for a faster reaction, the addition of higher catalyst loading of inexpensive OMS-2 would be preferable.
XPS of catalysts after reaction: hydrogenation of chloronitrobenzene
XPS spectra of OMS-2, Pt/OMS-2 and Pd/OMS-2 fresh and used catalysts are shown in Fig. 4. The Mn 2p3/2 photoelectron peak is positioned at 641.7 eV which can be assigned to the Mn4+ state (Fig. 4a). The O 1s photoelectron peak was composed of two features (Oα and Oβ) indicating the presence of oxygen species in a mixed valence state (Fig. 4b). The peak positioned at a binding energy of 529.2 eV is characteristic of lattice oxygen (O2−) and the peak positioned at 532.2 eV could be assigned to the surface oxygen ions with lower valence or in the form of hydroxyl, as shown in ref. 32 and 33. The corresponding concentration of Oα calculated from the relative peak areas [Oα × 100/(Oα + Oβ)] indicated that OMS-2 has a ∼16% lattice oxygen concentration which can be depleted and replenished with ease; however, the lattice oxygen concentration increases in the presence of metal to ∼33% for Pd/OMS-2 and ∼47% for Pt/OMS-2. Fig. 4c shows the peak fit of Pt 4f photoelectron spectral lines. In the case of the fresh Pt/OMS-2 catalyst, the 4f7/2 peak positioned at the binding energy of 74.8 eV was assigned to PtO2. In the case of the used Pt/OMS-2 catalyst, the 4f7/2 feature is shifted to a lower binding energy with 2 components fitted at 71.9 and 73.6 eV. The 4f7/2 peak positioned at the binding energy of 71.9 eV could be assigned to PtO and the 4f7/2 peak positioned at the binding energy of 73.6 eV could be assigned to PtO sites with high oxygen concentration.34 In fresh Pt/OMS-2, Pt metal is present as PtO2 on the catalyst surface resulting from calcination in air, but under the experimental conditions at 373 K in the presence of H2 1 MPa, Pt is reduced to Pt0 metal. In our previous work, we have shown that in situ pre-reduction of a catalyst in the liquid phase is a very effective technique to reduce Pt to its metallic state as evident from the Pt LIII edge HERFD-XANES spectrum of the catalyst.35Fig. 4d shows the peak fit of Pd 3d spectral lines. The 3d5/2 peak, positioned at a binding energy of 337.7 eV was assigned to PdO, while a small component at the binding energy of 335.3 eV can be assigned to the Pd0 state. In the case of the used Pd/OMS-2 catalyst, a Pd 3d5/2 peak was observed in a similar position to that of the fresh Pd/OMS-2 catalyst, positioned at a binding energy of 337.9 eV, but with significantly enhanced peak areas indicating a higher surface coverage of Pd nanoparticles, possibly due to redispersion of the Pd metal on the catalyst support. Increased Pd nanoparticle dispersion is also indicated from the decrease in the peak areas of Mn 2p3/2 features in the used Pd/OMS-2 catalysts in comparison to the fresh Pd/OMS-2 catalyst (Fig. 4a). This has been observed previously for supported Au catalysts on exposure to halogenated molecules.36 Therein, Au catalysts supported on a range of carbon and oxide supports showed the formation of dimers and trimers of gold stabilised by the halogen from large metal nanoparticles.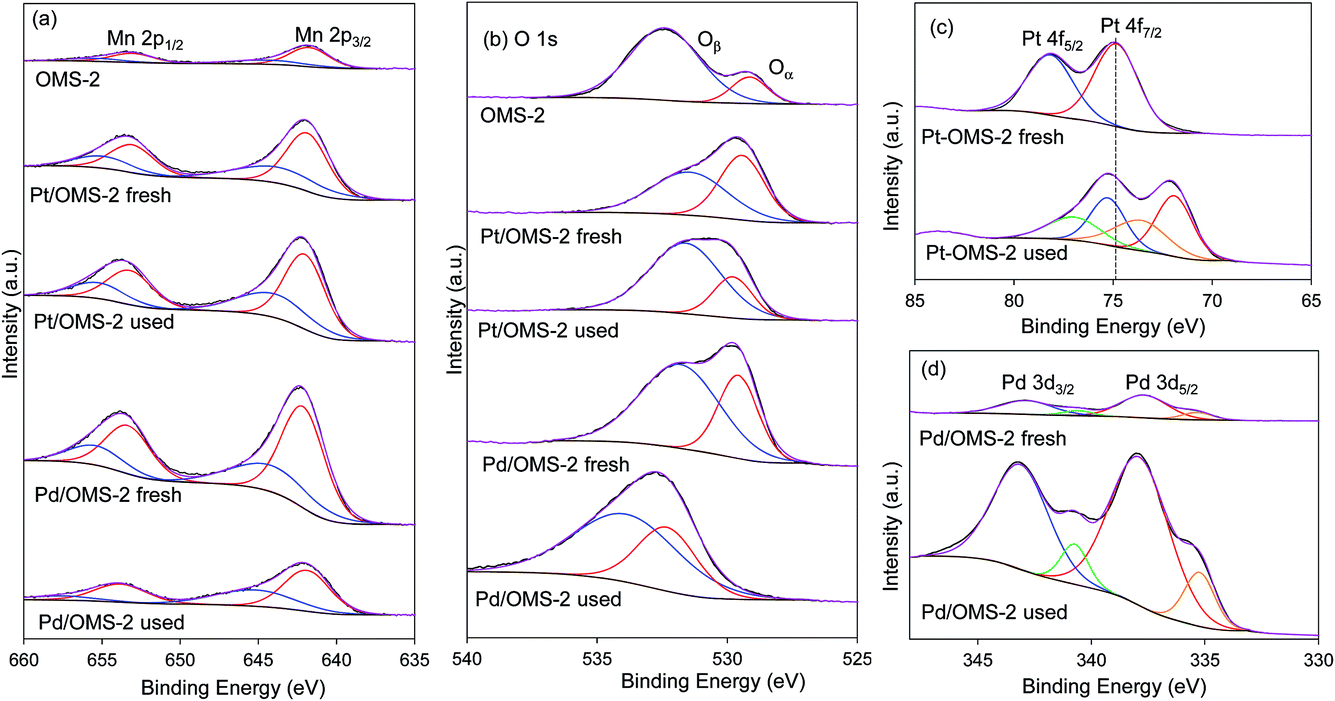 | ||
| Fig. 4 XPS of fresh and used Pt and Pd/OMS-2 catalysts for the hydrogenation of chloronitrobenzene at 25 °C and 2 bar H2. | ||
Mechanistic study
As the hydrogenation of chloronitrobenzene over OMS-2 and Pt/OMS-2 resulted in a large number of intermediate products which were not available commercially, the mechanistic study of this reaction was carried out using nitrobenzene. This was considered appropriate as negligible dehalogenation occurred over these catalysts meaning the halogen did not influence surface adsorption or catalysis.Hydrogenation of nitrobenzene
The reaction profiles for the hydrogenation of nitrobenzene over OMS-2, Pt/OMS-2 and Pd/OMS-2 at 25 °C and 2 bar H2 are shown in Fig. 5. The reaction rate is significantly faster over Pd/OMS-2 compared to Pt/OMS-2 and OMS-2 and, as found in the hydrogenation of chloronitrobenzene, the rate of reaction over Pt/OMS-2 is greater than the OMS-2 support. The major product for all catalysts is aniline. Over Pd/OMS-2, the only additional product identified in the liquid phase is nitrosobenzene. In contrast, for reactions over OMS-2 and Pt/OMS-2, coupling products were also observed. In each of these catalysts similar reaction profiles were found with aniline as well as azoxybenzene, and azo/hydrazobenzene and nitrosobenzene being formed. The similarity in the reaction profiles and selectivities for Pt/OMS-2 and OMS-2 again suggests that, as for hydrogenation of the halogenated reagents, the reaction occurs predominantly over OMS-2 while for the Pd catalyst, the reaction occurs predominantly on the metal. This switch in the site for hydrogenation has been suggested to be due to the relative adsorption strengths of the substrate and hydrogen on the metals and the support.37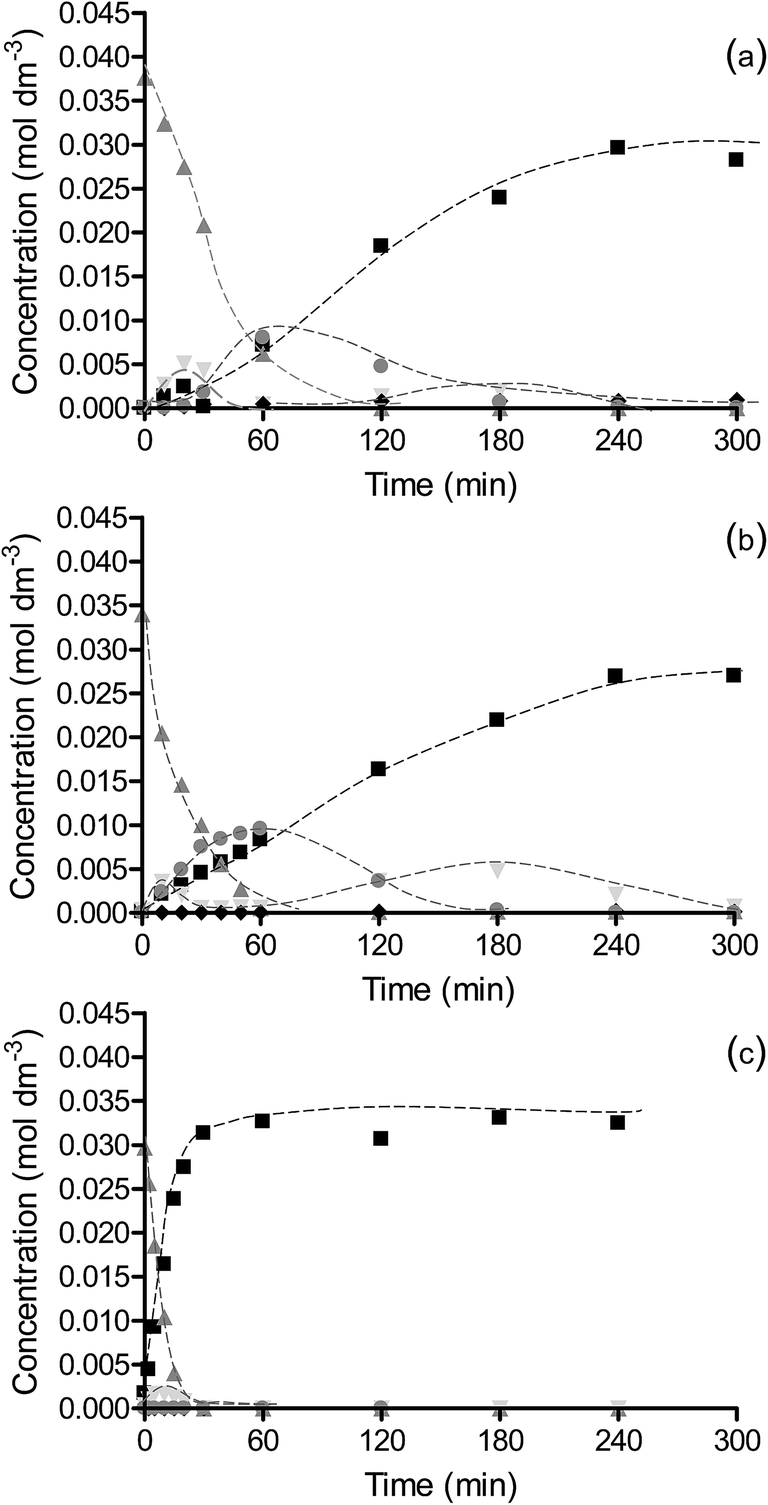 | ||
Fig. 5 Reaction profiles for (a) OMS-2, (b) Pt/OMS-2 and (c) Pd/OMS-2 for hydrogenation of nitrobenzene. Reactions were carried out using 0.03 g catalyst and 0.002 mol nitrobenzene at 2 bar H2 and 25 °C in 60 cm3 methanol. Nitrobenzene ( ), nitrosobenzene ( ), nitrosobenzene ( ), aniline ( ), aniline ( ), azoxybenzene ( ), azoxybenzene ( ) and azo/hydrazobenzene ( ) and azo/hydrazobenzene ( ). ). | ||
The differing reaction rates and product distribution suggest that the hydrogenation of nitrobenzene could be occurring through different pathways on Pd compared to the OMS-2 and Pt/OMS-2 catalysts. The two pathways are thought to be direct hydrogenation or hydrogenation via the formation of coupling products with the predominant route changing with the extent of conversion. At low conversions of nitrobenzene (∼10%), the only products observed in the reaction profiles for OMS-2, Pt/OMS-2 and Pd/OMS-2 were nitrosobenzene and aniline. No coupling products were observed at this conversion suggesting that at a high initial surface coverage of nitrobenzene, the direct hydrogenation of nitrobenzene through nitrosobenzene and phenylhydroxylamine to aniline is the major pathway. The selectivity to aniline is higher for Pd at 85.6% and comparable over OMS-2 and Pt/OMS-2 at 59.0% and 63.5%, respectively, suggesting a faster conversion of nitrosobenzene/phenylhydroxylamine intermediates to aniline over the Pd catalyst. As the conversion of nitrobenzene increased to 40%, the aniline selectivity over Pd/OMS-2 increased to 88.3%. In contrast, over Pt/OMS-2 and OMS-2, the coupled product azoxybenzene was formed with azo/hydrazobenzene also being observed for the support alone, giving selectivities to aniline of only 19.7% and 19.1%, respectively.
The formation of coupling products occurs by condensation of either two nitrosobenzene molecules or phenylhydroxylamine and nitrosobenzene. These processes are known to be dependent on the concentration of these species on the catalyst surface with higher concentrations enhancing the coupling reaction. The higher selectivity to nitrosobenzene and coupled products over OMS-2 suggests that this hydrogenation pathway to aniline is occurring on OMS-2. The selectivity at low nitrobenzene conversions suggests that direct hydrogenation can occur over all the catalysts but that the slower rate over OMS-2 and Pt/OMS-2 increases the surface concentration of intermediates, for example nitrosobenzene and phenylhydroxylamine, which can then go through the coupling hydrogenation route. Over palladium the reaction always proceeds through the direct hydrogenation route due to the reaction occurring rapidly on the metal. This is in contrast to the hydrogenation of chloronitrobenzene over Pd/OMS-2 where some coupling products were also observed, albeit at small levels (∼7% selectivity). In this case, as discussed previously, the rapid dehalogenation of chloronitrobenzene could result in chloride poisoning of the metal sites with hydrogenation, without dehalogenation, then occurring on the OMS-2 support through a coupling pathway as observed on OMS-2 alone.
Hydrogenation of intermediate products
Reaction of the intermediates nitrosobenzene, hydrazobenzene and azoxybenzene was studied to probe the rate and selectivity of the reactions over OMS-2 (Fig. 6). The reaction over Pt/OMS-2 and Pd/OMS-2 of the intermediates nitrosobenzene and hydrazobenzene are shown in Fig. S4 and S5,† respectively. The hydrogenation of nitrosobenzene, where initial concentrations are much higher than those observed during the hydrogenation of nitrobenzene, shows similar trends to that observed during nitrobenzene hydrogenation. Despite the higher concentration of nitrosobenzene, very little conversion to azoxybenzene was observed over Pd/OMS-2 but rather a rapid complete conversion to aniline. In contrast, the reaction of nitrosobenzene over OMS-2 and Pt/OMS-2 initially showed a fast reaction with high selectivity to azoxybenzene; however, the hydrogenation of nitrosobenzene slowed greatly and complete conversion of nitrosobenzene was not observed for Pt/OMS-2 or OMS-2. The main product initially was azoxybenzene which then formed azo/hydrazobenzene together with a slow formation of aniline. As expected, the Pt/OMS-2 catalyst showed faster hydrogenation of all intermediate products than the OMS-2 support. The main difference between the nitrobenzene and nitrosobenzene reaction profiles is the increased amount of coupled products formed in the latter case, due to the increased concentration of nitrosobenzene on the surface. Reaction of hydrazobenzene, while selectively only forming aniline, was slow and all catalysts were showed to deactivate (Fig. 6, S4 and S5†). This is in line with the slow formation of aniline through the coupled products route observed during hydrogenation of nitrobenzene and nitrosobenzene over OMS-2 and Pt/OMS-2 discussed previously.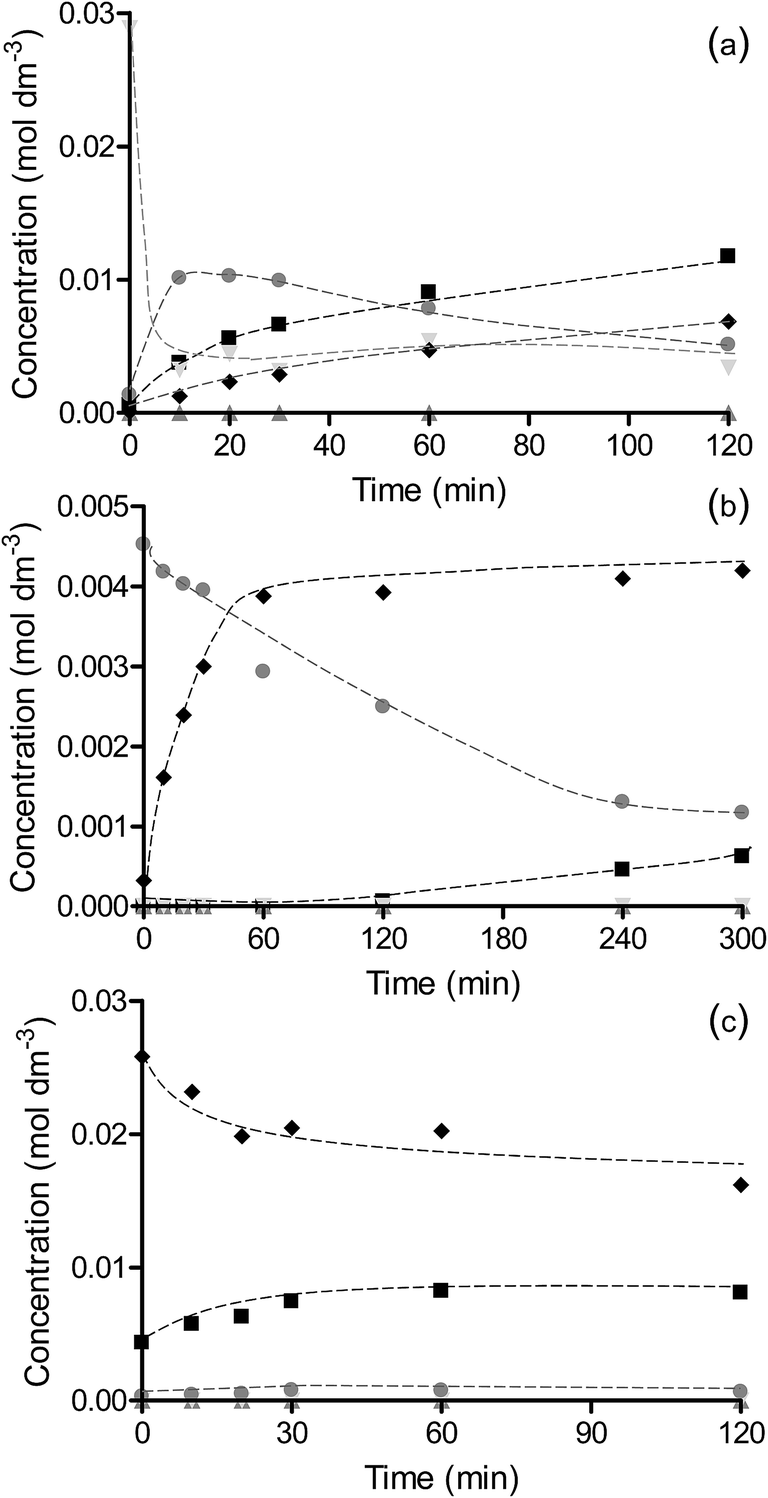 | ||
Fig. 6 Hydrogenation of reaction intermediates (a) nitrosobenzene, (b) azoxybenzene and (c) hydrazobenzene over OMS-2. Reaction of nitrosobenzene was carried out using 0.03 g catalyst and 0.002 mol nitrosobenzene at 2 bar H2 and 25 °C in 60 cm3 methanol. Reaction of azoxybenzene was carried out using 0.01 g catalyst and 0.0003 mol nitrobenzene at 2 bar H2 and 25 °C in 60 cm3 methanol. Reaction of hydrazobenzene was carried out using 0.03 g catalyst and 0.002 mol hydrazobenzene at 2 bar H2 and 40 °C in 60 cm3 methanol. Nitrosobenzene ( ), azo/hydrazobenzene ( ), azo/hydrazobenzene ( ), azoxybenzene ( ), azoxybenzene ( ) and aniline ( ) and aniline ( ). ). | ||
To further probe the reaction mechanism, reactions were carried out at higher temperatures. As previously described, at low temperatures, the reaction selectivity at low conversion of nitrobenzene (∼10%) followed direct hydrogenation to aniline with only aniline and nitrosobenzene products observed in the liquid phase. At higher conversions of nitrobenzene, the nitrosobenzene concentration increased and, due to the slow hydrogenation of this intermediate and/or poisoning of the catalyst, coupling occurs leading to the much slower hydrogenation route to aniline.
The rate of nitrobenzene hydrogenation over OMS-2 increases with increasing reaction temperature and interestingly, a change in the selectivity is also observed (Fig. 7 and S6†). At temperatures between 25 and 50 °C there is an initial formation of nitrosobenzene and a concomitant increase in coupled products as the concentration of nitrosobenzene increases. As the temperature of the reaction over OMS-2 is increased to 65 °C, there is significantly less azoxybenzene formed (Fig. 7). Increasing the reaction temperature results in a shift in the reaction pathway favouring the direct hydrogenation pathway over OMS-2. This is likely due to weaker adsorption of nitrosobenzene/an increased rate of hydrogenation of nitrosobenzene and hence lower surface concentration and reduced propensity to undergo coupling.
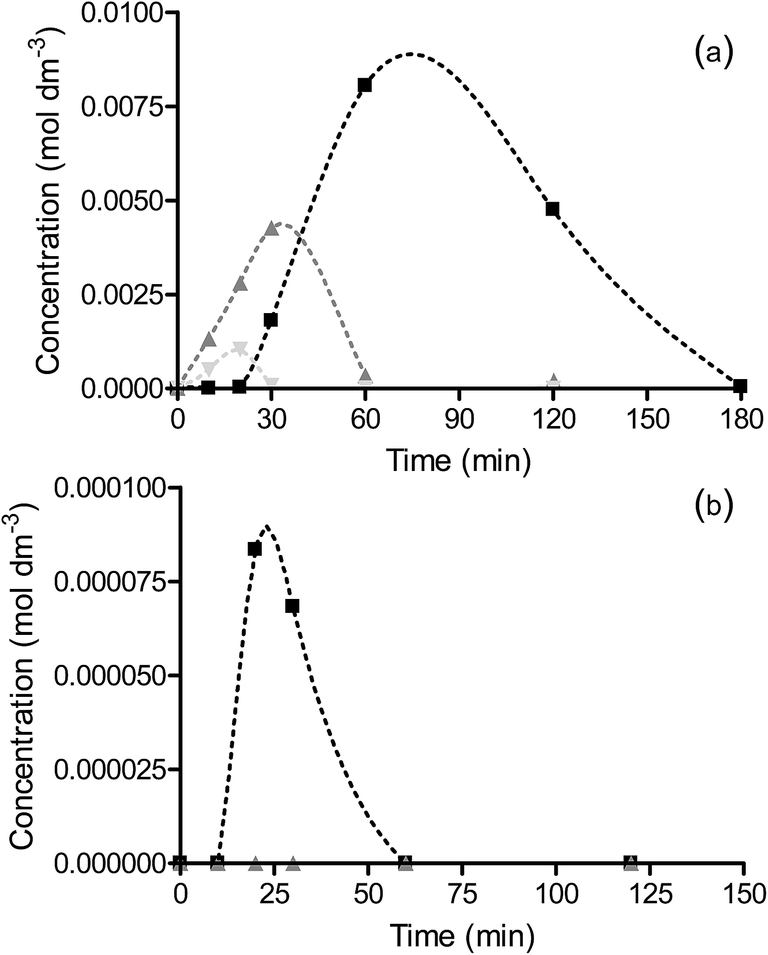 | ||
Fig. 7 Concentration of azoxybenzene formed during hydrogenation of nitrobenzene at 25 °C ( ), 50 °C ( ), 50 °C ( ) and 65 °C ( ) and 65 °C ( ) for (a) OMS-2 and (b) Pd/OMS-2. Reactions were carried out using 0.03 g catalyst and 0.002 mol nitrobenzene at 2 bar H2 and 25 °C in 60 cm3 methanol. ) for (a) OMS-2 and (b) Pd/OMS-2. Reactions were carried out using 0.03 g catalyst and 0.002 mol nitrobenzene at 2 bar H2 and 25 °C in 60 cm3 methanol. | ||
For reactions over Pt/OMS-2, the same trends are observed as for OMS-2 with the enhancement in direct hydrogenation which occurs at a lower temperature than for OMS-2, at 50 °C rather than 65 °C. The hydrogenation of nitrobenzene over Pd/OMS-2 at 65 °C compared to 25 °C shows a small increase in the reaction rate but no change in the product distribution with only nitrosobenzene and aniline formed with lower selectivity to nitrosobenzene with the increase in reaction temperature.
Conclusions
This study has shown the inherent hydrogenation activity of OMS-2 both on its own as well as a support for platinum group metals. In the case of OMS-2, little hydro-dehalogenation was found, providing a route for non-precious metal based catalytic materials. The rate of the reaction was enhanced by the incorporation of platinum on the surface which provided a more facile pathway for dissociative H2 adsorption with little change in the selectivity. In contrast, with the use of palladium supported on OMS-2, the reaction proceeds on the metal leading to significant dehalogenated products. The mechanism has been further probed using nitrobenzene as the exemplar molecule. In this case, over Pd/OMS-2 the direct hydrogenation pathway to aniline was preferred with few coupled products being formed. In contrast, over OMS-2 and Pt/OMS-2, both the direct and indirect (via the coupled products) pathways were found again consistent with the role of OMS-2 as the hydrogenation site in these catalysts.Acknowledgements
We acknowledge EPSRC for funding as part of the CASTech grant (EP/G011397/1) and the Department of Employment and Learning for a studentship (IM). Supporting data are openly available on Queen’s University, Belfast Research Portal http://pure.qub.ac.uk/portal/en/datasets.References
- H. Blaser, H. Steiner and M. Studer, ChemCatChem, 2009, 1, 210–221 CrossRef CAS.
- V. Kratky, M. Kralik, M. Mecarova, M. Stolcova, L. Zalibera and M. Hronec, Appl. Catal., A, 2002, 235, 225–231 CrossRef CAS.
- M. Liu, W. Yu and H. Liu, J. Mol. Catal. A: Chem., 1999, 138, 295–303 CrossRef CAS.
- I. Pogorelic, M. Filipan-Litvic, S. Merkas, G. Ljubic, I. Cepanec and M. Litvic, J. Mol. Catal. A: Chem., 2007, 274, 202–207 CrossRef CAS.
- S. Ichikawa, T. Seki and T. Ikariyaa, Adv. Synth. Catal., 2014, 356, 2643–2652 CrossRef CAS.
- S. Piña Jr, D. M. Cedillo, C. Tamez, N. Izquierdo, J. G. Parsons and J. J. Gutierrez, Tetrahedron Lett., 2014, 55, 5468–5470 CrossRef.
- J. Lyu, J. Wang, C. Lu, L. Ma, Q. Zhang, X. He and X. Li, J. Phys. Chem. C, 2014, 118, 2594–2601 CAS.
- M. A. Keane, ChemCatChem, 2011, 3, 800–821 CrossRef CAS.
- K. C. Christoforidis, E. Serestatidou, M. Louloudi, I. K. Konstantinou, E. R. Milaeva and Y. Deligiannakis, Appl. Catal., B, 2011, 101, 417–424 CrossRef CAS.
- C. M. King, R. B. King, N. K. Bhattacharyya and M. G. Newton, J. Organomet. Chem., 2000, 600, 63–70 CrossRef CAS.
- A. Scrivanti, B. Vicentini, V. Beghetto, G. Chessa and U. Matteoli, Inorg. Chem. Commun., 1998, 1, 246–248 CrossRef CAS.
- D. T. Ferrughelli and l. T. Horvath, J. Chem. Soc., Chem. Commun., 1992, 806–807 RSC.
- H. Min, S. Lee, M. Park, J. Hwang, H. M. Jung and S. Lee, J. Organomet. Chem., 2014, 755, 7–11 CrossRef CAS.
- R. Baumgartner, G. K. Stieger and K. McNeill, Environ. Sci. Technol., 2013, 47, 6545–6553 CAS.
- Y. Ukisu and T. Miyadera, J. Mol. Catal. A: Chem., 1997, 125, 135–142 CrossRef CAS.
- X. Wang, N. Perret and M. A. Keane, Chem. Eng. J., 2012, 210, 103–113 CrossRef CAS.
- F. Haber, Z. Elektrochem. Angew. Phys. Chem., 1898, 22, 506 Search PubMed.
- E. A. Gelder, S. D. Jackson and C. M. Lok, Chem. Commun., 2005, 522–524 RSC.
- G. Richner, J. A. van Bokhoven, Y. M. Neuhold, M. Makosch and K. Hungerbühler, Phys. Chem. Chem. Phys., 2011, 13, 12463–12471 RSC.
- P. H. Ho, S. C. Lee, J. Kim, D. Lee and H. C. Woo, Fuel Process. Technol., 2015, 131, 238–246 CrossRef CAS.
- W. K. Pang, V. K. Peterson, N. Sharma, C. Zhang and Z. Guo, J. Phys. Chem. C, 2014, 118, 3976–3983 CAS.
- N. Duan, S. L. Suib and C. O'Young, J. Chem. Soc., Chem. Commun., 1995, 1367–1368 RSC.
- C.-H. Chen and S. L. Suib, J. Chin. Chem. Soc., 2012, 59, 465–472 CrossRef CAS.
- V. D. Makwana, Y.-C. Son, A. R. Howell and S. L. Suib, J. Catal., 2002, 210, 46–52 CrossRef CAS.
- H. C. Genuino, S. Dharmarathna, E. C. Njagi, M. C. Mei and S. L. Suib, J. Phys. Chem. C, 2012, 116, 12066–12078 CAS.
- G. D. Yadav and R. K. Mewada, Catal. Today, 2012, 198, 330–337 CrossRef CAS.
- H. G. Manyar, B. Yang, H. Daly, H. Moor, S. McMonagle, Y. Tao, G. D. Yadav, A. Goguet, P. Hu and C. Hardacre, ChemCatChem, 2013, 5, 506–512 CrossRef CAS.
- G. D. Yadav and R. K. Mewada, Chem. Eng. J., 2013, 221, 500–511 CrossRef CAS.
- D. A. Shirley, Phys. Rev. B: Solid State, 1972, 5, 4709–4714 CrossRef.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603–619 CrossRef CAS.
- M. Arai, A. Obata, Y. Nishiyama and M. Arai, J. Catal., 1997, 166, 115–117 CrossRef CAS.
- R. Wang and J. Li, Environ. Sci. Technol., 2010, 44, 4282–4287 CrossRef CAS PubMed.
- L. S. Wei, J. H. Li and X. F. Tang, Catal. Lett., 2009, 127, 107–112 CrossRef CAS.
- C. R. Parkinson, M. Walker and C. F. McConville, Surf. Sci., 2003, 545, 19–33 CrossRef CAS.
- J. Sa, C. Kartusch, M. Makosch, C. Paun, J. A. van Bokhoven, E. Kleymenov, J. Szlachetko, M. Nachtegaal, H. G. Manyar and C. Hardacre, Chem. Commun., 2011, 47, 6590–6592 RSC.
- J. Sa, S. F. R. Taylor, H. Daly, A. Goguet, R. Tiruvalam, Q. He, C. J. Kiely, G. J. Hutchings and C. Hardacre, ACS Catal., 2012, 2, 552–560 CrossRef CAS; J. Sa, A. Goguet, S. F. R. Taylor, R. Tiruvalam, C. J. Kiely, M. Nachtegaal, G. J. Hutchings and C. Hardacre, Angew. Chem., Int. Ed., 2011, 50, 8912–8916 CrossRef PubMed.
- H. G. Manyar, R. Morgan, K. Morgan, B. Yang, P. Hu, J. Szlachetko, J. Sa and C. Hardacre, Catal. Sci. Technol., 2013, 3, 1497–1500 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5fd00227c |
| This journal is © The Royal Society of Chemistry 2016 |