 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Quantum chemistry of the oxygen evolution reaction on cobalt(II,III) oxide – implications for designing the optimal catalyst†
Craig P.
Plaisance
*a,
Karsten
Reuter
a and
Rutger A.
van Santen
*bc
aChair of Theoretical Chemistry and Catalysis Research Center, Technical University of Munich, D-85747, Garching, Germany. E-mail: craig.plaisance@tum.de
bInstitute for Complex Molecular Systems, Eindhoven University of Technology, 5612 AZ, Eindhoven, The Netherlands. E-mail: R.A.v.Santen@tue.nl
cLaboratory of Inorganic Materials Chemistry, Schuit Institute of Catalysis, Eindhoven University of Technology, 5612 AZ, Eindhoven, The Netherlands
First published on 5th January 2016
Abstract
Density functional theory is used to examine the changes in electronic structure that occur during the oxygen evolution reaction (OER) catalyzed by active sites on three different surface terminations of Co3O4. These three active sites have reactive oxo species with differing degrees of coordination by Co cations – a μ3-oxo on the (311) surface, a μ2-oxo on the (110)-A surface, and an η-oxo on the (110)-B surface. The kinetically relevant step on all surfaces over a wide range of applied potentials is the nucleophilic addition of water to the oxo, which is responsible for formation of the O–O bond. The intrinsic reactivity of a site for this step is found to increase as the coordination of the oxo decreases with the μ3-oxo on the (311) surface being the least reactive and the η-oxo on the (110)-B surface being the most reactive. A detailed analysis of the electronic changes occurring during water addition on the three sites reveals that this trend is due to both a decrease in the attractive local Madelung potential on the oxo and a decrease in electron withdrawal from the oxo by Co neighbors. Applying a similar electronic structure analysis to the oxidation steps preceding water addition in the catalytic cycle shows that analogous electronic changes occur during this process, explaining a correlation observed between the oxidation potential of a site and its intrinsic reactivity for water addition. This concept is then used to specify criteria for the design of an optimal OER catalyst at a given applied potential.
Introduction
The oxygen evolution reaction (OER), in which two molecules of water are oxidized to give molecular oxygen, generates the electrons needed for the photocatalytic or electrocatalytic production of transportation fuels by the reduction of water or CO2.1,2 Despite significant research in developing catalytic materials for carrying out this reaction, the most active materials known at this time are RuO2 and IrO2.3,4 Due to the high cost and scarcity of Ru and Ir, there is much interest in finding active catalysts for the OER based on the earth-abundant 3d transition metal oxides.3–9One of the 3d transition metal oxides, Co3O4, has recently received particular attention as a suitable OER catalyst.10–13 Studies by Frei and coworkers14 have shown that this material exhibits strong structure sensitivity, with less than 1/1000 of the exposed sites responsible for most of the observed activity. They postulate that this highly active site consists of two Co(IV) cations connected by bridging oxos that carries out O–O bond formation by nucleophilic addition of water to a terminal oxo. We recently used density functional theory (DFT) and a simplified kinetic model to show that this highly active site likely corresponds to a dual-Co site on a step edge of the (311) surface that forms the O–O bond by water addition to a bridging μ3-oxo, as shown in Scheme 1.15 The turnover frequency (TOF) calculated for this site at the experimental applied potential was within two orders of magnitude of the one measured by Frei and coworkers, and it was found that both water addition and O2 release are kinetically relevant, in agreement with the observation of a superoxo intermediate on the surface in experiments. Sites consisting of a single Co center with a terminal η-oxo and dual-Co sites on the (001) terrace were found to be several orders of magnitude less active by our kinetic model at the experimental applied potential.
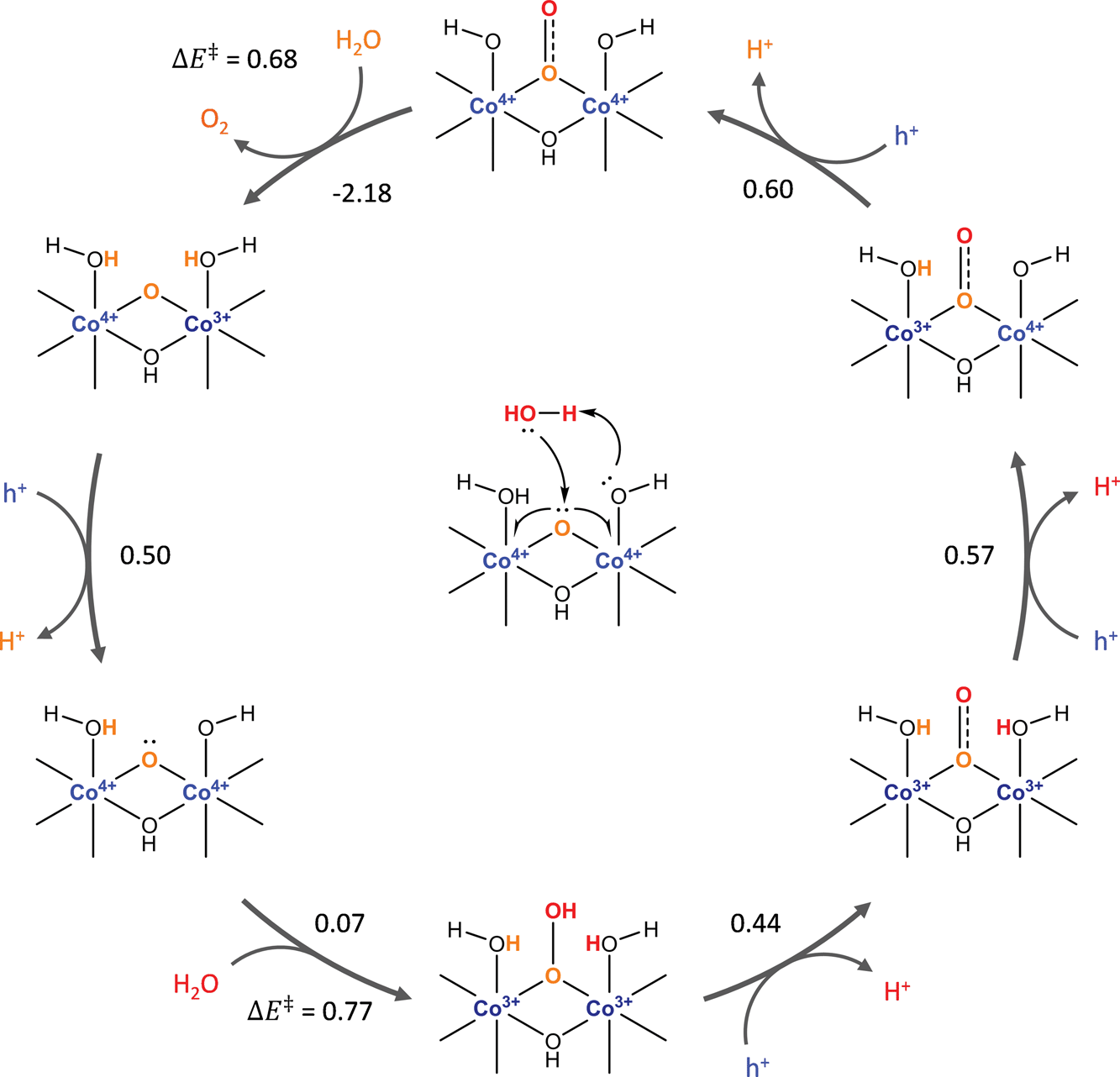 | ||
| Scheme 1 Catalytic cycle and energetics (eV at η = 0) of the OER for the dual-Co site on the (311) surface. Reproduced from ref. 15. | ||
One of the key results of our previous study was that the surface containing the most active site changes as the overpotential (applied potential with respect to the equilibrium potential of the OER) is increased. As shown in Fig. 1, at overpotentials below 0.77 V, the dual-Co μ3-oxo site on either the (311) or (001) surface was found to give the highest TOF, while at overpotentials higher than this, a single-Co η-oxo site on the B termination of the (110) surface was found to take over. We explained this behavior by noting that the single-Co site has a lower intrinsic activation barrier for water addition than the dual-Co site but requires a higher overpotential to oxidize to the active state for this reaction. When the overpotential is <0.77 V, the single-Co site exists in the inactive reduced state so that the dual-Co site has a higher TOF. At higher overpotentials, a significant fraction of the single-Co sites are oxidized to the active state so that this site dominates due to its higher intrinsic reactivity for water addition.
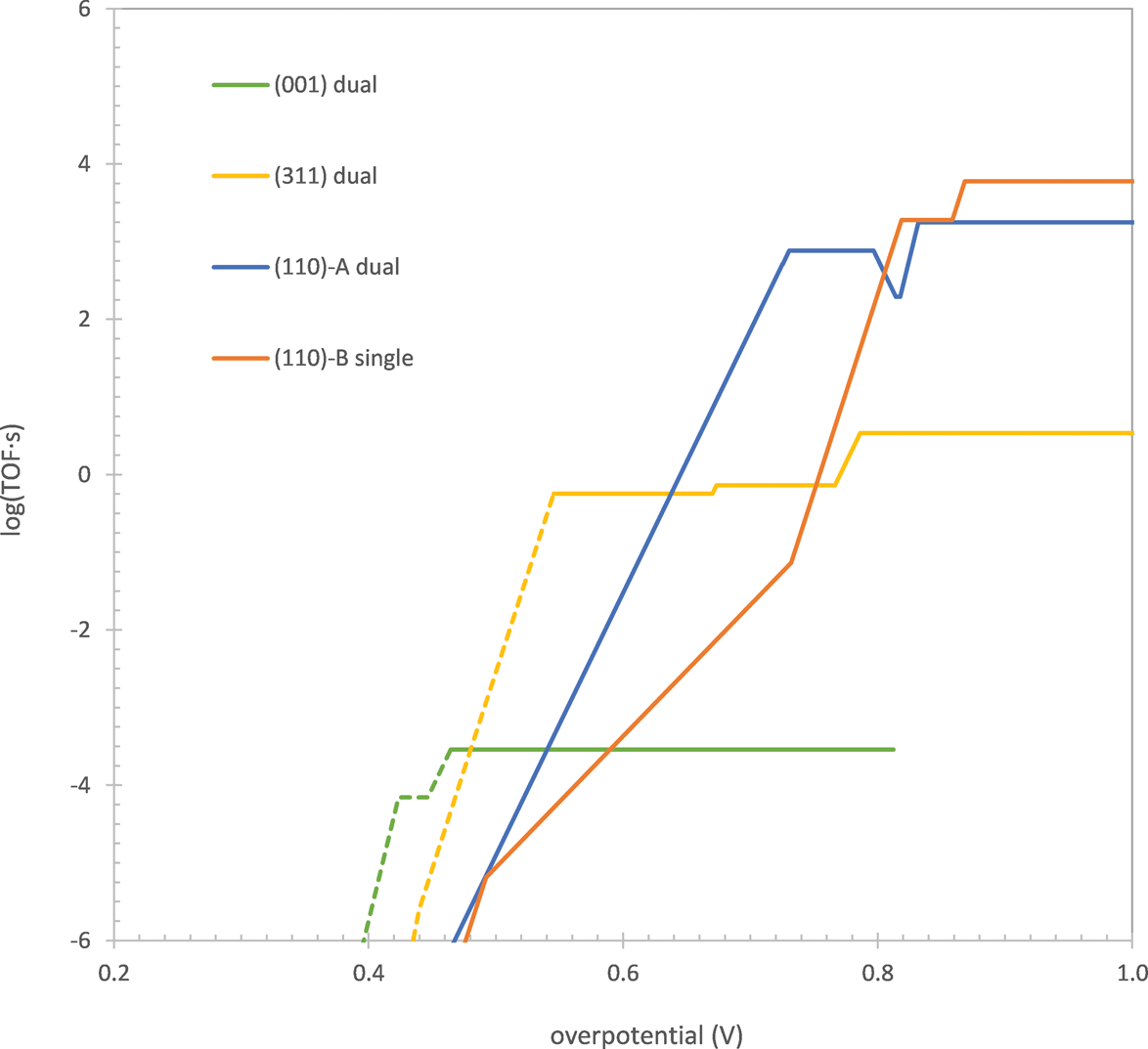 | ||
| Fig. 1 TOF of the OER on the μ3-oxo dual-Co sites on the (001) and (311) surfaces, the μ2-oxo dual-Co site on the (110)-A surface, and the η-oxo single-Co site on the (110)-B surface as a function of overpotential. Solid curves show the regions where H2O addition is rate limiting. Dashed curves show the regions where O2 release is rate limiting. Data for (001), (311), and (110)-B surfaces taken from ref. 15. | ||
Based on the comparison between the single-Co and dual-Co sites, we postulated that a correlation exists between the intrinsic reactivity of a site for water addition and the applied potential at which it is oxidized to the active state for this reaction. While we noted that water addition and the oxidation occurring prior to it are related by the fact that the Co center(s) oxidized in the prior step are reduced during water addition, we did not provide a rigorous quantum chemical justification for this correlation. Providing such a justification will be a central topic of the current manuscript. Additionally, we now identify a third active site on the A termination of the (110) surface (the polar (110) surface has two terminations, A and B) containing a μ2-oxo. This site is found to have an intrinsic water addition reactivity and oxidation potential intermediate to those of the μ3-oxo and η-oxo sites. It gives a higher TOF than the other sites at overpotentials between 0.64 and 0.80 V, as shown in Fig. 1. Thus, there appears to also be a correlation between the coordination of the oxo undergoing water addition and its intrinsic reactivity and oxidation potential.
The aim of this manuscript is to unravel the quantum chemical factors that underlie this relation between the structure of the active site and its water addition reactivity and use these insights to develop a set of design principles for the optimal active site at a given applied overpotential. First, we will examine the changes in electronic structure that occur during water addition to the μ3-oxo on the (311) surface, the μ2-oxo on the (110)-A surface, and the η-oxo on the (110)-B surface. It will be seen that analogous electronic changes occur on all three sites but that the energetics of these processes are very different. We then examine the quantum chemical details underlying the differences in these electronic changes and find that they arise primarily from differences in the energy to localize a hole on the oxo prior to O–O bond formation. The latter is related to the local coordination of the oxo by the local Madelung potential and the electron withdrawing properties of the Co neighbors. In order to study these quantum chemical details, we have developed several techniques for examining the electronic structure of plane wave DFT calculations. The O–O bond formation process that occurs during water addition is then compared to the process of adding a hydrogen atom to the same oxo, which is the reverse of one of the oxidation steps occurring prior to water addition. It is seen that these two steps involve similar changes in electronic structure which explains the correlation noted in our previous work between the intrinsic water addition reactivity of a site and the overpotential required to oxidize it to the active state for this reaction. Based on this realization, we finally identify the properties of an ideal active site giving the highest TOF at a given applied overpotential and compare the performance of this ideal site to the performances of the actual sites.
Theoretical methods
Density functional theory calculations
The quantum chemical calculations in this work are carried out using spin-polarized DFT in the generalized gradient approximation (GGA) using the Vienna Ab initio Simulation Package (VASP),16 the details of which are given in the ESI.† A particular issue in applying this method to the study of 3d transition metal oxides is an over-delocalization of the 3d electrons due to the presence of electron self-interaction in all GGA functionals.17 An effective and low-cost method to correct for this is by applying an on-site self-interaction correction to the 3d electrons.18–20 In this method, called GGA + U, the effective coulombic repulsion integral between the 3d electrons (U) must be specified. For this work, we use a value of U determined from a first-principles linear response method as detailed in our previous work.15All calculations were performed on periodic slabs separated by vacuum, not including the electrolyte which is present in the actual system. Although solvation of the transition states by the electrolyte were partially accounted for by the inclusion of one or two extra water molecules in the system (details given in the ESI†), this is a crude approximation and does not include the electrostatic screening effects of the electrolyte. This latter effect is important as it allows for charged surfaces and pathways involving sequential proton and electron transfer processes in addition to the concerted proton and electron transfer processes examined in this work.21 It is thus possible that a more accurate model of the electrolyte could lead to different results. However, our neglect of charged surface states is supported by experimental observations suggesting that all proton and electron transfers involved in the transition state and pre-equilibria are coupled.22
While standard methods have been used to locate minimum energy structures (details given in the ESI†), it was necessary for us to develop a special technique for finding the transition states for the water addition reaction. This is due to the fact that the transition state corresponds to the crossing of potential energy surfaces for two electronic states. As such, the transition state is not a saddle point on a single potential energy surface and cannot be located using the typical methods such as the nudged elastic band and dimer methods that require this. A detailed description of the method we use is given in the ESI† and was also used in our study in ref. 15.
Electronic structure analysis
Results and discussion
The goal of this work is to examine the quantum chemistry of the water addition reaction on Co3O4 to gain insight into the properties that lead to an OER catalyst having maximum activity. We first present a detailed examination of the mechanism of water addition to the (311) μ3-oxo, the (110)-A μ2-oxo, and the (110)-B η-oxo. Based on the computed mechanisms, we decompose the water addition reaction into a proton coupled electron transfer (PCET) step and an O–O bond formation step and find that the differences in intrinsic activity between the three sites are primarily due to differences in the O–O bond formation step. We next apply methods we have developed for examining the electronic structure based on constrained DFT calculations and quasiatomic orbitals to further examine the O–O bond formation step occurring during water addition. This shows that the energetic differences in the step between the three sites are primarily a result of differences in the energy to localize a hole on oxos in different coordination environments. A similar electronic structure analysis is then carried out for the hydrogen atom removal occurring in the oxidation step that precedes water addition and it is found that the energy of this process is related to the energy of O–O bond formation by the hole localization energy, which is common to both processes. Based on this correlation between oxidation potential and water addition activity, we finally propose criteria for the design of the optimal OER catalyst at a given applied overpotential.Mechanism and electronic transitions during water addition
The water addition reaction involves deprotonation of the attacking water by a basic oxygen atom on the active site and formation of an O–O bond between the resulting hydroxide and an oxo. In the process, the oxygen of the water and the oxo are both oxidized from O(−II) to O(−I) with the two electrons being transferred to holes on the Co center(s) of the active site. As will be seen in the following section, this reaction can be decomposed into two steps. The first step is a PCET step in which a proton and electron are transferred to the active site, forming a hydroxyl radical. The second step is the formation of the O–O bond between the hydroxyl and the oxo with a possible electron transfer from the oxo to a hole on one of the Co centers. It will be seen that the relative difficulties of these two steps are very different for the three active sites discussed below and this then leads to the differences in activity as a function of overpotential seen in Fig. 1.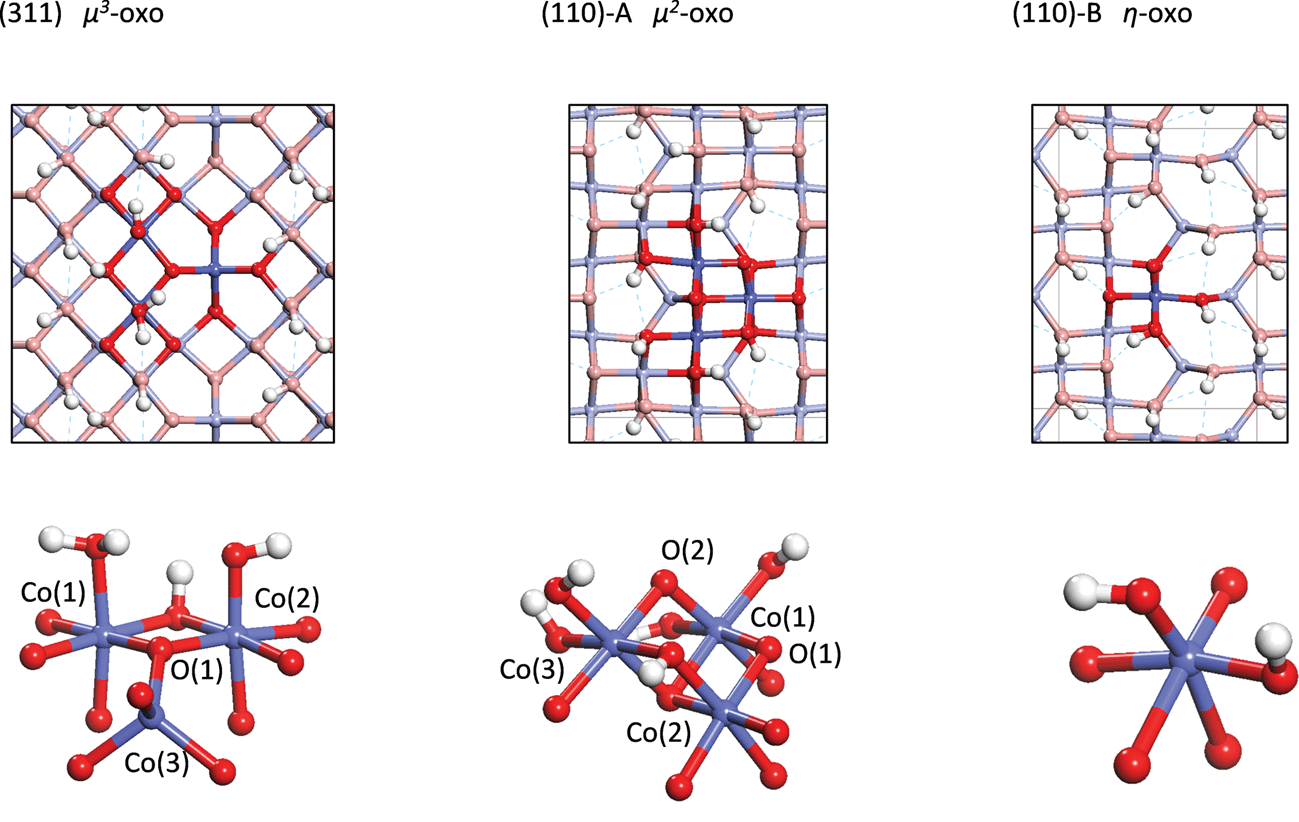 | ||
| Fig. 2 Top and perspective views of the structures of the active sites on the three surfaces (Co = blue, O = red, H = white spheres). | ||
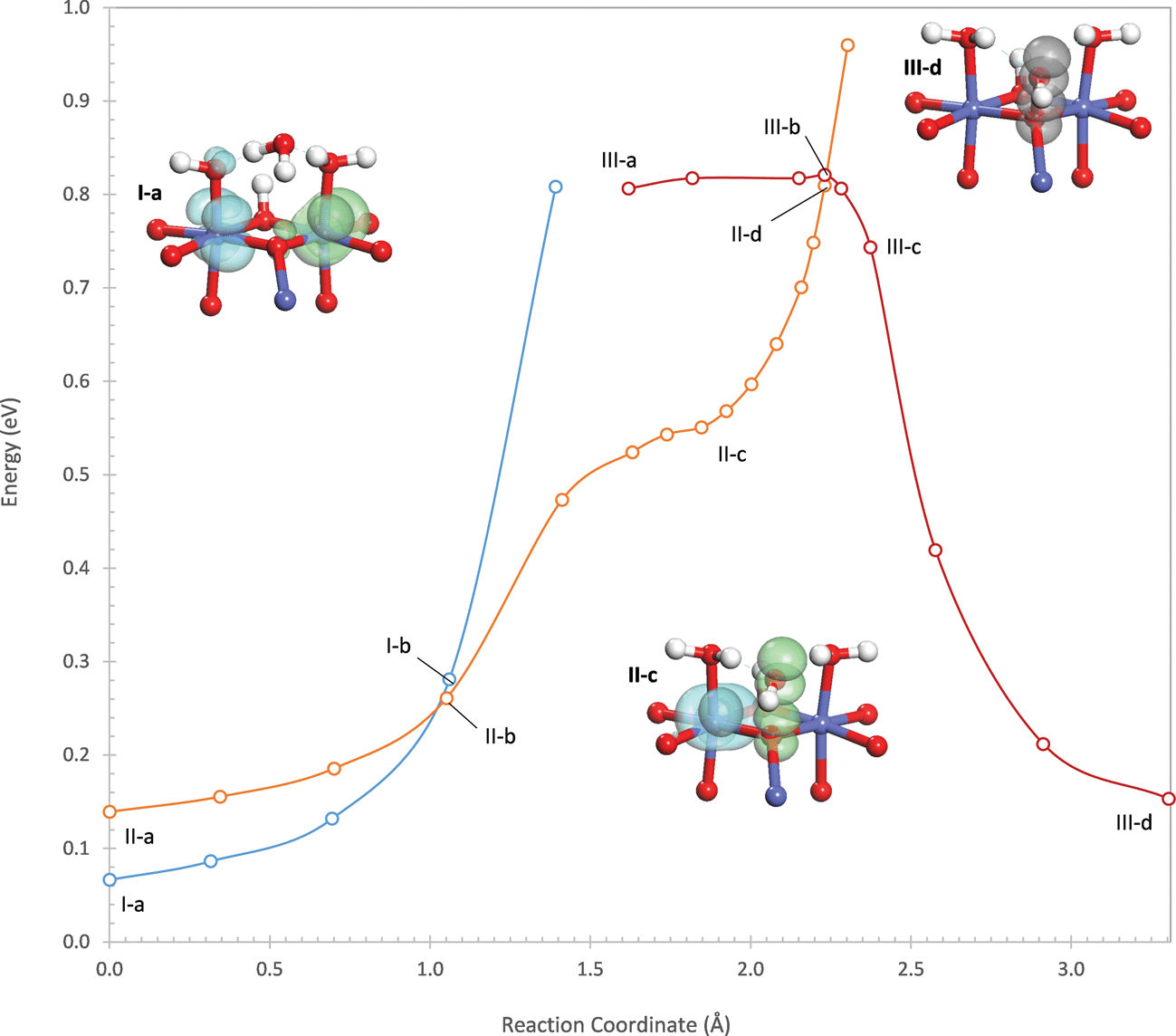 | ||
| Fig. 3 Potential energy profiles for the three electronic states involved in water addition to the μ3-oxo on the (311) dual-Co site. The insets show structures along the reaction pathway together with densities of the two holes involved in the reaction (green = spin-up, blue = spin-down, grey = both spins). Energies are relative to the oxidized state of the active site preceding water addition. Additional structures are shown in the ESI.† | ||
The reaction begins by transfer of a proton from the attacking water to an η-OH on the active site. This proton transfer is initially very unfavorable as can be seen by the sharp rise in energy at the start of the potential energy profile in Fig. 3. A second electronic state (II) exists in which the hole originally on the Co(1) center is delocalized into the O 2p band so that this Co is in the +III oxidation state. This state is initially higher in energy than state I, but the proton transfer is considerably more favorable in state II than in state I. This is because, as the proton is transferred, the hole in the O 2p band localizes into an occupied σ* orbital formed between lone pairs on the hydroxide (being formed from the deprotonating water) and the oxo, which can be seen in structures II-b and II-c of Fig. 3. This withdraws negative charge from the hydroxide, stabilizing it and facilitating proton transfer. As a result, the potential energy surface of state II eventually crosses below that of state I as the proton is transferred. This process is therefore a PCET. The crossing of the two potential energy surfaces occurs at a point that is 0.26 eV above the energy of the oxidized site.
Completion of the PCET results in the intermediate shown in structure II-c of Fig. 3, where one hole still resides on a Co(IV) (Co(2)) while the other is localized in the σ* orbital formed between the oxo and the hydroxyl. The hole in the σ* orbital has 89% of its density on the two oxygen atoms. This intermediate is not a local minimum on the potential energy surface but exists as an inflection point in the reaction profile in Fig. 3, 0.55 eV above the energy of the oxidized site. In the intermediate, the hydroxyl forms strong hydrogen bonds with the two water ligands coordinating the Co cations in the active site, with O–H distances of 1.52 and 1.77 Å. These hydrogen bonds withdraw 0.18 electrons from the hydroxyl – this quantity (and all further charge quantifications below) is determined using a population analysis discussed in the ESI† based on the quasiatomic orbital representation.
From this intermediate, water addition continues by formation of the O–O bond between the oxo and the hydroxyl. As the O–O bond distance decreases, the energy of state II increases sharply due to Pauli repulsion because of the single electron still occupying the σ* orbital. At an O–O distance of 1.96 Å, the potential energy surface of state II crosses above that of a third electronic state (III) in which the hole residing on Co(2) in state II is delocalized into the O 2p band. The crossing point is 0.27 eV higher in energy than the intermediate and 0.82 eV higher in energy than the oxidized site. In electronic state III, O–O bond formation occurs easily since, as the O–O distance decreases, the hole in the O 2p band localizes into the σ* orbital so that both holes now reside in the σ* orbital. In the product state, both holes are strongly localized in the σ* orbital with 97% of the hole density on the two oxygen atoms.
In the oxidized state prior to water addition, two of the Co cations (Co(1) and Co(2)) are in the +IV state while the third Co (Co(3)) is in the +III state when the overpotential is <0.81 V and in the +IV state when it is greater (discussed in more detail in the ESI†). The oxidation of Co(3) at 0.81 V is coupled with deprotonation of the bridging hydroxyl. A hole resides in a 3d t2g orbital on each of the two Co cations (Co(1) and Co(2)) coordinated by the bridging oxo (O(1)) which forms the O–O bond. These holes, shown in structure I-a of Fig. 4, have opposite spins as required for the water addition reaction. The hole on Co(2) is well localized in a 3d t2g orbital, with 87% of the hole density residing on the Co cation. The hole on Co(1) is significantly delocalized onto the two bridging oxo ligands coordinating it, with only 66% of the hole density residing on the Co cation.
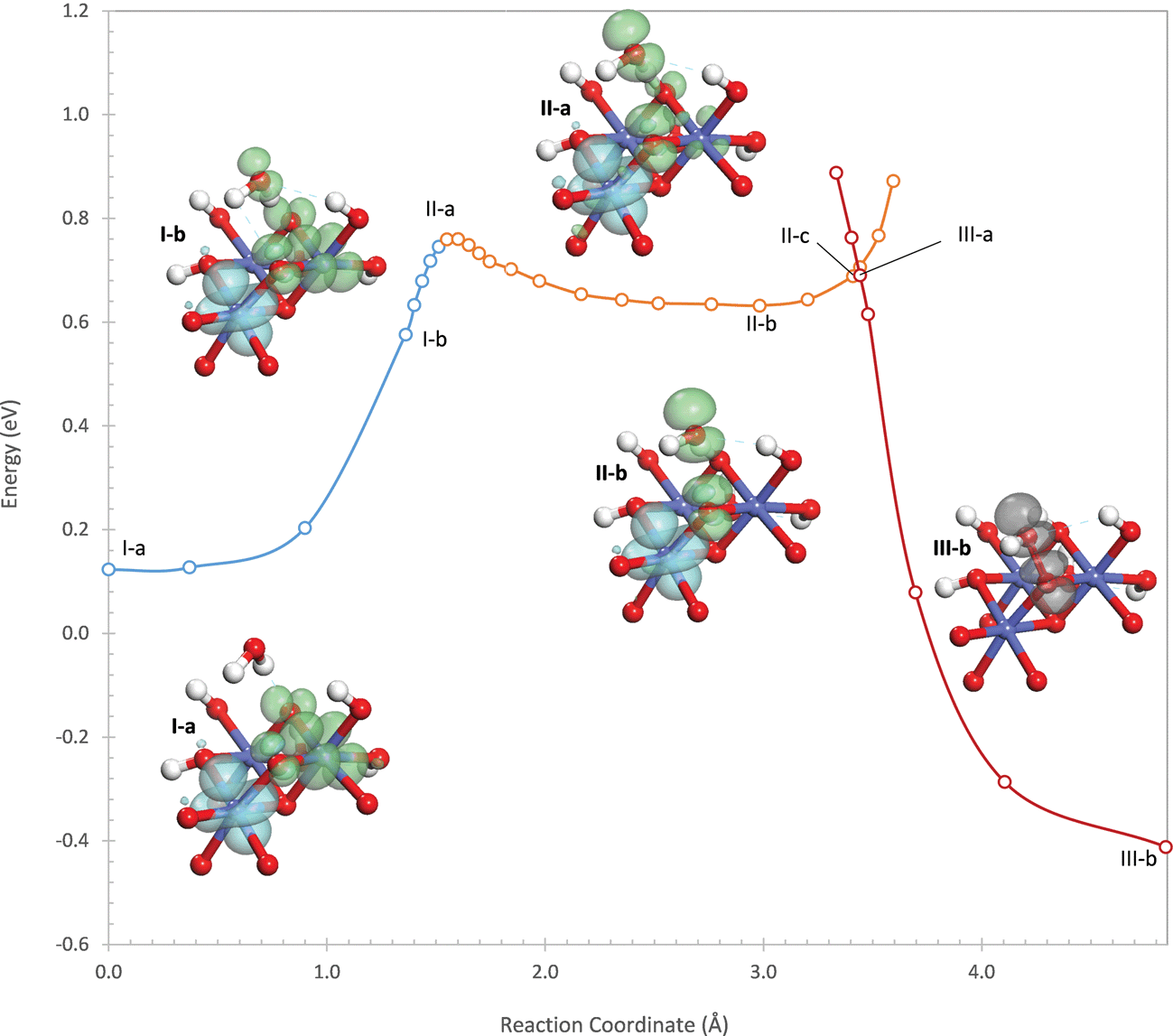 | ||
| Fig. 4 Potential energy profiles for the three electronic states involved in water addition to the μ2-oxo on the (110)-A dual-Co site. The insets show structures along the reaction pathway together with densities of the two holes involved in the reaction (green = spin-up, blue = spin-down, grey = both spins). Energies are relative to the oxidized state of the active site preceding water addition. Additional structures are shown in the ESI.† | ||
Water addition initiates by the transfer of a proton from the attacking water to one of the μ2-oxos O(2). The potential energy profile is shown in Fig. 4 where it can be seen that, unlike the (311) site, the proton transfer on the (110)-A site does not involve a crossing between two potential energy surfaces. Instead, the hole transfers smoothly from Co(1) to the oxygen on the water and the bridging oxo O(1). A transition state occurs at the point where only 9% of the hole density remains on Co(1), labeled II-a in Fig. 4. The transition state is 0.76 eV higher in energy than the initial oxidized site. In this transition state, the proton is in transit between the hydroxyl and the oxo.
As the proton transfer completes, an intermediate (II-b) is formed in which the hole becomes almost completely localized in a σ* orbital formed between the hydroxyl and the oxo, with 96% of the hole density on these two atoms. Thus, an electron is transferred from the σ* orbital to an empty orbital centered on a Co(IV) so that this first step is a PCET. The energy of the intermediate is 0.63 eV above the energy of the oxidized site prior to water addition. Unlike the intermediate on the (311) site which was identified as an inflection point on the potential energy surface, this intermediate is a true local minimum. As with the intermediate on the (311) site, this intermediate also forms two hydrogen bonds with nearby hydroxo ligands. These hydrogen bonds are weaker than they were on the (311) site, having O–H distances of 1.87 and 2.02 Å and involving the withdrawal of 0.08 electrons from the hydroxyl (compared to 0.18 electrons on the (311) site). The weaker hydrogen bonds are the likely reason that the intermediate requires more energy to form from the oxidized site than during water addition on the (311) site.
Water addition proceeds from the intermediate II-b by shortening of the O–O bond between the hydroxyl and the oxo. As with the (311) site, this leads to a sharp increase in energy due to increased Pauli repulsion resulting from the electron remaining in the σ* orbital. A second electronic state (III) also exists in which the hole on the remaining Co(IV) (Co(2)) is transferred to the σ* orbital. From this state, O–O bond formation proceeds easily without any barrier since both holes now reside in the σ* orbital. The point where the potential energy surfaces of the two electronic states cross is only 0.07 eV higher in energy than the intermediate, much lower than on the (311) site where the crossing point was 0.27 eV higher in energy than the intermediate. This leads to an activation barrier of 0.70 eV with respect to the initial oxidized site, which is lower than the barrier for the PCET. As shown in the ESI,† however, interactions with additional water molecules that mimic the solvent phase reduce the barrier for the PCET by 0.18 eV while reducing the barrier for O–O bond formation by 0.11 eV. Thus, in the presence of solvent, the two transition states will likely have nearly the same energy.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O is in a triplet state. If we assign the holes in the π* orbitals to the Co cation, then it is in the +V oxidation state and the oxo is in the −II oxidation state.
O is in a triplet state. If we assign the holes in the π* orbitals to the Co cation, then it is in the +V oxidation state and the oxo is in the −II oxidation state.
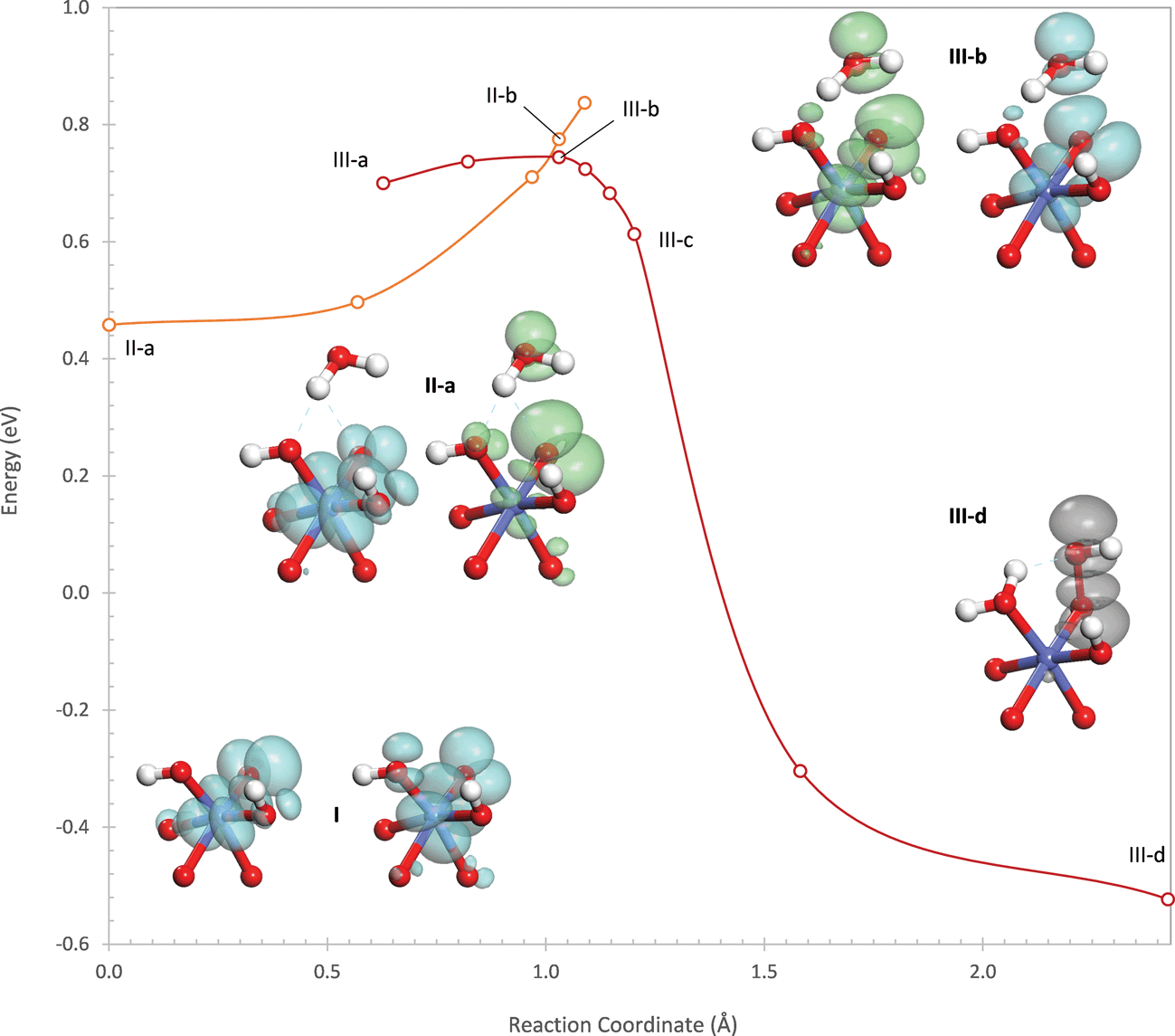 | ||
| Fig. 5 Potential energy profiles for the three electronic states involved in water addition to the η-oxo on the (110)-B single-Co site. The insets show structures along the reaction pathway together with densities of the two holes involved in the reaction (green = spin-up, blue = spin-down, grey = both spins). Energies are relative to the oxidized state of the active site preceding water addition. Additional structures are shown in the ESI.† | ||
Since formation of the O–O bond requires two holes of opposite spin, the CoO must first transition to a singlet state that is 0.18 eV higher in energy than the triplet state. In the singlet state, the spin-up hole localizes onto the oxo while the spin-down hole localizes onto the Co, seen in structure II-a of Fig. 5. The spin-up hole has 78% of its density on the oxo, 11% on the water, and only 4% on the Co while the spin-down hole has 79% of its density on the Co and 12% on the oxo. This is best described qualitatively as an O(−I) bound to a Co(IV). Despite the formal transfer of an electron from the oxo to the Co during the spin transition, the actual electron populations on both change by less than 0.02 electrons. This is due to the fact that the holes were initially in π* orbitals that had nearly equal density on the Co and the oxo.
Like the other two sites, water addition begins by transfer of a proton from the water to the η-OH ligand, leading to an initial increase in energy (Fig. 5) since the electrophilic Co(IV) coordinated by the η-OH likely makes this ligand a weak base. A second electronic state (III), initially higher in energy, exists in which both holes reside in orbitals centered on the oxo (52% of the hole density) with significant delocalization onto the Co (28%) and the water (10%) as shown in structures III-a and III-b in Fig. 5. The proton transfer proceeds easily and with almost no activation barrier from this second electronic state (III) so that the potential energy surfaces of the two electronic states cross when the proton is halfway between the hydroxyl and the oxo. The crossing point is 0.56 eV higher in energy than the oxidized site in the singlet state and represents the transfer of an electron from the oxo and water to the Co(IV), reducing it to Co(III) – thus, as with the other two sites, the proton transfer is a PCET.
As the proton is transferred, the holes in the final electronic state localize into the σ* orbital formed from the oxo and the hydroxyl. Unlike on the other two sites where the σ* orbital still contained one electron, the empty σ* orbital on the (110)-B site allows the O–O bond to form concertedly with the transfer of the proton. This leads to a much lower proton transfer barrier and the absence of a barrier for O–O bond formation. As a result, the intrinsic barrier of 0.56 eV for this site from the singlet oxidized state is the lowest of all three sites. The actual barrier is 0.18 eV higher since the site is initially in the lower energy triplet state. As shown in the ESI,† however, interactions with additional water molecules that mimic the solvent phase lower the barrier substantially so that this site has the lowest intrinsic water addition barrier, even when starting in the triplet state.
Quantum chemical analysis of water addition
It was seen in the last section that the three different oxo species — μ3-oxo on the (311) surface, μ2-oxo on the (110)-A surface, and η-oxo on the (110)-B surface — possess very different reactivities in the water addition reaction. This was seen, in particular, to be related to the ease with which the oxo forms an O–O bond with the hydroxyl radical formed during the initial PCET process. In this section, we show that the O–O bond formation process is responsible for the differences in water addition energetics on the different sites and that this is due to differences in the cost of localizing a hole on the oxo. The hole localization process is examined using the constrained DFT method we have developed in the quasiatomic orbital formalism and is shown to be related to the coordination of the oxo by Co cations via the local Madelung potential and electron withdrawal effects.In order to separate the PCET and O–O bond formation processes occurring during the water addition reaction, we decompose it into the two separate steps shown in Fig. 6. The first step is the transfer of a proton and electron from a molecule of water in vacuum to the active site, leaving a hydroxyl radical in vacuum. The proton is transferred to a hydroxo ligand on the active site and the electron is transferred to a Co(IV) center, reducing it to Co(III). This step corresponds to the PCET observed in the actual reaction mechanism. In the second step, the hydroxyl radical produced in the first step forms an O–O bond with an oxo on the active site.
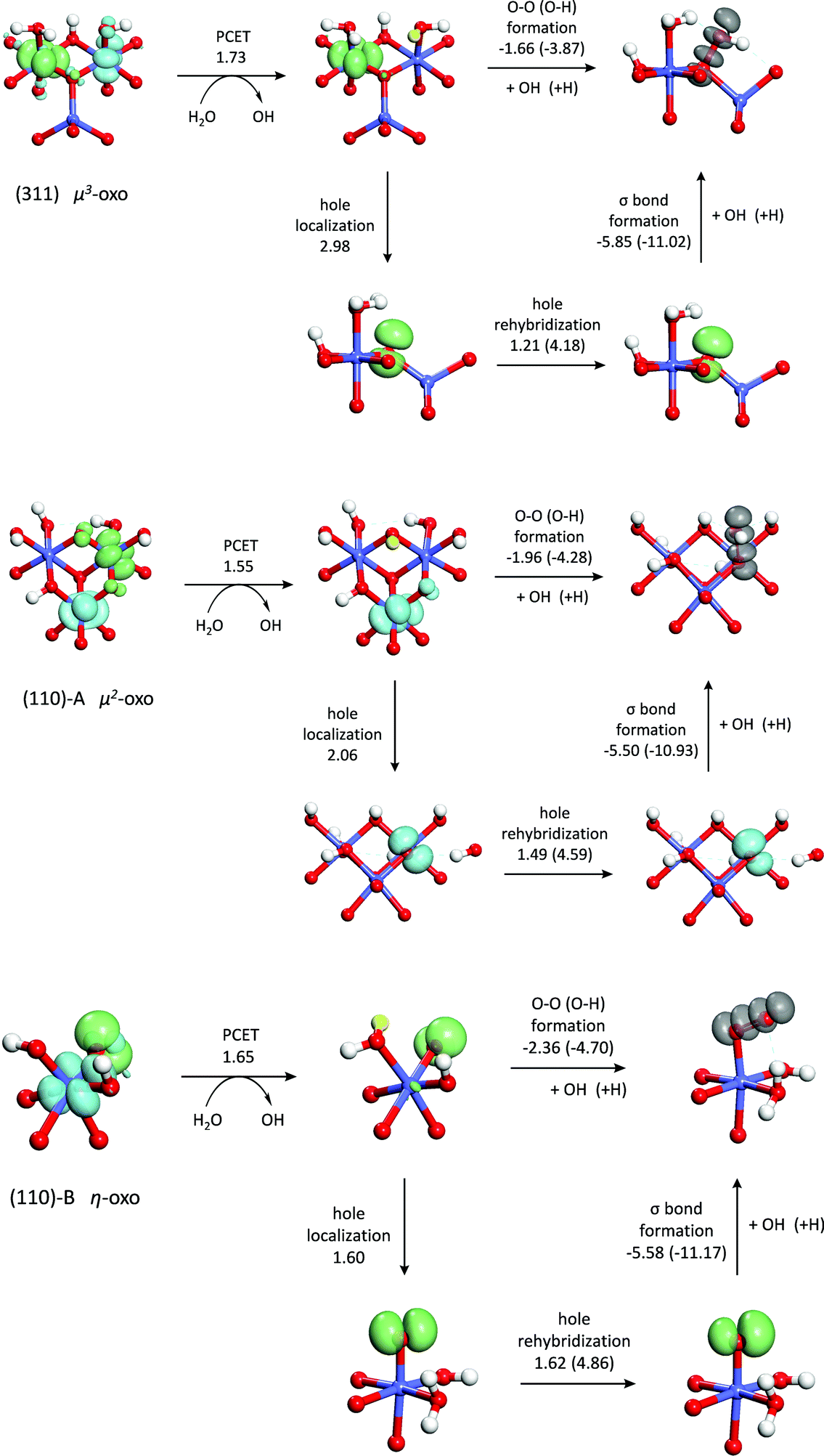 | ||
| Fig. 6 Energy decomposition of the water addition reaction on the three active sites along with the density of the hole(s) at each step (green = spin-up, blue = spin-down, grey = both spins). The energy decomposition of hydrogen atom addition is also shown (energies in parentheses). All energies are in eV. | ||
The energies of the two steps shown in Fig. 6 confirm that it is the O–O bond formation step rather than the PCET step that is responsible for the differences in reactivity between the sites. This step is most exothermic (−2.36 eV) on the (110)-B η-oxo site which has the highest reactivity. The energy of this step is significantly less exothermic (−1.96 eV) on the less reactive (110)-A μ2-oxo site and even less (−1.66 eV) on the least reactive (311) μ3-oxo site. Although the PCET step also accounts for a large part of the difference in reaction energies between the (311) μ3-oxo site and the (110)-A μ2-oxo site, this step is more endothermic on the more reactive (110)-B η-oxo site than the less reactive (110)-A μ2-oxo site.
From the above analysis, it appears that as the coordination of the oxo decreases, the O–O bond formation step becomes more exothermic. To better understand this relationship, we examine the changes in the electronic structure accompanying the O–O bond formation process. Prior to the bond formation on both dual-Co sites, there are four lone pairs localized on the oxo, while on the single-Co site, there are three lone pairs on the oxo and one singly-occupied O 2p orbital. These orbitals are shown in the ESI.† Following O–O bond formation, one of the lone pairs on the oxo and the singly occupied orbital on the hydroxyl transform into a doubly occupied σ bonding orbital and an unoccupied σ* antibonding orbital (also shown in the ESI†). Formation of a doubly occupied σ orbital and an unoccupied σ* orbital from a lone pair on the oxo on the dual-Co sites and a singly occupied orbital on the hydroxyl requires removal of one electron. This electron is placed into an empty 3d t2g orbital on the neighboring Co(IV), thus formally transferring a hole from the Co to the oxo. On the single-Co site, the hole already resides on the oxo, although it is somewhat delocalized onto the Co. This suggests that O–O bond formation can be thought of as consisting of two steps – first, the hole is transferred from the Co(IV) and localizes on the oxo to form a singly occupied hybrid orbital. This is followed by formation of the σ bond with the singly occupied hybrid orbital on the hydroxyl. By analyzing these two steps separately, it is possible to further identify the differences in this process between the three active sites.
In order to separate the hole localization and σ bond formation steps of the O–O formation process, an intermediate state is defined in which the hole is completely localized into an orbital on the oxo. To model this intermediate state, the constrained DFT method based on quasiatomic orbitals is used to freeze the shape and occupation of the orbital containing the hole. The singly occupied orbital h containing the hole is defined as a linear combination of the single 2s and three 2p QOs on the oxo.
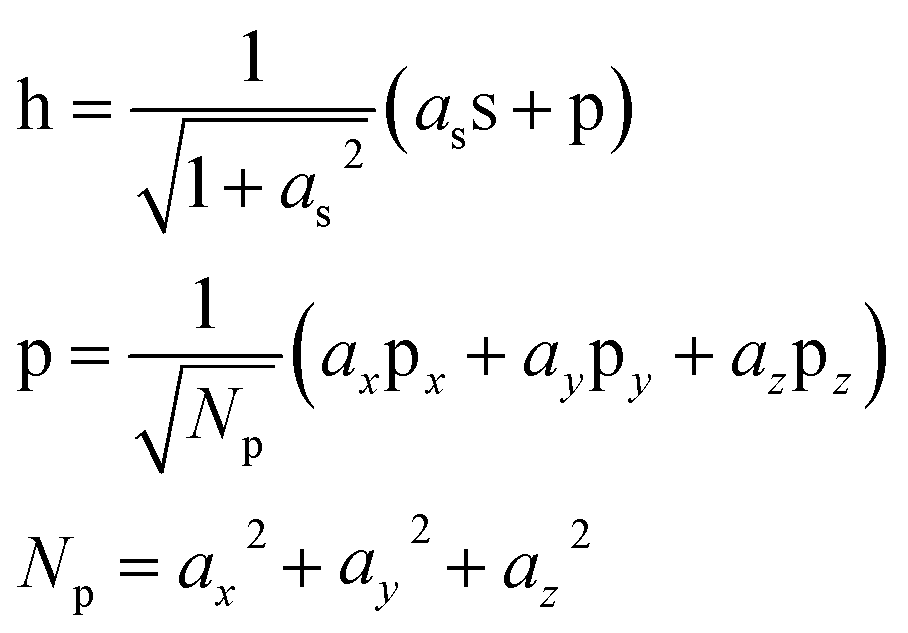 | (1) |
The expansion coefficients of the orbital containing the hole are determined by minimizing the total energy of the intermediate electronic state. The resulting orbitals (shown in Fig. 6) contain very little 2s character due to the much lower energy of the 2s orbital compared to the 2p orbitals (13–14 eV). The energy required to localize the hole on the oxo, defined as the energy difference between the intermediate electronic state and the initial electronic state, is seen to be highest for the μ3-oxo and lowest for the η-oxo, consistent with the trends in the O–O bond formation energy and the overall water addition reaction energy. This indicates that the differences in hole localization energy are responsible for most of the activity differences between the three sites. The least reactive μ3-oxo requires 2.98 eV to localize the hole. In contrast, the most reactive η-oxo requires only 1.60 eV since the hole is already mostly localized on the oxo following the PCET.
Although the hole localization energy seems to explain the trends in the O–O bond formation energy and the water addition reactivity, it overestimates these differences. The differences in O–O bond formation energy between the μ3-oxo and μ2-oxo and the μ2-oxo and η-oxo are 0.30 eV and 0.40 eV while the differences in hole localization energy are 0.91 eV and 0.47 eV, respectively. This is due to the fact that the hole on the oxo has almost entirely 2p character, while the hybrid forming the O–O σ bond has a significant contribution from the 2s orbital (7–8%). This higher 2s character is due to the increased concentration of the hybrid towards the hydroxyl fragment upon mixing with the 2s orbital which leads to increased orbital overlap and a stronger O–O bond. Although the energy to localize the hole in an orbital with significant 2s character is higher, the energy released by formation of the O–O σ bond in the next step is greater.
In order to quantify this rehybridization energy, a second intermediate electronic state is determined in which as in eqn (1) is set to 0.3, which is approximately the value of as in the hybrid orbitals forming the σ bond (0.28–0.30). As with the first intermediate, the expansion coefficients for the 2p part are determined by minimizing the total energy. Interestingly, the rehybridization energy, shown in Fig. 6, is highest for the η-oxo and lowest for the μ3-oxo, reducing the differences in hole localization energy so that they are closer to the differences in O–O bond formation energy. This explains the overestimation of these differences when localizing the hole into an orbital with almost entirely 2p character. The reason for these differences in rehybridization energy can be seen in Fig. 6 which shows the change in hole density upon rehybridization. On the (311) surface, this has the effect of shifting the hole density away from the positively charged Co cations, leading to reduced repulsion and greater donation of electron density from the orbitals not containing the hole. A similar effect is seen for the μ2-oxo on the (110)-A surface due to polarization of the hole away from a nearby proton. The effect is much weaker, however, because the proton is rather far away from the oxo (2.24 Å). No such effect exists for the η-oxo on the (110)-B surface.
Hole transfer energy
By decomposing the energy of water addition to an oxo into components related to PCET, hole localization, and σ bond formation, it was seen that most of the difference in reactivity between the μ3-, μ2-, and η-oxo sites is due to the energy to localize a hole on the oxo. This energy was seen to be highest on the least reactive μ3-oxo and lowest on the most reactive η-oxo. In this section, we further analyze the hole localization process in order to determine why the energy decreases as the coordination of the oxo decreases.To analyze the hole localization process, we carry out additional constrained orbital calculations for the process shown in Fig. 7. This process consists of the transfer of a hole from a singly occupied 3d orbital on the Co(IV) to a doubly occupied 2p orbital on the oxo. The constrained orbital on the oxo is the 2p component of the hybrid containing the hole in the intermediate state in eqn (1).
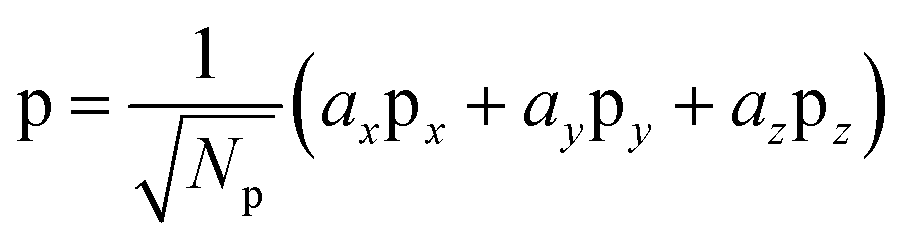 | (2) |
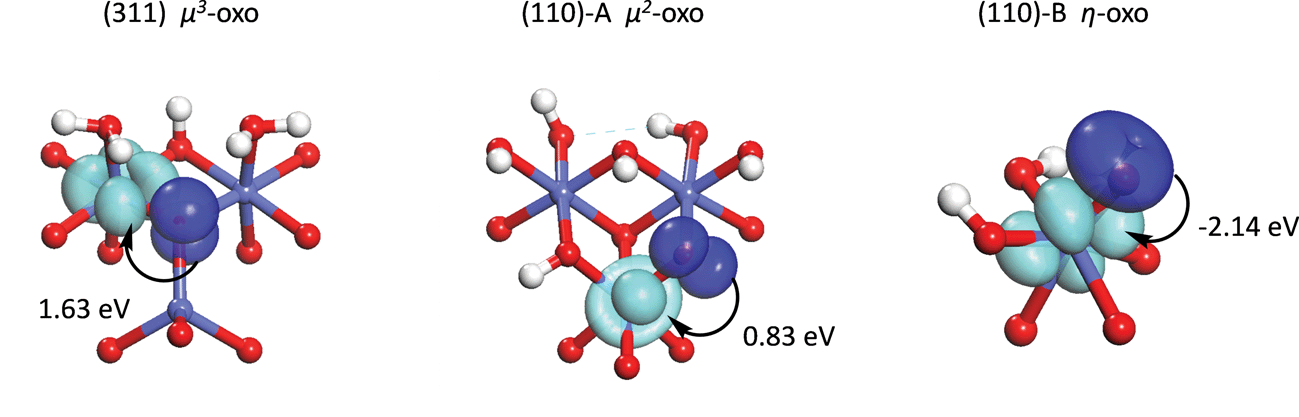 | ||
| Fig. 7 Orbitals used on the three sites to model the hole transfer process from a 3d orbital on a Co cation (light blue) to a 2p orbital on the oxo (dark blue). Energies of the hole transfer process are given for each active site. | ||
The constrained orbital on the Co is, likewise, a linear combination of the 3d quasiatomic orbitals on the Co cation
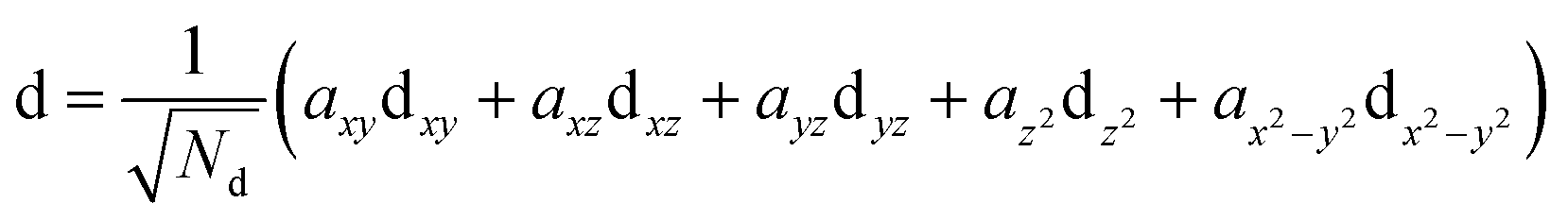 | (3) |
The energy to transfer the hole from the 3d orbital on the Co(IV) to the 2p orbital on the oxo in the constrained system (shown in Fig. 7) follows the same trend as the energy to localize the hole on the oxo in the actual system, being highest for the μ3-oxo and lowest for the η-oxo. The hole transfer energy in the constrained system is lower than in the actual system, however, due to delocalization of the lone pair on the oxo in the initial state of the actual system. Constraining the lone pair to reside completely on the 2p orbital on the oxo raises the energy of the initial state considerably. The delocalization of the lone pair in the initial state is greater on the less coordinated oxos so that the differences in hole transfer energy in the constrained system overestimate the differences in hole localization energy in the actual system. The hole transfer energy is even negative for the η-oxo since the hole is more stable on the oxo than on the Co in the actual system. Nevertheless, the hole transfer process in the constrained system does provide insight into the differences in hole localization energy in the actual system as will be seen below.
The differential energy to transfer the hole from the Co to the oxo is given by the difference in the energies of the two orbitals containing the holes
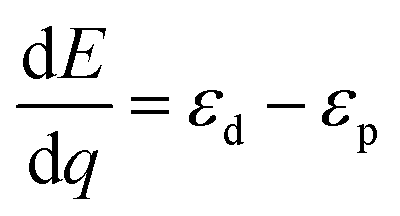 | (4) |
We make the approximation that the orbital energies depend linearly on the electron population (nCo and nO) and magnetic moment (mCo and mO) of the respective atoms, with the slopes of the relationships related to the Coulomb integral U and the exchange integral J
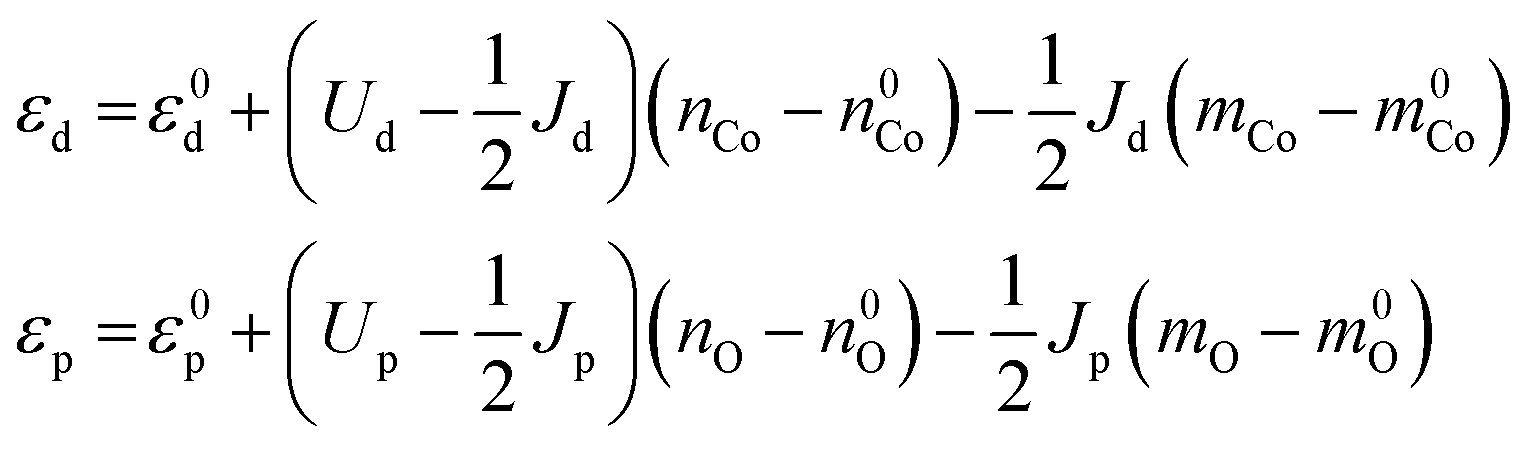 | (5) |
We make the further approximation that the electron populations and magnetic moments of the Co and oxo depend linearly on the occupancy of the constrained 3d and 2p orbitals, respectively. For convenience, the reference populations (n0Co and n0O) and magnetic moments (m0Co and m0O) are taken from the state where half of the hole resides in each of the two orbitals (q = 1/2). The resulting quantities are given by:
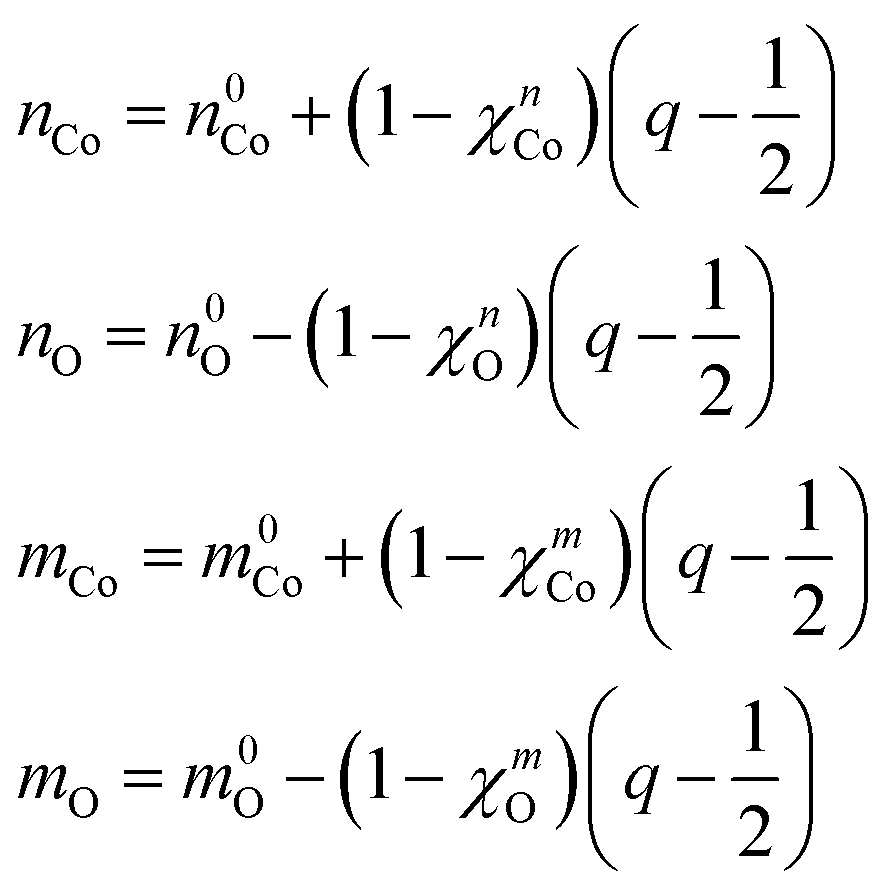 | (6) |
In these expressions, χ is the screening constant for the electron population and magnetic moment of the respective atom which accounts for changes in the occupancies of the non-constrained atomic orbitals as the occupancy of the constrained orbital varies. Substituting eqn (5) and (6) into eqn (4) and integrating from q = 0 to q = 1 gives the overall hole transfer energy of
| ΔE = ε0d − ε0p | (7) |
So the hole transfer energy is equal (in the linear approximation we have made) to the difference in the two orbital energies when half of the hole is residing on each orbital.
We would now like to address why the difference in orbital energies increases as the coordination of the oxo increases. In general, there are two possible causes for this. The first is the increase in Madelung potential on the oxo as the number of positively charged Co neighbors increases and the second is the decrease in electron population on the oxo due to donation to an increasing number of Co neighbors. In order to separate these two components, we first evaluate the Madelung potential by plotting the energy of the constrained orbital (referenced to the unscreened energy of the Co 3d orbital as explained in the ESI†) with respect to the electron population of the oxo. The electron population on the oxo is varied by changing the occupancy of the constrained orbital. The result is shown in the ESI† where it can be seen that the curve for the η-oxo has the highest energy and the μ3-oxo has the lowest energy. At an electron population of 6.6 electrons, the orbital energies on the μ3-, μ2-, and η-oxo sites are −5.89, −2.72, and 0.96 eV, respectively. This confirms that the Madelung potential becomes significantly more attractive as the number of Co neighbors increases, making it more difficult to transfer an electron from the oxo to the Co.
The electron withdrawing ability of the Co neighbors is more difficult to evaluate. It is not appropriate to quantify it by the electron population on the oxo since this quantity is strongly correlated with the Madelung potential – a more repulsive Madelung potential will cause more electrons to be donated to the Co neighbors even when their electron withdrawing ability is unchanged. Therefore, we quantify the electron withdrawing ability by plotting the number of electrons donated with respect to the unscreened orbital energy of the constrained 2p orbital on the oxo (referenced to the unscreened energy of the Co 3d orbital as explained in the ESI†). The unscreened orbital energy is determined from the self-consistent orbital energy by subtracting off the contribution due to the decrease in electron population from donation to the Co neighbors
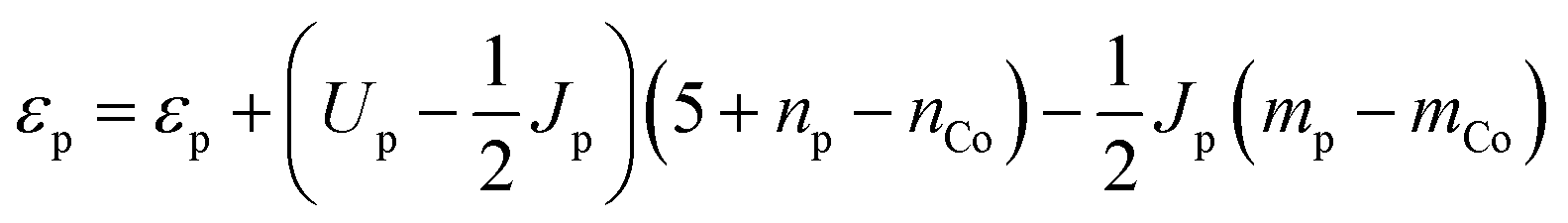 | (8) |
The plot in Fig. 8 shows that electron withdrawal by the Co neighbors is nearly equal for the μ3- and μ2-oxos, but is much weaker for the η-oxo. The weaker electron withdrawal for the η-oxo is expected since it has only one Co neighbor. One would, however, naively expect that the electron withdrawal from the μ3-oxo should be greater than from the μ2-oxo since it has one more Co neighbor. It turns out that the extra Co(II) cation coordinated to the μ3-oxo is not a very strong electron acceptor. This can be seen by comparing the number of electrons donated by the oxo to each Co neighbor at a given value of the unscreened orbital energy. To compute this quantity, we first determine the delocalized lone pairs ![[small phi, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e2.gif) i by projecting the three non-constrained quasiatomic orbitals ϕi of the oxo onto the density matrix P
i by projecting the three non-constrained quasiatomic orbitals ϕi of the oxo onto the density matrix P
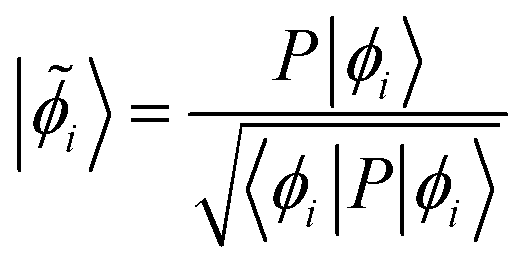 | (9) |
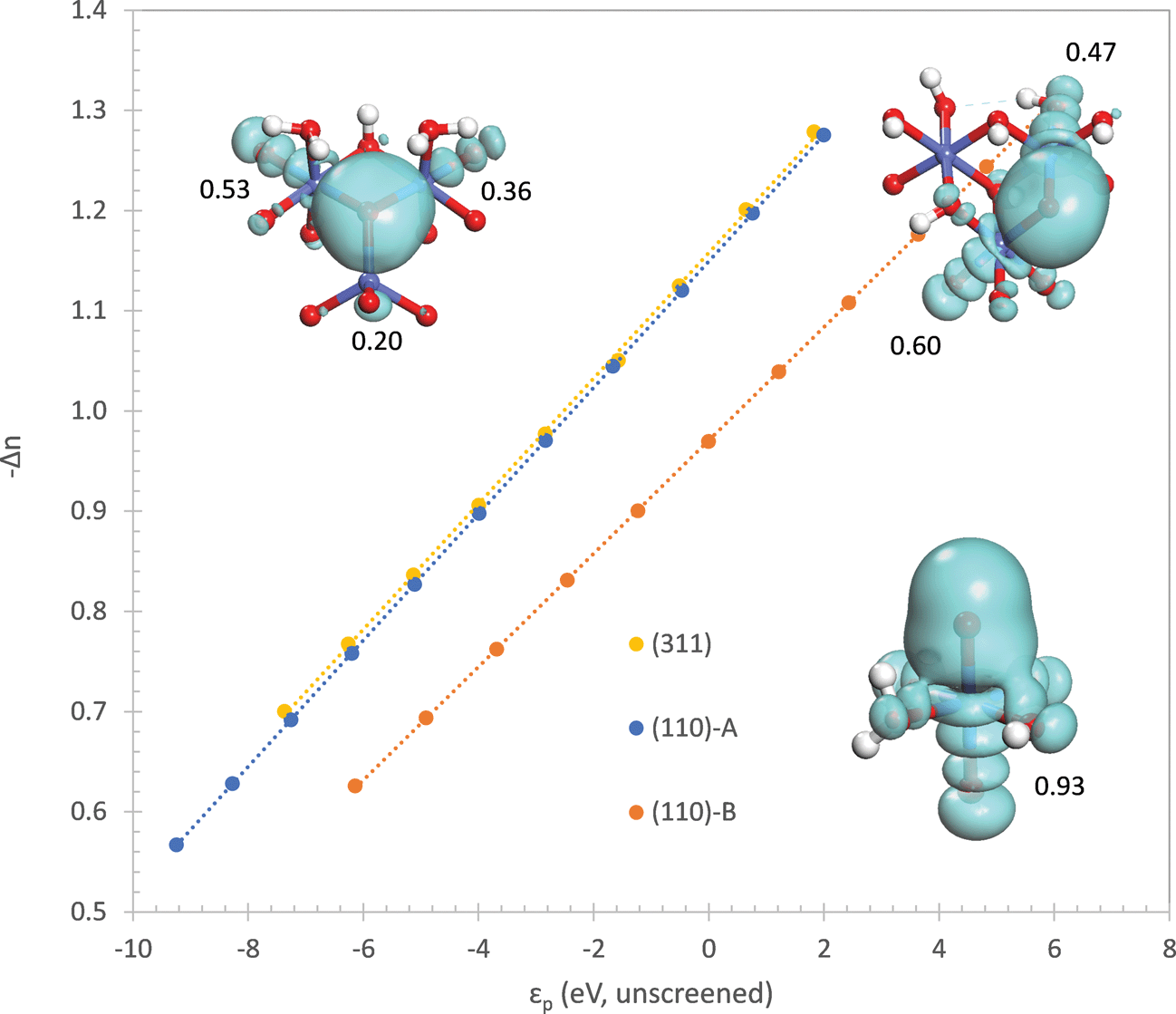 | ||
| Fig. 8 Number of electrons donated from lone pairs on the oxo to neighboring atoms as a function of the unscreened energy of the 2p orbital containing the hole on the oxo (see text). The total density distribution of the delocalized lone pairs is shown for each site along with the number of electrons donated to each Co neighbor. | ||
We then calculate the population of this orbital on each Co neighbor
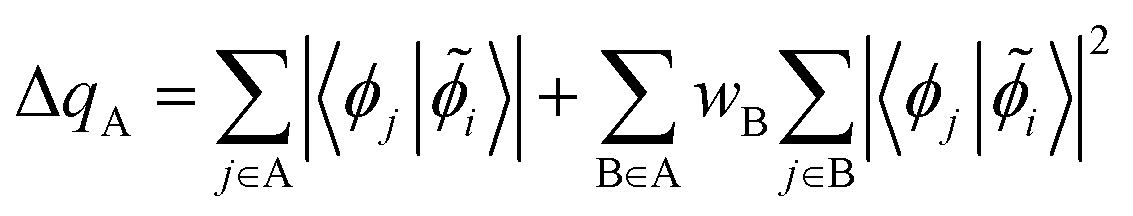 | (10) |
 in eqn (10). The results of this analysis are given in Fig. 8 where we can see that only 0.20 electrons are donated to the Co(II) coordinated to the μ3-oxo. This confirms that the Co(II) is a weak electron acceptor compared to the Co(III) and Co(IV) cations which withdraw 0.36 and 0.53 electrons, respectively.
in eqn (10). The results of this analysis are given in Fig. 8 where we can see that only 0.20 electrons are donated to the Co(II) coordinated to the μ3-oxo. This confirms that the Co(II) is a weak electron acceptor compared to the Co(III) and Co(IV) cations which withdraw 0.36 and 0.53 electrons, respectively.
The above analysis makes it clear that the main effect of coordinating the oxo by an additional Co(II) cation in the (311) surface is to increase the attractive Madelung potential on the oxo. This lowers the energy of the orbitals on the oxo and increases the energy needed to localize a hole in one of these orbitals. The presence of the Co(II) cation coordinating the oxo does not lead to a significant increase in electron withdrawal from the oxo. In contrast, the additional Co(III) coordinating the μ2-oxo on the (110)-A surface leads to significantly greater electron withdrawal compared to the η-oxo on the (110)-B surface coordinated by only a single Co(IV). As with the Co(II) coordinated to the μ3-oxo on the (311) surface, this Co(III) also increases the attractive Madelung potential on the μ2-oxo compared to the η-oxo on the (110)-B surface. Thus, localization of the hole on the μ2-oxo requires significantly more energy than localization on the η-oxo due to both an increase in the attractive Madelung potential on the μ2-oxo as well as increased electron withdrawal by the Co(III) cation.
Quantum chemical relationship between water addition and oxidation of the active site
Now that we have developed a profound quantum chemical understanding of the O–O bond formation process occurring during water addition, we apply a similar approach to understanding the oxidation step that occurs in the catalytic cycle (Scheme 1) prior to water addition and which determines the overpotential at which the site becomes active. It will be seen that the energetics of the two processes are both largely controlled by the energy to localize a hole on the oxo. This then, is responsible for the correlation observed between the intrinsic reactivity of a site for water addition and the overpotential at which it becomes oxidized to the active state that we observed in our previous work.15The relationship between oxidation and O–O bond formation can be seen by examining the oxidation process in the same context as O–O bond formation. The latter process was seen to proceed by localization of a hole on the oxo followed by formation of a σ bond with a hydroxyl radical. Likewise, as shown in Fig. 9, the oxidation process can be thought of as involving cleavage of an O–H σ bond to give a hydrogen atom and a hole on the resulting oxo which then transfers to a neighboring Co cation. Thus, the reverse of the oxidation process can be thought of as O–H bond formation between an oxo and a hydrogen atom, analogous to the O–O bond formation between the oxo and a hydroxyl. If we decompose the O–H bond formation process in the same way as the O–O bond formation process in Fig. 6, we see that the hole localization energy is exactly the same for the two processes. The rehybridization energy is much higher for O–H bond formation compared to O–O bond formation since the resulting hybrid has significantly higher 2s character (as ≈ 0.6 vs. as ≈ 0.3) although the trend is still similar, with the rehybridization energy increasing as coordination of the oxo decreases. The σ bond formation energy is much larger than the O–O bond formation energy but is similar for the three different sites and thus is not the main reason for the differences in O–H bond formation energy.
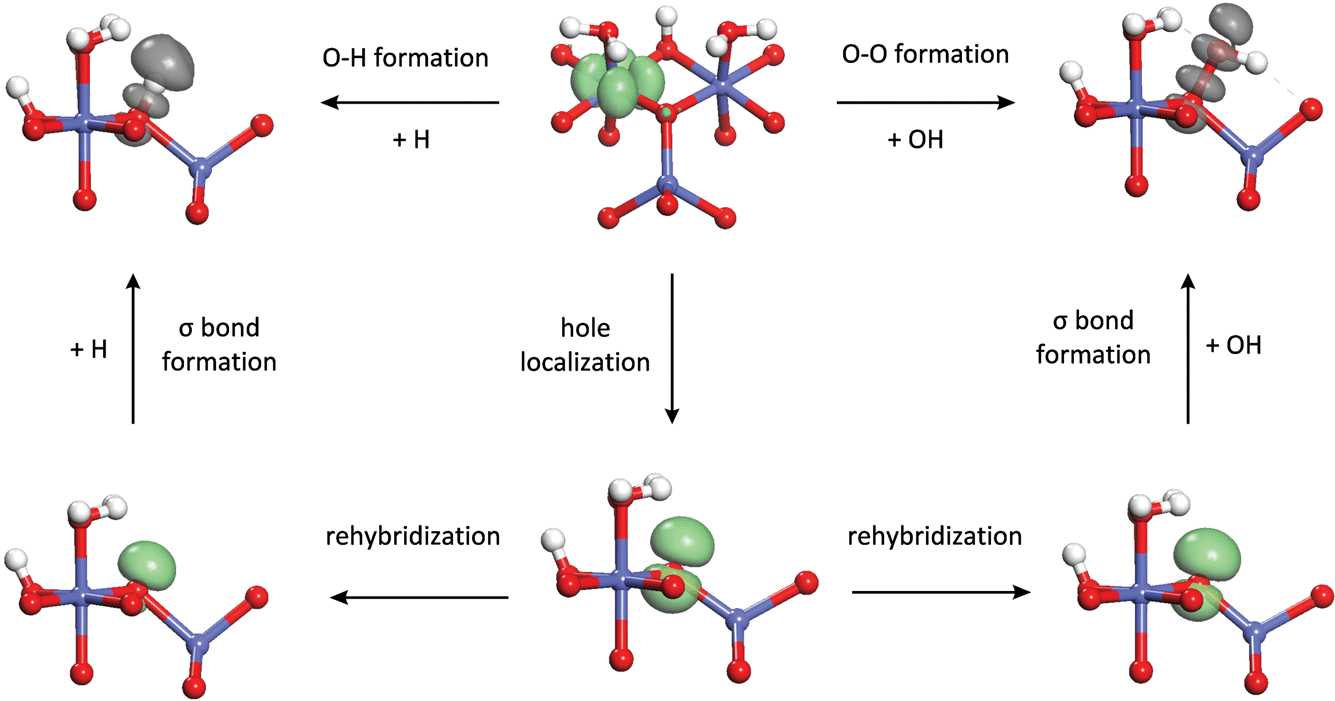 | ||
| Fig. 9 Comparison of the changes in electronic structure occurring during O–O bond formation during water addition and O–H bond formation during the reverse of the oxidation step preceding water addition. | ||
From the comparison between the O–O and O–H bond formation processes, it can be concluded that the correlation between O–H bond formation (reverse of oxidation) energy and O–O bond formation energy is based on the similar requirements for localization of a hole on the oxo in both processes. The hole localization requires the most energy on the (311) μ3-oxo so that O–O bond formation is least favorable on this site but oxidation (O–H bond cleavage) is easier than on the other sites. As a result, the (311) μ3-oxo is the most active site at low overpotentials, as shown in Fig. 1. The hole localization requires less energy on the (110)-A μ2-oxo and (110)-B η-oxo but oxidation is harder so that these sites lead to higher TOFs than the (311) site at high overpotentials. A similar result was observed by Busch et al. who found a roughly linear correlation between the endothermicity of the oxidation step and the exothermicity of the subsequent O–O bond formation step on a binuclear model catalyst containing different 3d transition metal centers.27 Such a correlation was also found by Man et al. between the binding energies of hydroxyl and hydroperoxyl species to oxide surfaces, leading to similar behavior.28
Design principles for the ideal OER active site at a given overpotential
We would now like to apply the quantum chemical understanding we have developed concerning the water addition reaction catalyzed by different active sites on Co3O4 to gain insights into how to design the optimal OER catalyst. From the results showing the TOF as a function of applied overpotential in Fig. 1, it is obvious that it is difficult to say what is the optimal active site since this changes with overpotential. By going to higher overpotentials, one can in general achieve higher TOFs but at the cost of electrochemical efficiency. Thus, one will always have to consider the tradeoff between activity and efficiency. Here, we will focus on the design of the optimal catalyst (the one giving the highest TOF) at a given overpotential.To calculate the TOF as a function of overpotential, we assume that water addition is the rate limiting step (which is true at overpotentials above 0.53 V (ref. 15)) and that the active site exists predominantly in one particular state, the dominant resting state i, which has a lower free energy than all of the other possible states. We also make the assumption that water addition occurs through a single transition state that proceeds from a state j and that state j (the active state) is in quasi-equilibrium with the dominant resting state i. The TOF is then given by the following expression15
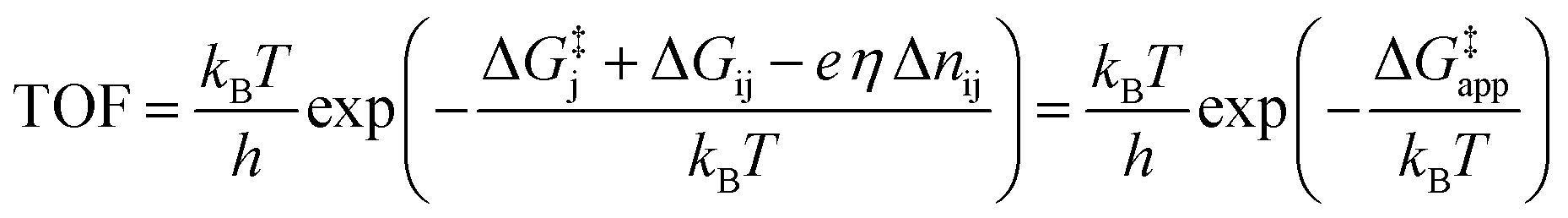 | (11) |
For simplicity, we consider a model consisting of the four possible states of the active site depicted in Scheme 2. The first of these states (0) corresponds to the active state j. In the next two states, one of the Co(IV) cations in the dual-Co active sites is reduced to Co(III) or the Co(V) in the single-Co site is reduced to Co(IV) and a proton is added to either the basic oxygen that deprotonates the water during the PCET part of water addition (1) or the oxo undergoing water addition (2). In the remaining state (3), both Co(IV) cations in the dual-Co site or the Co(V) in the single-Co site are reduced to Co(III) and protons are added to both the oxo and the basic oxygen.
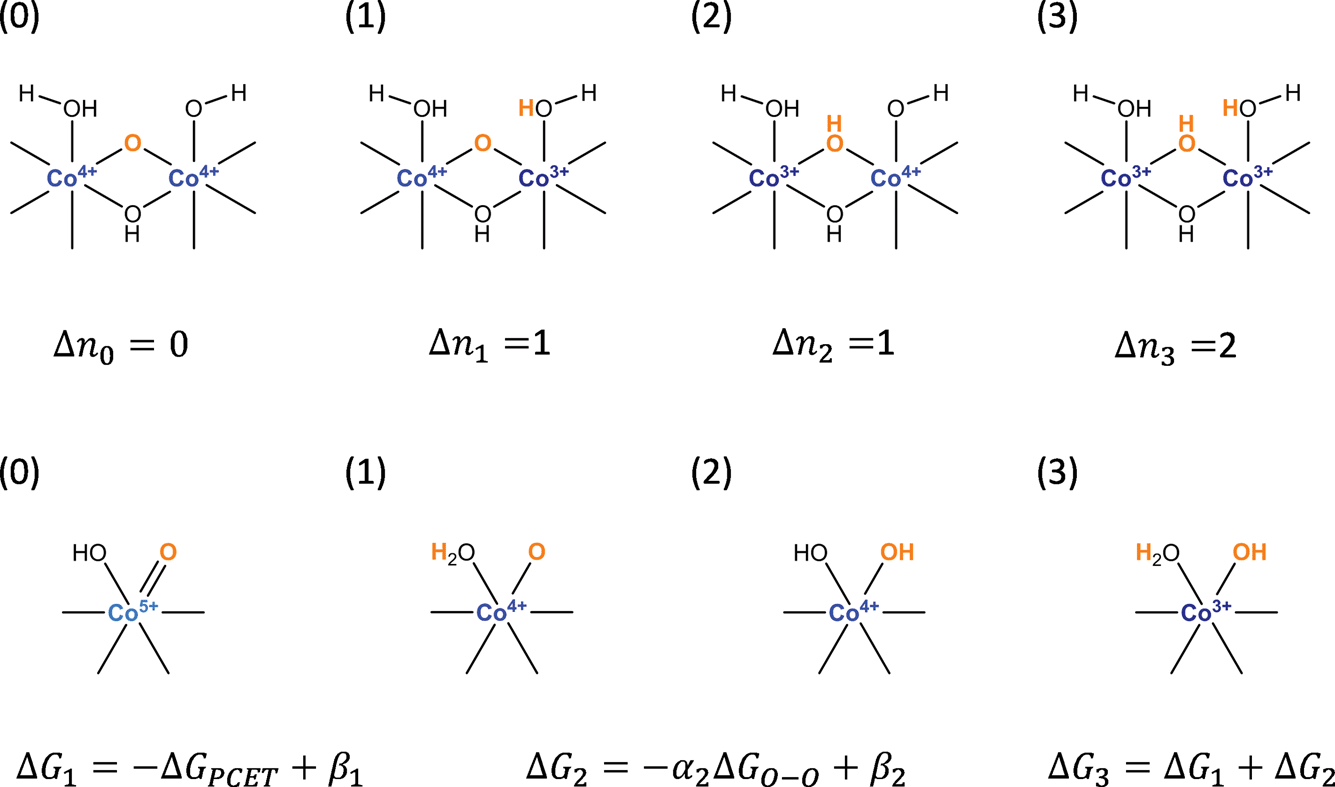 | ||
| Scheme 2 States involved in the two oxidation steps preceding water addition along with definitions of parameters appearing in eqn (17). | ||
The free energy differences ΔGij and values of Δnij in eqn (11) for the four states are given in Table 1. In defining the free energy difference ΔG3 between state (3) and the active state (0) we have made the further assumption that the oxidations occurring between states (1) or (2) and the active state are additive so that ΔG3 = ΔG1 + ΔG2. With these definitions, the apparent activation free energy in eqn (11) can be written as
| ΔG‡app = ΔG‡j + max{(ΔG1 − eη), 0} + max{(ΔG2 − eη), 0} | (12) |
| (001) | (311) | (110)-A | (110)-B | (110)-Bf | |
|---|---|---|---|---|---|
| a η ox,1 = ΔG1/e. b η ox,2 = ΔG2/e. c η ox,id = (ηox,1 + ηox,2)/2. d η ox,corr = ηox + ΔG0/e. e Actual calculated oxidation potential. f More oxidized (110)-B surface. | |||||
| η ox,1 | 0.58 | 0.50 | 0.68 | 0.58 | 0.64 |
| η ox,2 | 0.25 | 0.21 | 0.63 | 1.04 | 1.01 |
| η ox,id | 0.41 | 0.36 | 0.66 | 0.81 | 0.83 |
| η ox,corr | 0.66 | 0.50 | 0.73 | 1.00 | 1.05 |
| η ox | 0.42 | 0.50 | 0.73 | 0.82 | 0.87 |
From this expression, we can see that if one of the two oxidations converting states (1) and (2) to the active state has a positive free energy, then the active site will exist predominantly in the reduced state and the free energy to oxidize this state to the active state must be added to the intrinsic activation barrier. If both oxidation free energies are positive, then the active site will exist predominantly in state (3) and both oxidation free energies must be added to the intrinsic activation barrier.
We now make an approximation that the intrinsic free energy activation barrier is linearly related to the free energy of the water addition reaction ΔGrxn by a Brønsted–Evans–Polanyi relationship
| ΔG‡ = α0ΔGrxn + β0 | (13) |
A plot showing this relationship is included in the ESI† and gives values of α0 = 0.42 and β0 = 0.68 eV. As shown previously, the reaction free energy of water addition is the sum of the PCET free energy ΔGPCET and the O–O bond formation energy ΔGO–O
| ΔGrxn = ΔGPCET + ΔGO–O | (14) |
The PCET process is simply the reverse of the oxidation process occurring between state (1) and the active state so that
| ΔGPCET = −ΔG1 + β1 | (15) |
| ΔGO–O = −α2ΔG2 + β2 | (16) |
This is based on the relationship between the two quantities discussed earlier and a plot showing the relationship is included in the ESI.† The correlation is not excellent, but there is definitely a trend which leads to values of the parameters in eqn (16) of α2 = 1.00 and β2 = −1.34 eV. Substituting eqn (13)–(16) into eqn (12) finally gives
| ΔG‡app/e = −α′1ηox,1 − α′2ηox,2 + β + max{(ηox,1 − η), 0} + max{(ηox,2 − η), 0} | (17) |
| ηox,1 = ΔG1/e |
| ηox,2 = ΔG2/e |
| α′1 = α0 = 0.42 |
| α′2 = α0α2 = 0.42 |
| β = β0 + α0β1 + α0β2 = 1.05 eV |
This expression allows us to define the optimal active site at a given overpotential, which is illustrated in Fig. 10. When both of the oxidation potentials of the active site (ηox,1 and ηox,2) are less than the overpotential, the site exists predominantly in the active state so that the last two terms on the RHS of eqn (17) are zero. In this case, increasing either of the oxidation potentials is correlated with a decrease in the intrinsic activation barrier by the first two terms on the RHS of eqn (17) so that the TOF increases. If one or both of the oxidation potentials is greater than the overpotential, then the active site will exist predominantly in a reduced state so that the oxidation energy must be added to the intrinsic activation barrier. Since α′1 and α′2 are both less than unity, the increase in the last two terms on the RHS of eqn (17) corresponding to an increase in the oxidation potentials will dominate the decrease in intrinsic activation barrier in the first two terms, leading to a decrease in the TOF. This means that the active site giving the maximum TOF at a given overpotential, the Sabatier maximum, will have both oxidation potentials exactly equal to the overpotential.
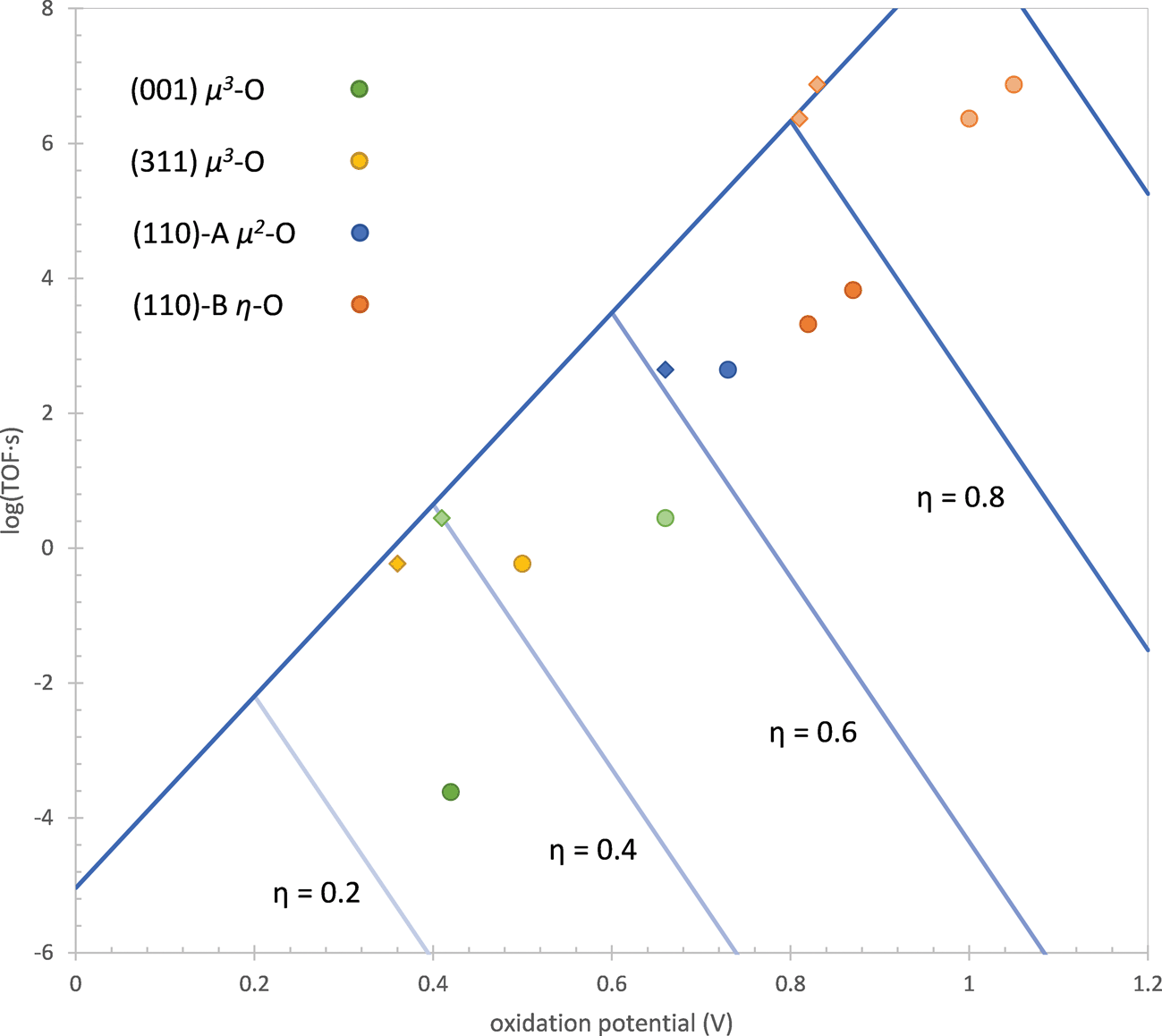 | ||
| Fig. 10 TOF of water addition as a function of the oxidation potential of the active site at different overpotentials η (V) for the ideal site based on eqn (17). The TOF and oxidation potentials of the actual sites are also shown (dark colored circles). Light colored circles for the (001) and (110)-B sites show the TOF and oxidation potentials when the lower energy state preceding the active state is not considered. Diamonds show the performance of the sites if both oxidation potentials depicted in Scheme 2 are equal. Two active states of the site on the (110)-B surface are shown, one operating at overpotentials above 0.86 V and the other operating at overpotentials lower than this. | ||
Fig. 10 also shows the performance of the active sites on the three surfaces examined in this work plus the dual-Co site on the (001) surface examined in our previous work15 in comparison to the ideal performance. The values of the oxidation potentials used to determine these results are given in Table 1. The oxidation potential is taken as the potential at which the active site is first oxidized to the active state for water addition (the first point where the TOF in Fig. 1 becomes flat with respect to overpotential) and the TOF is calculated at this overpotential. One can see that all of the real active sites are less active than the ideal active site. There are two causes of this, the first being that the two oxidation potentials are not equal and the second being the existence of a state that has the same degree of oxidation as the active state but has a lower free energy. The first of these affects all four active sites while the second affects only the (001) and (110)-B sites. We will now discuss these causes of lower activity in more detail.
For the sites on the (001) and (110)-B surfaces, the lower activity is partially due to the existence of a state that has the same degree of oxidation as the active state but has a lower free energy. As a result, the intrinsic activation barrier increases by an amount equal to the free energy difference between these two states. Since the two states have the same degree of oxidation, increasing the overpotential has no effect on this free energy difference. On the (001) site, the lower energy state has the proton from the μ3-OH of the active state attached to the η-OH instead (shown in the ESI†). The free energy of this state is 0.24 eV below that of the active state. This was discussed in detail in our previous work15 and is due to the high acidity of the μ3-OH. The lower energy state on the (110)-B site has the CoO fragment in the triplet state rather than the active singlet state, which is lower in energy by 0.18 eV. In Fig. 10, points for these two sites are also plotted in which the lower energy state has been removed. This is done by increasing the oxidation potentials and decreasing the activation barriers by the energy difference ΔG0 between the two states (0.24 eV for the (001) site and 0.18 eV for the (110)-B site). This moves the points for these two sites closer to the ideal activity.
Even after eliminating the performance decrease due to the presence of lower energy states for the (001) and (110)-B sites, the activities for these sites and the other two sites are still about 2–3 orders of magnitude lower than the ideal activity. This is caused by the two oxidation potentials depicted in Scheme 2 and listed in Table 1 not being equal. The overpotential at which the site becomes oxidized to the active state is determined by the higher of these two potentials. If one of them is lower than the other, then the intrinsic activation barrier is not as low as it would be if the lower oxidation potential was equal to the higher oxidation potential. This is similar to the concept put forward in the purely thermodynamic model of the OER by Rossmeisl, Nørskov, and others29 where it is proposed that the optimal catalyst will have the same oxidation potential for all four oxidations occurring during the reaction. It is interesting that this aspect of the simpler model still appears in our more complete kinetic model.
Fig. 10 shows the activities of the four sites at an oxidation potential equal to the average of the two values ηox,1 and ηox,2 in Table 1 (called the ideal oxidation potential, ηox,id). Except for the (110)-A site, all of the other sites have activities very close to the ideal activity, demonstrating that this is indeed the primary cause of the lower activity. The (110)-A site has an activity that is still about one order of magnitude lower than the ideal activity, possibly due to steric effects since the bridging oxo and hydroxo ligands on this site are more rigid than the terminal hydroxo and water ligands on the other sites.
Conclusions
We have examined the mechanism and quantum chemistry of the nucleophilic addition of water to oxo species on three different surface terminations of Co3O4. This particular elementary step was examined because it was found to be the rate controlling step on these sites over the largest range of applied overpotentials in our previous study.15 We find that the intrinsic activation barrier and reaction energy of water addition decreases as the coordination of the oxo decreases, being highest for a μ3-oxo on the (311) dual-Co site and lowest for an η-oxo on the (110)-B single-Co site, with a μ2-oxo on the (110)-A dual-Co site lying in between. Water addition occurs on all three sites by the coupled transfer of a proton and electron (PCET) from the attacking water to a basic oxygen and a Co(IV) on the active site followed by formation of an O–O σ bond between the resulting hydroxyl and the oxo. Differences in the energetics of the O–O bond formation step are responsible for the differences in intrinsic reactivity between the three sites, with this step being more difficult on the two dual-Co sites due to the required transfer of a hole from a Co(IV) center to the oxo prior to O–O bond formation. On the single-Co site, the hole already resides on the oxo so that O–O bond formation occurs concertedly with the PCET step. Furthermore, O–O bond formation following the PCET has a considerable activation barrier on the μ3-oxo of the (311) site while it has only a small activation barrier on the μ2-oxo of the (110)-A site.To determine the origin of the more favorable O–O bond formation step on sites with a less coordinated oxo, we examined the energy required to localize a hole in a 2p orbital on the oxo on each of the sites. This energy was found to be highest on the least active (311) μ3-oxo and lowest on the most active (110)-B η-oxo, with the (110)-A μ2-oxo lying in between. By examining the orbital energies in a model process involving the transfer of a hole from a Co 3d orbital to an O 2p orbital, we conclude that the higher hole localization energy on the μ3-oxo compared to the μ2-oxo is due to a more attractive Madelung potential on the μ3-oxo from the additional Co(II) cation coordinating it. The lower hole localization energy on the η-oxo compared to the μ2-oxo is also due to a more attractive Madelung potential on the more highly coordinated μ2-oxo as well as greater electron withdrawal from the oxo by the two Co neighbors of the μ2-oxo compared to the single Co neighbor of the η-oxo.
We used the same technique to examine the hydrogen atom removal occurring during oxidation of the active site prior to water addition in order to identify the quantum chemical origin of the correlation seen between oxidation potential and intrinsic activity for water addition. It is seen that the hydrogen atom addition occurring during the reverse of the oxidation process involves similar changes in electronic structure as the hydroxyl radical addition occurring during O–O bond formation. Both processes involve localization of a hole on the oxo followed by σ bond formation. The correlation between the energies of the two processes is related to the hole localization energy, which is common for both.
Based on this correlation between intrinsic water addition reactivity and oxidation potential, we investigate the properties leading to the most active catalyst at a given applied potential. We find that for the ideal active site at a given applied potential, the oxidation potentials of the two steps preceding water addition must be equal to each other and to the applied potential. All of the real active sites examined in this work have sub-optimal activity due to the two oxidation potentials being unequal and, on some sites, the existence of an inactive state that has the same degree of oxidation but a lower free energy than the active state. This suggests a possible strategy for engineering an active site with improved activity.
Acknowledgements
C. P. acknowledges partial support from NRSC-Catalysis and the Institute for Complex Molecular Systems at the Technical University of Eindhoven and the Solar Technologies Go Hybrid initiative of the State of Bavaria. We also acknowledge NWO for access to supercomputing facilities.References
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110(11), 6446–6473 CrossRef CAS PubMed.
- A. Marshall, B. Børresen, G. Hagen, M. Tsypkin and R. Tunold, Energy, 2007, 32(4), 431–436 CrossRef CAS.
- E. Fabbri, A. Habereder, K. Waltar, R. Kötz and T. J. Schmidt, Catal. Sci. Technol., 2014, 4(11), 3800–3821 CAS.
- W. T. Hong, M. Risch, K. a. Stoerzinger, A. Grimaud, J. Suntivich and Y. Shao-horn, Energy Environ. Sci., 2015, 8, 1404–1427 CAS.
- M. E. G. Lyons and M. P. Brandon, J. Electroanal. Chem., 2010, 641(1–2), 119–130 CrossRef CAS.
- M. Dinca, Y. Surendranath and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2010, 107(23), 10337–10341 CrossRef CAS PubMed.
- J. Suntivich, K. J. May, H. a. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334(6061), 1383–1385 CrossRef CAS PubMed.
- D. G. Nocera, Acc. Chem. Res., 2012, 45(5), 767–776 CrossRef CAS PubMed.
- M. Zhang and H. Frei, Catal. Lett., 2015, 145(1), 420–435 CrossRef CAS.
- F. Jiao and H. Frei, Angew. Chem., Int. Ed., 2009, 48(10), 1841–1844 CrossRef CAS PubMed.
- D. A. Lutterman, Y. Surendranath and D. G. Nocera, J. Am. Chem. Soc., 2009, 131(11), 3838–3839 CrossRef CAS PubMed.
- M. Bajdich, M. García-mota, A. Vojvodic, J. K. Nørskov and A. T. Bell, J. Am. Chem. Soc., 2013, 135(36), 13521–13530 CrossRef CAS PubMed.
- M. García-mota, M. Bajdich, V. Viswanathan, A. Vojvodic, A. T. Bell and J. K. Nørskov, J. Phys. Chem. C, 2012, 116(39), 21077–21082 Search PubMed.
- M. Zhang, M. de Respinis and H. Frei, Nat. Chem., 2014, 6(4), 362–367 CrossRef CAS PubMed.
- C. P. Plaisance and R. A. van Santen, J. Am. Chem. Soc., 2015, 137(46), 14660–14672 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54(16), 11169–11186 CrossRef CAS.
- A. J. Cohen, P. Mori-Sanchez and W. Yang, Science, 2008, 321(5890), 792–794 CrossRef CAS PubMed.
- V. I. Anisimov and O. Gunnarsson, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 43(10), 7570–7574 CrossRef CAS.
- A. I. Liechtenstein, V. I. Anisimov and J. Zaanen, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 52(8), R5467–R5470 CrossRef CAS.
- V. I. Anisimov, F. Aryasetiawan and A. I. Lichtenstein, J. Phys.: Condens. Matter, 1997, 9(4), 767–808 CrossRef CAS.
- M. T. M. Koper, Phys. Chem. Chem. Phys., 2013, 15(5), 1399–1407 RSC.
- Y. Surendranath, M. W. Kanan and D. G. Nocera, J. Am. Chem. Soc., 2010, 132(14), 16501–16509 CrossRef CAS PubMed.
- X. Qian, J. Li, L. Qi, C.-Z. Wang, T.-L. Chan, Y.-X. Yao, K.-M. Ho and S. Yip, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78(24), 245112 CrossRef.
- A. E. Reed and F. Weinhold, J. Chem. Phys., 1985, 83(4), 1736 CrossRef CAS.
- F. Weinhold and C. R. Landis, Valency and Bonding: A Natural Bond Orbital Donor–Acceptor Perspective, Cambridge University Press, 2005 Search PubMed.
- J. P. Foster and F. Weinhold, J. Am. Chem. Soc., 1980, 102(24), 7211–7218 CrossRef CAS.
- M. Busch, E. Ahlberg and I. Panas, Catal. Today, 2013, 202, 114–119 CrossRef CAS.
- I. C. Man, H.-Y. Su, F. Calle-Vallejo, H. A. Hansen, J. I. Martínez, N. G. Inoglu, J. Kitchin, T. F. Jaramillo, J. K. Nørskov and J. Rossmeisl, ChemCatChem, 2011, 3(7), 1159–1165 CrossRef CAS.
- J. Rossmeisl, Z.-W. Qu, H. Zhu, G.-J. Kroes and J. K. Nørskov, J. Electroanal. Chem., 2007, 607(1–2), 83–89 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5fd00213c |
| This journal is © The Royal Society of Chemistry 2016 |