 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hydrothermal synthesis of bi-functional nanostructured manganese tungstate catalysts for selective oxidation†
Xuan
Li
ab,
Thomas
Lunkenbein
a,
Jutta
Kröhnert
a,
Verena
Pfeifer
ac,
Frank
Girgsdies
a,
Frank
Rosowski
bd,
Robert
Schlögl
a and
Annette
Trunschke
*a
aDepartment of Inorganic Chemistry, Fritz-Haber-Institut der Max-Planck-Gesellschaft, Faradayweg 4-6, 14195, Berlin, Germany. E-mail: trunschke@fhi-berlin.mpg.de
bUniCat-BASF Joint Lab, Technische Universität Berlin, 10623, Berlin, Germany
cHelmholtz-Zentrum Berlin für Materialien und Energie GmbH, Elektronenspeicherring BESSY II, Albert-Einstein-Str. 15, 12489 Berlin, Germany
dBASF SE, Process Research and Chemical Engineering, Heterogeneous Catalysis, Carl-Bosch-Straße 38, 67056, Ludwigshafen, Germany
First published on 8th December 2015
Abstract
The mechanism of C–H activation in selective oxidation reactions of short-chain alkane molecules over transition metal oxides is critically affected by the balance of acid–base and redox sites at the surface of the catalyst. Using the example of manganese tungstate we discuss how the relative abundance of these sites can be controlled via synthetic techniques. Phase-pure catalysts composed of the thermodynamic stable monoclinic MnWO4 phase have been prepared using hydrothermal synthesis. Variation of the initial pH value resulted in rod-shaped nano-crystalline MnWO4 catalysts composed of particles with varying aspect ratio. The synthesis products have been analysed using transmission electron microscopy, X-ray diffraction, infrared, and photoelectron spectroscopy. In situ Raman spectroscopy was used to investigate the dissolution–re-crystallization processes occurring under hydrothermal conditions. Ethanol oxidation was applied to probe the surface functionalities in terms of acid–base and redox properties. Changes in the aspect ratio of the primary catalyst particles are reflected in the product distribution induced by altering the fraction of acid–base and redox sites exposed at the surface of the catalysts in agreement with the proposed mechanism of particle growth by re-crystallization during ageing under hydrothermal conditions.
Introduction
Metal oxides are widely used as heterogeneous catalysts in the synthesis of chemicals, and in energy conversion and storage applications.1–4 High performance, selectivity, and stability of oxides in heterogeneous catalysis are bound to the homogeneity of the solid.5 In complex reactions, such as selective oxidation of hydrocarbons, multi-functionality is necessarily required and can be achieved either chemically or by nano-structuring.6,7Solvothermal techniques have been efficiently used in the preparation of metastable phases or oxides with particular morphological properties.8–13 However, the underlying preparation strategies are often based on experience and parameter variation. Targeted design of metal oxide catalysts with predictable functionalities needs a deeper understanding of the chemistry in precursor solutions and during nucleation, growth, and ageing.
In situ Raman spectroscopy has proven beneficial in the investigation of condensation reactions of transition metal oxide species occurring at elevated temperature and pressure inside an autoclave.14,15 Our recent study concerning the speciation of molybdates in aqueous media revealed that the molecular structure of the oxo-anions in the temperature range between 100 and 200 °C differs significantly from that at room temperature at comparable concentration and pH values.15 Implicit understanding of the known condensation chemistry of molybdates under ambient conditions is, therefore, not beneficial in terms of knowledge-based planning of the hydrothermal synthesis of molybdenum oxide-based catalysts. Likewise, systematic in situ spectroscopic studies of the synthesis of complex metastable mixed oxide phases, like the so-called M1 phase of MoVTeNb oxide, resulted in the development of modular preparation techniques that yield the desired phase more effectively in shorter synthesis time with improved catalytic properties.14
Herein we present in situ Raman spectroscopic and transmission electron microscopy studies that provide insight into the mechanism of particle growth and re-crystallization during the synthesis of the thermodynamic stable monoclinic manganese tungstate phase in wolframite-type structure (ICSD 67906).16 Nano-structuring of manganese tungstate features a feasible strategy to control bi-functional properties of the mixed metal oxide with implications on the selectivity in oxidation reactions applied to upgrade alkanes. We demonstrate the viability of such a strategy by studying rod-shaped nano-crystalline MnWO4 catalysts consisting of particles with varying aspect ratio (AR). The particle morphology was controlled via the concentration of OH− ions under hydrothermal synthesis conditions. Oxidation of ethanol was chosen to probe the surface chemistry of the synthesized materials in a catalytic reaction. The substrate molecule may undergo multiple pathways depending on the nature of the active sites at the catalyst surface. Generally, the alcohol is oxidized to the aldehyde at redox active centres whereas dehydration of ethanol to ethylene or the formation of diethyl ether indicate surface sites that facilitate acid/base reactions.
Experimental
Hydrothermal synthesis
The hydrothermal synthesis of MnWO4 was performed in an analytical autoclave HPM-PT-040 (Premex Reactor GmbH) described before,15 adopting a synthesis method that has been reported previously.16 In the first step, a 0.2 M aqueous solution of Mn(NO3)2 (Mn(NO3)2·4H2O, 98%, Roth) was added to a 0.2 M aqueous solution of Na2WO4 (Na2WO4·2H2O, 99%, Sigma Aldrich) while stirring, leading to a mixed solution of pH = 6.7. Subsequently, the pH of the mixed solution was adjusted to 6.3, 8.0, 9.1, and 9.9 by adding appropriate amounts of 0.1 M HNO3 (64–66%, Sigma Aldrich) or 0.1 M NaOH (98%, Alfa Aesar). The mixtures were transferred to the autoclave and the temperature was raised from 20 °C to 180 °C at a rate of 5 °C min−1. The synthesis temperature was kept at 180 °C for 12 h. During hydrothermal synthesis the pH was recorded using a pH probe (ZrO2 probe Model A2 and Ag/AgCl reference electrode, both with a 1/2” outer tubing made from Hastelloy C-276; Corr Instruments). The pH probes were calibrated by use of four buffer solutions at the given reaction temperatures prior to the experiments. At the same time the Raman spectra of the synthesis gels were recorded using a Raman probe (RAMAN RXN1, immersion optic 1/4” OD (HC-276); Kaiser Optical Systems). After cooling down the gel at a rate of 5 °C min−1, the products of the hydrothermal synthesis were filtered by centrifugation and washed twice with de-ionized water (MilliPore®). In the final step, the solids were dried in a muffle furnace in air at 80 °C for 12 h. Depending on the pH value before hydrothermal treatment, yellowish to brownish solids were collected. The solids were annealed in argon (flow rate 50 mL min−1) at 400 °C (heating rate 5 °C min−1) for 2 h using a rotary tube furnace (XERION) resulting in five phase-pure MnWO4 catalysts with different mean aspect ratios (AR) characterized by the identification numbers 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 112 (AR = 1.5), 19113 (AR = 1.7), 19114 (AR = 3.2), 19251 (AR = 3.9), and 19116 (AR = 5.1). The catalysts are called ARx.x, where x.x corresponds to the mean aspect ratio of the particles in the materials after thermal activation as determined by analysis of transmission electron microscopy images.
112 (AR = 1.5), 19113 (AR = 1.7), 19114 (AR = 3.2), 19251 (AR = 3.9), and 19116 (AR = 5.1). The catalysts are called ARx.x, where x.x corresponds to the mean aspect ratio of the particles in the materials after thermal activation as determined by analysis of transmission electron microscopy images.
Characterization of catalysts
Transmission electron microscopy (TEM) studies were conducted using a Philips CM200 FEG transmission electron microscope operating at 200 kV. High resolution TEM (HRTEM) and high resolution high angle annular dark field scanning transmission electron microscopy (HAADF-STEM) were performed using a Cs corrected FEI TITAN 80–300 operated at 300 kV. TEM samples were prepared by drop deposition from ethanolic suspensions onto lacey-carbon coated Cu grids, and by cross section preparation.Field emission scanning electron microscopy (FESEM) was carried out with a Hitachi S4800 instrument operating at 5 kV.
The X-ray diffraction (XRD) measurements were performed in Bragg–Brentano geometry on a Bruker AXS D8 Advance theta/theta diffractometer, using Ni filtered Cu Kα radiation and a position sensitive LynxEye silicon strip detector. The XRD data were evaluated by whole powder pattern fitting according to the Rietveld method as implemented in the TOPAS software [version 4.2, copyright 1999–2009 Bruker AXS]. During the routine fitting, which uses an isotropic peak width model (i.e. the diffraction profile widths are described as a smooth function of the diffraction angle, independent of hkl), systematic peak profile mismatches of varying degrees were observed. With the anisotropic crystallite shape observed by electron microscopy in mind, we developed an appropriate anisotropic (i.e., hkl dependent) peak width model. An efficient model was obtained by modifying the phenomenological model published by Stephens.17 Due to the macro language implemented in TOPAS, user defined peak models can be implemented easily. The original Stephens model, which was derived to describe anisotropic strain broadening, did not work well with our data. Since we expected anisotropic crystallite size to be the predominant peak broadening factor in our case, we replaced the angular dependent term tan(θ) (representing strain) of the original Stephens model with a 1/cos(θ) (i.e., size) term, while retaining the hkl dependent expression. In addition to a good overall fit, this modified model allowed us to obtain the nominal crystallite size for different crystal directions. It should be noted that such values represent volume weighted average lengths of unit cell columns, LVol–IB. This includes averaging over parallel columns of different lengths within crystallites (shape dependent), as well as averaging over different crystallites of possibly different sizes (size distribution dependent). Thus, the reported LVol–IB values cannot be directly compared to the physical dimensions of discrete crystallites as, e.g., observed by electron microscopy. Nevertheless, the XRD derived dimensions may be considered to represent the (volume weighted) average crystallite morphology.
The specific surface area was measured in a volumetric N2 adsorption device (Autosorb-6-B, Quantachrome) at the temperature of liquid nitrogen. The sample was degassed under dynamic vacuum at a temperature of 300 °C for 2 h prior to adsorption. The relative N2 pressure was varied and 11 data points were measured. The linear range of the adsorption isotherm (p/p0 = 0.05–0.3) was considered to calculate the specific surface area according to the BET method.
Ethanol oxidation was performed at atmospheric pressure in a feed composed of 30.2 mL of N2, 2.8 mL of O2 and 1.2 mL of CH4. The feed was passed through a saturator at 15 °C to achieve a concentration of 4 vol% ethanol. A laboratory quartz U-tube fixed bed reactor (4 mm inner diameter, 6 mm outer diameter, 26 cm length) was used, which contained 200 mg of the catalyst previously pressed and sieved to a particle size of 250–355 μm. Gas analysis was performed using online gas chromatography (GC 6890A, Agilent) equipped with two channels. A combination of two capillary columns (GS-Carbonplot and Plot Mole Sieve 5A) in connection with a thermal conductivity detector (TCD) was used to analyse the permanent gases CO2, O2, N2 and CO. A combination of two capillary columns (HP-FFAP and HP Plot Q) connected to a flame ionization detector (FID) was applied to analyse alkanes, olefins and oxygenates.
Transmission Fourier transform infrared spectroscopy (FTIRS) measurements were carried out using a Varian 670 spectrometer equipped with a MCT detector. The spectra were recorded at a resolution of 4 cm−1 accumulating 512 scans. Self-supported wafers (area weight of 23–29 mg cm−2) were transferred into an IR cell that was connected to a vacuum system, in which residual pressures of ca. 1 × 10−6 mbar can be employed. Prior to the adsorption of gases, the catalysts were heated under dynamic vacuum at 300 °C for 1 h. A reference spectrum of the solid was taken after cooling down to 40 °C. Then, the sample chamber was charged with 7 mbar equilibrium pressure of NH3 at 40 °C. After 30 min, spectra were recorded applying the spectrum of the pretreated MnWO4 as background. The Brønsted acid site density was calculated applying the extinction coefficient 16 cm μmol−1.18
Near-ambient-pressure X-ray photoelectron spectroscopy (NAP-XPS) was conducted at the end station of the ISISS beam line at BESSY II/HZB (Helmholtz-Zentrum Berlin, Germany). Details of the setup have been published previously.19 For the XPS measurements, Mn 2p, O 1s, and W 4f core level spectra were collected at constant kinetic energies (KE) of the photoelectrons of 150 eV and 750 eV, resulting in inelastic mean free paths (IMFP) of the excited photoelectrons of ∼0.6 nm (150 eV, denominated as surface) and ∼1.6 nm (750 eV, denominated as deep). The experiments were performed at 300 °C and a total pressure of 0.25 mbar in a 1/1.1 O2/He mixture with a total gas flow of 4.2 sccm.
Results and discussion
Phase formation and particle growth under hydrothermal conditions
The preparation of monoclinic manganese tungstate composed of the wolframite-type structure (ICSD 67906) was performed by hydrothermal synthesis for 12 hours at 180 °C in aqueous medium. The formation of the solid within the autoclave was monitored using in situ Raman spectroscopy. Fig. 1 shows the spectra (Fig. 1a) taken during the synthesis of a slightly acidulated synthesis gel (pH of the starting mixture 6.3, Fig. 1b) and a mixture in which the pH has been adjusted to 9.1 (Fig. 1c) by addition of NaOH solution (Fig. 1d). Reaction between manganese nitrate and sodium tungstate occurs immediately after mixing the precursor solutions at room temperature as is evident from the Raman spectra. The Raman spectrum of the Mn precursor solution shows only the nitrate peak at 1048 cm−1 and no peaks due to Mn–O stretching vibrations, since divalent manganese ions are present under these conditions as an octahedral aquo complex. In the Raman spectrum of the sodium tungstate solution peaks at 933, 837, and 324 cm−1 occur due to the presence of dissolved [WO4]2− ions. The peaks belong to the four Raman active fundamental vibrational modes (A, +E, +2F2) of an undistorted tetrahedron (Td symmetry).20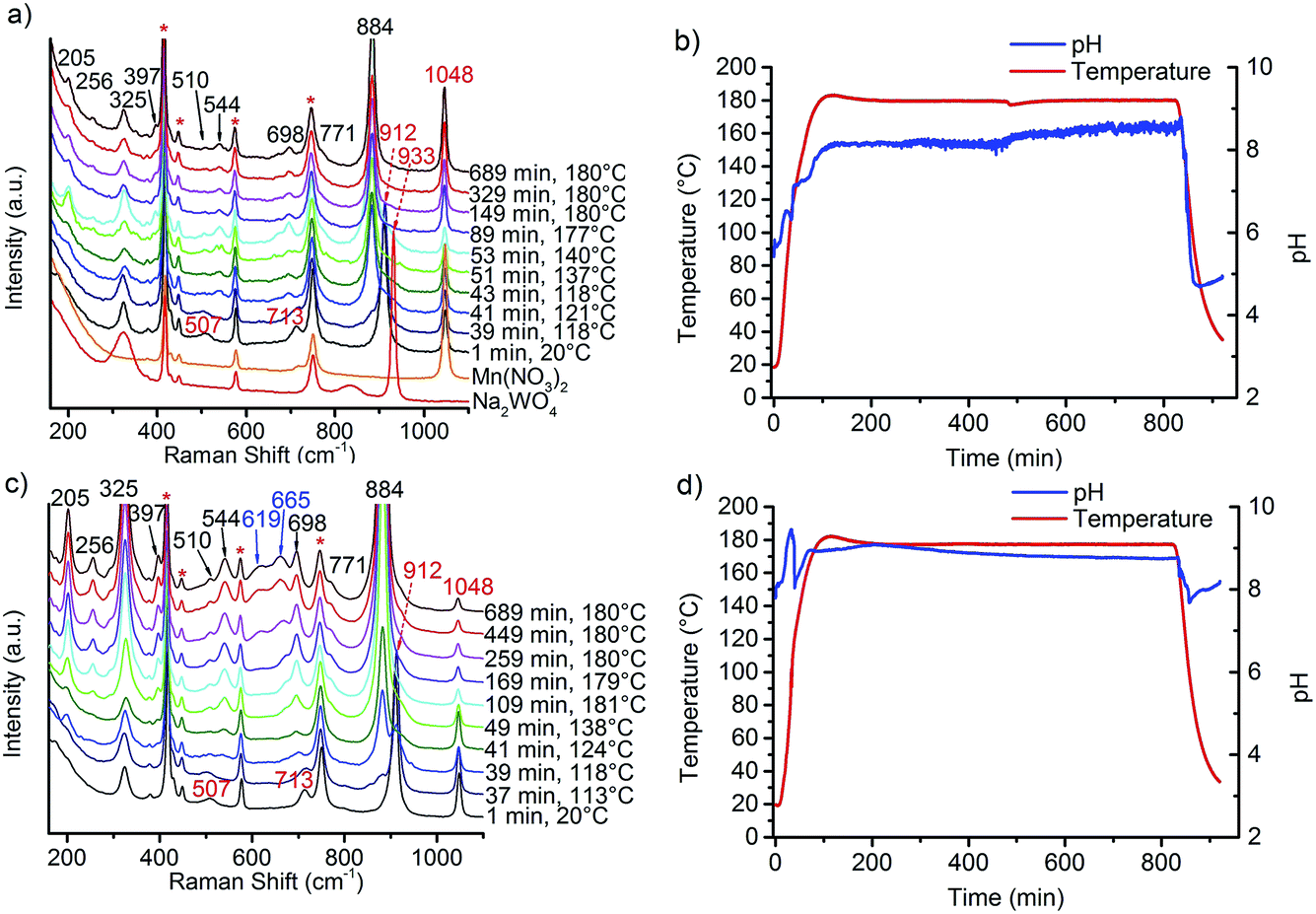 | ||
| Fig. 1 In situ Raman spectra recorded during the synthesis of the catalysts (a) AR1.5 and (c) AR3.9 and the corresponding profiles of temperature and pH during synthesis of (b) AR1.5 and (d) AR3.9; the symbol * in the Raman spectra indicates the bands of the sapphire window of the Raman probe. | ||
By mixing the two precursor solutions, the peak at 837 cm−1 disappears immediately, peaks at 507 and 713 cm−1 emerge, and the peak at 933 cm−1 is shifted to 912 cm−1 suggesting reaction between the manganese nitrate and sodium tungstate solution at room temperature. The bands are attributed to the formation of a partially crystalline product of unknown crystal structure that exhibits a layered-like morphology (Fig. S1†), indicated also by a peak in the XRD patterns at small angles that suggests regular stacking with a d spacing of 0.8 nm. The intermediate contains Mn and W in a molar ratio close to 1. During heating, bands at 205, 256, 325, 397, 510, 544, 698, 771, and 884 cm−1 indicate phase formation of MnWO4 above 100 °C that is completed at 120 °C.21,22 It has to be noted at this point that the Raman spectra measured by using the immersion probe originate from contributions of dispersed nanoparticles as well as from molecular species dissolved in the mother liquor. The phase formation is additionally indicated by a sharp drop of the pH in the temperature range from 110–120 °C. The pH at 180 °C rises above 8 during the synthesis of all catalysts. The average value increases with increasing starting pH and amounts to 8.1 in the synthesis of the catalysts AR1.5 and AR1.7, to 9.0 in the synthesis of the catalysts AR3.2 and AR3.9, and to 9.3 in the synthesis of catalyst AR5.1. The regulation of the initial pH involves changes in the ionic strength of the mother liquor from 1.20 mol L−1 for catalyst AR1.5 to 1.22 mol L−1 for catalyst AR5.1. The interpretation of the evolution of the pH values during hydrothermal synthesis is not straightforward, since the measured value results from superimposing condensation and hydrolysis reactions. All peaks observed at 180 °C, except weak bands at 619 and 665 cm−1, belong to the normal modes of crystalline MnWO4.21,22 The extra peaks are tentatively attributed to manganese hydroxide species as will be discussed below. Irrespective of the initial pH value, crystallization occurs at 120 °C (Fig. 1) implying that the crystallization process is determined by thermodynamics, since the monoclinic wolframite-type P2/c structure is the thermodynamic stable phase under the applied synthesis conditions.16 According to the XRD, all synthesis products are phase-pure materials (Fig. S2†). Thermal treatment in inert gas at 400 °C does not change the phase composition of the products (Fig. S3†).
The morphology of nano-sized MnWO4 can be tuned kinetically by adjusting the chemical potential during synthesis within the autoclave. The structural alterations are induced by different dissolution–recrystallization rates of the involved manganese tungstate species. These rates are predominantly controlled by the starting pH value of the synthesis gel. Thus, the shape of the primary MnWO4 nanoparticles in the hydrothermal product crucially depends on the starting pH and varies from cube-like particles (starting pH = 6.3) to anisotropic nano-rods (starting pH = 9.9) (Fig. 2). These changes are reflected in the aspect ratio of the MnWO4 nanoparticles, which can be obtained by measuring the length and width of the nanoparticles from the TEM images (Fig. 2, S4, and S5,†Table 1). Before catalytic testing, the as-synthesised materials have to be thermally treated above the reaction temperature of the catalytic reaction. Annealing in argon at 400 °C has no significant influence on the shape and size of the nanoparticles (Fig. 2), but the specific surface areas are slightly reduced (Table 1). Electron microscopy indicates a defect-rich structure and preferential growth of the rods along the <001> axis. Normally, crystallization and particle growth are induced by minimization of the free energy of the system. At the given chemical potential this observation suggests that the {001} surface is a high-energy surface. Thus, growing along <001> avoids its exposure to the environment. In addition, the fraction of surfaces with lower energies increases significantly, which improves the stabilization of the nanoparticle. In fact, analysis of the HAADF-STEM images of AR1.5 and AR5.1 allows for the allocation of specific facets of the particles, including the {100}, {010} and {110} crystal planes as illustrated in Fig. 3.
 | ||
| Fig. 2 Electron microscopy images of the as-synthesized (top row) and thermally treated (bottom row) nanostructured MnWO4 materials AR1.5 (a) and (f), AR1.7 (b) and (g), AR3.2 (c) and (h), AR3.9 (d) and (i), and AR5.1 (e) and (j); uncoloured TEM images are presented in the ESI (Fig. S4†). | ||
| Catalyst | Catalyst IDa | pHb | Surface area of hydrothermal productc (m2 g−1) | Surface area of catalysta (m2 g−1) | Number of particlesd | Mean particle lengthd (nm) | Mean particle diameterd (nm) | Mean aspect ratiod | D <100> (nm) | D <010> (nm) | D <001> (nm) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a Required to identify different batches of catalyst synthesis, catalyst after thermal activation at 400 °C for 2 hours in argon. b pH value adjusted before the hydrothermal synthesis. c After drying at 80 °C. d Number average values analysed based on TEM images as shown in Fig. 2, S4 and S5. e Based on XRD of the catalysts (Fig. S3). | |||||||||||
| AR1.5 | 19112 |
6.3 | 27.7 | 25.9 | 132 | 53.8 | 35.7 | 1.5 | 27.1 | 27.3 | 41.4 |
| AR1.7 | 19113 |
6.7 | 26.1 | 22.9 | 124 | 59.0 | 35.1 | 1.7 | 27.2 | 26.3 | 46.8 |
| AR3.2 | 19114 |
8.0 | 23.3 | 22.1 | 93 | 104 | 32.6 | 3.2 | 28.5 | 25.6 | 70.3 |
| AR3.9 | 19251 |
9.1 | 25.7 | 24.0 | 125 | 119 | 30.8 | 3.9 | 27.2 | 22.1 | 65.3 |
| AR5.1 | 19116 |
9.9 | 31.0 | 28.7 | 131 | 122 | 24.4 | 5.1 | 24.0 | 17.3 | 55.4 |
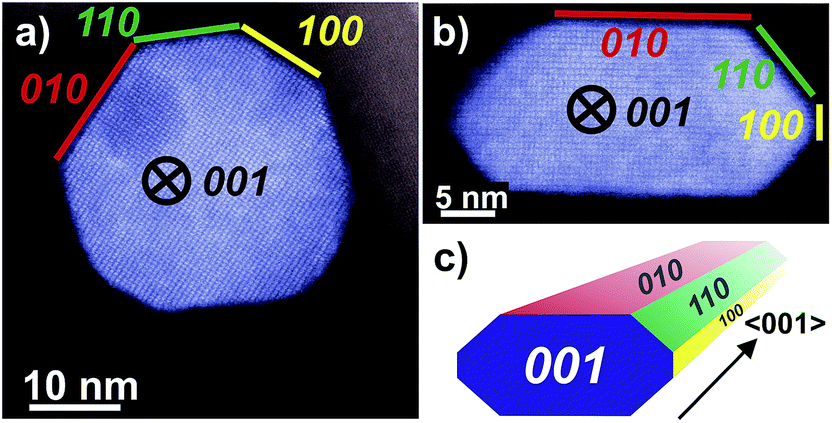 | ||
| Fig. 3 HAADF-STEM images of MnWO4 nanoparticles viewed along <001> with different aspect ratios: (a) AR1.5, (b) AR5.1 and (c) perspective model for a typical faceted nanoparticle. The original uncoloured images including Fast Fourier transform analysis are given in the ESI, Fig. S6.† | ||
The dissolution–recrystallization processes occurring during ageing under hydrothermal conditions in the autoclave at 180 °C are illustrated in Fig. 4. The dissolution of [WO4]2− ions and the hydrolysis of [Mn(H2O)6]2+ species by deprotonation of water ligands is facilitated in a basic environment in the presence of a high concentration of OH− ions. Anisotropic growth is imaginable when the local structure of the WO6 chains at different planes at the surface of the MnWO4 crystals is taken into consideration. At the {010} crystal planes, edge sharing –O–W–O– bonds are exposed that can be easily attacked by OH− ions leading to dissolution of [WO4]2− species. Hydrolysis is hindered at other planes, for example at {100} and {110}, due to geometric reasons. Only half of the –O–W–O– bridges in a WO6 octahedron are exposed at these planes. Therefore, the WO4 units cannot be dissolved. Free [WO4]2− species could condense with W–O(H) groups at the high-energy {001} basal planes propagating the tungsten oxide zigzag chains by forming new edge sharing O–W–O bonds (Fig. 4). Divalent manganese ions may interact with negatively charged tungstate chains and are in this way incorporated into the structure. With increasing OH− concentration, the dissolution–recrystallization process is enhanced and nanoparticles with increased aspect ratio are formed as evidenced by particle size analysis (Fig. S5,†Table 1). The statistical analysis based on the TEM images is in agreement with the average particle diameter found by XRD. With increasing initial pH value, the (010) peaks in the XRD patterns (Fig. S3†) become more broadened, providing additional support for smaller crystallite sizes along the <010> direction with increasing pH (Table 1). During dissolution–recrystallization, hydrolysis of [Mn(H2O)6]2+ species leads to the formation of Mn(OH)x species at the surface of the catalysts as indicated by an increasing intensity of the two bands at 619 and 665 cm−1 in the Raman spectra recorded during hydrothermal synthesis that are tentatively attributed to surface manganese oxide–hydroxide species.23 The appearance of the corresponding bands is more distinct in reaction mixtures that contain a higher concentration of OH− ions (compare Fig. 1a and c). However, the concentration of dissolved tungsten oxide and manganese species is apparently rather low, since no peaks due to such species in solution are detectable by Raman spectroscopy using the immersion probe.
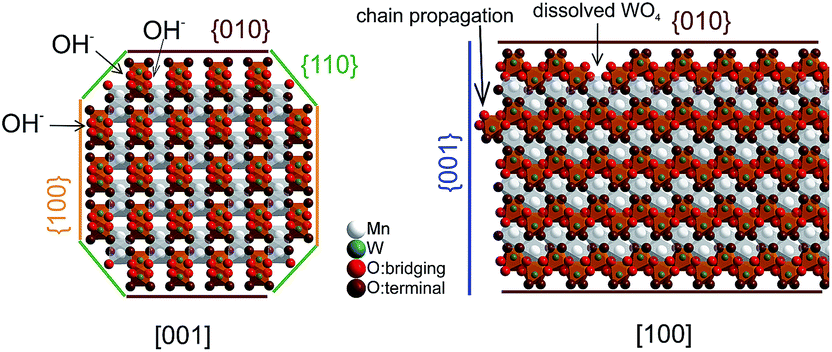 | ||
| Fig. 4 Schematic representation of the proposed anisotropic mechanism of particle growth. | ||
Ethanol oxidation
The side products ethylene and diethyl ether in the oxidation of ethanol to acetaldehyde reveal the presence of extra acid/base functionalities at the catalyst surface of an oxidation catalyst. Ethanol oxidation has been performed over all catalysts within the reaction temperature range from 280 to 310 °C achieving conversion of ethanol from 5 to 15% (Fig. S7†). Under these conditions acetaldehyde as well as ethylene are formed over all catalysts, but the selectivity is different for the catalysts with different aspect ratios of their primary particles. In Fig. 5 the product distribution is compared at 10% ethanol conversion. The ethylene selectivity is quite high over the catalyst AR1.5, but decreases as the aspect ratio increases.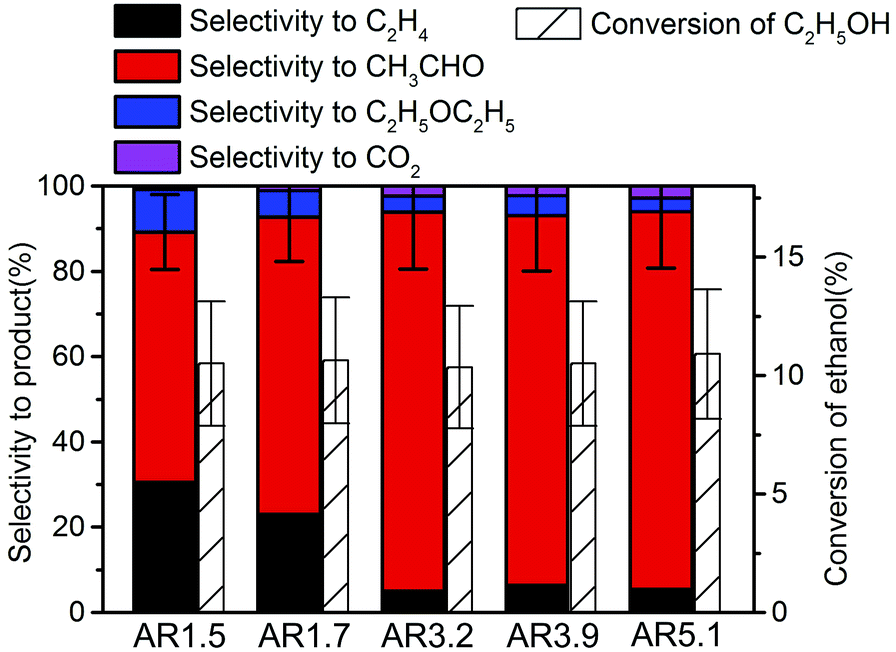 | ||
| Fig. 5 Selectivity in ethanol oxidation over the nanostructured MnWO4 catalysts at 10% ethanol conversion. | ||
The selectivity to acetaldehyde decreases with increasing temperature and conversion, whereas ethylene selectivity rises (Fig. S7†). At constant temperature the selectivity to acetaldehyde decreases in general slightly with time on stream, whereas ethylene selectivity is quite constant or even increases (Fig. S7†) suggesting no structural relations between redox and acid–base sites.
The specific rate of the acid-catalysed dehydration reaction decreases with increasing aspect ratio (Fig. 6). In contrast, the specific redox reaction rate exhibits a flat maximum at the aspect ratio 3.2. It should be noted at this point that the rates are integral rates. Conclusions about the intrinsic activity are not possible from these values. However, the apparent activation energy of ethanol oxidation is quite constant (48 ± 6 kJ mol−1) suggesting that not the nature, but the number of active sites changes in the current catalyst series.
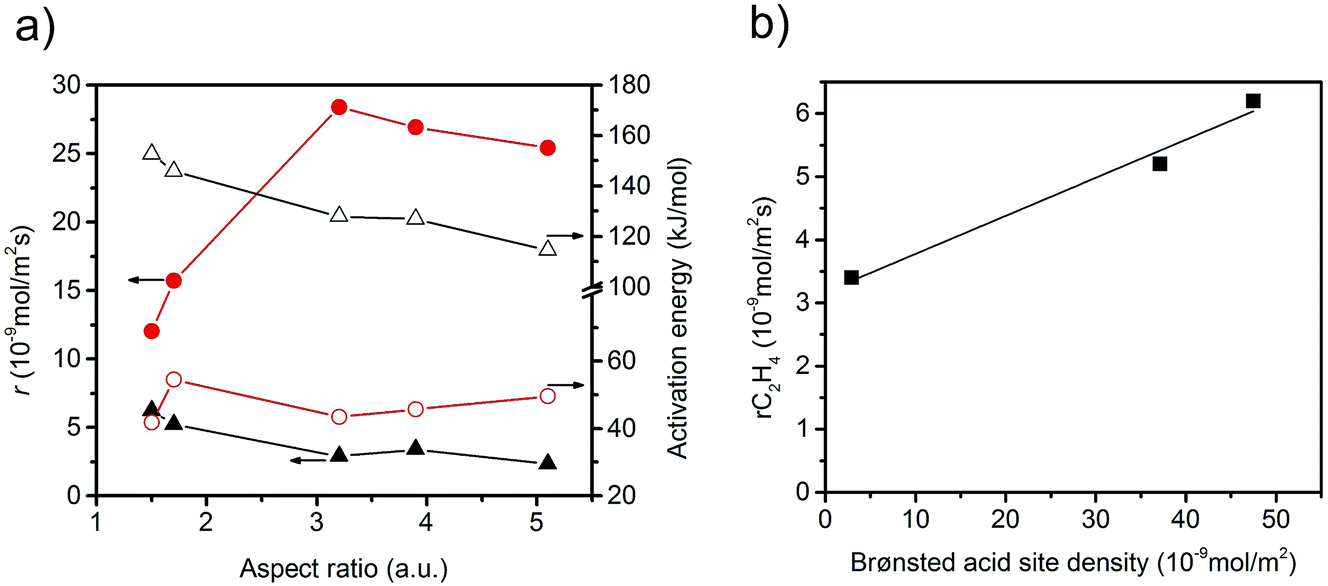 | ||
| Fig. 6 (a) Specific reaction rates measured at T = 310 °C and normalized to surface area, and apparent activation energy as a function of the aspect ratio; red solid circle: acetaldehyde formation rate; red open circle: apparent activation energy of acetaldehyde formation; black solid triangle: ethylene formation rate; black open triangle: apparent activation energy of ethylene formation; (b) rate of formation of ethylene as a function of Brønsted acid site density at the catalyst surface determined by ammonia adsorption and specific surface area measurements. | ||
The observed trends may be related to a change in the relative fraction of acid–base and redox species at the catalyst surface that may be, again, related to differences in the termination of the catalyst particles within the current series. The presence of a defect rich MnOx chain-like 2D over-layer, particularly, at the {010} termination of particles in the catalyst AR5.1 has been verified using electron microscopy and photoelectron spectroscopy.24 These MnOx chains might bear oxygen defects, which are believed to provide the active sites in the activation of propane. The same manganese oxide sites might be relevant for the oxidation of ethanol in the present experiments as well. Due to the preferential growth of the particles along the <001> direction, the relative abundance of {010} planes that host these manganese oxide sites for oxidation catalysis may increase with increasing aspect ratio (Fig. 3). Therefore, an aspect ratio above three is reflected in increased integral rates of the oxidation reaction compared to the integral rates of the acid-catalysed dehydration reaction over the corresponding catalysts. The sites responsible for dehydration may be, in contrast, preferentially located at the two ends of the rods, which are terminated by {001} crystal planes. According to structural considerations, Brønsted acid sites might be preferentially located at these planes (Fig. S8†). Therefore, the two catalysts AR1.5 and AR1.7 exhibit enhanced acid–base functionalities, which is in agreement with the shape (Fig. 2 and 3a) and the proposed dissolution–recrystallization mechanism (Fig. 4 and S8†).
Experimentally, the acidity of the catalysts has been studied using infrared spectroscopy. After heating to the reaction temperature of ethanol oxidation (300 °C) under vacuum, three bands located at 3470, 3400, and 3366 cm−1 are observed in the O–H stretching region on top of a broad feature (Fig. 7a). The latter is attributed to hydroxy groups that undergo hydrogen bonding or strongly adsorbed water molecules. A residual band is observed at 1642 cm−1 that might be due to the bending mode of molecular water, but discrimination of this band from the overtones and combination vibrations of MnWO4 is difficult. Based on the very low frequency and the unusual narrow bandwidth, the three sharp bands at 3470, 3400, and 3366 cm−1 are tentatively assigned to hydroxy groups that form well-ordered hydrogen bonding networks at the surface. Further investigations are necessary to clarify the origin of these bands. To investigate the acidity of the hydroxy groups, NH3 was adsorbed. Unfortunately, the bands of the adsorbed ammonia in the N–H stretching vibration region between 3400 and 3000 cm−1 overlap with the hydroxy bands due to the low frequency of the latter (Fig. 7a). Therefore, the consumption of particular OH species in the reaction with ammonia cannot be monitored. But weak acidity is confirmed by the formation of a weak but distinct band at 1439 cm−1 due to the asymmetric deformation vibration of ammonium ions formed by reaction of ammonia with Brønsted acid sites at the surface of the catalysts AR1.5, AR1.7, and AR3.9 (Fig. S9†). No clear indications regarding redox processes of adsorbed ammonia molecules have been observed in the spectra. The concentration of acid sites at the surface of catalyst AR5.1 was below the detection limit. The small number of acid sites (in the range from 0 for AR5.1 to 1.2 μmol g−1 for AR1.5) suggests that most of the OH groups observed in the infrared spectra of the catalysts after dehydroxylation at 300 °C under vacuum (Fig. 7a) are nonacidic in nature. Essentially all the OH groups observed at the surface of catalyst 5.1 are not able to protonate ammonia. Therefore, the band at 3366 cm−1 that represents the dominant hydroxy species at the surface of catalyst AR5.1 is tentatively assigned to Mn–OH groups. A sharp band below 3500 cm−1 has been attributed to Mn–OH in the layered Mn2+ hydroxide Mn2(OH)2SO4.25 The surface concentration of acid sites correlates with the formation rate of ethylene (Fig. 6b), demonstrating the involvement of these sites in the dehydration of ethanol. The non-zero intercept of the fitting line in Fig. 6b suggests that Lewis acid sites may be involved in the dehydration of ethanol as well. Pathways leading to ethylene from ethanol adsorbed on Ce4+ have been discussed.26 Similar reaction routes might also be possible at coordinatively unsaturated manganese or tungsten sites that are present at the surface of the catalysts as it becomes evident from ammonia adsorption (Fig. S9†). The peak at 1591 cm−1 may arise from the asymmetric bending vibration of ammonia molecules adsorbed at Lewis acid sites.
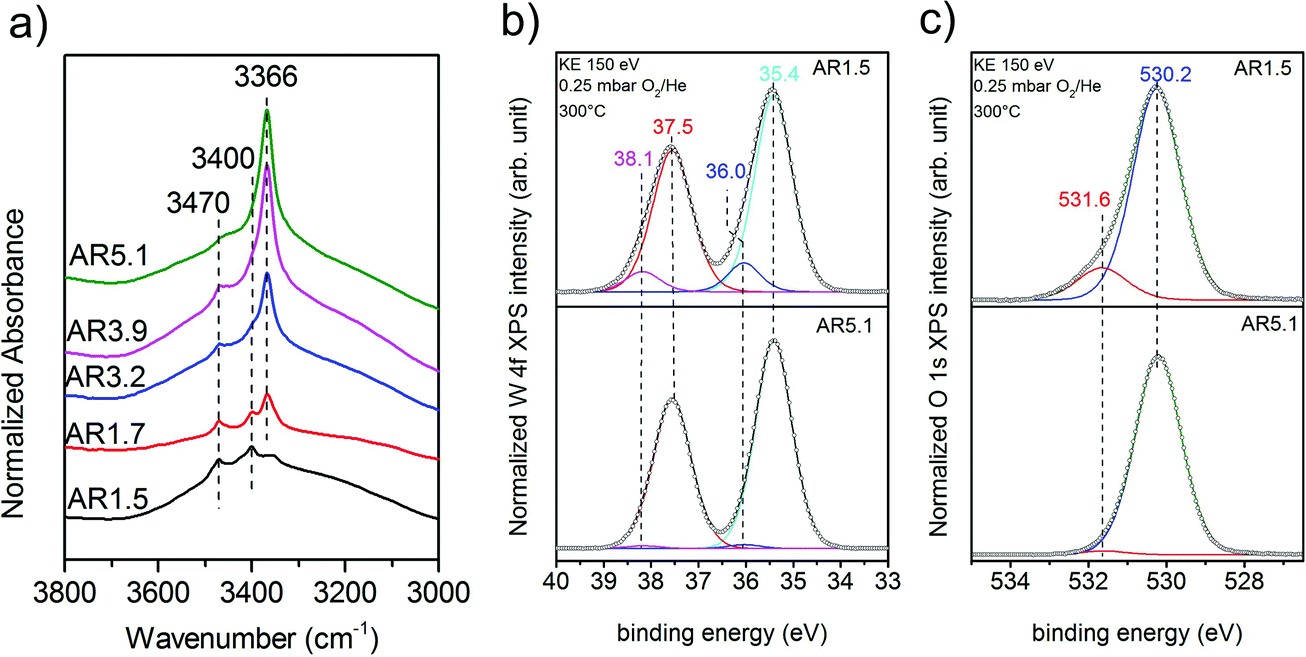 | ||
| Fig. 7 (a) Infrared spectra in the region of OH stretching vibrations after thermal treatment of the catalyst in the infrared cell under vacuum at 300 °C; the measurement was performed under vacuum at 40 °C; W 4f spectra (b), and O 1s spectra (c) of the catalysts AR1.5 and AR5.1 measured using synchrotron-based near ambient pressure X-ray photoemission spectroscopy (NAP-XPS) at an inelastic mean free path (IMFP) of ca. 0.6 nm in 0.25 mbar in O2/He at a total gas flow of 4.2 sccm at 300 °C. | ||
A high concentration of hydroxy groups at the surface of the catalyst AR1.5 was also confirmed by NAP-XPS measured in the presence of oxygen at 300 °C. The O 1s core level spectra of the catalysts AR1.5 and AR5.1 are shown in Fig. 7c. The main peak at 530.2 eV is assigned to lattice oxygen, while the component at 531.6 eV is attributed to hydroxy groups. Fig. 7c shows that catalyst AR1.5 comprises a higher concentration of hydroxy groups than catalyst AR5.1. A detailed analysis of the W 4f core level reveals that also the W 4f7/2 and W 4f5/2 doublet (Fig. 7b) can be deconvoluted into two contributions. The low binding energy doublet at 35.4 and 37.5 eV is in good agreement with the binding energy values reported in the literature for metal tungstates.27,28 The high binding energy doublet at 36.0 and 38.1 eV is more pronounced in the AR1.5 catalyst than in the AR5.1 catalyst, in line with the higher hydroxyl concentration observed in the O 1s spectrum of the AR1.5 catalyst. Furthermore, the contribution of the high binding energy W 4f doublet as well as the hydroxide component in the O 1s spectrum is decreasing with increasing probing depth (compare Fig. 7b and c, S10a and b†). Therefore, we tentatively assign the high binding energy doublet in the W 4f spectrum to modified tungsten atoms bearing hydroxy groups at the surface of the catalyst. Thus, the XPS measurements indicate that more W–OH groups are present at the surface of the catalyst containing primary particles with a low aspect ratio (AR1.5). These results are complementary to the characterization of the catalysts using ammonia adsorption. The acid sites probed using FTIRS of adsorbed ammonium ions are attributed to W–OH groups at the surface of the catalyst, which is also plausible in terms of the acid–base chemistry of tungsten compared to manganese. Mn–OH groups are apparently not resolved by XPS, neither in the O 1s spectra (Fig. 7c), nor in the Mn 2p spectra (Fig. S11†). The discrepancy between the FTIRS and XPS concerning the presence of hydroxy groups at the surface of the two catalysts might be due to the fact that the Mn–OH contributions are enveloped by M − O contributions in the O 1s (Fig. 7c) and Mn 2p (Fig. S11†) spectra, or due to differences in the pretreatment procedures before IR (vacuum, 300 °C) and XPS (0.25 mbar O2 at 300 °C) measurements, respectively.
In summary, the variation of the initial pH in the hydrothermal synthesis of nanostructured MnWO4 affects the dissolution–recrystallization kinetics of the freshly formed particles during ageing at 180 °C. Although these dissolution and recrystallization processes are responsible for anisotropic particle growth, more important is that ageing provides the basis for the formation of the catalytically active sites. With increasing pH under hydrothermal conditions the fraction of acid W–OH groups at the surface of the final catalyst decreases and, consequently, acid–base catalysed side reactions during ethanol oxidation are increasingly suppressed, which is reflected in enhanced selectivity to acetaldehyde with increasing starting pH (Fig. 5).
Selectivity is the major issue in oxidation catalysis.6,29 Activation of C–H bonds in saturated hydrocarbons is challenging because the formed reaction products and intermediates easily undergo consecutive and parallel reactions. Minimization of rates of undesired pathways requires co-ordinated design of bulk electronic properties and surface dynamics of oxidation catalysts.30 C–H bond activation may involve multiple mechanisms including carbenium or carbonium intermediates and homolytic splitting of C–H bonds at metal oxide surface functional groups under formation of radical species.31,32 Model calculations, generally based on small cluster models, favour the homolytic pathway over transition metal oxide catalysts.33 However, Lewis acid sites in terms of coordinatively unsaturated metal cations and Brønsted acid sites may be present at the surface of a transition metal oxide under reaction conditions as well. Brønsted acid sites are particularly expected since water is an unavoidable coproduct in oxidation reactions and, as demonstrated in the current example, the dehydroxylation temperature is often above the reaction temperature.
Whereas acidity at the surface of nanostructured MnWO4 catalysts is attributed to the presence of W–OH groups and coordinatively unsaturated metal cations, the origin of the redox activity is not that straightforward. During particle growth under hydrothermal conditions the formation of defects may occur. In the course of dissolution and recrystallization, chemical defects in terms of cationic vacancies or OH− groups at anionic positions are generated that may be related to structural defects.34 Establishing relations between defect chemistry and redox activity requires further kinetic studies and more comprehensive characterization of the catalysts, which is currently under way.
Conclusions
Nanostructured MnWO4 catalysts have been prepared using hydrothermal synthesis. The aspect ratio of the primary MnWO4 particles increases with increasing pH during ageing at 180 °C under hydrothermal conditions. Electron microscopy revealed that the particles grow along the <001> axis. A mechanism of particle growth by dissolution re-crystallization is proposed that leads to an increase in the fraction of terminating {010} planes with increasing aspect ratio. At the same time, the fraction of {001} planes, which are terminated by W–OH groups, is decreasing. These changes are reflected in the selectivity patterns of the ethanol oxidation reaction that probes both, redox and acid–base sites. Metastable structures are required to achieve catalytic activity over MnWO4. Solid-state synthesis provides theses structures only on a limited scale. The implementation of chemical defects as a basis for the catalytic activity of MnWO4 succeeded via variation of the chemical potential under mild hydrothermal conditions.Acknowledgements
This work was conducted in the framework of the BasCat collaboration between BASF SE, TU Berlin, FHI, and the cluster of excellence “Unified Concepts in Catalysis” (UniCat http://www.unicat.tu-berlin.de). X.L. acknowledges the Berlin International Graduate School of Natural Sciences and Engineering (BIG NSE) as part of UniCat for financial support. The authors thank Maike Hashagen and Jasmin Allan for technical assistance. We thank the HZB staff for their continual support of the electron spectroscopy activities of the FHI at BESSY II.Notes and references
- I. E. Wachs and K. Routray, ACS Catal., 2012, 2, 1235–1246 CrossRef CAS.
- E. W. McFarland and H. Metiu, Chem. Rev., 2013, 113, 4391–4427 CrossRef CAS PubMed.
- W. Ueda, J. Jpn. Pet. Inst., 2013, 56, 122–132 CrossRef CAS.
- R. Schlögl, Angew. Chem., Int. Ed., 2015, 54, 3465–3520 CrossRef PubMed.
- A. Trunschke, in Chemical Energy Storage, ed. R. Schlögl, Walter de Gruyter GmbH, Berlin/Boston, 2013, ch. 4.2, pp. 277–301 Search PubMed.
- R. Schlögl, Top. Catal., 2011, 54, 627–638 CrossRef.
- K. Amakawa, Y. V. Kolen'ko, A. Villa, M. E. Schuster, L.-I. Csepei, G. Weinberg, S. Wrabetz, R. Naumann d'Alnoncourt, F. Girgsdies, L. Prati, R. Schlögl and A. Trunschke, ACS Catal., 2013, 3, 1103–1113 CrossRef CAS.
- M. Rajamathi and R. Seshadri, Curr. Opin. Solid State Mater. Sci., 2002, 6, 337–345 CrossRef CAS.
- C. S. Cundy and P. A. Cox, Microporous Mesoporous Mater., 2005, 82, 1–78 CrossRef CAS.
- M. K. Devaraju and I. Honma, Adv. Energy Mater., 2012, 2, 284–297 CrossRef CAS.
- C. Perego and R. Millini, Chem. Soc. Rev., 2013, 42, 3956–3976 RSC.
- G. R. Patzke, Y. Zhou, R. Kontic and F. Conrad, Angew. Chem., Int. Ed., 2011, 50, 826–859 CrossRef CAS PubMed.
- W. Shi, S. Song and H. Zhang, Chem. Soc. Rev., 2013, 42, 5714–5743 RSC.
- M. Sanchez Sanchez, F. Girgsdies, M. Jastak, P. Kube, R. Schlögl and A. Trunschke, Angew. Chem., Int. Ed., 2012, 51, 7194–7197 CrossRef CAS PubMed.
- J. Noack, F. Rosowski, R. Schlögl and A. Trunschke, Z. Anorg. Allg. Chem., 2014, 640, 2730–2736 CrossRef CAS.
- S. H. Yu, B. Liu, M. S. Mo, J. H. Huang, X. M. Liu and Y. T. Qian, Adv. Funct. Mater., 2003, 13, 639–647 CrossRef CAS.
- P. Stephens, J. Appl. Crystallogr., 1999, 32, 281–289 CrossRef CAS.
- A. A. Budneva, E. A. Paukshtis and A. A. Davydov, React. Kinet. Catal. Lett., 1987, 34, 63–67 CrossRef CAS.
- A. KnopGericke, E. Kleimenov, M. Hävecker, R. Blume, D. Teschner, S. Zafeiratos, R. Schlögl, V. I. Bukhtiyarov, V. V. Kaichev, I. P. Prosvirin, A. I. Nizovskii, H. Bluhm, A. Barinov, P. Dudin and M. Kiskinova, in Advances in Catalysis, ed. C. G. Bruce and K. Helmut, Academic Press, 2009, vol. 52, pp. 213–272 Search PubMed.
- F. Gonzalez-Vilchez and W. P. Griffith, J. Chem. Soc., Dalton Trans., 1972, 1416–1421, 10.1039/dt9720001416.
- L. H. Hoang, N. T. M. Hien, W. S. Choi, Y. S. Lee, K. Taniguchi, T. Arima, S. Yoon, X. B. Chen and I.-S. Yang, J. Raman Spectrosc., 2010, 41, 1005–1010 CrossRef CAS.
- M. N. Iliev, M. M. Gospodinov and A. P. Litvinchuk, Phys. Rev. B: Condens. Matter Mater. Phys, 2009, 80, 212302 CrossRef.
- F. Kapteijn, A. D. Vanlangeveld, J. A. Moulijn, A. Andreini, M. A. Vuurman, A. M. Turek, J. M. Jehng and I. E. Wachs, J. Catal., 1994, 150, 94–104 CrossRef CAS.
- X. Li, T. Lunkenbein, V. Pfeifer, M. Jastak, P. K. Nielsen, F. Girgsdies, A. Knop-Gericke, F. Rosowski, R. Schlögl and A. Trunschke, Angew. Chem., Int. Ed., 2016, 55, 4092–4096 CrossRef CAS PubMed.
- M. B. Salah, S. Vilminot, T. Mhiri and M. Kurmoo, Eur. J. Inorg. Chem., 2004, 2272–2276 CrossRef.
- M. Li, Z. Wu and S. H. Overbury, J. Catal., 2013, 306, 164–176 CrossRef CAS.
- S. Muthamizh, R. Suresh, K. Giribabu, R. Manigandan, S. Praveen Kumar, S. Munusamy and V. Narayanan, J. Alloys Compd., 2015, 619, 601–609 CrossRef CAS.
- T.-D. Nguyen, D. Mrabet, T.-T.-D. Vu, C.-T. Dinh and T.-O. Do, CrystEngComm, 2011, 13, 1450–1460 RSC.
- J.-M. M. Millet, Top. Catal., 2006, 38, 83–92 CrossRef CAS.
- M. Eichelbaum, M. Hävecker, C. Heine, A. M. Wernbacher, F. Rosowski, A. Trunschke and R. Schlögl, Angew. Chem., Int. Ed., 2015, 54, 2922–2926 CrossRef CAS PubMed.
- C. Coperet, Chem. Rev., 2009, 110, 656–680 CrossRef PubMed.
- R. A. van Santen, I. Tranca and E. J. M. Hensen, Catal. Today, 2015, 244, 63–84 CrossRef CAS.
- X. Rozanska, R. Fortrie and J. Sauer, J. Am. Chem. Soc., 2014, 136, 7751–7761 CrossRef CAS PubMed.
- Y. Chabre and J. Pannetier, Prog. Solid State Chem., 1995, 23, 1–130 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: SEM, TEM, XRD, FTIR, DRIFTS, XPS. See DOI: 10.1039/c5fd00191a |
| This journal is © The Royal Society of Chemistry 2016 |