
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffect of chemical structure on the sonochemical degradation of perfluoroalkyl and polyfluoroalkyl substances (PFASs)†
Nerea Abad
Fernandez
ab,
Lucia
Rodriguez-Freire
ac,
Manish
Keswani
*c and
Reyes
Sierra-Alvarez
a
aDepartment of Chemical and Environmental Engineering, The University of Arizona, P.O. Box 210011, Tucson, Arizona, USA
bDepartment of Chemical Engineering and Environmental Technology, EII, Sede Mergelina, Valladolid University, Valladolid, Spain
cDepartment of Materials Science and Engineering, The University of Arizona, P.O. Box 210012, Tucson, Arizona, USA. E-mail: manishk@email.arizona.edu; Fax: +520 621 8059; Tel: +520 270 4361
First published on 5th September 2016
Abstract
Perfluoroalkyl surfactants include chemicals characterized by a fully fluorinated carbon chain (hydrophobic and oleophobic tail) bound to a hydrophilic head (a carboxyl or sulfonic group). These compounds are toxic and highly resistant to chemical/biological attack, and some are known to be bio-accumulative. This study investigates the sonochemical degradation at 500 kHz of different carboxylic and sulfonic perfluoroalkyl and polyfluoroalkyl substances (PFASs, 1.7 mM total organic fluorine) to assess the effect of chain length, functional head group, and substituents (–CH2–CH2– moiety and ether group) on the degradation rate. Under these conditions, the rates of defluorination determined for two widely used perfluoroalkyl substances, perfluorooctanesulfonate (PFOS) and perfluorooctanoic acid (PFOA), were 3.5 to 3.7 μM F− min−1, respectively. The degradation rate of perfluoroalkyl sulfonates decreased with the perfluorocarbon chain length as indicated by the 1.3 and 1.9-fold lower defluorination rates for perfluorohexane- and perfluorobutane sulfonate than that of PFOS. A similar trend was observed during the sonolysis of perfluoroalkyl carboxylate analogs with 6, 5 or 3 carbon atoms which had 1.1-, 1.8-, and 2.3-fold lower defluorination rates, respectively, than that of PFOA. Furthermore, perfluoroalkyl compounds appeared more amenable to sonolysis than the polyfluoroalkyl analogues with the same number of C atoms (defluorination rate of PFOS/6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 fluorotelomer sulfonate ≈ 2.3). The results demonstrate that sonolysis is a promising approach to treat PFASs in aqueous streams. Furthermore, they underscore that the chemical structure of PFASs has a marked effect on the rate at which they undergo sonochemical degradation.
:2 fluorotelomer sulfonate ≈ 2.3). The results demonstrate that sonolysis is a promising approach to treat PFASs in aqueous streams. Furthermore, they underscore that the chemical structure of PFASs has a marked effect on the rate at which they undergo sonochemical degradation.
Water impactPerfluoroalkyl and polyfluoroalkyl substances (PFASs) are highly persistent, toxic pollutants that are receiving increasing attention due to their widespread environmental distribution. Conventional treatment methods have proven ineffective to treat these hazardous contaminants due to their high resistance to chemical and biological attack. This study demonstrates that sonochemical treatment at 500 kHz has promise as a remediation technique to degrade a range of PFASs with varying carbon chain length and functional groups. Furthermore, this study provides new experimental data confirming that the rate of sonolysis is strongly dependent on the chemical structure of the PFASs. |
Introduction
Fluorinated surfactants are widely used chemicals which may be classified as either perfluorinated, in which all hydrogen atoms are substituted by fluorine atoms, or as partially fluorinated, in which some carbon atoms are bound to hydrogen atoms.1 Perfluoroalkyl chemicals are a group of compounds with varying carbon chain length and functional groups, which according to the United States Environmental Protection Agency (US-EPA) can be broadly categorized into perfluoroalkyl sulfonates (PFSAs) and perfluoroalkyl carboxylic acids (PFCAs). Polyfluoroalkyl substances, which are defined as fluorinated compounds having at least one perfluoroalkyl moiety CnF2n+1−, are related compounds that have the potential to be transformed abiotically or biotically into perfluoroalkyl substances.2Perfluoroalkyl- and polyfluoroalkyl substances (PFASs) have a wide variety of applications, such as non-stick polymers, water and stain proof coatings for paper and textiles, oxidative protective coatings on metals, inert surfactants for semiconductor etching, aqueous film-forming foams (AFFFs), thermally stable lubricants preservatives, fluoropolymer and fluoroelastomer production, surface treatment, food packing, hydraulic oil for airplanes, cosmetics, floor wax, polish, paints and lacquers.3,4 Perfluorooctane sulfonate (PFOS, C8F17SO3−) and perfluorooctanoic acid (PFOA, C7F15COOH) are two widely used PFASs which have been detected in surface water, groundwater, sediments, sewage treatment effluents and sludge, landfill leachate and drinking water in various parts of the world.5–8 Moreover, they are distributed across the globe and found in animals living in pristine environments (e.g., polar bears) in the most remote locations.9 The total emissions of PFOA over the time period 1950–2010 have been estimated to be in the range of 2600–5050 tons.10 Over the same time period, 450–2700 tons of PFOS have been released. PFOS, its salts and perfluorooctane sulfonyl fluoride (PFOS-F) have been classified as PBTs (persistent, bioaccumulative and toxic chemicals) by the Stockholm Convention in May 2009,11 since they are extremely persistent in the environment, bioaccumulative in wildlife and humans, and toxic to laboratory animals and wildlife.6,12–15 PFOA, its salts and PFOA-related compounds are currently under consideration to be included in the PBT list. In December 2002, EPA significant new use rules (SNURs) allowed the continuation of a few, limited and highly technical applications of PFOS-related substances where no known alternatives are available. Lastly, in May 2016, EPA set a new lifetime health advisory (HA) of 70 ng L−1 PFOS and PFOA combined.16
Organic perfluorination imparts these compounds with unique physical properties such as chemical and thermal stability,17 a greater surface activity,1 and a higher oxidative resistance.3 The low carbon–fluorine (C–F) bond polarizability gives them both hydrophobic and oleophobic characters.18 PFASs are also stable to attack by acids, bases, oxidants, and reductants, since the C–F bond is the strongest among organics.1 This stability makes them recalcitrant toward most conventional degradation technologies. Treatment technologies such as reverse osmosis, nano-filtration and activated carbon can remove perfluorochemicals from water.3 However, incineration of the concentrated waste is required for complete destruction of the fluorochemicals. Even advanced oxidation processes (AOPs), which utilize the hydroxyl radical, are relatively ineffective for PFOA and PFOS destruction, requiring very stringent conditions such as very high temperature and/or pressures to provide some degradation.3 In contrast, sonolysis is a promising treatment to degrade perfluorinated surfactants.17,19,20 Acoustic irradiation of liquid induces cavitation, a process during which preexisting gas cavities in the liquid oscillate or collapse in a periodically changing pressure field created by sound waves.21 Transient bubble collapses generate average vapor/gas temperatures near 5000 K and much higher bubble vapor/gas core temperatures (>10000 K),22 resulting in hydroxyl radical (HO˙) formation by the thermal decomposition of water.23 The heat energy unleashed by cavitation yields thermal decomposition of PFAS molecules to their inorganic constituents (fluoride (F−), sulfate (SO42−), CO, and CO2).17 Sonolysis of perfluorinated surfactants is believed to occur primarily at the water–cavitating bubble interface via pyrolytic reactions. PFSAs and PFCAs have very low vapor pressures24 and, therefore, a low tendency to partition into the vapor phase of the bubbles.
Previous sonochemical studies have mainly focused on the degradation of PFOS and PFOA due to their considerable industrial importance and wide application. Although PFOS and PFOA are the most studied PFASs, other perfluoroalkyl chemicals with chain lengths varying from C2–C12 have also been detected in different environmental samples, even in remote polar regions.12,25,26 Furthermore, AFFF are made of a wide variety of fluorinated chemicals and their treatment poses a serious challenge.27,28 Thus, there is a gap in understanding the effect of the chemical structure in their sonochemical degradation.
The present work investigated the sonochemical degradation under 500 kHz sound frequency of various classes of PFASs, including several PFSA and PFCA surfactants with varying perfluorocarbon chain length, a perfluorinated ether (perfluoro(2-ethoxyethane)sulfonic acid (PFEES)), and a fluorotelomer compound (6:2 fluorotelomer sulfonate (6:2 FTS)), with the objective to understand the effect of the chemical structure of PFASs on the effectiveness of the sonochemical treatment.
Materials and methods
Chemicals
Fig. 1 and Table 1 show the chemical structures and key physicochemical properties of the different PFASs, respectively. Perfluoro(2-ethoxyethane)sulfonic acid (PFEES, CAS # 113507-82-7; 97% purity) was purchased from Alfa-Aesar (Ward Hill, MA, USA). PFOA (CAS # 335-67-1; 96% purity), the potassium salt of PFOS (CAS # 2795-39-3; ≥98% purity), perfluorohexane carboxylic acid (PFHxA, CAS # 307-24-4; ≥97% purity), perfluoropropionic acid (PFPrA; CAS # 422-64-0; 97% purity), perfluoropentanoic acid (PFPA, CAS # 2706-90-3; 97% purity), sodium perfluorobutane sulfonate (PFBS, CAS # 375-73-5; 97% purity), and perfluorohexanesulfonic acid potassium salt (PFHxS, CAS # 3871-99-6; ≥98% purity) were procured from Sigma-Aldrich, Co. (St. Louis, Mo, USA). 6:2 Fluorotelomer sulfonic acid (6:2 FTS, CAS # 27619-97-2; 98% purity) was purchased from SynQuest Labs, Inc. (Alachua, FL, USA). Ultrapure water obtained from a MilliQ Plus UV system with a minimum resistance of 18 MΩ cm−1 was used for preparation of all solutions for the degradation experiments. Sodium fluoride (99%) and the total ionic strength adjustment buffer (TISAB II) used during fluoride measurements were purchased from Fisher Scientific Inc. (New Jersey, USA). Compressed argon was provided by Cryogenics and Gas Facility (The University of Arizona, Tucson, AZ, USA).
 | ||
| Fig. 1 Chemical structures and acronyms of the different perfluoroalkyl and polyfluoroalkyl chemicals (PFASs) studied. “n” represents the number of carbons in the perfluorocarbon chain. (1) Perfluoroalkyl sulfonates (PFSAs), (2) perfluoroalkyl carboxylic acids (PFCAs), (3) 6:2 fluorotelomer sulfonate (6:2 FTS), and (4) perfluoro(2-ethoxyethane) sulfonic acid (PFEES). | ||
| Compound | Abbreviation | Structural formula | Water solubility (μM) | Vapor pressurea (kPa) | Henry's constant (dimensionless) | pKa |
|---|---|---|---|---|---|---|
| All properties determined at 25 °C; N/A = not available. a Vapor pressure determined for the undissociated species. b Modeled; (a) Giesy and Kannan (2002);9 (b) Kwan (2001);52 (c) Campbell et al. (2009);20 (d) Wang et al. (2011);44 (e) ENVIRON International Corporation (2014).53 | ||||||
| Perfluorooctanesulfonate | PFOS | C8F17SO3− | 1.7 × 104–3.3 × 104 (a) | 0.33 (a) | 0.01 (a) | −3.41 (d)b |
| Perfluorohexanesulfonate | PFHxS | C6F13SO3− | N/A | N/A | 4.20 × 10−3 (d)2 | −3.45 (d)b |
| Perfluorobutanesulfonate | PFBS | C4F9SO3− | N/A | N/A | 2.60 × 10−3 (d)2 | −3.94 (d)b |
| Perfluorooctanoate | PFOA | C8F15O2− | 5.3 × 105 (c) | 1.53–1.85 (b) | 0.09 (a) | 0.90 (d)b |
| Perfluorohexanoate | PFHxA | C6F11O2− | ≪1.7 × 103 (e) | 0.11–0.12 (e)b | N/A | 0.84 (e) |
| Perfluoropentanoate | PFPA | C5F9O2− | N/A | 2.16–3.24 (b) | N/A | 0.81 (d)b |
| Perfluoropropionate | PFPrA | C3F5O2− | N/A | 3.93 (b) | N/A | N/A |
| 6:2 Fluorotelomer sulfonate |
6:2 FTS |
C8H5F13SO3− | N/A | N/A | N/A | N/A |
| Perfluoro(2-ethoxyethane) sulfonate | PFEES | C4F9SO4− | N/A | N/A | N/A | N/A |
Sonochemical experiments
A multi-frequency cubical reactor custom made by Weber Ultrasonics (Clarkston, MI, USA) was used. The stainless steel reactor (23 cm × 23 cm × 23 cm) was fitted with one transducer assembly on each wall with operating frequencies in the range of 25 to 500 kHz. A generator was used to provide the transducer with an input power of 200 W (∼8 W cm−2).All the experiments were carried out in a beaker containing a 200 mL of PFAS solution. A rectangular glass container (20 cm × 6 cm × 6 cm and 0.3 cm wall thickness) was suspended 1 cm from the 500 kHz transducer surface. The experimental solution contained in the beaker was partially covered with Parafilm® and bubbled with argon for 20 min prior to the experiment and a blanket of this gas was maintained throughout the experiment. The beaker was placed in the sonochemical reactor, which contained cooling water circulated through a heat exchanger to maintain the temperature of the experimental solution constant at 30–35 °C during exposure to the acoustic field. A scheme of the experimental set-up can be seen in Fig. 2. Samples were taken periodically for 3 hours and the fluoride concentration was analyzed. An experiment was performed to evaluate the sonochemical degradation of PFOS over an extended time (360 min). Additional measurements of sulfate and total organic carbon (TOC) were also performed for selected experiments. The solution pH and temperature were recorded during the experiments.
 | ||
| Fig. 2 Experimental set-up utilized to perform the sonochemical degradation experiments under 500 kHz. The experimental set-up at 1 MHz is the same, but with the PTC sonochemical reactor instead of the Weber reactor (represented in this figure). | ||
Analytical methods
The fluoride ion concentration was determined using an ion selective electrode (ISE) provided by Thermo Scientific Inc. (Beverly, MA, USA). The ISE was calibrated using standards prepared with varying concentrations of sodium fluoride (10−6 to 10−1 M). A total ion strength adjustment buffer (TISAB II) solution was added to the samples to ensure high ionic strength for potentiometric measurements. Equal volumes (5 mL) of samples and buffer were mixed and stirred before fluoride ion measurements.The concentration of sulfate ions in aqueous samples was measured using an ion-chromatography system Dionex IC-3000 (Sunnyvale, CA, USA) fitted with a Dionex IonPac AS18 analytical column (4 mm × 250 mm) and a AG18 guard column (4 mm × 50 mm). The eluent (KOH) concentration was 17 mM.
The TOC in the solution as a function of sonication time was measured as an indicator of the degree of mineralization. A Shimadzu total organic carbon analyzer (VCSH model, Shimadzu, Columbia, MD, USA) was used for TOC analysis as non-purgeable organic carbon (NPOC). A calibration curve using potassium hydrogen phthalate (KHP) concentrations in the appropriate range was generated prior to each round of sample measurement. Samples (2 mL) were added to vials containing 8 mL of water adjusted to pH 2 with concentrated hydrochloric acid (HCl). One standard (15 mL) with a known concentration of PFOS (464 μM) was always measured to ensure the protocol was working. The method was validated by comparing the measured with the theoretical TOC concentration in solutions containing known PFOS concentrations. As shown in Fig. S1 in the ESI,† the TOC recovery was measured to be 93% of the theoretical value. Previous studies have also demonstrated that the TOC analysis was able to measure 81–100% (91% average) of the carbon content in four fluorinated test substances (PFOS, PFOA, pentafluoropropanol, and trifluoroacetate).29
Results and discussion
PFOS and PFOA degradation
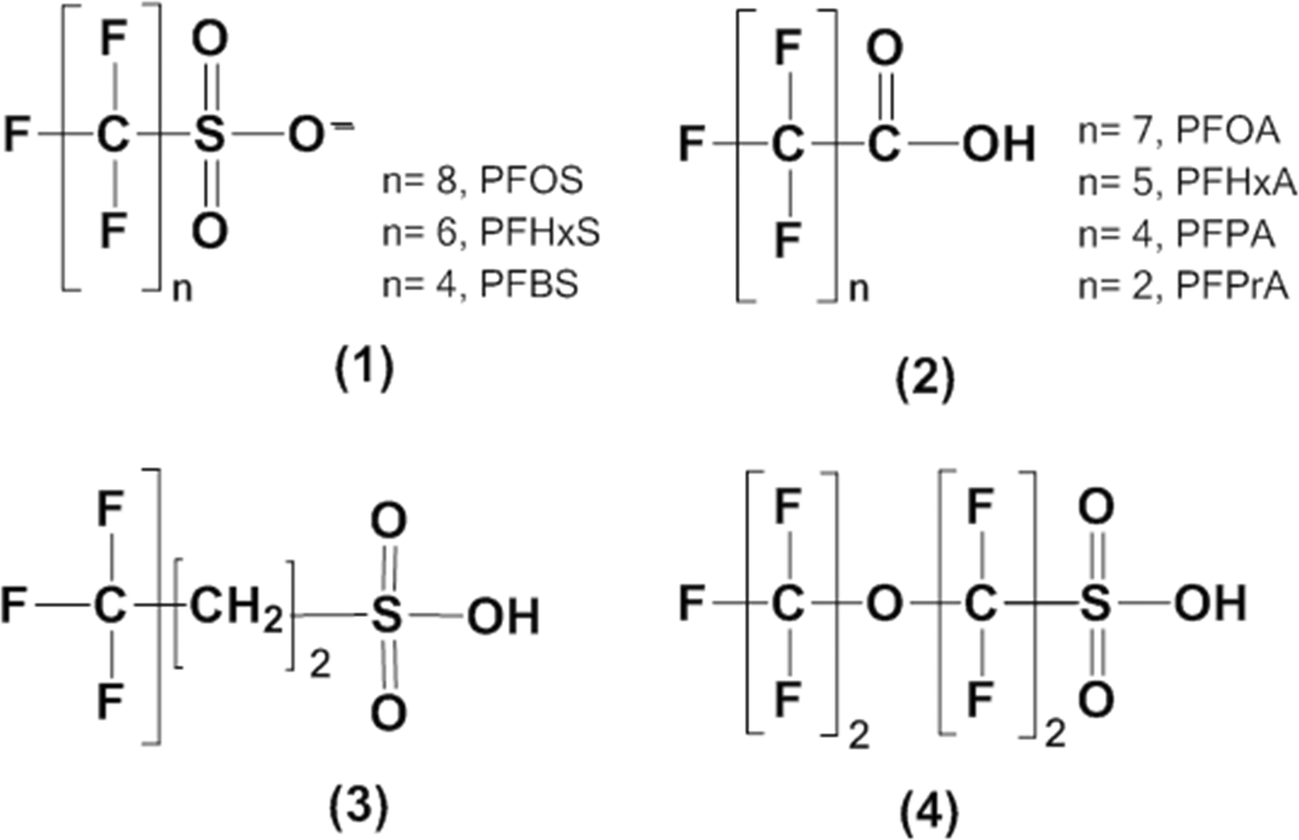
The sonochemical degradation of two widely used PFASs, PFOS and PFOA, with the same carbon chain length (8 C) was investigated. Fig. 3A and 4A plot the concentration of fluoride released as a function of sonication time in experiments with PFOS (100 μM) and PFOA (113 μM), respectively. PFOS and PFOA defluorinated at a similar rate under 500 kHz. After an initial lag phase of approximately 30 min, a steady release of fluoride with time was observed with a rate of 3.5 to 3.7 μM F− min−1, which is indicative of PFOS and PFOA defluorination. The concentration of fluoride released after 180 min was 640 μM for PFOS (37.6% of the total initial fluorine content of 1.7 mM) and 670 μM for PFOA (39.8% defluorination). When the sonochemical degradation time was extended to 360 min, 79.2 ± 5.51% of the total fluorine was released at the end of the experiment, and the defluorination rate (3.7 μM min−1) was constant during this time (Fig. S2 in the ESI†). An increase in the concentration of dissolved sulfate with sonication time at a rate of 9.6 10−2 μM SO42− min−1 was also observed in the experiments with PFOS (Fig. 3B), which indicates cleavage of the C–S bond and oxidation of the terminal sulfonic group. The concentration of sulfate released after 180 min was 18 μM, which corresponds to 17.8% of the total theoretical sulfur content in the added PFOS.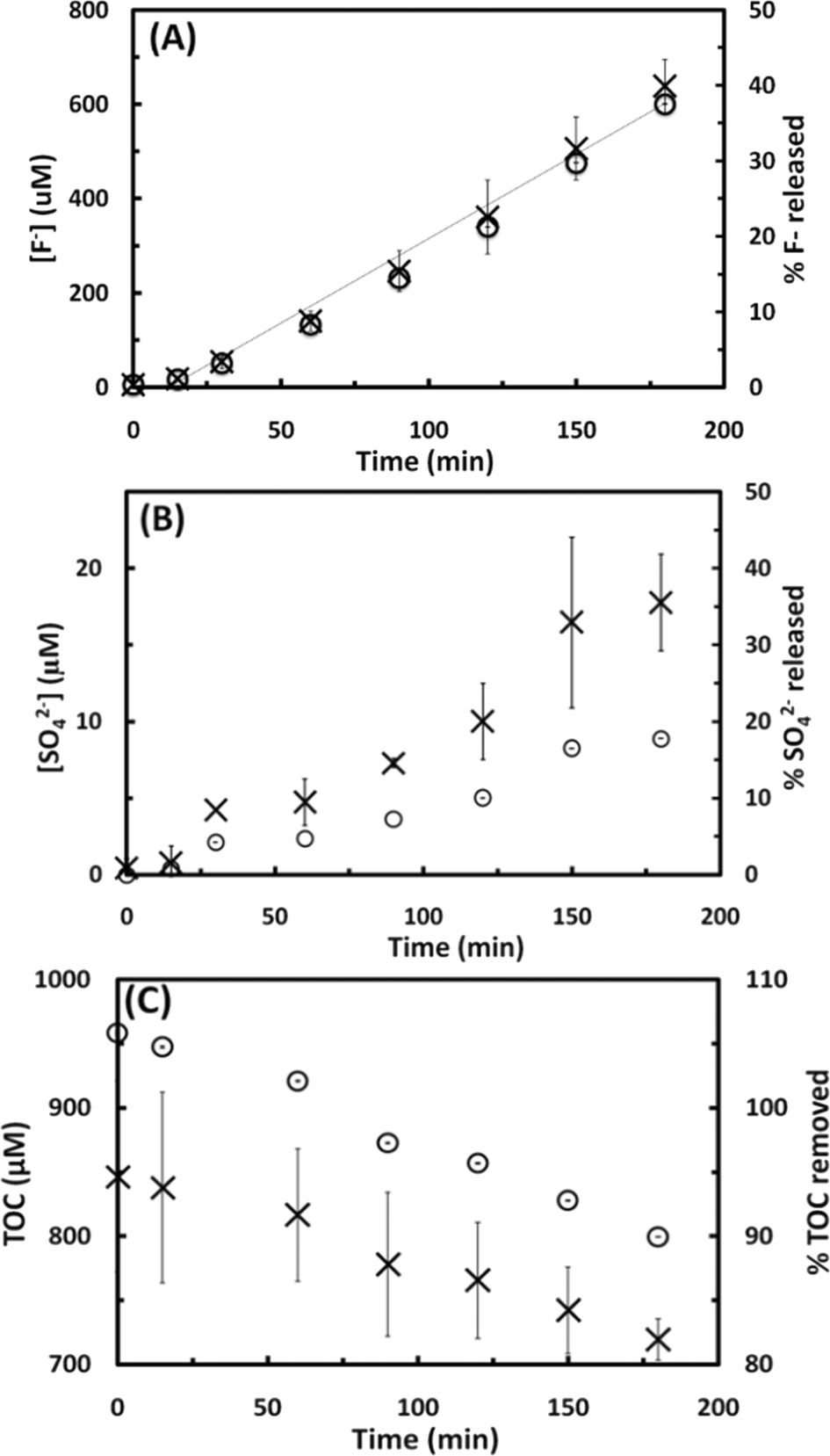 | ||
| Fig. 3 Degradation of PFOS (100 μM) exposed to 500 kHz sound field for 180 min. Release of fluoride (x) and percentage fluoride released (○) (panel A); release of sulfate (x) and percentage sulfate released (○) (panel B); and removal of total organic carbon (TOC) (x) and percentage TOC removed (○) (panel C) as a function of sonication time. The release of F− followed a zero-order reaction rate with the equation: [F−]t = [F−]0 + kt, where [F−]0 = 41.78 μM is the F− concentration at time zero, k = 3.575 μM min−1 is the reaction rate constant, with R2 = 0.982. | ||
 | ||
| Fig. 4 Degradation of PFOA (113 μM) exposed to 500 kHz sound field for 180 min. Release of fluoride (x), and percentage fluoride released (○) (panel A), and the removal of total organic carbon (TOC) (x) and percentage TOC removed (○) (panel B) as a function of treatment time. The release of F− followed a zero-order reaction rate with the equation: [F−]t = [F−]0 + kt, where [F−]0 = 36.00 μM is the F− concentration at time zero, k = 3.674 μM min−1 is the reaction rate constant, with R2 = 0.98. | ||
If PFOS is completely mineralized, the expected ratio between the molar concentration of F− and SO42− released (Δ[F−]/Δ[SO42−]) is 17; however, the experimentally measured Δ[F−]/Δ[SO42−] ratio was 35.3. This observation was also reported in a previous study investigating the extended sonochemical degradation of 100 μM PFOS under 1 MHz frequency.19 The higher Δ[F−]/Δ[SO42−] ratio suggests that defluorination is easier than the removal of the sulfonic acid head group. In this respect, it is interesting to note that Moriwaki et al. (2005)30 observed the formation of shorter chain PFASs with 6 and 7 carbons following sonolysis of PFOS (20 μM) at a frequency of 200 kHz. In contrast with these results, Vecitis et al. (2008)17 reported higher rates of sulfate production than that of fluoride formation during the sonochemical degradation studies with PFOS (10 μM) at 618 kHz, suggesting preferential cleavage of the C–S bond under the experimental conditions utilized in their study. These discrepancies could be related to the difference in the concentration used in the study performed by Vecitis and coworkers and in the current study (10 μM vs. 100 μM, respectively), and to the different sound frequencies utilized (500 kHz vs. 618 kHz, respectively). Previous studies have confirmed that the sonolytic degradation of PFOS is influenced by the frequency applied.19
In addition to the concentration of F− released and SO42− concentration, the mineralization of PFOS and PFOA was also studied by measuring the change in the TOC concentration in the solution as a function of the sonication time, as shown in Fig. 3C and 4B, respectively. The TOC concentration decreased from the initial 846 to 719 μM (15.0% of TOC removal) in the case of PFOS, and from 871 to 485 μM (44.3% of TOC removal) in the case of PFOA after 180 min of sonication, following a zero-order reaction at an average rate of 0.7 and 1.7 μM min−1 for PFOS and PFOA, respectively. These results, together with the sulfur and fluorine released, indicate that sonochemical treatment can mineralize both compounds. The higher mineralization rate observed for PFOA than PFOS could be explained by the different nature of their degradation intermediates and products. PFOA could produce shorter chain perfluoro- and polyfluoroalkyl carboxylates while PFOS could release both perfluoro- and polyfluroroalkyl carboxylates and sulfonates. The difference in degradation rates among PFCAs and PFSAs will be investigated in the next subsection. Finally, the pH decreased from 5.7 to 3.1 in the case of PFOS, and from 4.1 to 3.4 for PFOA in 180 min, which is likely due to the production of hydrofluoric acid (pKa (HF) = 3.2) and PFAS intermediates, and subsequent consumption of hydroxyl ions.31
From the results obtained, it can be concluded that acoustic cavitation is effective in the degradation of aqueous solutions of PFOS and PFOA. The rate of defluorination observed in this study for PFOS (100 μM) and PFOA (113 μM) followed zero-order kinetics, with R2 values of 0.98 (Fig. 3A and 4A), which is in agreement with previous observations reporting that at high initial PFAS concentrations (>40–100 μM TOF, depending on the frequency used) saturation concentration is reached and the sonochemical degradation of both compounds is not concentration dependent.18,19 This observation has been attributed to saturation of bubble–water interface sites where sonochemical degradation of perfluoroalkyl surfactants is believed to occur via a pyrolytic mechanism. At lower PFAS concentrations, however, sonochemical degradation appears to follow pseudo-first order kinetics.17,19,30
Chain length and functional group effect on the sonochemical degradation of PFASs
The sonochemical degradation of various PFASs (Fig. 1) was investigated under 500 kHz sound frequency to understand the effect of changes in their chemical structure on the effectiveness of the sonochemical treatment. The compounds investigated included several PFSAs and PFCAs with perfluorocarbon chain length ranging from 3 to 6 as well as two other related compounds, a perfluorinated ether (PFEES, a 4-C compound), and a sulfonated fluorotelomer (6:2 FTS, C6F13CH2CH2SO3−) containing two non-fluorinated C atoms (Fig. 1). The initial concentrations used and rates of defluorination during the degradation of the various PFASs are listed on Table 2.
| Group | Compound | MW (g mol−1) | Degradation rate (μM F− min−1) | Fluoride released (after 180 min) | |
|---|---|---|---|---|---|
| (%) | (μM) | ||||
| PFAS | PFOS | 499.1 | 3.5 ± 0.3 | 37.6 ± 3.3 | 638.0 ± 56.6 |
| PFHxS | 399.1 | 2.6 ± 0.5 | 27.6 ± 5.7 | 469.0 ± 96.2 | |
| PFBS | 299.1 | 1.8 ± 0.2 | 19.1 ± 2.2 | 323.5 ± 37.5 | |
| PFCA | PFOA | 413.1 | 3.7 ± 0.3 | 39.8 ± 2.8 | 671.0 ± 48.1 |
| PFHxA | 313.1 | 3.5 ± 0.1 | 37.9 ± 1.3 | 644.0 ± 21.2 | |
| PFPA | 263.1 | 2.5 ± 0.4 | 26.5 ± 4.2 | 449.5 ± 71.4 | |
| PFPrA | 163.0 | 1.6 ± 0.2 | 17.1 ± 2.0 | 290.0 ± 33.9 | |
| Other fluorochemicals | 6:2 FTS |
428.2 | 1.5 ± 0.1 | 16.2 ± 0.6 | 275.0 ± 11.3 |
| PFEES | 316.1 | 3.9 ± 0.5 | 41.9 ± 4.8 | 711.5 ± 81.3 | |
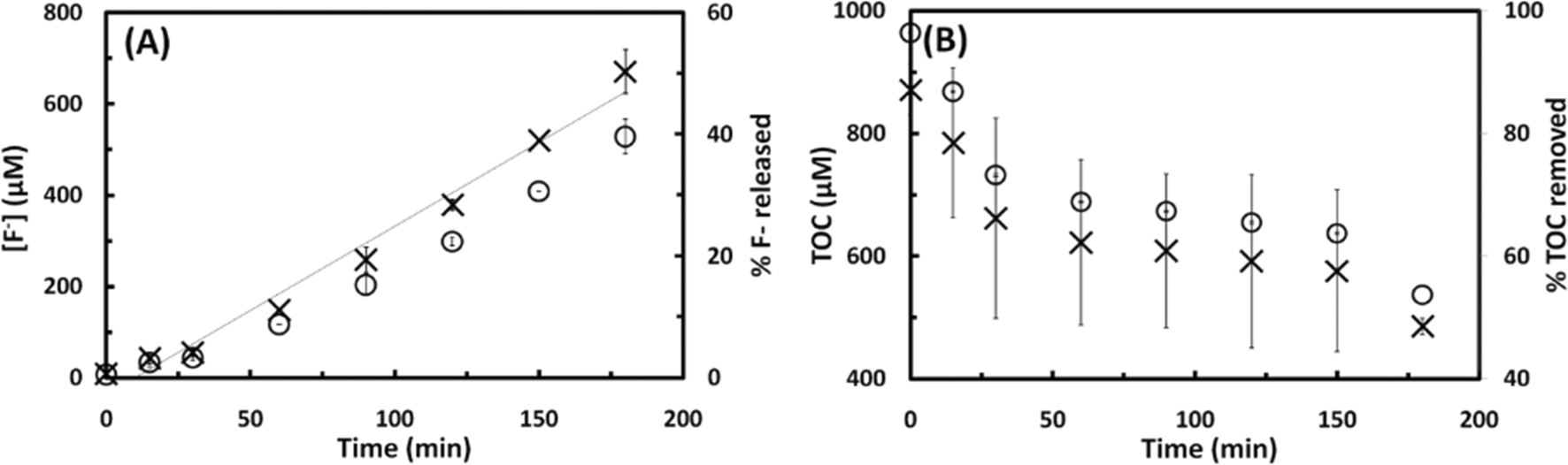
The fluoride released upon the degradation of the different PFSAs as a function of the sonication time is shown on Fig. 5A. The rate of defluorination determined for these analogues followed zero-order kinetics and increased with increasing number of perfluorocarbon atoms. The defluorination rate determined for PFOS, a compound with 8 C atoms, was 3.5 μM min−1, which is 1.3- and 1.9-fold higher than the rates calculated for the PFSA analogues with 6 and 4 C atoms (2.6 and 1.8 μM min−1 for PFHxS and PFBS, respectively). Similarly, the defluorination of the PFCAs proceeded according to zero-order kinetics and the rate generally increased with increasing number of perfluorocarbon atoms (Fig. 5B). The rate of fluoride release determined for PFOA (8 C) was 1.1-, 1.9- and 2.3-fold higher than those calculated for PFHxA (6 C, 3.5 μM min−1), PFPA (5 C, 2 μM min−1), and PFPrA (3 C, 1.6 μM min−1). A summary of the results obtained for the PFSAs and PFCAs tested is shown in Fig. 6. The observed increase in the rate of defluorination can be attributed to an increase in the hydrophobic character of these perfluoroalkyl surfactants with increasing alkyl chain length,32,33 which in turn would be expected to increase the affinity of the molecule for the water–bubble interface, resulting in reduced pyrolytic degradation. Numerous theoretical studies have shown that, within a homologous series of linear PFASs with the same functional group, the octanol–water (Kow) and air–water partition coefficients (Kaw) are expected to increase exponentially with increasing perfluorinated chain length of 2–12 carbons.20,34,35 The increasing lipophilicity of PFCAs and PFSAs with increasing perfluoroalkyl chain has been attributed to the increase in van der Waals interactions and free-energy costs for creating a cavity in the solvent associated with the addition of a CF2 group,35 and the strong electron-withdrawing effect of the perfluoroalkyl chain on the oxoanion headgroup.36 Although differences in Kow for PFASs with a given perfluorinated chain length are small,35 a recent experimental study has demonstrated that enrichment of PFSAs and PFCAs at the water–air interface of microdroplets increases asymptotically with the number of perfluorinated carbons (n) reaching values that are about 2× higher for PFSAs than for PFCAs above n ∼ 9.37 The hyperbolic, rather than the predicted exponential, increase of Kaw with n was ascribed to the onset of conformational restrictions to interfacial enrichment above n ∼ 4.
 | ||
| Fig. 5 Effect of the fluorocarbon chain length on the rate of perfluoroalkyl sulfonate (PFSA) and perfluoroalkyl carboxylate (PFCA) degradation under a sound frequency of 500 kHz. Panel A: fluoride release from 100 μM PFOS (x), 114 μM PFHxS (□), and 189 μM PFBS (○), and percentage fluoride released from 100 μM PFOS (▲), 114 μM PFHxS (■), and 189 μM PFBS (●) as a function of time. Panel B: fluoride release from 113 μM PFOA (Δ), 154 μM PFHxA (○), 189 μM PFPA (□), and 340 μM PFPrA (x); and percentage fluoride released from 113 μM PFOA (▲), 154 μM PFHxA (●), 189 μM PFPA (■), and 340 μM PFPrA (♦) as a function of treatment time. | ||
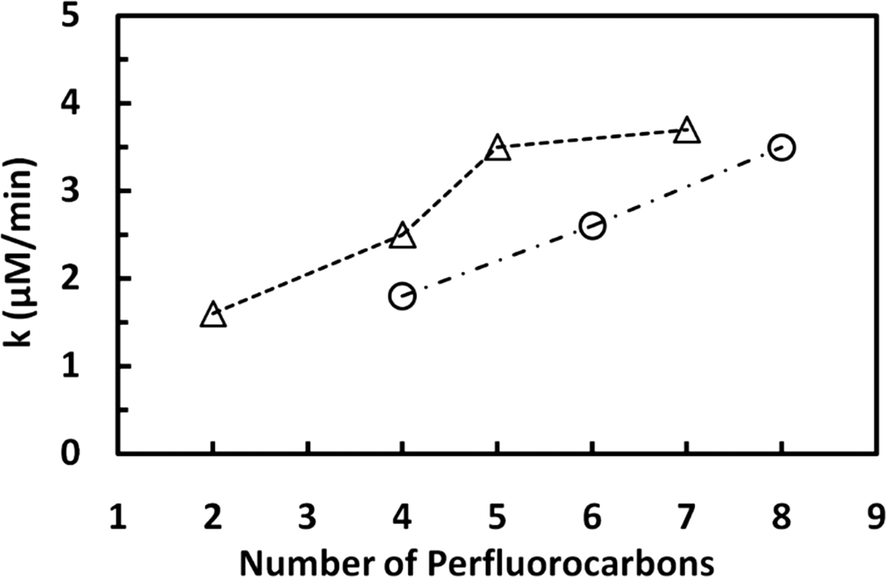 | ||
| Fig. 6 Effect of perfluorocarbon chain length on the susceptibility of perfluoroalkyl carboxylates (Δ) and perfluoroalkyl sulfonates (○) to sonochemical degradation at an acoustic frequency of 500 kHz. k is the pseudo-first order rate determined from the fluoride release vs. time data for the various compounds. The trend lines have R2 values of 0.83 and 0.99, respectively. | ||
These results together with those discussed in the previous section indicate that the hydrophilic functional group has a weak effect on the rate of PFAS degradation, with carboxylates degrading slightly faster than sulfonates with the same perfluorocarbon chain length (Fig. 6). For example, the rates of defluorination determined for PFBS and PFPA were 1.8 to 2.0 μM F− min−1, respectively. In studies performed at much lower concentrations of PFASs, Campbell et al. (2009)20 also observed that the sonolytic kinetics of PFHxA (0.32 μM) was faster than that of PFHxS (0.23 μM) at frequencies ranging from 202 to 1060 kHz. Although PFCAs are less hydrophobic and surface active than PFSAs with the same number of carbon atoms,32 the latter study attributed the higher defluorination rates observed for the PFCAs to their lower thermal activation energies.
Effect of the degree of fluorination on sonochemical degradation
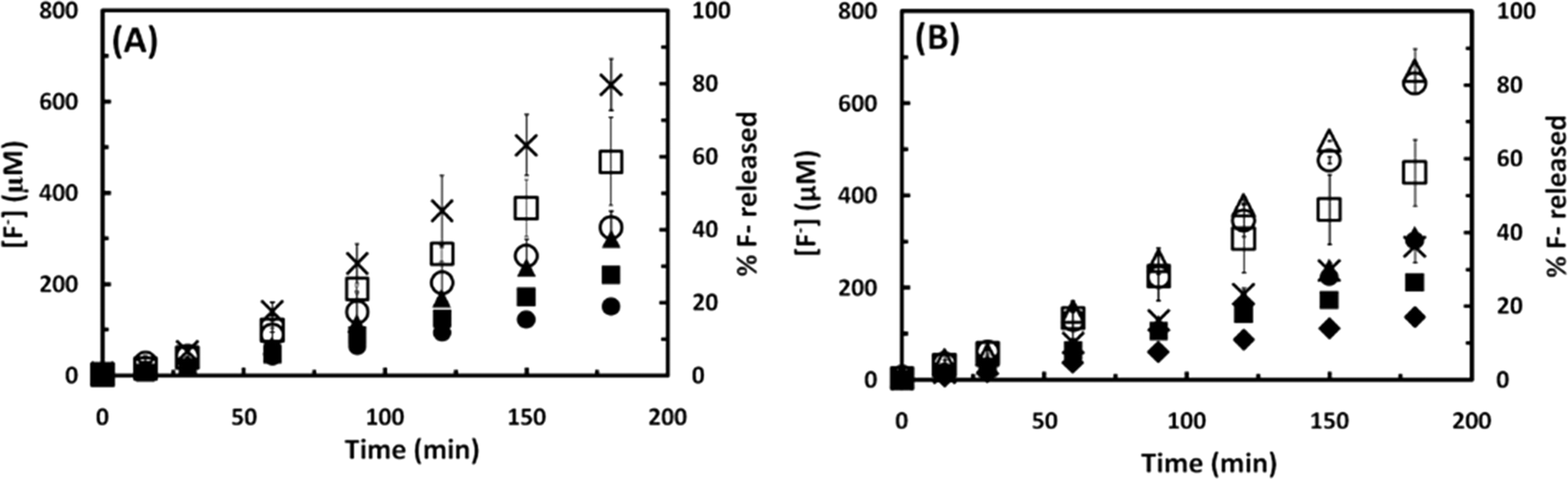
The fluorotelomer 6:2 FTS (C8F17(CH2)2SO3H, Fig. 1) was selected as an example of a polyfluoroalkyl sulfonate to examine the impact of the presence of CH2 moieties on the rate of sonochemical attack. Surfactants based on six-carbon fluorotelomers are important chemicals which have largely replaced PFOS and PFSA-based surfactants in numerous applications.38,39 Fluorotelomer sulfonates are also formed as degradation products of fluorotelomer sulfonyl surfactants which are widely used in AFFFs.40,41 The sonochemical degradation of 6:2 FTS (130 μM) and PFOS (100 μM), two fluorinated sulfonates with an identical number of C atoms but different number of F atoms, was compared. Similar to the results obtained for the PFCAs and PFSAs, the defluorination of 6:2 FTS followed zero-order kinetics (after ∼30 min of initial lag) at a rate of 1.5 μM min−1, which is 2.3-fold lower than the rate determined for PFOS under similar conditions. The pH of the solution decreased from 5.0 to 3.6 at the end of the experiment. The lower degradation rate of 6:2 FTS than that of PFOS seems to be related to its chemical structure (Fig. 1). Polyfluorinated chemicals, such as 6:2 FTS, are susceptible to chemical and microbial attack leading to the formation of shorter chain length perfluoro- and polyfluoroalkyl carboxylates.38,42–44 The formation of shorter PFCAs is likely to result in a decrease of the rate of sonolysis since, as discussed in the previous section, the shorter chain length compounds are less hydrophobic and do not adsorb as effectively on the bubble–liquid interface. 6:2 FTS itself is also considerable less hydrophobic than PFOS (Kow values of 2.66 and 4.49 estimated for 6:2 FTS and PFOS, respectively, using EPI-SUITE), a factor that could also contribute to the lower degradation rates determined. The lower lipophilicity (lower Kow and Kaw) of fluorotelomer acids than that of their perfluorinated analogues has been attributed to the decrease in the electron-withdrawing effect of the perfluorinated chain on the oxyanion head group caused by CH2 moieties and the resulting increase in the polarity of the sulfonate or carboxylate group.36
The sonochemical degradation of PFEES (CF3CF2–O–(CF2)2SO3−, 183 μM) was also studied. PFEES is a perfluorinated ether sulfonate (Fig. 1) with superior mechanical properties and high chemical and thermal stability, which is used as a PFOS substitute in a number of applications including as a cracking catalyst and in proton exchange membranes for fuel cells.45 PFEES contains an ether bond linking two perfluoroalkyl chains, one of which has a terminal sulfonate group. Fig. 7 compares the defluorination rate observed during the sonochemical degradation of PFEES to that obtained for PFOS. The rate of defluorination determined in experiments with PFEES, 3.9 μM min−1, was very similar to that observed for PFOS but 2.2-fold higher than that of PFBS, a PFSA with the same number of perfluorocarbon atoms. The rate of degradation is also higher than anticipated based on the low hydrophobicity of the compound (Kow = 2.25, estimated using EPI-SUITE). The higher defluorination rates observed for PFEES may be due to the susceptibility of the compound to attack by hydroxyl radicals (OH˙) produced during the sonochemical processes. Several studies have demonstrated that hydroxyl radicals attack both the main- and the side chains in PFEES causing the rupture of the C–O–C ether bond and loss of the sulfonic group from the side chain.45–48 In contrast, hydroxyl radicals seem to play a minor role in the sonochemical degradation of PFASs as indicated by the ineffectiveness of advanced oxidation processes (AOPs) that rely on OH˙ attack such as UV-ozonation or Fenton's reaction (a process based on H2O2 and Fe2+) to destroy PFOS and PFOA.30,49,50
 | ||
| Fig. 7 Effect of fluorination degree on the degradation rate of various fluorochemicals under a sound frequency of 500 kHz. Fluoride released from 100 μM PFOS (x), 130 μM 6:2 FTS (○), and 183 μM PFEES (Δ), and percentage fluoride released from 100 μM PFOS (♦), 130 μM 6:2 FTS (●), and 183 μM PFEES (▲) as a function of sonication time. | ||
Application of sonochemical degradation
The scalability of a sonochemical process depends on the acoustic frequency and transducer power density. At lower ultrasonic frequency (20–100 kHz) and higher power density (>10 W cm−2), a conical bubble structure is known to form close to the transducer surface,51 which restricts the bubble activity in the vicinity of the transducer making the process less scalable. At higher megasonic frequency (>2 MHz) and lower power density, although the process is scalable, it generates milder cavitation, which may adversely affect the extent of mineralization of the contaminants and result in a less effective process. Therefore, a choice of an intermediate acoustic frequency (0.5–1.0 MHz) with optimum power density (such as the one selected for this study) will offer the benefit of both scalability and performance for remediation of large volumes of a wide range of organic pollutants.Conclusions
This study demonstrates that perfluoroalkyl compounds (i.e., PFSAs, PFCAs, perfluorosulfonate ethers) as well as polyfluoroalkyl compounds (i.e., fluorotelomer) are susceptible to sonochemical degradation as evidenced by the observed release of fluoride, loss of total organic carbon, and, in the case of PFSAs, release of sulfate, when their aqueous solutions were irradiated at a frequency of 500 kHz. The degradation rate increased significantly with increasing perfluoroalkyl chain length. In contrast, the head group had a weak effect on the rate of PFAS degradation, with carboxylates degrading slightly faster than sulfonates with the same perfluorocarbon chain length. Furthermore, the rate of defluorination was also found to increase with the degree of fluorination as confirmed by the 2.6-fold higher rate determined for PFOS than that of a fluorotelomer compound with the same number of carbon atoms (6:2 FTS). The observed enhancement in the susceptibility of PFASs to sonolytic decomposition with increasing perfluorocarbon chain length and fluorination degree is likely related to the contribution of these chemical features in increasing compound hydrophobicity and, thereby, the compound concentration adsorbed at the interface of acoustic cavitation bubbles where pyrolytic degradation is believed to take place. These results indicate that sonochemical treatment is a promising method for the treatment of aqueous streams containing PFASs.
Acknowledgements
The work presented here was supported by the Air Force Civil Engineering Center, under the project number FA8903-13-C-0011.References
- C. A. Moody and J. A. Field, Environ. Sci. Technol., 2000, 34, 3864–3870 CrossRef CAS.
- R. C. Buck, J. Franklin, U. Berger, J. M. Conder, I. T. Cousins, P. de Voogt, A. A. Jensen, K. Kannan, S. A. Mabury and S. P. J. van Leeuwen, Integr. Environ. Assess. Manage., 2011, 7, 513–541 CrossRef CAS PubMed.
- C. D. Vecitis, H. Park, J. Cheng, B. T. Mader and M. R. Hoffmann, Front. Environ. Sci. Eng. China, 2009, 3, 129–151 CrossRef CAS.
- J. Seow, Department of Environment and Conservation Western Australia, http://www.hemmingfire.com/news/get_file.php3/id/287/file/Seow_WADEC_PFCs_Firefighting_Foam_final_version_7June2013.pdf, 2013, p. 76.
- D. Skutlarek, M. Exner and H. Farber, Environ. Sci. Pollut. Res., 2006, 13, 299–307 CrossRef CAS PubMed.
- S. Fujii, C. Polprasert, S. Tanaka, N. P. H. Lien and Y. Qiu, J. Water Supply: Res. Technol.--AQUA, 2007, 56, 313–326 CrossRef CAS.
- M. P. Krafft and J. G. Riess, Curr. Opin. Colloid Interface Sci., 2015, 20, 192–212 CrossRef CAS.
- A. B. Lindstrom, M. J. Strynar and E. L. Libelo, Environ. Sci. Technol., 2011, 45, 7954–7961 CrossRef CAS PubMed.
- J. P. Giesy and K. Kannan, Environ. Sci. Technol., 2002, 36, 146A–152A CrossRef CAS PubMed.
- A. G. Paul, K. C. Jones and A. J. Sweetman, Environ. Sci. Technol., 2008, 43, 386–392 CrossRef.
- POPRC, Persistent Organic Pollutants Review Committee (POPRC). Recommendations 405 for listing Chemicals. Secretariat of the Stockholm Convention, http://chm.pops.int/Convention/POPs-ReviewCommittee/Chemicals/tabid/243/language/en-US/Default.aspx, 2015.
- M. Haukas, U. Berger, H. Hop, B. Gulliksen and G. W. Gabrielsen, Environ. Pollut., 2007, 148, 360–371 CrossRef CAS PubMed.
- M. Saez, P. de Voogt and J. R. Parsons, Environ. Sci. Pollut. Res., 2008, 15, 472–477 CrossRef CAS PubMed.
- C. Lau, K. Anitole, C. Hodes, D. Lai, A. Pfahles-Hutchens and J. Seed, Toxicol. Sci., 2007, 99, 366–394 CrossRef CAS PubMed.
- F. M. Hekster, R. Laane and P. de Voogt, Reviews of Environmental Contamination and Toxicology, Vol 179, 2003, vol. 179, pp. 99–121 Search PubMed.
- EPA, Lifetime Health Advisories and Health Effects Support Documents for Perfluorooctanoic Acid and Perfluorooctane Sulfonate, EPA–HQ–OW–2014–0138; FRL–9946–91–OW, 2016.
- C. D. Vecitis, H. Park, J. Cheng, B. T. Mader and M. R. Hoffmann, J. Phys. Chem. A, 2008, 112, 4261–4270 CrossRef CAS PubMed.
- C. D. Vecitis, H. Park, J. Cheng, B. T. Mader and M. R. Hoffmann, J. Phys. Chem. C, 2008, 112, 16850–16857 CAS.
- L. Rodríguez-Freire, R. Balachandrana, R. Sierra-Alvarez and M. Keswani, J. Hazard. Mater., 2015, 300, 662–669 CrossRef PubMed.
- T. Y. Campbell, C. D. Vecitis, B. T. Mader and M. R. Hoffmann, J. Phys. Chem. A, 2009, 113, 9834–9842 CrossRef CAS PubMed.
- M. A. Oturan and J.-J. Aaron, Crit. Rev. Environ. Sci. Technol., 2014, 44, 2577–2641 CrossRef CAS.
- E. B. Flint and K. S. Suslick, Science, 1991, 253, 1397 CrossRef CAS PubMed.
- P. Chowdhury and T. Viraraghavan, Sci. Total Environ., 2009, 407, 2474–2492 CrossRef CAS PubMed.
- S. Rayne and K. Forest, J. Environ. Sci. Health, Part A: Toxic/Hazard. Subst. Environ. Eng., 2009, 44, 1145–1199 CrossRef CAS PubMed.
- K. Prevedouros, I. T. Cousins, R. C. Buck and S. H. Korzeniowski, Environ. Sci. Technol., 2005, 40, 32–44 CrossRef.
- P. C. Rumsby, C. L. McLaughlin and T. Hall, Philos. Trans. R. Soc., A, 2009, 367, 4119–4136 CrossRef CAS PubMed.
- B. J. Place and J. A. Field, Environ. Sci. Technol., 2012, 46, 7120–7127 CrossRef CAS PubMed.
- W. J. Backe, T. C. Day and J. A. Field, Environ. Sci. Technol., 2013, 47, 5226–5234 CrossRef CAS PubMed.
- A. L. L. Bourgeois, Biodegradability of fluorinated fire-fighting foams, MS thesis, Worcester Polytechnic Institute, May 2014 Search PubMed.
- H. Moriwaki, Y. Takagi, M. Tanaka, K. Tsuruho, K. Okitsu and Y. Maeda, Environ. Sci. Technol., 2005, 39, 3388–3392 CrossRef CAS PubMed.
- Y. Qu, C. J. Zhang, P. Chen, Q. Zhou and W. X. Zhang, Chemosphere, 2014, 107, 218–223 CrossRef CAS PubMed.
- J. M. Conder, R. A. Hoke, W. De Wolf, M. H. Russell and R. C. Buck, Environ. Sci. Technol., 2008, 42, 995–1003 CrossRef CAS PubMed.
- M. Kim, L. Y. Li, J. R. Grace and C. Yue, Environ. Pollut., 2014, 196, 462–472 CrossRef PubMed.
- H. P. H. Arp and K. U. Goss, Environ. Sci. Technol., 2009, 43, 8542–8547 CrossRef CAS PubMed.
- Z. Y. Wang, M. MacLeod, I. T. Cousins, M. Scheringer and K. Hungerbuhler, Environ. Chem., 2011, 8, 389–398 CrossRef CAS.
- P. Jing, P. J. Rodgers and S. Amemiya, J. Am. Chem. Soc., 2009, 131, 2290–2296 CrossRef CAS PubMed.
- E. Psillakis, J. Cheng, M. R. Hoffmann and A. J. Colussi, J. Phys. Chem. A, 2009, 113, 8826–8829 CrossRef CAS PubMed.
- X. Yang, J. Huang, K. Zhang, G. Yu, S. Deng and B. Wang, Environ. Sci. Pollut. Res., 2014, 21, 4634–4642 CrossRef CAS PubMed.
- R. A. Hoke, B. D. Ferrell, T. Ryan, T. L. Sloman, J. W. Green, D. L. Nabb, R. Mingoia, R. C. Buck and S. H. Korzeniowski, Chemosphere, 2015, 128, 258–265 CrossRef CAS PubMed.
- B. Krol, K. Prochaska and Ł. Chrzanowski, Fire Technol., 2012, 48, 173–181 CrossRef.
- M. K. Moe, S. Huber, J. Svenson, A. Hagenaars, M. Pabon, M. Trumper, U. Berger, D. Knapen and D. Herzke, Chemosphere, 2012, 89, 869–875 CrossRef CAS PubMed.
- D. A. Ellis, J. W. Martin, A. O. De Silva, S. A. Mabury, M. D. Hurley, M. P. S. Andersen and T. J. Wallington, Environ. Sci. Technol., 2004, 38, 3316–3321 CrossRef CAS PubMed.
- E. F. Houtz and D. L. Sedlak, Environ. Sci. Technol., 2012, 46, 9342–9349 CrossRef CAS PubMed.
- N. Wang, J. Liu, R. C. Buck, S. H. Korzeniowski, B. W. Wolstenholme, P. W. S. Folsom and L. M. Sulecki, Chemosphere, 2011, 82, 853–858 CrossRef CAS PubMed.
- M. Danilczuk, L. Lina, S. Schlicka, S. J. Hamrockb and M. S. Schabergb, J. Power Sources, 2011, 196, 8216–8224 CrossRef CAS.
- A. M. Dreizler and E. Roduner, Fuel Cells, 2012, 12, 132–140 CrossRef CAS.
- L. Ghassemzadeh and S. Holdcroft, J. Am. Chem. Soc., 2013, 135, 8181–8184 CrossRef CAS PubMed.
- T. Ishimoto, R. Nagumo, T. Ogura, T. Ishihara, B. Kim, A. Miyamoto and M. Koyama, J. Electrochem. Soc., 2010, 157, B1305–B1309 CrossRef CAS.
- H. Hori, E. Hayakawa, H. Einaga, S. Kutsuna, K. Koike, T. Ibusuki, H. Kiatagawa and R. Arakawa, Environ. Sci. Technol., 2004, 38, 6118–6124 CrossRef CAS PubMed.
- H. F. Schroder and R. J. W. Meesters, J. Chromatogr. A, 2005, 1082, 110–119 CrossRef PubMed.
- B. Dubus, C. Vanhille, C. Campos-Pozuelo and C. Granger, Ultrason. Sonochem., 2010, 17, 810–818 CrossRef CAS PubMed.
- W. C. Kwan, https://tspace.library.utoronto.ca/bitstream/1807/16278/1/MQ62941.pdf, 2001.
- E. I. Corporation, http://www.fluorocouncil.com/PDFs/Assessment-of-POP-Criteria-for-Specific-Short-Chain-Perfluorinated-Alkyl-Substances.pdf, 2004.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ew00150e |
| This journal is © The Royal Society of Chemistry 2016 |