
Nitrate removal from water using a nanopaper ion-exchanger
Andreas
Mautner
*ab,
Henry A.
Maples
a,
Houssine
Sehaqui
c,
Tanja
Zimmermann
c,
Uxua
Perez de Larraya
d,
Aji P.
Mathew
e,
Chi Yan
Lai
f,
Kang
Li
f and
Alexander
Bismarck
*ab
aPolymer & Composite Engineering (PaCE) Group, Institute for Materials Chemistry & Research, University of Vienna, Währingerstr. 42, A-1090 Vienna, Austria. E-mail: andreas.mautner@univie.ac.at; alexander.bismarck@univie.ac.at
bPolymer & Composite Engineering (PaCE) Group, Dept. of Chemical Engineering, Imperial College London, South Kensington Campus, London, SW7 2AZ, UK
cApplied Wood Materials Laboratory, Empa – Federal Laboratories for Materials Science and Technology, Überlandstrasse 129, CH-8600 Dübendorf, Switzerland
dCemitec, Polígono Mocholí, Plaza Cein 4, E-31110 Noain, Navarra, Spain
eDivision of Materials Science, Department of Engineering Sciences and Mathematics, Luleå University of Technology, S-97187 Luleå, Sweden
fDept. of Chemical Engineering, Imperial College London, South Kensington Campus, London, SW7 2AZ, UK
First published on 17th August 2015
Abstract
Nitrates seriously affect drinking water quality. We herein present a process for the efficient removal of nitrates from water using a nanopaper ion-exchanger, which can be operated in flow-through conditions. The nanopaper ion-exchanger was produced from nanofibrillated cellulose obtained from fibre sludge, a paper-production waste stream, using a simple paper-making process. The cellulose nanofibrils were modified with quaternary trimethylammonium groups. The performance of these cationic nanopaper ion-exchangers was assessed with respect to their permeance and nitrate adsorption. Nitrates could be successfully captured onto the cationic nanopaper and thus rejected from contaminated water during dynamic filtration experiments. The ion-exchange nanopaper had adsorption capacities in the range of commercial available adsorbers but with the advantage of reduced contact time.
Water impactNitrates are a major fraction in fertilizers, utilized worldwide to increase crop yield. The accumulation of nitrates in the environment is problematic as it causes eutrophication and acidification. Additionally, high concentrations of nitrates in potable water cause adverse health effects and therefore, the removal of nitrates is of great importance. Currently this is done by approaches such as adsorption, reverse osmosis and biological denitrification, each of which exhibits particular drawbacks. We here present the development of ion-exchange nanopapers from a renewable material derived from an industrial waste stream – fibre sludge, a waste product of the paper industry. Our method combines the advantages of membrane processes and an adsorbent resulting in effective adsorption and thus removal of nitrates in a continuous process. |
Introduction
The accumulation of nitrates in fresh water constitutes a serious threat to human health.1,2 Most of the nitrates are derived from effluents of the agricultural industry, stemming from fertilizers and manure run-off,3 as well as leakage from septic systems, landfill leachate and unsafe disposal of untreated sanitary and industrial waste.4,5 NOx air stripping waste from air pollution control devices is a further source of nitrates.4 Due to the high water solubility of nitrates, via the process of seepage, nitrates are leached into ground water wells or rivers, which constitute the main sources of drinking water in most regions of our planet.6 Accordingly, apart from eutrophication processes,5 the contamination of drinking water is a big problem. Even though moderate nitrate concentrations, below 15 mg L−1, are not considered harmful to humans,7 higher concentrations may cause adverse health effects such as methemoglobinemia8 and the formation of carcinogenic nitrosamines in the human body.9 In due course, the concentration of nitrates in potable water must be limited and not exceed 50 mg L−1, as suggested by the WHO.10 This has already become a law, e.g. in the EU (Nitrates Directive (91/676/EEC)) and the United States.3 However, in regions with hot and dry climate, especially arid and semi-arid zones,5 these limits are already very hard to reach due to the extensive use of fertilizers and water shortage; hence, solutions to this problems are therefore urgently needed. Thus, in the field of water treatment operations for providing clean potable water, addressing nitrates is an important task.Nowadays, the problem of high nitrate concentrations in potable water is usually addressed by chemical denitrification,11 often supported by (photo)catalytic processes.12 In addition, adsorbents,6,13 for instance ion exchange resins14 or renewable materials such as chitosan,15 reverse osmosis (RO)16 and nanofiltration (NF)17 as well as microbiological denitrification are used.18 Combining biological denitrification with membrane technology has led to the development of membrane bioreactors.19 Electroreduction and electrocoagulation20 as well as electrodialysis21 have also been used for the removal of nitrates. Some of these processes are already successfully commercialized. However, there are several drawbacks inherent in these processes. For example, adsorbent materials have to be applied batchwise and thus no continuous process is possible.4 Additionally, the problem of safe disposal of saturated adsorbent materials still poses problems.6 Efficient nitrate removal is possible by membrane operations such as RO and NF which are continuous operations, including back-wash procedures, which constitutes a big advantage.2,22 Yet, RO and NF membranes, unfortunately, suffer from an inherently low permeability and require pressures up to 50 bar, thus necessitating a lot of energy to achieve reasonable effectiveness in terms of water purification.23 In addition, most of these membranes are made from synthetic polymers, requiring solvents and chemicals during their manufacture,24 or ceramics, which have even higher energy requirements compared to polymer membranes.25 Membranes from natural resources, e.g. regenerated cellulose or cellulose derivatives, such as cellulose acetate, also require considerable amounts of chemicals, solvents and energy for their production.26
Nanopapers have been recently introduced as an alternative to synthetic polymers for use as membranes in tight aqueous ultrafiltration and organic solvent nanofiltration processes, providing the advantage of a clean and simple nanopaper production process.27 Nanopapers are produced from cellulose nanofibrils (CNF), i.e. cellulose fibrils with diameters in the nm range and lengths around single-digit μm, prepared by mechanical disintegration of pulp fibers from wood or agricultural residues, via a simple and scalable paper-making filtration process.28 However, even though nanopapers are used for aqueous ultrafiltration processes close to NF, the permeability of these membranes is relatively low and the rejection rates for ions are limited.27 Furthermore, nanopapers lack suitable functional groups for adsorption of nitrate ions.
In recent years, it was demonstrated that CNF are capable of adsorbing different kinds of ions, e.g. heavy metal ions or cationic dyes, via electrostatic interaction between functional groups and ions in static conditions.29,30 The introduction of certain functional groups, such as phosphates, acid or ammonium groups, greatly improved the amount of adsorbed ions.30,31 Since the production of CNF not necessarily requires chemicals or solvents, a natural renewable material is at hand, having the capacity to adsorb pollutants from aqueous solutions.32,33 It was recently demonstrated that ammonium functionalized cellulose has potential for the removal of nitrates from contaminated water.34,35 The introduction of quaternary ammonium groups onto CNF also tackles the problem of membrane fouling, which is a major concern in the field of membrane technology, since ammonium groups improve anti-fouling properties, due to increased hydrophilicity and positive charges.36
In this work, we combine the advantages of a natural adsorbent and membrane filtration processes for the development of a nanopaper ion-exchanger intended for the adsorption of nitrates in a continuous filtration process. We produced nanopapers using cellulose nanofibrils prepared from fibre sludge, a waste stream in the paper industry, functionalized with ammonium groups. These nanopapers were subsequently used to remove nitrate ions in a filtration process. The evolution of an adsorption material into a continuously working nanopaper ion-exchanger constitutes a step forward in tackling the problem of nitrate accumulation in water resources.
Experimental
Materials
Glycidyltrimethylammonium chloride (GTMAC), NaOH, KOH, HCl, KCl, AgNO3 and NaNO3 were purchased from Aldrich and used without further purification. Deionized water was used for permeance measurements.Characterization of cationic CNF
Manufacturing of unmodified/cationic nanopapers
Nanopapers were produced according to a previously reported method.27,39,40 First, the initial consistency of cationic or unmodified CNF was adjusted to 0.3 wt% by water addition. This suspension was subsequently blended (Breville VBL065-01, Oldham, UK) for 2 min to produce a homogeneous CNF-in-water suspension. These suspensions were then vacuum-filtered onto a cellulose filter paper (VWR 413, 5–13 μm pore size, Lutterworth, UK) until a wet filter cake was formed. The wet filter cake was wet-pressed under a weight of 10 kg between blotting papers (3MM Chr VWR, Lutterworth, UK) for 5 min to further remove excess water. These wet filter cakes, which had a measured moisture content of approximately 85%, were then consolidated and dried in a hot-press (25-12-2H, Carver Inc., Wabash, USA) under a compression weight of 1 t for 1 h at 120 °C by sandwiching the wet filter cakes between fresh blotting papers and metal plates.Hybrid nanopapers, consisting of mixtures of modified and unmodified CNF, were prepared by mixing and blending diluted suspensions of equal amounts of CNF-0 and CNF-A, respectively, at 0.3 wt%. No aggregation of oppositely charged CNF was observed. This was followed by filtering the hybrid-suspension mixture, pressing and hot-pressing as described above.
From both types of CNF and a mixture of them, nanopapers with grammages of 30 and 50 g m−2 (gsm) – corresponding to thicknesses of 30 and 50 μm, respectively – were prepared.
Characterization of nanopapers
The nitrate adsorption of our nanopapers was determined by passing a 5 mmol L−1 aqueous solution of NaNO3 (pH 5.8) through the nanopapers. The permeate was collected in fractions that enabled analysis using ion-chromatography; fractions of 2 to 20 mL were diluted by a factor of 10. The permeate fractions were analyzed using ion chromatography (882 Compact IC plus with 863 Compact Autosampler, Metrohm, Herisau, Switzerland). From the volume of each permeate fraction and its measured nitrate concentration, the mass of nitrate ions adsorbed per unit nanopaper area was calculated and plotted against the volume of permeate. Finally, the total weight and amount of rejected nitrate ions was determined and the ratio of the mass of adsorbed nitrate to the total mass of cationic CNF was calculated.
Results and discussion
Modification of CNF
Both modified and unmodified CNF were produced from fibre sludge, a waste product of the pulp and paper industry. Unmodified CNF were produced by mechanical grinding of sludge dispersed in water.32 For the production of cationic CNF functionalized with quaternary ammonium groups, the sludge was first modified with glycidyltrimethylammonium chloride (GTMAC) and then mechanically disintegrated, similar to a method previously reported.31,37,41 A scheme of the reaction procedure is shown below (Scheme 1).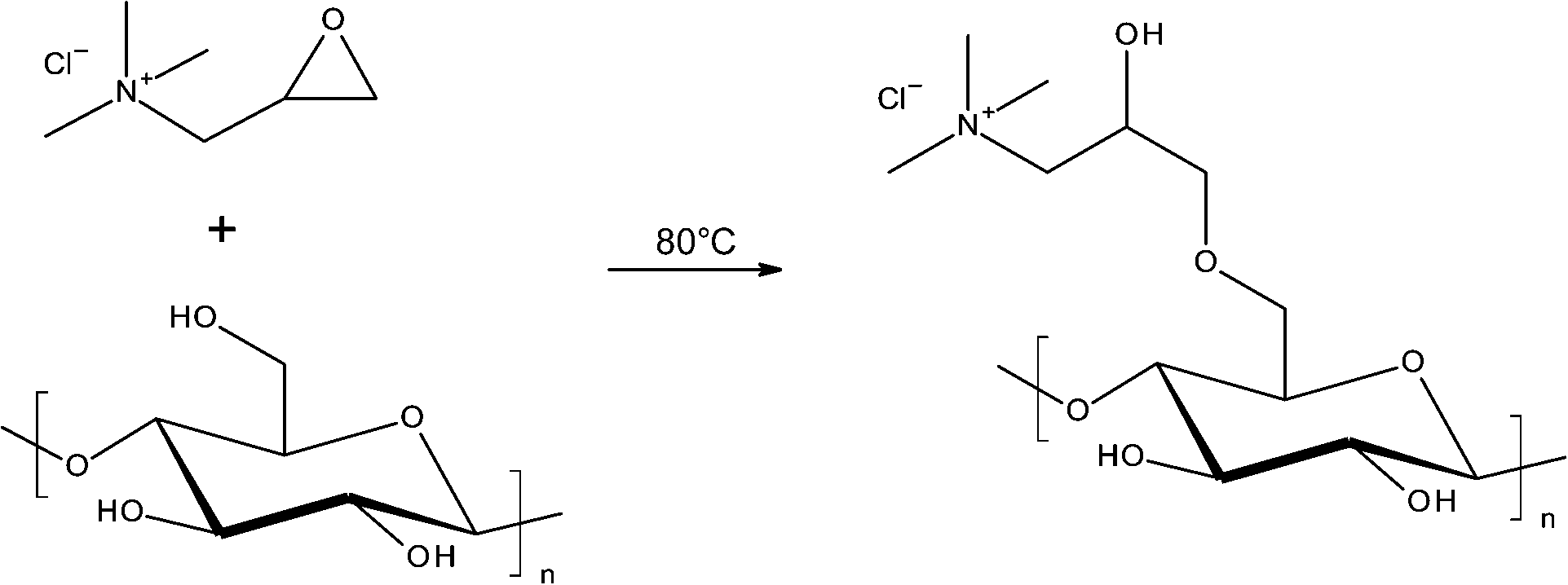 | ||
| Scheme 1 Functionalization of CNF with quaternary ammonium groups. | ||
The success of the reaction was confirmed by means of FT-IR-spectroscopy and conductometric titration. The presence of a new absorption band/shoulder at 1480 cm−1, corresponding to the methyl groups of the quaternized ammonium in the FT-IR spectra of cationic CNF, confirmed the introduction of the quaternary ammonium groups.31 The amount of ammonium groups grafted onto the cellulose fibrils, as determined by conductometric titration, was 0.38 mmol g−1. This corresponds to a DS of 0.07, which is in the range of DS reported before.31,37,41
Cationic modification will influence the surface charge of CNF; ζ-potential measurements (Fig. 1) showed that the surface charge reversed due to the presence of cationic ammonium groups. Unmodified CNF exhibit a negative ζ-potential between −4 and −21 mV over the whole pH range examined and a plateau around −20 mV at high pH, indicating that the surface is acidic as all dissociable functional groups are fully deprotonated.39 With decreasing pH, the ζ-potential increases due to the protonation of functional groups. The extrapolated isoelectric point (iep), at which ζ = 0, is at pH 2. On the other hand, CNF-A exhibit a positive ζ-potential between 33 and 54 mV, which is slightly higher than previously reported.37,41 This charge reversal is due to the introduction of cationic ammonium groups on the surface of the nanofibrils and should eventually enable adsorption of anions.
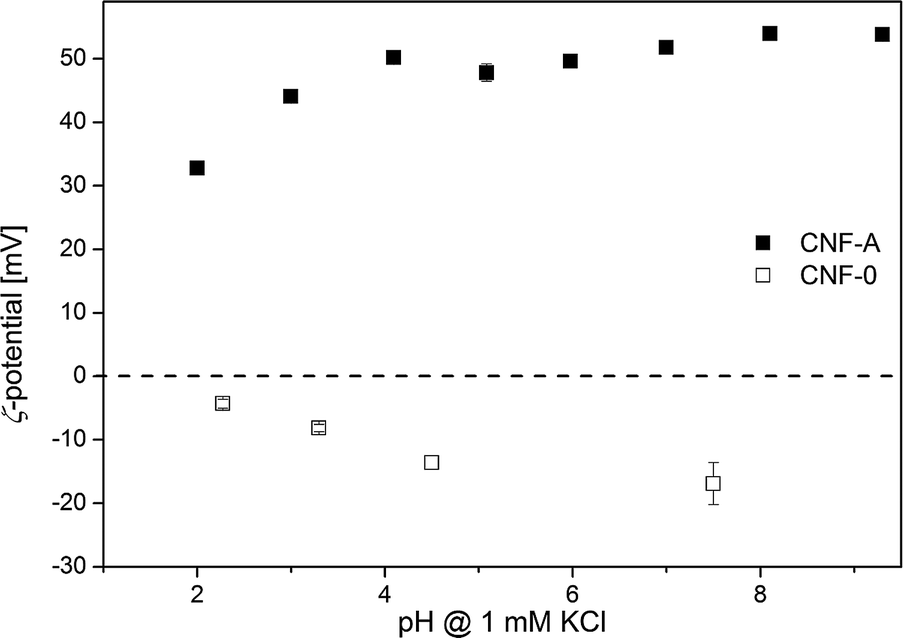 | ||
| Fig. 1 ζ-potential of CNF-A and CNF-0 as a function of pH. | ||
In the SEM images (Fig. 2) of CNF, single nanofibrils as well as agglomerates of fibrils can be seen. The diameter of the CNF-A fibrils was in the range of 10 to 100 nm, with an average of 23 nm. For CNF-0, a similar result was found.32 However, the average fibril diameter for CNF-0 ranged from 30 to 40 nm. The BET surface area of CNF-A and CNF-032 after supercritical drying was determined to be 115 m2 g−1 and 112 m2 g−1, respectively. Thus, no significant difference was detected between modified and unmodified CNF.
 | ||
Fig. 2 SEM images of CNF-0 (left) and CNF-A (right), magnification: 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000×. 000×. | ||
 | ||
| Fig. 3 Pictures (top) and SEM images (bottom, magnification: 10000×) of CNF-A (left) and CNF-H (right) nanopapers with 30 gsm/30 μm thickness. | ||
The effectiveness of adsorbents is most prominently affected by their surface charge. The ζ-potential provides information about the types of moieties present and their relative concentrations on a material's surface. In addition, it shows whether the functional groups attached to the CNF have survived the nanopaper production process, as cleavage of functional groups at enhanced temperatures might have taken place. The ζ-potential of the nanopapers as a function of pH was measured for unmodified, modified and hybrid nanopapers (Fig. 4).
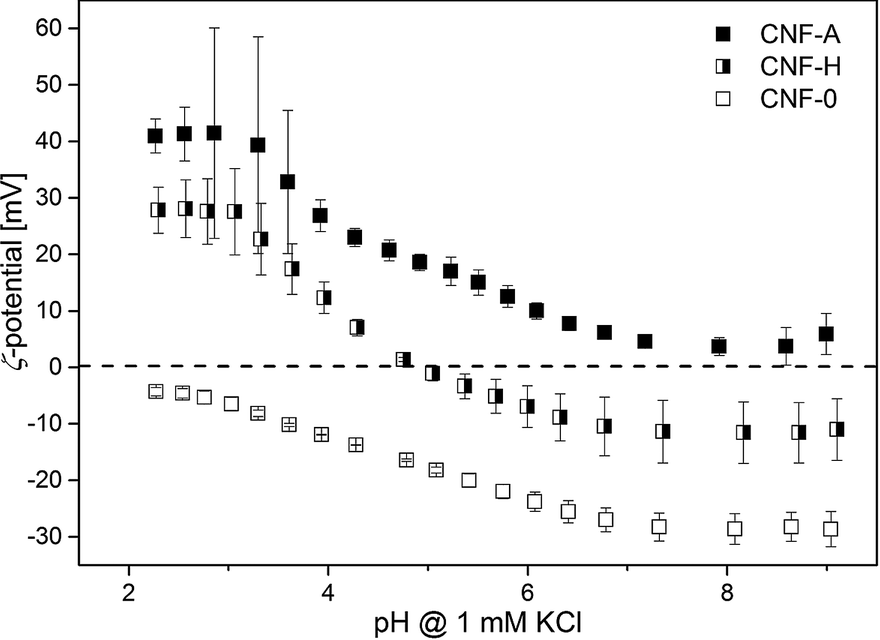 | ||
| Fig. 4 Streaming ζ-potential of CNF-A, CNF-0 and CNF-H nanopapers as a function of pH. | ||
Over the whole pH range examined, the unmodified CNF-0 nanopapers exhibit, as expected, a negative ζ-potential due to the presence of some carboxyl groups, such as uronic acids, originating from the residual hemicelluloses in the pulp with a small amount attributable to cellulose oxidation during the pulping operation.41 The trend is similar to that of nanofibrils alone (Fig. 1), with an extrapolated iep at pH 2. At low pH for cationic nanopapers, the ζ-potential values are 40 mV, similar to the ζ-potential of individual CNF-A nanofibrils. At a pH of 3.5 the acid groups present in the unmodified part of the CNF-A start to dissociate, leading to an overall charge reduction on the surface layer of the CNF-A nanopaper interacting with the flow, due to the low ammonium group content of 0.38 mmol g−1. Contrary to electrophoretic measurements on fibrils in suspension, in which the concentration of hemicellulosic uronic acid groups is limited and hardly detectable, during streaming potential measurements the ζ-potential is determined over the complete surface area of the nanopaper. Accordingly, all uronic acid groups contribute to the overall ζ-potential, eventually reducing it at high pH. It is striking to see that the behaviour of CNF-H is a combination of CNF-0 and CNF-A; when the unmodified CNF has a low surface charge, the ammonium moiety dominates the electrokinetic behaviour. As soon as the acid groups of unmodified cellulose dissociate, around pH 3.3, which corresponds to the pKa of uronic acids,42 the CNF-0 behaviour dominates and the ζ-potential drops. Furthermore, the proximity of the acid and ammonium groups on the CNF on or near the surface of the nanopaper results in charge–charge interaction which further screens the overall charge and so the ζ-potential drops, explaining the deviation from CNF-H to the average of ζ = f(pH) of CNF-A and CNF-0.
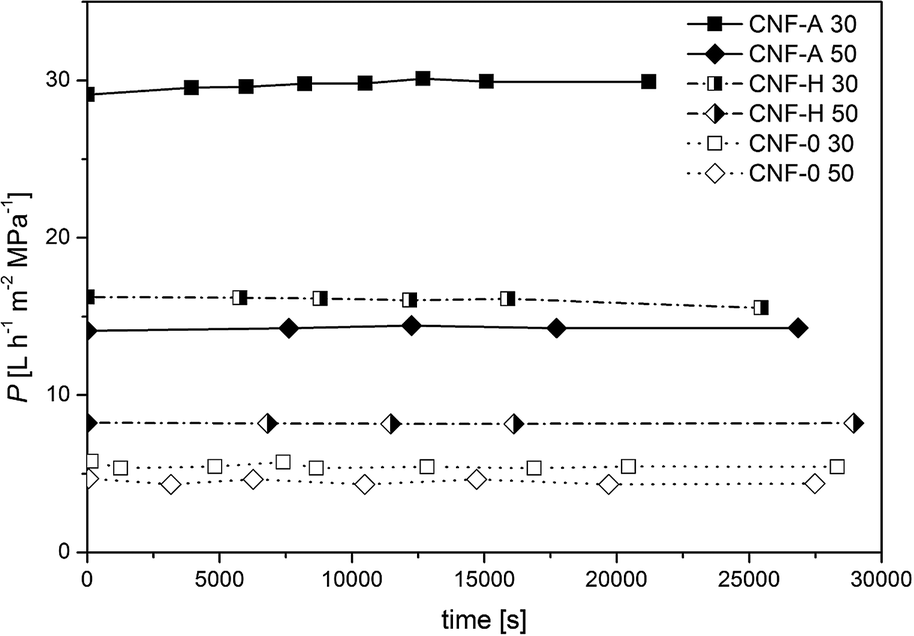 | ||
| Fig. 5 Permeance as a function of filtration time of CNF-A, CNF-0 and CNF-H nanopapers with grammages of 30 and 50 gsm, respectively. | ||
Irrespective of the grammage, the nanopapers produced solely from CNF-A exhibited a much higher permeance as compared to those produced from unmodified CNF (CNF-0). The CNF-A nanofibrils had lower fibril diameters than CNF-0 and therefore, the network density is influenced, causing the higher permeance of CNF-A nanopapers. Hybrid nanopapers, produced from mixtures of modified and unmodified CNF, exhibited, as expected, permeances in between those of nanopapers produced from modified and unmodified CNF. As expected the permeance of the nanopapers is very dependent on the paper grammage and thus the thickness of the nanopapers: thicker nanopapers (i.e. higher grammage) have lower permeances compared to thinner ones, which is common for nanopapers.27
To demonstrate the usefulness of cationic CNF as ion-exchange nanopaper for the removal of nitrates from water, filtration experiments were conducted. The adsorption of nitrates onto ammonium groups has been demonstrated in previous static experiments,35 in which during a certain incubation time adsorption takes place. However, during real-time filtration this period is significantly reduced to seconds or even fractions of seconds. Thus, in order to explore whether adsorption is also feasible during these short time intervals, dynamic adsorption experiments were performed.
The determination of the dynamic adsorption characteristics of CNF-A and CNF-H nanopapers was performed using a 5 mmol L−1 aqueous solution of sodium nitrate, equivalent to 310 mg L−1, which is a loading corresponding to heavily nitrate contaminated water. As preliminary batch-adsorption tests demonstrated hardly any adsorption of nitrates on CNF-0, no data for CNF-0 nanopapers were acquired. The nanopapers were loaded into a dead-end cell and the nitrate solution was forced through it. The permeate fractions were collected and analyzed by ion chromatography. From the determination of the nitrate concentration of the permeate fractions, the amount of nitrate that has been removed from the particular fraction has been calculated, related to the active membrane area used and plotted vs. the permeate volume (Fig. 6).
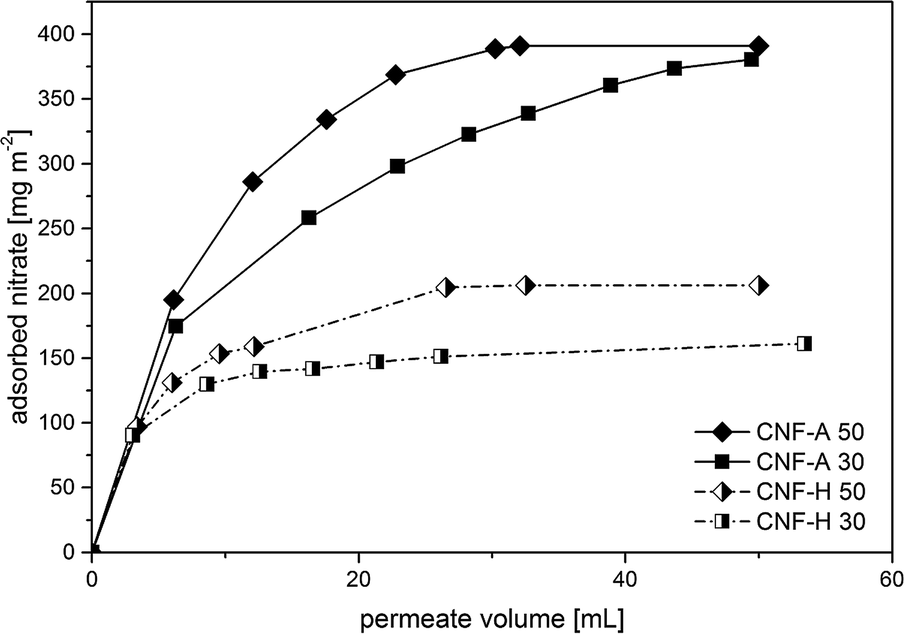 | ||
| Fig. 6 Mass of adsorbed nitrate per unit filtration area vs. permeate volume for CNF-A & CNF-H nanopapers with 30 and 50 gsm, respectively. The error of each individual point corresponds to the experimental error of the measurement which is equal to 2%. | ||
As can be seen, the initial slopes of the curves, which corresponds to the initial adsorption of nitrates onto the nanopapers, are almost identical. After reaching saturation with adsorbed nitrate, the curve levels off. This value is obviously about two times higher for pure CNF-A nanopapers (380 and 390 mg m−2 for CNF-A 30 and CNF-A 50, respectively) as compared to CNF-H nanopapers (160 mg m−2 and 205 mg m−2 for CNF-H 30 and CNF-H 50, respectively). This can be explained by the number of ammonium groups on the surface of the nanopapers, which is statistically two times higher for the pure CNF-A nanopapers, as can be seen in the results of the ζ-potential measurements (Fig. 4). It is disappointing to see that if thicker papers are used no increased adsorption was observed, which can be explained by the fact that quaternary ammonium groups interact with residual uronic acid groups in the paper bulk due to the proximity of the nanofibrils. There they are lost for nitrate adsorption. Accordingly, surface groups dominate the nitrate removal process as they are accessible. Therefore, it seems appropriate to assume that a series of thin nanopaper ion-exchangers will result in more effective nitrate removal. The final saturation level is a measure of the overall amount of nitrate that can be adsorbed. In the case of pure cationic CNF-A nanopapers, this level is significantly – around two times – higher as compared to the hybrid nanopapers. However, the total mass of modified CNF-A in the nanopaper slightly influences the amount of nitrate that can be adsorbed for pure CNF-A nanopapers, and the amount of adsorbed nitrates is only slightly higher for nanopapers with grammages of 50 gsm compared to 30 gsm. This is maybe due to the higher contribution of surface adsorption of nitrate compared to the adsorption of nitrate in the bulk of the nanopaper.
Eventually, the value of nitrate adsorbed by the nanopaper at the saturation level can be related to the amount of active adsorption agent, i.e. cationic CNF, in the nanopapers resulting in a value for the mass of adsorbed nitrate per mass of adsorbent (Fig. 7). The result of the ratio of adsorbed nitrates per mass of cationic CNF, i.e. the adsorption capacity, suggests a slightly higher contribution of adsorption taking place on the surface of the nanopapers as well, since a slightly higher value is obtained for the pure 30 gsm nanopaper as compared to the 50 gsm nanopaper (12.7 mg g−1vs. 7.8 mg g−1, respectively), which is comparable to commercially available or currently researched adsorbents.4 Exemplarily, char coal from industrial residues achieved adsorption values of around 1.5 mg g−1.43 However, the contact time in our experiments was several seconds, whereas in common batchwise adsorption contact times of several hours up to numerous days are frequently in use.4,43 Hybrid nanopapers, containing statistically only half of the ammonium groups on the surface as compared to the amount in pure cationic nanopapers, exhibit a nitrate adsorption capacity of 10.7 mg g−1 and 8.3 mg g−1 for both 30 and 50 gsm CNF-H nanopapers, respectively. The lower adsorption capacity of the hybrid nanopaper could be due to the fact that some positive charges of cationic CNF are engaged in electrostatic interaction with the unmodified CNF along with the already mentioned lower concentration of ammonium groups on the surface of the nanopapers. In addition, the overall ζ-potential at the pH of the NaNO3 (pH 5.8) solution is negative, resulting in lower adsorption affinity towards negatively charged nitrate ions.
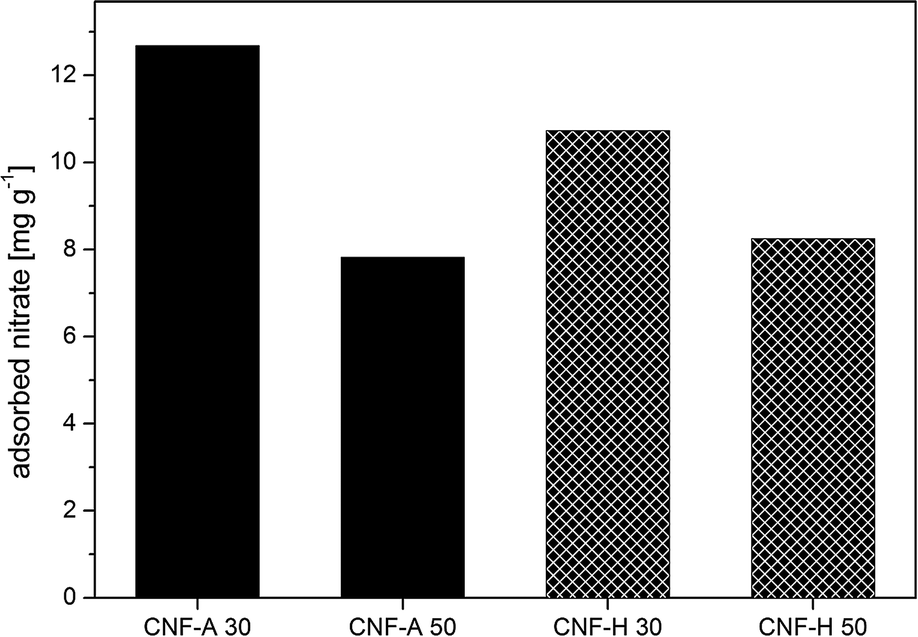 | ||
| Fig. 7 Mass of adsorbed nitrate per unit mass of the active adsorbent agent for CNF-A & CNF-H nanopapers with 30 and 50 gsm, respectively. | ||
Conclusions
Cellulose nanofibrils were functionalized with quaternary ammonium groups by modifying fibre sludge, an industrial waste stream, with glycidyltrimethylammonium chloride followed by mechanical disintegration. FT-IR spectroscopy, conductometric titration and ζ-potential measurements confirmed the success of the modification. From these cationic CNF, nanopapers, intended to be used as ion-exchange nanopapers for the capture of nitrates, were manufactured using a simple filtration paper-making process. The permeance of the modified nanopapers was dependent on the grammage and thus on the thickness of the nanopaper; thicker nanopapers had a higher grammage and were less water permeable. Nanopapers made of cationic CNF exhibited a higher permeance as compared to nanopapers made of unmodified CNF, which was explained by the lower fibril diameter of cationic CNF-A. Finally, the nanopapers were shown to adsorb nitrates under dynamic conditions, i.e. while contaminated water was permeating the nanopapers in dynamic filtration experiments. It was found that nanopapers containing ammonium moieties were able to adsorb nitrates up to 390 mg per m2 filtration area, equivalent to an adsorption capacity of more than 12 mg of nitrate per g of active adsorption agent. These experiments also showed that the contribution of nitrate adsorption by quaternary ammonium groups on the surface of the nanopaper is probably higher than that of functional groups in the bulk of the nanopapers. We showed that nanopapers containing cationic cellulose nanofibers are efficient as ion exchange nanopapers allowing for nitrate removal from contaminated water. It can be concluded that an even higher concentration of ammonium groups on the surface of thin nanopapers should result in an even higher efficiency; i.e. exhibit a higher nitrate “rejection”.Acknowledgements
The authors greatly acknowledge the funding provided by the EU FP7 project NanoSelect (Grant No. 280519) and Processum for providing the raw materials.References
- E. Eroglu, V. Agarwal, M. Bradshaw, X. Chen, S. M. Smith, C. L. Raston and K. Swaminathan Iyer, Green Chem., 2012, 14, 2682 RSC.
- M. A. Shannon, P. W. Bohn, M. Elimelech, J. G. Georgiadis, B. J. Marinas and A. M. Mayes, Nature (London, U. K.), 2008, 452, 301 CrossRef CAS PubMed.
- C. Della Rocca, V. Belgiorno and S. Meriç, Desalination, 2007, 204, 46 CrossRef CAS.
- A. Bhatnagar and M. Sillanpää, Chem. Eng. J., 2011, 168, 493 CrossRef CAS.
- K. Riha, G. Michalski, E. Gallo, K. Lohse, P. Brooks and T. Meixner, Ecosystems, 2014, 17, 1 CrossRef.
- A. Bhatnagar, E. Kumar and M. Sillanpää, Chem. Eng. J., 2010, 163, 317 CrossRef CAS.
- P. Santamaria, J. Sci. Food Agric., 2006, 86, 10 CrossRef CAS.
- H. H. Comly, JAMA, J. Am. Med. Assoc., 1987, 257, 2788 CrossRef CAS.
- I. T. Vermeer, D. M. Pachen, J. W. Dallinga, J. C. Kleinjans and J. M. van Maanen, Environ. Health Perspect., 1998, 106, 459 CrossRef CAS PubMed; J. M. van Maanen, I. J. Welle, G. Hageman, J. W. Dallinga, P. L. Mertens and J. C. Kleinjans, Environ. Health Perspect., 1996, 104, 522 CrossRef PubMed.
- WHO, Guidelines for Drinking-water Quality, 4th edn, 2011 Search PubMed.
- O. S. G. P. Soares, J. J. M. Órfão and M. F. R. Pereira, Appl. Catal., B, 2011, 102, 424 CrossRef CAS.
- N. Barrabés and J. Sá, Appl. Catal., B, 2011, 104, 1–5 CrossRef CAS; C. L. Constantinou, C. N. Costa and A. M. Efstathiou, Catal. Today, 2010, 151, 190 CrossRef; M. Shand and J. A. Anderson, Catal. Sci. Technol., 2013, 3, 879 Search PubMed; C. P. Theologides, P. G. Savva and C. N. Costa, Appl. Catal., B, 2011, 102, 54 CrossRef.
- H. Demiral and G. Gündüzoğlu, Bioresour. Technol., 2010, 101, 1675 CrossRef CAS PubMed.
- S. Ebrahimi and D. J. Roberts, J. Hazard. Mater., 2013, 262, 539 CrossRef CAS PubMed.
- S. Chatterjee, D. S. Lee, M. W. Lee and S. H. Woo, J. Hazard. Mater., 2009, 166, 508 CrossRef CAS PubMed; S. Chatterjee and S. H. Woo, J. Hazard. Mater., 2009, 164, 1012 CrossRef PubMed.
- J. J. Schoeman, Water SA, 2009, 35, 721 CrossRef CAS.
- A. H. Mahvi, M. Malakootian, A. Fatehizadeh and M. H. Ehrampoush, Desalin. Water Treat., 2011, 29, 326 CrossRef CAS.
- A. Kapoor and T. Viraraghavan, J. Environ. Eng., 1997, 123, 371–380 CrossRef CAS; J. Y. Park and Y. J. Yoo, Appl. Microbiol. Biotechnol., 2009, 82, 415 CrossRef PubMed.
- E. J. McAdam and S. J. Judd, Desalination, 2006, 196, 135–148 CrossRef CAS.
- A. S. Koparal and Ü. B. Öğütveren, J. Hazard. Mater., 2002, 89, 83 CrossRef CAS PubMed.
- L. J. Banasiak and A. I. Schäfer, J. Membr. Sci., 2009, 334, 101 CrossRef CAS.
- R. J. Petersen, J. Membr. Sci., 1993, 83, 81 CrossRef CAS.
- M. Elimelech and W. A. Phillip, Science, 2011, 333, 712 CrossRef CAS PubMed.
- K. P. Lee, T. C. Arnot and D. Mattia, J. Membr. Sci., 2011, 370, 1 CrossRef CAS.
- K. Li, Ceramic Membranes for Separation and Reaction, John Wiley & Sons, Ltd., Chichester, UK, 2007 Search PubMed.
- M. Henmi, K. Nakatsuji, T. Ichikawa, H. Tomioka, T. Sakamoto, M. Yoshio and T. Kato, Adv. Mater., 2012, 24, 2238 CrossRef CAS PubMed; A. A. Mungray and Z. V. P. Murthy, Ionics, 2012, 18, 811 CrossRef.
- A. Mautner, K. Y. Lee, P. Lahtinen, M. Hakalahti, T. Tammelin, K. Li and A. Bismarck, Chem. Commun., 2014, 50, 5778 RSC; A. Mautner, K.-Y. Lee, T. Tammelin, A. P. Mathew, A. J. Nedoma, K. Li and A. Bismarck, React. Funct. Polym., 2015, 86, 209 CrossRef CAS.
- L. Hu, G. Zheng, J. Yao, N. Liu, B. Weil, M. Eskilsson, E. Karabulut, Z. Ruan, S. Fan, J. T. Bloking, M. D. McGehee, L. Wagberg and Y. Cui, Energy Environ. Sci., 2013, 6, 513 CAS.
- A. Mohammadi, M. M. Lakouraj and M. Barikani, React. Funct. Polym., 2014, 83, 14 CrossRef CAS; A. Nematollahzadeh, A. Shojaei and M. Karimi, React. Funct. Polym., 2015, 86, 7 CrossRef; F. Liu, B.-r. Ma, D. Zhou, L.-J. Zhu, Y.-Y. Fu and L.-X. Xue, React. Funct. Polym., 2015, 86, 191 CrossRef; E. S. Abdel-Halim, React. Funct. Polym., 2013, 73, 1531 CrossRef.
- P. Liu, H. Sehaqui, P. Tingaut, A. Wichser, K. Oksman and A. Mathew, Cellulose, 2014, 21, 449 CrossRef CAS; H. Sehaqui, U. de Larraya, P. Liu, N. Pfenninger, A. Mathew, T. Zimmermann and P. Tingaut, Cellulose, 2014, 21, 2831 CrossRef.
- A. Pei, N. Butchosa, L. A. Berglund and Q. Zhou, Soft Matter, 2013, 9, 2047 RSC.
- M. Jonoobi, A. P. Mathew and K. Oksman, Ind. Crops Prod., 2012, 40, 232 CrossRef CAS.
- D. Klemm, F. Kramer, S. Moritz, T. Lindstroem, M. Ankerfors, D. Gray and A. Dorris, Angew. Chem., Int. Ed., 2011, 50, 5438 CrossRef CAS PubMed.
- S. Hamoudi, A. El-Nemr, M. Bouguerra and K. Belkacemi, Can. J. Chem. Eng., 2012, 90, 34 CrossRef CAS.
- A. Sowmya and S. Meenakshi, Chem. Eng. J., 2014, 257, 45 CrossRef CAS.
- F. Liu, B.-r. Ma, D. Zhou, L.-J. Zhu, Y.-Y. Fu and L.-x. Xue, React. Funct. Polym., 2015, 86, 191 CrossRef CAS; V. Kochkodan, D. J. Johnson and N. Hilal, Adv. Colloid Interface Sci., 2014, 206, 116 CrossRef PubMed.
- M. Hasani, E. D. Cranston, G. Westman and D. G. Gray, Soft Matter, 2008, 4, 2238 RSC.
- H. Sehaqui, Q. Zhou, O. Ikkala and L. A. Berglund, Biomacromolecules, 2011, 12, 3638 CrossRef CAS PubMed.
- K.-Y. Lee, T. Tammelin, K. Schulfter, H. Kiiskinen, J. Samela and A. Bismarck, ACS Appl. Mater. Interfaces, 2012, 4, 4078 CAS.
- H. Sehaqui, A. Liu, Q. Zhou and L. A. Berglund, Biomacromolecules, 2010, 11, 2195 CrossRef CAS PubMed; M. Henriksson, L. A. Berglund, P. Isaksson, T. Lindström and T. Nishino, Biomacromolecules, 2008, 9, 1579 CrossRef PubMed.
- A. Olszewska, P. Eronen, L.-S. Johansson, J.-M. Malho, M. Ankerfors, T. Lindström, J. Ruokolainen, J. Laine and M. Österberg, Cellulose, 2011, 18, 1213 CrossRef CAS.
- R. Kohn and P. Kovác, Chem. Zvesti, 1978, 32, 478 Search PubMed; H. M. Wang, D. Loganathan and R. J. Linhardt, Biochem. J., 1991, 278, 689 CrossRef CAS PubMed.
- P. C. Mishra and R. K. Patel, J. Environ. Manage., 2009, 90, 519 CrossRef CAS PubMed; K. Mizuta, T. Matsumoto, Y. Hatate, K. Nishihara and T. Nakanishi, Bioresour. Technol., 2004, 95, 255 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2016 |