
Silica nanoparticle-generated ROS as a predictor of cellular toxicity: mechanistic insights and safety by design†‡
Sean E.
Lehman
 a,
Angie S.
Morris
ab,
Paul S.
Mueller
a,
Aliasger K.
Salem
b,
Vicki H.
Grassian
a and
Sarah C.
Larsen
*a
a,
Angie S.
Morris
ab,
Paul S.
Mueller
a,
Aliasger K.
Salem
b,
Vicki H.
Grassian
a and
Sarah C.
Larsen
*a
aDepartment of Chemistry, University of Iowa, Iowa City, IA 52242, USA. E-mail: sarah-larsen@uiowa.edu; Tel: + 319 335 1346
bDivision of Pharmaceutics and Translational Therapeutics, College of Pharmacy, University of Iowa, Iowa City, IA 52242, USA
First published on 10th November 2015
Abstract
Evaluating toxicological responses of engineered nanomaterials such as silica nanoparticles is critical in assessing health risks and exposure limits. Biological assays can be used to evaluate cytotoxicity of individual materials, but specific nano-bio interactions—which govern its physiological response—cannot currently be predicted from materials characterization and physicochemical properties. Understanding the role of free radical generation from nanomaterial surfaces facilitates understanding of a potential toxicity mechanism and provides insight into how toxic effects can be assessed. Size-matched mesoporous and nonporous silica nanoparticles in aminopropyl-functionalized and native forms were investigated to analyze the effects of porosity and surface functionalization on the observed cytotoxicity. In vitro cell viability data in a murine macrophage cell line (RAW 264.7) provides a model for what might be observed in terms of cellular toxicity upon an environmental or industrial exposure to silica nanoparticles. Electron paramagnetic resonance spectroscopy was implemented to study free radical species generated from the surface of these nanomaterials and the signal intensity was correlated with cellular toxicity. In addition, in vitro assay of intracellular reactive oxygen species (ROS) matched well with both the EPR and cell viability data. Overall, spectroscopic and in vitro studies correlate well and implicate production of ROS from a surface-catalyzed reaction as a predictor of cellular toxicity. The data demonstrate that mesoporous materials are intrinsically less toxic than nonporous materials, and that surface functionalization can mitigate toxicity in nonporous materials by reducing free radical production. The broader implications are in terms of safety by design of nanomaterials, which can only be extracted by mechanistic studies such as the ones reported here.
Nano impactThis study develops a more complete understanding of the correlation between surface-generated free radicals and observed cellular toxicity. In vitro assays are heavily implemented for assessing risks and toxicity of engineered nanomaterials, so it is critical to understand mechanisms that are responsible for observed toxic responses. The fundamental result demonstrates that surface-generated radicals such as hydroxyl can be directly correlated to observed cellular toxicity. The literature strongly suggests ROS as an ultimate toxicant for nanoparticles, which our study reaffirms. We implemented a multi-faceted approach to understanding how silica nanoparticles generate radical species in biological systems, and ultimately aim to provide a deeper understanding of how safer materials can be designed for biomedical and environmental applications as an important component of sustainable nanotechnology. |
Introduction
Evaluating the toxicity of nanomaterials, commonly referred to as nanotoxicity, is a rapidly growing field of research.1,2 As engineered nanomaterials diversify in type and synthetic complexity, exposures to these materials will increase in both direct (biomedical) and indirect (environmental) pathways. Porous nanomaterials such as mesoporous silica are undergoing rapid development for a variety of applications such as drug delivery, catalysis, biomedical imaging, and environmental remediation.3–6 There is a growing body of literature aimed at assessing risks of nanomaterials. However, correlations between specific nanomaterial properties and toxicity in biological systems remain lacking.Current research focuses on in vitro methodologies to determine the biological impact of nanomaterials. In the case of silica nanomaterials, current studies focus on different types of silica, such as fumed silicas, porous silicas, and nonporous materials. Porous silica nanomaterials exhibit very low cytotoxicity against a variety of cell lines.7 The synthesis of each material gives it a unique chemical surface that influences how it interacts with biological systems. These nano-bio interactions ultimately control the reactions, interactions, and fate of these materials. Despite the lack of systematic studies, several themes have emerged that appear to be central paradigms for understanding and predicting silica nanoparticle toxicity. Two of these factors are the surface silanol density and the production of reactive oxygen species (ROS).8–10
The primary structure of silica nanoparticles consists of tetrahedrally-bonded Si–O moieties that are amorphously condensed into a network. Porous materials possess pore-voids around which the tetrahedra are condensed, but in the nonporous variant there are only amorphous silica tetrahedra present. On the surface of the material there are free silanol moieties (Si–OH), which are analogous to alcohol functionalities. Work in the field has shown that these surface silanols are important factors that control interactions with biological systems.11 Surface silanols are negatively charged at physiological pH (as silicate anion Si–O−), and thus can interact with biological components such as cell membranes via electrostatic interactions.12 Furthermore, the silanol oxygen is nucleophilic; making it mechanistically viable to attack electrophilic carbonyl groups, which are present in protein molecules. Due to the nature of the hydrolysis and condensation of the precursor molecules, each type of material has different densities of surface silanols, as well as different types in terms of their coordination at the surface.13 While surface silanols are important in understanding toxicity, their reaction with biological macromolecules can screen them from interacting with other molecules in biological compartments.
Generation of ROS from the silica surface has long been held to be a dominating factor in toxicity. For some time, it was believed that ROS were produced via Fenton-like chemistry.14 More recent work implicates ROS production at the nanoparticle surface as a factor. In the case of fumed silica, Zhang et al. showed that the cleavage of strained siloxane rings were responsible for the production of ROS from the silica nanoparticle surface.9 It has been shown that nanoparticle-derived ROS can overcome the natural cellular defense mechanism, glutathione-mediated redox chemistry. Production of species such as hydroxyl radical results in destructive chemical modification of lipids and DNA, as well as conversion to organic peroxynitrite (R–ONOO−) via reaction with nitric oxide, as previously described.15
Increased intracellular ROS leads to oxidative stress in the cell. Oxidative stress has deleterious effects if sustained for extended periods of time or at high levels for acute periods. Damage to the cellular machinery can result in apoptosis, or programmed cell death. This has been observed to happen upon exposure to nanomaterials. This has been investigated, and oxidative stress has been measured through a variety of methodologies. Some work has suggested there is a band gap-redox potential relation that correlates with oxidative stress upon exposure to metal oxide nanoparticles.16 Some work has shown that some nanoparticles like ceria (CeO2) actually protect against oxidative stress, and that materials such as titania (TiO2) do not seem to have any oxidative effect in certain cell lines.17 As mentioned above, the cellular defense against oxidative stress is redox chemistry mediated by glutathione, which the cell keeps reserves of and regenerates as necessary. When cells are exposed to nanoparticles that induce oxidative stress, these glutathione reserves can be depleted, as has been measured. Once the reserves are depleted, the ROS generated can damage cellular components ultimately leading to cell death.18
In terms of sustainable nanotechnology, a natural extension of these insights is that chemical modification of the surface may be able to mitigate the toxicity of silica nanomaterials by reducing ROS formed. This has been verified experimentally, and overall functionalization of the surface does seem to decrease cellular toxicity in in vitro models.19,20 Surface functional groups added to the surface vary widely, from small alkylamine groups to large non-ionic molecules such as polyethylene glycol (PEG). It has been postulated, but not clearly demonstrated, that this decrease in cytotoxicity arises from a decrease in total surface silanol density, as well as modification of the overall particle surface charge. However, investigation into the mechanism by which surface modification mitigates toxicity is still ongoing, as it is not fully understood.21
The study presented here explores the fundamental relationship between surface properties and the toxicological response of engineered silica nanoparticles, and how this insight can be used to inform and guide “safety by design” approaches for nanomaterials. Particles used were size-matched at approximately 50 nm in diameter. In terms of biological applications (such as drug delivery/biomedical imaging), the size of less than 100 nm is critical to prevent rapid clearance from the systemic circulation by the reticuloendothelial system.22 Wormhole-type mesoporous silica and nonporous silica were synthesized to examine the effects of porosity on cytotoxicity and free radical production. The materials were further functionalized with a small organic amine, which is commonly used to increase the overall surface charge as well as enable further chemical modification. Thus, the synergistic effects of porosity and surface functionality could be evaluated for amorphous silica nanoparticles, with the goal of correlating the physicochemical properties to observed, in vitro cellular toxicity.
Experimental methods
Materials synthesis
Mesoporous silica nanoparticles (MS) with a particle size of approximately 50 nm were synthesized according to the procedure described previously.23,24 In the synthesis of wormhole-type silica, 33 mL 25% aqueous cetyltrimethylammonium chloride (CTAC), 33 mL absolute ethanol, and 200 mL Millipore (18.2 MΩ cm) water were combined and stirred at room temperature for approximately 10 minutes. Triethanolamine (13 mL) was added and the solution was allowed to stir for an hour before being heated to 60 °C. Tetraethylorthosilicate (20.4 mL, TEOS, Sigma) was then added to the rapidly stirring solution at a rate of about 2 mL min−1 and the solution was stirred at 60 °C for 2.5 hours. The solution was cooled, the material centrifuged out, and then washed in triplicate with water. The samples were dried overnight, then the template was removed by calcination in air at 600 °C for 6 hours.Stöber-type nonporous silica (NPS) was prepared following a modified procedure from the literature.25 In this synthesis no surfactant was used, and 120 mL absolute ethanol was mixed with 6.0 mL of 28% aqueous ammonia and stirred for 5 minutes. TEOS was again used as the silicon source and 4.0 mL was added at room temperature. The reaction mixture was stirred at room temperature for 24 hours and then centrifuged to obtain the products, which were washed in triplicate with water and dried at 60 °C overnight to give the Stöber silica material.
Surface functionalization
Functional groups of interest were covalently attached to the nanoparticle surface using a post-synthesis grafting method. Functionalization with amine groups was carried out by refluxing a mixture of 4 g of aminopropyltriethoxysilane (APTES, Sigma) with 1.00 g of silica nanoparticles in 60 mL of toluene for 48 h. Then, the reaction mixture was centrifuged, washed with dichloromethane three times and dried overnight at 80 °C. The functionalized mesoporous and nonporous materials are referred to as MS@APTES and NPS@APTES, respectively.Material characterization
Silica nanomaterials were characterized using electron microscopy, nitrogen adsorption isotherms, and thermogravimetric analysis. TEM images were acquired using a JEOL JEM-1230 transmission electron microscope. Nitrogen adsorption experiments were conducted using a Nova 1200 Nitrogen Adsorption Instrument (Quantachrome). Approximately 100 mg of powder was dried at 120 °C under vacuum overnight. A seven-point BET isotherm and a 50-point adsorption/desorption isotherm in a liquid nitrogen bath were obtained, using pure nitrogen gas as the adsorbate. Surface area was calculated using BET (Brunauer–Emmett–Teller) method. Pore diameter and volume were calculated using BJH (Barrett–Joyner–Halenda) methodology, using the desorption branch of the isotherm. The samples were evaluated by thermogravimetric analysis using a TA Q5000 TGA instrument with a linear heating rate of 5 °C min−1. Samples were heated from room temperature to ~800 °C under a flow of nitrogen. Mass loss during the run was used to approximate the loading of the organic functional group. Zeta potential measurements were conducted on a Malvern Zetasizer. The materials (~3–5 mg) were dispersed in 1.5 mL of 10 mM sodium phosphate, pH = 7.4 and sonicated for 15 min prior to measurement. All samples were analyzed in triplicate, and the average ± SD is reported for each of the nanomaterials.Electron paramagnetic resonance (EPR) spectroscopy
EPR was employed to measure reactive oxygen species released from the nanoparticle surface, adapting a previously published method.9 Samples were prepared at 5% weight dispersion in 500 μL final volumes. A solution containing hydrogen peroxide at 200 mM final concentration and the spin-trap 5,5-dimethyl-1-pyrroline N-oxide (DMPO) at a final concentration of 25 mM was added to the powdered materials, mixed, and then equilibrated at room temperature for 15 min prior to measurement on a Bruker EMX CW EPR spectrometer operating at a Larmor frequency of 9.76 GHz (X-band). A flat TM100 cell was used to hold the samples and four scans were co-added to give the final spectra. Quantification was facilitated by using 3-carboxy-PROXYL as an external standard. Double integration of the standard and sample spectra gave areas that were used to calculate the concentration of each radical species.Cell culture and viability assay
Murine leukemia macrophage cells (RAW264.7) were maintained in RPMI-1640 medium (Gibco, Life Technologies) supplemented with 10% fetal bovine serum (Atlanta Biologics), 10 mM HEPES (Gibco), 1 mM sodium pyruvate (Gibco), 1 mM Glutamax (Gibco), and 50 μg mL−1 gentamycin sulfate (Cellgro). Cells were incubated at 37 °C and 5% CO2.RAW264.7 cells were seeded in 96-well plates at 1.0 × 104 cells per well and incubated for 24 hours at 37 °C and 5% CO2. The medium was removed from the wells and replaced with 200 μL of particle suspension in fresh RPMI medium. Cells were exposed to particle treatments for 4, 24 or 48 hours after which the treatments were removed and fresh medium was added. MTS reagent (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) or CellTiter 96 Aqueous One Solution reagent (Promega) was then added to the medium in the wells and incubated at 37 °C for 1–4 hours depending on the rate of formazan production. Before spectrophotometry, the 96-well plates were centrifuged at 500 × g for 20 minutes; the supernatant of each well was removed and added to a new 96-well plate. This step was performed to avoid any unwanted scattering from the particles during the absorbance measurement. The absorbance of the supernatant was collected at 420 nm using a SpectraMax Plus 384 microplate reader (Molecular Devices). Percent relative cell viability was calculated by normalizing treated cells to an untreated control sample. Medium and MTS reagent without cells served as a method blank for all samples.
In vitro ROS assay in RAW 264.7 cell line
RAW264.7 cells were seeded in 60 mm culture dishes at a density of 2 × 105 cells per dish and incubated at 37 °C and 5% CO2 for 24 hours. After the initial incubation, the medium was aspirated and replaced with fresh medium. Then, 200 μL of particle suspension in medium were added at a final concentration of 50 μg mL−1. After 24 hours of treatment, the medium was aspirated and the cells were removed from the culture dish using trypsin. After five minutes fresh medium was added to the trypsinized cells; the cells were washed several times from the dishes and collected in centrifuge tubes. The cells were washed twice with PBS (Gibco) containing 5 mM sodium pyruvate (Gibco) and centrifuged at 230 × g for 5 minutes in between washes. Then, each sample was resuspended in 1.00 mL of the PBS/pyruvate solution and stained with dihydroethidium (DHE) (Molecular Probes, Life Technologies) dissolved in dimethyl sulfoxide (DMSO, Aldrich). The final concentration of DHE was 10 μM. To account for any background fluorescence, a negative control sample consisted of untreated cells with the same volume of DMSO added as in the DHE stained samples. Antimycin A (Aldrich) was added to untreated cells at a final concentration of 10 μM to serve as a positive control followed by immediate staining with DHE using the exact same conditions as in the nanoparticle treated samples. All samples were incubated for 40 minutes after the addition of DHE (or DMSO for the negative control) at 37 °C after which they were placed on ice and analyzed for DHE fluorescence using a FACScan flow cytometer (Becton Dickinson Immunocytometry Systems) using 488 nm excitation and measuring the emission at 585 nm. The mean fluorescence intensity of three samples was used to compare the relative generation of intracellular superoxide.Results and discussion
Material design and physical characterization
The materials used in this study were wormhole-type (WO) mesoporous silica and nonporous silica nanoparticles, approximately 50 nm in diameter. The WO mesoporous silica material has a network of pores that is less ordered than that of other mesoporous silica materials, such as MCM-41 and SBA-15. The pores of WO-type silica form a network of voids that interconnect and interrupt each other throughout the material. The materials were characterized using traditional physical characterization methods. Electron microscopy images and physical characterization data can be seen in Fig. 1 and Table 1, respectively. | ||
| Fig. 1 Transmission electron microscopy images of materials used in this study. Wormhole-type (WO) mesoporous silica (L) and nonporous (Stöber) silica (R). The scale bar in each image is 50 nm. | ||
| Sample | Diameter (nm) | Surface area (m2 g−1) | Pore volume (mL g−1) | Pore diameter (nm) | Functional group loading (mmol g−1) | ζ Potential (mV) pH = 7.4, 10 mM sodium phosphate |
|---|---|---|---|---|---|---|
| Mesoporous silica (MS) | 49 (±5) TEM | 1100 (±44) | 0.56 (±0.03) | 3.100 (±0.007) | N/A | −39 (±2) |
| MS@APTES | — | 700 (±28) | 0.152 (±0.009) | 3.100 (±0.007) | 4.283 (0.0009) | 1.9 (±0.2) |
| Nonporous silica (NPS) | 47 (±7) TEM | 66 (±3) | N/A | N/A | N/A | −49 (±3) |
| NPS@APTES | — | 42 (±2) | N/A | N/A | 0.582 (0.001) | 9.6 (±0.7) |
| Min-U-Sil (α-quartz) | 270 (±21) DLS | 7.7 (±0.3) | N/A | N/A | N/A | −67 (±3) |
The surface area and pore volume decreases upon functionalization, as previously observed and reported in the literature.26 The nonporous and mesoporous silica nanomaterials were size-matched so that variations in porosity and surface functionalization could be correlated with cytotoxocity.
In vitro cellular toxicity
The toxicity of these materials was evaluated using a standard cell viability assay against a murine macrophage cell line (RAW 264.7). As immune cells, macrophages respond to the presence of nanomaterials in a biological system. Macrophages also mitigate inflammation responses via cytokine induction and other cellular pathways.27 Additionally, previous work has demonstrated a clear ability of these cell types to uptake nanoparticles by phagocytosis increasing the local concentration inside the cellular compartment.28 In order to assess the time- and concentration-dependent cytotoxicity effects, an array of dosages were implemented ranging from 10–200 μg mL−1. Cell viability was monitored spectroscopically as a function of time at time intervals of 4, 24, and 48 hours. The results at 48 hours can be seen in Fig. 2. Data for Min-U-Sil, the positive control, are also included for comparison. The results at 4 and 24 hours for all materials, including the Min-U-Sil, are included in the supplemental information. | ||
| Fig. 2 Cell toxicity data in RAW 264.7 cells, 48 h post-exposure. The bare nonporous and mesoporous materials show both time- and concentration-dependent behavior. Min-U-Sil data, also at 48 h, is shown as the positive control for comparison. The data corresponding to 4 and 24 h post-exposure is available in the SI. | ||
In terms of concentration effects, there is a marked effect of concentration on viability for the bare nonporous silica. At the highest concentration tested (200 μg mL−1) effectively all cells for that dosage were no longer viable at 48 hours post-exposure. There is also some slight concentration-dependent effect on cell viability from the bare mesoporous silica, but not nearly as apparent as that for the nonporous material. The two functionalized materials showed very little observed toxicity at the concentrations tested. This is consistent with previous studies which implicate that surface functionalization lowers toxicity by modification of surface charge and reduction in the number of free surface silanols.29 The only clear time-dependent effect was again observed in the nonporous silica, which showed a clear trend over time in terms of cell viability.
Previous work in the field has looked at effects of porosity in the interaction of silica nanoparticles with RAW 264.7 macrophage cells. Yu, et al., for example, observed similar trends, with the functionalized porous and nonporous materials showing mitigated toxicological responses across the dosage range evaluated.30 Using propidium iodide staining, it was observed that nonporous silica nanoparticles instigated an approximately four-fold larger disruption to the macrophage cell membrane than the mesoporous nanoparticles. A caveat is that the materials used in the previous study were slightly larger than 100 nm, and so their size regime effects cannot be directly compared to our data. Still, Maurer-Jones, et al. have shown for smaller particles (~25 nm) elevated hemolytic activity for the nonporous material across a similar concentration range.8 The particles in our study, which are of intermediate size, then fit directly into this previous work but with an added dimension of the effects of functionalization on the observed toxicity.
EPR spectroscopy and quantification of free radical species
As a means of assessing ROS produced from the surface, electron paramagnetic resonance (EPR) spectroscopy was employed. EPR has been used previously to assess radical formation in many different systems of interest, including in vivo studies. Here, it has been used to detect and quantify hydroxyl radical (HO˙) produced from the homolytic cleavage of hydrogen peroxide:| H2O2 → 2˙OH | (1) |
This has been previously carried out in a study by Zhang et al. in which they looked at the processing pathway dependence of the material on its toxicological effects.9 Zhang's work focused primarily on fumed silica, whereas our study focuses instead on colloidal silica prepared via hydrothermal synthesis. In Zhang's work, the observed toxicity was attributed to the formation and cleavage of strained siloxane rings on the surface, leading to ROS formation. Due to the hydrothermal nature of the synthesis of the materials used in this study, any strained rings would have been broken during the synthetic process and subsequent washing steps. However, it is still reasonable to expect that ROS may form from surface-catalyzed homolytic cleavage of hydrogen peroxide.
In these EPR studies, H2O2 was implemented to mimic a cellular reserve of molecules that may undergo chemical modification by interaction with the nanoparticle surface. Furthermore, work in the field has elucidated the effect nanoparticle exposures can have on oxidative stress in biological systems. Natural redox-active protection mechanisms can quickly be overcome by an influx of ROS species via uncontrolled chemical reactions in intracellular or extracellular compartments. Due to the reactive nature of the hydroxyl radical, direct detection, even using highly sensitive EPR spectroscopy, is not feasible.
However, a common method to observe and quantify free radical species that evade direct detection is enabled by using a spin-trap. These are commonly utilized and have great application to studying complex biological systems. The spin traps most commonly used are nitrone spin traps like 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO), or DMPO which form a spin adduct with the hydroxyl or other radical species (Fig. 3).
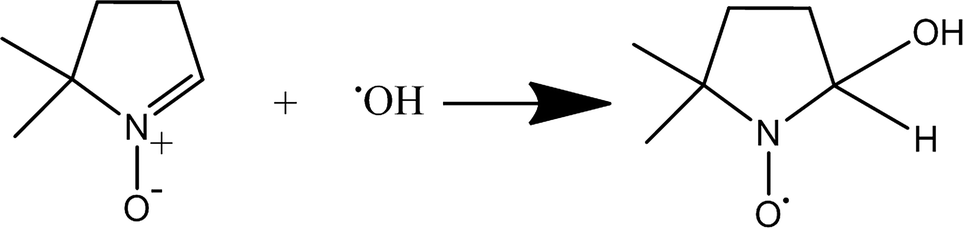 | ||
| Fig. 3 Reaction between DMPO molecule and free hydroxyl to give DMPO-HO· spin adduct which is detectable by EPR spectroscopy. The unpaired electron primarily localizes on the oxygen atom. | ||
The half-life of these spin adducts is quite long, on the order of tens of minutes to hours.31 Therefore, detection and quantification of the radical signals is possible by implementing a spin trap. The DMPO-HO˙ spin adduct gives a characteristic 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:2:1 spectrum centered at approximately 3490 Gauss in the X-band (νL = 9.79 GHz) region. This four line spectrum arises from splitting of the peaks via coupling of the unpaired electron to the nearby nitrogen as well as the hydrogen at the beta position, with measured hyperfine coupling constants of aN = 14.9 G and aH = 14.9 G, matching those reported in the literature.32
:2:2:1 spectrum centered at approximately 3490 Gauss in the X-band (νL = 9.79 GHz) region. This four line spectrum arises from splitting of the peaks via coupling of the unpaired electron to the nearby nitrogen as well as the hydrogen at the beta position, with measured hyperfine coupling constants of aN = 14.9 G and aH = 14.9 G, matching those reported in the literature.32
EPR spectra for the detected hydroxyl radical spin adduct for the materials can be seen in Fig. 4. As a standard convention, these are reported as the derivative of the signal with respect to the magnetic field strength (in Gauss).
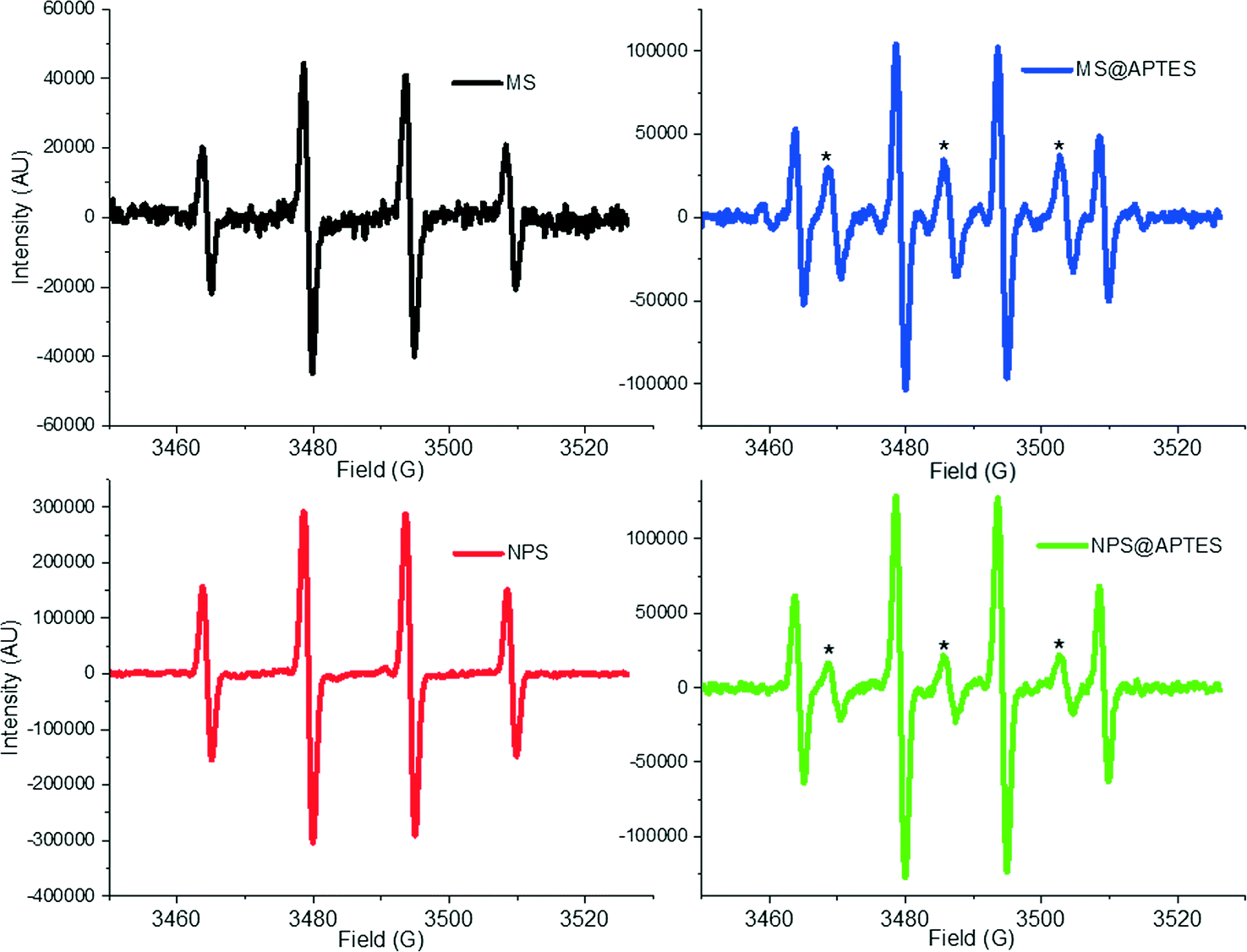 | ||
| Fig. 4 EPR spectra of the materials to quantify hydroxyl radical (HO˙) produced by surface-catalyzed decomposition of H2O2. The DMPO-HO˙ spin adduct is characterized by the four line spectrum shown of intensity 1:2:2:1, and the asterisk (*) indicates aminoxyl radical generated by oxidation of the amine functionality. | ||
It is of particular interest that additional peaks are present in the amine-functionalized samples. The peaks in between the four signals corresponding to the DMPO spin adduct have been assigned to aminoxyl (IUPAC recommended name for [R2N–O˙] [R2N˙+–O−]) radical. This is not surprising, as it is easy to imagine that due to the high peroxide concentration the amine has become oxidized to form an aminoxyl radical, which is more stable than free hydroxyl radical. The structure of the molecule would arise from oxidation of the APTES such that it has the structure similar to R–NHO˙ with the unpaired electron on the oxygen being detected by the EPR measurement. This type of aminoxyl radical formation is noted in the literature to occur under oxidizing conditions.33 From the EPR spectrum of the functionalized materials, hyperfine coupling constants of aN = 17.1 G are calculated for the 1:1:1 aminoxyl, which match well with other aminoxyl radicals (aN = 16.9 G) observed in aqueous solution by others.34 The aminoxyl radical has been simulated using the EasySpin simulation package and compared to the experimental data, which is available in the ESI† (Fig. S6).35 This comparison shows agreement between the simulated and experimental aminoxyl EPR spectrum.
Double integration followed by comparison with an external standard, in this case 2-carboxy-PROXYL, facilitates quantification of the hydroxyl species in solution. The signals were integrated twice to give first an absorption lineshape and then finally an integrated intensity which was compared to a 2-carboxy-PROXYL solution of known concentration to calculate the absolute radical concentration. A plot of the absolute radical concentration for both the hydroxyl and aminoxyl radical species for each material is given in Fig. 5.
 | ||
| Fig. 5 Absolute radical quantification from EPR spectroscopy conducted in water with DMPO spin trap and added H2O2. The type of radical is differentiated by color according to the legend. | ||
Mostly notably the concentration of hydroxyl radical in the bare nonporous silica is the highest of all measured samples, and is significantly higher when compared to its mesoporous analogue. This is surprising when you consider the approximate 20-fold difference in their surface areas. One would expect much more radical to form on the mesoporous silica surface, yet the opposite is observed, with a seven-fold larger concentration from the nonporous compared to the porous material. Additionally, due to an approximate 60% pore void volume in the mesoporous material, there are many more particles per same unit of mass. In the case of the bare materials, a volumetric calculation based on assumptions of silica density from the literature gives values of 1.9 × 1016 particles per g for the mesoporous material, and 6.9 × 1015 particles per g for the nonporous material.36 This is approximately a factor of three difference in particles per gram, yet the nonporous material shows an almost seven-fold larger concentration of radicals based on the EPR measurement.
The surface area of the material is expected to dictate the total amount of radical produced, with materials having larger surface area capable of producing more free radicals than materials of lower surface area. It can therefore be assumed that materials such as mesoporous silica will generate more free radicals than nonporous materials on the same mass unit basis. In order to account for this trend, the absolute concentration has been converted to radical production in terms of the pmol radical per m2 surface area. This radical production can be seen in Fig. 6, showing the radical production as a function of radical type and material.
 | ||
| Fig. 6 Radical production as a function of material and radical type. These values are obtained by normalizing for the different surface areas of each material as determined by BET adsorption isotherm. | ||
Due the very high surface area of the mesoporous material, the radical production per unit area is very low. Comparing the bare materials, a two order-of-magnitude difference between mesoporous and nonporous silica was observed. Also of note is that amine-functionalization decreases hydroxyl radical production in the nonporous silica. One result that remains puzzling is that functionalization of the mesoporous material results in an increase in total radical production. This is not expected, and is the opposite of the trend observed in the nonporous silica samples.
A final issue of note is that normalization of the radical concentration to radical production gives the positive control, Min-U-Sil, a large production of radical per surface area. Min-U-Sil, or α-quartz, is very toxic as a crystalline material which is thoroughly noted in the toxicological literature, and why it was implemented in these studies as a positive control.37 The EPR data demonstrate that a large amount of radicals can arise from a very small surface area of the material (~7 m2 g−1) for quartz. This implies that perhaps it is its capability to produce large amounts of free radicals that results in such potent toxic effects in biological systems. A caveat is that the quartz used in these studies is approximately five times the size of the other materials, which were deliberately size-matched. So the toxicological effect of the quartz cannot be isolated as separate from its larger physical size.
In vitro assessment of ROS species in RAW 264.7 cell line
The EPR spectroscopic measurements, while quantitative and informative, are somewhat limited by the fact that they are carried out in a simplified chemical environment. While it enables extraction and isolation of radical production as a surface catalyzed-process, a toxicological response occurs in the complex chemical environment of the cell. This is why an in vitro assay was carried out to quantify intracellular ROS species, using intracellular superoxide (O2˙−) concentration as an estimate of total intracellular ROS, and ultimately as a measure of oxidative stress. Cellular metabolic processes, in particular electron transport chain processes, generate ROS as a by-product of ATP production. In the terminal step of the electron transport chain, cytochrome c oxidase (Complex IV) reduces molecular oxygen to water. In a small proportion of these reactions, the oxygen is only partially reduced to give the superoxide radical, which then reacts to give other ROS species like hydroxyl radical and peroxynitrite (ONOO−). Nature has evolved mechanisms to deal with this source of superoxide radical via the superoxide dismutase family of enzymes which are among the most efficient enzymes known, being diffusion-limited in their catalytic capacity.38 Immune cells can also implement an oxidative burst as a means of destroying foreign pathogens.The key is to extract the increase in intracellular ROS that arises from the nanoparticle rather than endogenous species. The results of this experiment are shown in Fig. 7.
 | ||
| Fig. 7 In vitro ROS assay for each material, negative control, and the antimycin A positive control. The mean fluorescence intensity is proportional to total intracellular ROS. | ||
The negative control in this case is cells in the absence of nanoparticles, which gives the baseline response of physiological superoxide concentration. Antimycin A served as a positive control, as it inhibits cytochrome c oxidase, uncoupling the electron transport chain and leading to a buildup of intracellular superoxide. The silica nanomaterials, as seen in the data, show very little increase in intracellular ROS, with the exception of the bare nonporous silica material. The nonporous silica nanoparticles seems to induce intracellular formation of ROS in the RAW 264.7 macrophage cells. The other materials show very little elevation in intracellular ROS, which correlates with the cell viability results.
By using particle cell uptake values reported in the literature, and using the calculated radical productions for each material obtained via the EPR measurements, the true intracellular concentration of hydroxyl radical from the mesoporous and nonporous silica nanomaterials can be approximated. Typical values for these size silica nanoparticles give uptake values of around 15000 particles per cell for the RAW 264.7 cell line.39 Using this uptake value, the calculated radical production (Fig. 6), and the typical cellular volume (~2 pL), the intracellular concentration of [HO˙] the bare mesoporous and nonporous materials can be calculated to be approximately 800 and 16000 pM, respectively (see ESI† for full calculation). Since the concentration of hydrogen peroxide used in the EPR experiments is much higher than a true in vivo concentration, this can be used to correct the above concentrations to give realistically expected values (assuming no reaction with other molecules). The values obtained when this correction is employed are 4.2 × 10−16 and 7.9 × 10−15 M [HO˙] for the mesoporous and nonporous material, respectively. Compared to the typical in vivo concentration (~1 × 10−15 M) this gives a calculated [HO˙]/in vivo [HO˙] ratio of 0.42 for the mesoporous and 7.9 for the nonporous material.40
An elevation of eight times the normal concentration of hydroxyl radical can certainly cause oxidative damage and induce apoptosis. This is especially true if the radicals are generated rapidly, thereby overcoming natural ROS defense mechanisms such as glutathione-mediated redox chemistry.29 Increased cellular concentrations of ROS results in increased oxidative stress for the cell.41 We implicate increased intracellular ROS measured here as leading to oxidative stress in the cells. It is well-known from literature that increased oxidative species (i.e. ROS) results in oxidative modifications to biological macromolecules, such as lipid oxidation and protein degradation.42 Cellular response to this is to initiate signaling cascades that result in apoptosis.43 Therefore, increased ROS can result in observed cell death via a cascade of biological signaling pathways which destroy cells damaged by excessive oxidative reactions.
Combining cell viability, EPR spectroscopy, and intracellular ROS
Taken together, the data suggests that the elevated toxic response of the bare nonporous silica as understood from the cell viability assay is directly correlated to the high level of intracellular ROS produced upon exposure. This correlates well with the quantitative EPR results, while the other materials show very low cytotoxicity by cell viability as well as low intracellular ROS. In the case of the nonporous silica, the data are strongly suggestive that the observed toxicity can be directly correlated to the ROS produced. The data also strongly suggest that functionalization of nonporous silica reduces its toxicological response by reducing the number of free radicals formed at the surface of the material. This can be attributed to two factors. First, the number of surface silanols is reduced due to surface functionalization, which occurs through addition of the APTES moiety. The second factor is due to the presence of the functional group sterically blocking the approach of the hydrogen peroxide molecules to the surface-silicon atoms. In the functionalized material, the free molecular motion of the APTES molecule likely restricts the approach of the hydrogen peroxide oxygen, which is required for the surface-catalyzed reaction to occur. Certainly some peroxide molecules will be able to begin the homolytic reaction via the initial rate-determining step (RDS) of the bonding and/or coordination of the peroxide oxygen to the surface silicon. However, the overall rate of this step can be assumed to be much lower in the functionalized material as the APTES moiety hinders the approach of the peroxide molecules, since the APTES is relatively larger in size.A blank EPR experiment carried out on just a solution of DMPO with added H2O2 (in the absence of the nanomaterials) gives a very small signal, which leads to the conclusion that the production of the radicals is surface-catalyzed. Furthermore, other experiments carried out on the nanomaterials in water without H2O2 demonstrates no measurable signal of hydroxyl radical. Thus, we suggest that the reaction to generate hydroxyl radical is surface-catalyzed, and that it proceeds without the siloxane ring-breaking mechanism suggested by Zhang, et al. (vide supra). There has been some other work in the field investigating free radical production in titania (TiO2) from cleavage of hydrogen peroxide.44 The nanoparticle surface thus enhances the rate of homolytic cleavage of the hydrogen peroxide molecule. A proposed mechanism for the homolytic reaction at the silica nanoparticle surface is given in Fig. 8. The surface sites of the silica are free silanol (Si–OH) moieties. As mentioned previously, silanol moieties are fairly acidic functional groups (pKa ≈ 3.0). At physiological pH (7.4) these groups are uniformly deprotonated to give the silicate anion, possessing negative charge.
Modification of these groups via functionalization can change both the surface charge and the total acidity. In the case of the APTES functionalization utilized in this study, addition of the amine functionality serves to increase the surface charge through the ammonium cation formed at physiological pH; yet the functionalized material is also more basic in nature as some of the silanols have been removed via functionalization and the amine itself is fairly basic (pKa = 10.6). This would produce a more basic solution when compared to the bare materials. It is a well-established phenomenon that hydrogen peroxide is more stable in acidic than basic solutions. Therefore, the EPR data here shows the opposite trend from the expectation in that the bare nonporous material shows greater degree of the breakdown of H2O2 to products (i.e. hydroxyl radical) than the functionalized equivalent. The mechanism proposed here is that surface-coordination of the hydrogen peroxide lowers the activation energy and provides a more stable transition state, enhancing the rate for the homolytic reaction. If this hypothesis is correct, then the predominant mechanism for production of the hydroxyl radical is not in the bulk solvent (which would result from acid- or base-catalyzed homolysis) but is dominated by interactions between the hydrogen peroxide molecules and the silica surface.
In the mesoporous silica, the functionalized material seems to, at least from the EPR measurements, give rise to more free radicals in its functionalized form. This is perhaps due to different coordination of the hydrogen peroxide to the silica surface. It is possible that the amine functionality (which will be positively charged at most pH values) is better able to electrostatically interact with the hydrogen peroxide molecule in the pores of the mesoporous material. In contrast, in the nonporous material, only the external surface is available for bonding. The proposed model (Fig. 8) idealizes bonding between the peroxide oxygen and the silica silicon atom as an initial step. Overall, more of these atoms are freely accessible in the nonporous silica as they are all at the external surface. Most of the surface area of the mesoporous material is located within the pores themselves.
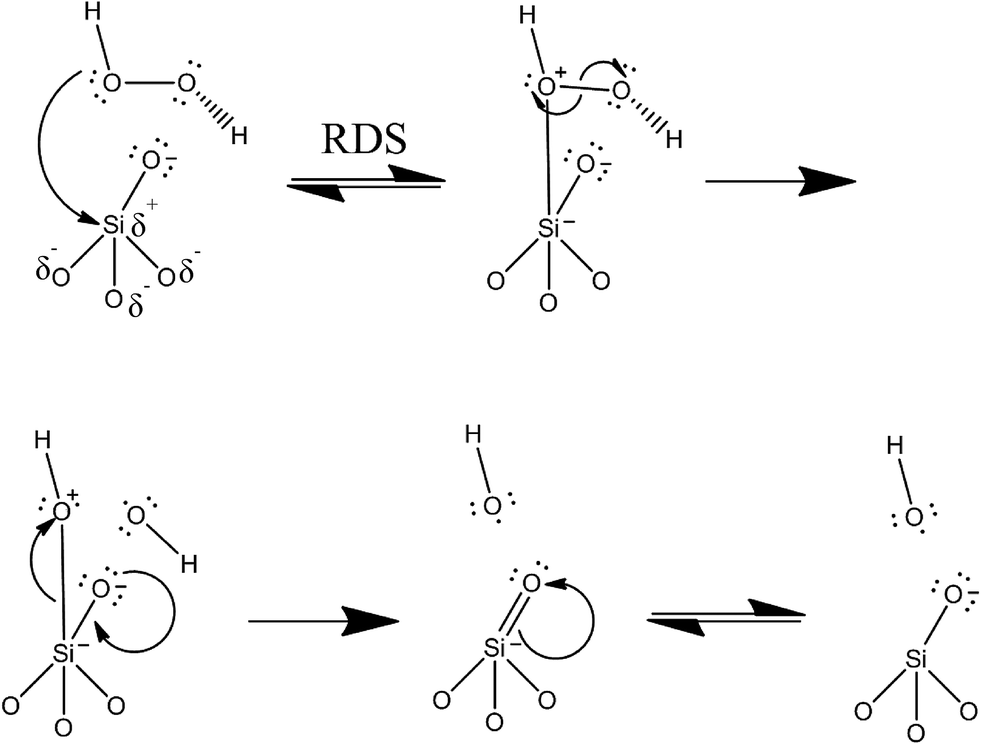 | ||
| Fig. 8 Proposed mechanism for homolytic cleavage of H2O2via surface-catalyzed reaction with silica nanoparticle surface. Surface catalysis leads to formation of two hydroxyl radicals (HO˙). The reaction is initiated by the partial positive charge on the central silicon atom due to the withdrawing effect of the attached oxygen atoms. | ||
The hydrodynamic diameter reported in literature for hydrogen peroxide is 0.5 nm which is small enough to fit into the pores of the mesoporous silica (dpore ~ 3 nm).45 However, flux through the pores is limited due to the nature of the porous material structure. Molecular flux (J) in one dimension is given by Fick's First Law:
 | (2) |
Here D is the molecular diffusion coefficient in units of m2 s−1, C is the concentration and x is distance. This applies to bulk systems but in the case of the inner pores of the mesoporous silica one must employ an effective diffusion coefficient, Deff, which accounts for the flux through the porous network. This can be reasonably approximated by:
 | (3) |
The system then becomes diffusion-limited as the molecules are forced to undergo flux through the pores to react at the liquid–solid interface. The motion of the molecules is isotropic, so only a concentration gradient drives them through the porous network. This gives rise to decreased mass transport, and perhaps explains why the mesoporous material gives so much lower ROS than that of the nonporous material. The approximated effective diffusion coefficient described above can account for the sevenfold difference in concentration between the bare porous and nonporous materials. Even though total surface area is larger, the effective surface area “seen” per unit time by the H2O2 molecules is much lower in the porous material. In addition, due to the physically constrained nature of the pore diameter, one can envision that the recombination rate of initially formed hydroxyl radicals is much higher than in free solution as their residence time in the total porous network must be much longer than the bulk analogue. The diffusion rate inside the pores ultimately controls the amount radical generated per unit time as almost all (>95%) of the surface area is contained within the porous network. The porosity then has a direct and dramatic effect on the total amount of ROS (hydroxyl radical) generated and thus measured by the EPR spectroscopy described above. This observation supports the working hypothesis of a surface-catalyzed reaction generating free hydroxyls in solution, which is again matched well by the measurement of in vitro ROS shown here.
As stated previously, the observation that functionalization of the mesoporous material seems to increase radical production is very puzzling. A possible explanation for the overall increase in radical production for the functionalized mesoporous silica is through a combination of diminished diffusion through the pore and the nature of the functionalized surface. The silica–water interface is an important and often studied area. These studies implicate a Stern layer that shows little change due to the highly hydrophilic surface. This exploration in interfacial water structure and dynamics leads to the presence of an increased viscosity near the interface that manifests as a viscosity gradient.47 In effect this gives a layer of adsorbed water that only slowly exchanges with bulk solvent. This could also limit diffusion of the hydrogen peroxide to the surface. If the surface is functionalized, however, one can imagine that the addition of the aminopropyl moieties can disrupt this Stern layer, enhancing flux across the surface. This would then result in an increase in the amount of hydrogen peroxide that is cleaved by interaction with the surface. Furthermore, inside the constrained pores of the mesoporous silica this effect would be further enhanced due to the diminished flux, as described above. So, perhaps it is a combination of diminished diffusion and a fairly rigid Stern layer on the silica surface which accounts for these results. However, the source of this effect is unclear at the present time.
Conclusions
Combined, the data enable a strong correlation between ROS derived from surface-catalyzed reactions and observed cellular toxicity. Porosity has a major effect on the production of ROS from the surface and amine-functionalization in the nonporous material decreases the amount of free radical generated at the solid–liquid interface. We conclude that the mesoporous silica shows less toxicity than the nonporous silica due to decreased the amount of free radicals generated at the solid–liquid interface; and that this ultimately derives from the total porosity of the material influencing radical production and diffusion to the surface. We invoke a flux-based argument to account for the observed differences, as the surface appears to dictate the radical production. Surface amine-functionalization seems to mitigate radical production by steric hindrance (primarily in the nonporous material) and perhaps also by producing a more rapidly exchanging Stern layer at the interface (in the mesoporous material). Finally, we imply that safety by design can be implemented when the nanomaterials surface effects are considered in synthesis, chemical modification and application.Acknowledgements
The authors gratefully acknowledge financial assistance from the National Institute for Environmental Health Sciences through the University of Iowa Environmental Health Sciences Research Center, NIEHS/NIH P30 ES05605. The ESR Facility at the University of Iowa, including Prof. Garry Buettner, is also acknowledged for assistance with EPR spectroscopy. Financial assistance for the EPR Facility is provided in part by the National Cancer Institute of the National Institutes of Health under Award P30CA086862. A. S. M. acknowledges support from an Institutional National Research Service Award (NRSA T90) in Oral Health Research from the National Institute of Dental and Craniofacial Research. S. E. L. wishes to thank Prof. J. Leddy at UI for helpful discussions concerning molecular diffusion in confined chemical systems.References
- L. Q. Chen, L. Fang, J. Ling, C. Z. Ding, B. Kang and C. Z. Huang, Chem. Res. Toxicol., 2015, 28, 501–509 CrossRef CAS PubMed.
- A. B. Seabra, A. J. Paula, R. de Lima, O. L. Alves and N. Durán, Chem. Res. Toxicol., 2014, 27, 159–168 CrossRef CAS PubMed.
- C.-C. Huang, C.-Y. Tsai, H.-S. Sheu, K.-Y. Chuang, C.-H. Su, U.-S. Jen, F.-Y. Cheng, C.-H. Su, H.-Y. Lei and C. S. Yeh, ACS Nano, 2011, 5, 3905–3916 CrossRef CAS PubMed.
- S. Nayab, A. Farrukh, Z. Oluz, E. Tuncel, S. R. Tariq, H. ur Rahman, K. Kirtchhoff, H. Duran and B. Yameen, ACS Appl. Mater. Interfaces, 2014, 6, 4408–4417 CAS.
- C. M. A. Partlett, D. W. Bruce, N. S. Hondow, A. F. Lee and K. Wilson, ACS Catal., 2011, 1, 636–640 CrossRef.
- G. Yang, S. Gai, F. Qu and P. Yang, ACS Appl. Mater. Interfaces, 2013, 5, 5788–5796 CAS.
- T. Asefa and Z. Tao, Chem. Res. Toxicol., 2012, 25, 2265–2284 CrossRef CAS PubMed.
- M. A. Maurer-Jones, Y.-S. Lin and C. L. Haynes, ACS Nano, 2010, 4, 3363–3373 CrossRef CAS PubMed.
- H. Zhang, J. Am. Chem. Soc., 2012, 134, 15790–15804 CrossRef CAS PubMed.
- J. Duan, Y. Yu, Y. Li, Y. Yu, Y. Li, X. Zhou, P. Huang and Z. Sun, PLoS One, 2013, 8 Search PubMed.
- A. Rimola, D. Costa, M. Sodupe, J.-F. Lambert and P. Ugliengo, Chem. Rev., 2013, 113, 4216–4313 CrossRef CAS PubMed.
- M. T. A. Samri, A. V. Biradar, A. R. Alsuwaidi, G. Balhaj, S. Al-Hammadi, S. Shehab, S. Al-Salam, S. Tariq, T. Pramathan, S. Benedict, T. Asefa and A.-K. Souid, Int. J. Nanomed., 2012, 7, 3111–3121 Search PubMed.
- V. Murashov, M. Harper and E. Demchuk, J. Occup. Environ. Hyg., 2006, 3, 718–723 CrossRef CAS PubMed.
- M. Ghiazza, M. Polimeni, I. Fenoglio, E. Gazzano, D. Ghigo and B. Fubini, Chem. Res. Toxicol., 2010, 23, 620–629 CrossRef CAS PubMed.
- C. Murray, E. L. Derro, T. D. Sechler and M. I. Lester, Acc. Chem. Res., 2009, 42, 419–427 CrossRef CAS PubMed.
- H. Zhang, Z. Ji, T. Xia, H. Meng, C. Low-Kam, R. Liu, S. Pokhrel, S. Lin, X. Wang, Y.-P. Liao, M. Wang, L. Li, R. Rallo, R. Damoiseaux, D. Telesca, L. Mädler, Y. Cohen, J. I. Zink and A. E. Nel, ACS Nano, 2012, 6, 4349–4368 CrossRef CAS PubMed.
- T. Xia, M. Kovochich, M. Liong, L. Mädler, B. Gilbert, H. Shi, J. I. Yeh, J. I. Zink and A. E. Nel, ACS Nano, 2008, 2, 2121–2134 CrossRef CAS PubMed.
- T. Xia, M. Kovochich, J. Brant, M. Hotze, J. Sempf, T. Oberley, C. Sioutas, J. I. Yeh, M. R. Wiesner and A. E. Nel, Nano Lett., 2006, 6, 1794–1807 CrossRef CAS PubMed.
- A. Yildirim, E. Ozgur and M. Bayindir, J. Mater. Chem. B, 2013, 1, 1909–1920 RSC.
- V. Marzaioli, J. A. Aguilar-Pimentel, I. Weichenmeier, G. Luxenhofer, M. Wiemann, R. Landsiedel, W. Wohlleben, S. Eiden, M. Mempel, H. Behrendt, C. Schmidt-Weber, J. Gutermuth and F. Alessandrini, Int. J. Nanomed., 2014, 9, 2815–2832 Search PubMed.
- J. L. Townson, Y.-S. Lin, J. O. Agola, E. C. Carnes, H. S. Leong, J. D. Lewis, C. L. Haynes and C. J. Brinker, J. Am. Chem. Soc., 2013, 135, 16030–16033 CrossRef CAS PubMed.
- X. He, H. Nie, K. Wang, W. Tan, X. Wu and P. Zhang, Anal. Chem., 2008, 80, 9597–9603 CrossRef CAS PubMed.
- K. Möller, J. Kobler and T. Bein, Adv. Funct. Mater., 2007, 17, 605–612 CrossRef.
- X. Huang, L. Li, T. Liu, N. Hao, H. Liu, D. Chen and F. Tang, ACS Nano, 2011, 5, 5390–5399 CrossRef CAS PubMed.
- W. Stöber, A. Fink and E. Bohn, J. Colloid Interface Sci., 1968, 26, 62–69 CrossRef.
- S. E. Lehman, Y. Tataurova, P. S. Mueller, S. V. Santhana Mariappan and S. C. Larsen, J. Phys. Chem. C, 2014, 118, 29943–29951 CAS.
- J. Y. Lee, Y.-J. Kim, H. J. Kim, Y.-S. Kim and W. Park, Molecules, 2012, 17, 5404–5411 CrossRef CAS PubMed.
- T. Kusaka, M. Nakayama, K. Nakamura, M. Ishimiya, E. Furusawa and K. Ogasawara, PLoS One, 2014, 9 Search PubMed.
- J. Ahmad, M. Ahamed, M. J. Akhtar, S. A. Alrokayan, M. A. Siddigui, J. Musarrat and A. A. Al-Khedhairy, Toxicol. Appl. Pharmacol., 2012, 259, 160–168 CrossRef CAS PubMed.
- T. Yu, A. Malugin and H. Ghandehari, ACS Nano, 2011, 5, 5717–5728 CrossRef CAS PubMed.
- F. A. Villamena, C. M. Hadad and J. L. Zweier, J. Phys. Chem. A, 2003, 107, 4407–4414 CrossRef CAS.
- S. Sanglioglu, C. M. Williams, L. Samavati, N. S. Butler, G. Wang, P. B. McCray, T. C. Ritchies, G. W. Hunninghake, E. Zandi and J. F. Englehardt, J. Biol. Chem., 2001, 276, 30188–30198 CrossRef PubMed.
- J. Lind and G. Merényi, J. Phys. Chem. A, 2006, 110, 192–197 CrossRef CAS PubMed.
- G. R. Buettner, B. D. Scott, R. E. Kerber and A. Mugge, Free Radical Biol. Med., 1991, 11, 69–70 CrossRef CAS PubMed.
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42–55 CrossRef CAS PubMed.
- Y.-S. Lin and C. L. Haynes, J. Am. Chem. Soc., 2010, 132, 4834–4842 CrossRef CAS PubMed.
- C. Michel, S. Herzog, C. de Capitani, P. Burkhardt-Holm and C. Pietsch, PLoS One, 2014, 9, 1–10 Search PubMed.
- I. Fridovich, Annu. Rev. Biochem., 1995, 64, 97–112 CrossRef CAS PubMed.
- A. Kurtz-Chalot, J.-P. Klein, J. Pourchez, D. Boudard, V. Bin, O. Sabido, L. Marmuse, M. Cottier and V. Forest, Biomed. Microdevices, 2015, 17, 1–12 CrossRef CAS PubMed.
- M. Giorgio, M. Trinei, E. Migliaccio and P. G. Pelicci, Nat. Rev. Mol. Cell Biol., 2007, 8, 722–728 CrossRef CAS PubMed.
- H. Zhang, T. Chen, J. Jiang, Y.-S. Wong, F. Yang and W. Zheng, J. Agric. Food Chem., 2011, 59, 8683–8690 CrossRef CAS PubMed.
- J. Imlay, Annu. Rev. Microbiol., 2003, 57, 395–418 CrossRef CAS PubMed.
- M. Ponniah, E. E. Billett and L. A. De Girolamo, Chem. Res. Toxicol., 2015, 28, 1693–1703 CrossRef CAS PubMed.
- Y. Du and J. Rabani, J. Phys. Chem. B, 2006, 110, 6123–6128 CrossRef CAS PubMed.
- F. Fogolari, A. Corazza, S. Toppo, S. C. E. Tosatto, P. Viglino, F. Ursini and G. Esposito, J. Biomed. Biotechnol., 2012, 2012, 1–9 CrossRef PubMed.
- K. P. Saripalli, R. J. Serne, P. D. Meyer and B. P. McGrail, Groundwater, 2002, 40, 346–352 CrossRef CAS.
- H. Zhang, A. A. Hassanali, Y. K. Shin, C. Knight and S. J. Singer, J. Chem. Phys., 2011, 134 Search PubMed.
Footnotes |
| † The authors declare no competing financial interests. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c5en00179j |
| This journal is © The Royal Society of Chemistry 2016 |