
Size distribution and sources of humic-like substances in particulate matter at an urban site during winter
Seungshik
Park
* and
Se-Chang
Son
Department of Environment and Energy Engineering, Chonnam National University, 77 Yongbong-Ro, Buk-gu, Gwangju 500-757, Korea. E-mail: park8162@chonnam.ac.kr; Fax: +82-62-530-1859
First published on 12th November 2015
Abstract
This study investigates the size distribution and possible sources of humic-like substances (HULIS) in ambient aerosol particles collected at an urban site in Gwangju, Korea during the winter of 2015. A total of 10 sets of size-segregated aerosol samples were collected using a 10-stage Micro-Orifice Uniform Deposit Impactor (MOUDI), and the samples were analyzed to determine the mass as well as the presence of ionic species (Na+, NH4+, K+, Ca2+, Mg2+, Cl−, NO3−, and SO42−), water-soluble organic carbon (WSOC) and HULIS. The separation and quantification of the size-resolved HULIS components from the MOUDI samples was accomplished using a Hydrophilic–Lipophilic Balanced (HLB) solid phase extraction method and a total organic carbon analyzer, respectively. The entire sampling period was divided into two periods: non-Asian dust (NAD) and Asian dust (AD) periods. The contributions of water-soluble organic mass (WSOM = 1.9 × WSOC) and HULIS (=1.9 × HULIS-C) to fine particles (PM1.8) were approximately two times higher in the NAD samples (23.2 and 8.0%) than in the AD samples (12.8 and 4.2%). However, the HULIS-C/WSOC ratio in PM1.8 showed little difference between the NAD (0.35 ± 0.07) and AD (0.35 ± 0.05) samples. The HULIS exhibited a uni-modal size distribution (@0.55 μm) during NAD and a bimodal distribution (@0.32 and 1.8 μm) during AD, which was quite similar to the mass size distributions of particulate matter, WSOC, NO3−, SO42−, and NH4+ in both the NAD and AD samples. The size distribution characteristics and the results of the correlation analyses indicate that the sources of HULIS varied according to the particle size. In the fine mode (≤1.8 μm), the HULIS composition during the NAD period was strongly associated with secondary organic aerosol (SOA) formation processes similar to those of secondary ionic species (cloud processing and/or heterogeneous reactions) and primary emissions during the biomass burning period, and during the AD period, it was only associated with SOA formation. In the coarse mode (3.1–10 μm), it was difficult to identify the HULIS sources during the NAD period, and during the AD period, the HULIS was most likely associated with soil-related particles [Ca(NO3]2 and CaSO4) and/or sea-salt particles (NaNO3 and Na2SO4).
Environmental impactThe results from size-segregated aerosol measurements of HULIS concentrations at an urban site during winter indicate that the HULIS-C (on a carbon basis)/WSOC ratio in fine particles showed little difference between the non-Asian dust (NAD) and AD periods, but the HULIS showed uni-modal during the NAD and bimodal size distributions during AD, respectively. Also the HULIS in the fine mode was strongly associated with both secondary organic aerosol (SOA) formation processes and primary biomass burning emissions during the NAD period, while it was associated with the SOA formation during the AD period. |
Introduction
The organic compounds that account for a significant amount of ambient particulate matter1 can be categorized into water-insoluble and water-soluble fractions and may affect the radiative balance of the Earth.2,3 Moreover, water-soluble organic compounds can act as cloud condensation nuclei to facilitate cloud formation. The water-insoluble organic carbon fraction is primarily generated as a result of the incomplete combustion of fossil fuels and biomass materials1,4 and is obtained by extraction with n-hexane, dichloromethane, acetone,5 or benzene.6 In contrast, the water-soluble organic carbon (WSOC) fraction is emitted directly from primary combustion sources, or formed in the atmosphere through transformation processes.7 The primary sources of WSOC include biomass burning (BB) emissions,1,8–12 traffic emissions (with small contributions),12–15 and residual oil combustions.16Solid phase extraction techniques can be used to further separate WSOC into more hydrophilic and more hydrophobic fractions.17–19 Humic-like substances (HULIS) are an important class of substances in the hydrophobic WSOC fraction,20,21 and these consist of a mixture of high molecular weight compounds22 with the ability to suppress the surface tension of aerosol and fog water.23,24 The physical and chemical properties of HULIS are similar to those of humic and fulvic acids that naturally occur in terrestrial and aquatic environments.20,25,26 They are thought to consist of polycyclic aromatic and aliphatic structures with oxygenated functional groups, such as hydroxyl, carboxyl, carbonyl, nitrate, and nitroxy organosulfate groups.18,27–29 HULIS play an important role in the hygroscopic growth and cloud condensation nuclei formation of aerosols;24,30,31 significantly contribute to light absorption;32,33 affect the aqueous-phase oxidation of organic pollutants;34 promote the production of reactive oxygen species under simulated physiological conditions.35
HULIS accounts for a significant fraction of WSOC in ambient aerosols, and numerous studies have shown that BB emissions16,21,33,36–39 and secondary formation processes16,21,39 are important sources of HULIS in ambient aerosol particles. In addition, marine aerosols40 and residual oil combustion16 have also been identified as possible sources of HULIS. Typically, the fraction of HULIS-C (on a carbon basis) in WSOC tends to increase in ambient samples due to the contribution of BB emissions,21,38,41 and this fraction has also been found to be higher in the winter than in the summer, possibly due to the increase in BB activities in the winter.16,39 Previous studies have shown that the fraction of HULIS-C in ambient PM2.5 is highly variable, accounting for 9–72% of the total WSOC.16,20,21,36,39,42,43 Also, BB emissions have a lower fraction (∼30%) of HULIS-C in WSOC in PM2.5.21,41
The size distribution of the chemical constituents in aerosol particles can provide important information on their sources, formation, and evolution in air. Size-resolved WSOC has been extensively measured in ambient environments44–49 and in biomass burning emissions in controlled chambers in laboratories.50–52 The size distributions of the WSOC in ambient aerosols and fresh/aged BB emissions mostly exhibit a dominant droplet mode peaking at 0.55–1.0 μm.45,46,52,53 Until now, HULIS measurements have been mainly made for bulk aerosol particles, such as TSP, PM10, and PM2.5 in various sampling environments. However, a limited amount of information is available on the size distribution of HULIS in ambient aerosols. In one example from a rural site in Southern China, the size distribution of ambient HULIS showed a dominant droplet mode peaking at 0.63–0.87 μm.21 Droplet-mode HULIS was attributed to secondary formation through cloud-processing and heterogeneous reactions or aerosol-phase reactions, and to the growth of fresh BB particles.
There is a lack of data on the size distribution of HULIS in ambient air, especially for East Asia (including Korea), and the chemical characteristics and sources/processes of size-resolved HULIS from these regions are poorly understood. Thus, an investigation of the size distribution of ambient HULIS particles can improve our understanding of the sources and formation processes of HULIS as a hydrophobic part of WSOC. To this end, this study collected 48 h size-segregated aerosol samples from an urban site in Gwangju, Korea during February 2015, and the samples were analyzed in terms of mass, water-soluble inorganic ions, total WSOC, and HULIS. This study aims to determine the mass size distribution of HULIS as well as to explore the likely sources and formation processes for size-resolved HULIS. In addition, differences in the size distribution and sources of HULIS for non-Asian dust and Asian dust samples are also discussed.
Experimental
Measurements of size-segregated ambient aerosol samples
A 10-stage Micro-Orifice Uniform Deposit Impactor (MOUDI, MSP 110; MSP Corp., MN) was used to collect size-segregated ambient aerosol samples between February 3 and February 28, 2015, from an urban site (35°11′N, 126°54′E) in Gwangju, Korea. The sampling site was located on the rooftop of a three-storey building (54.3 m above sea level) in a university, about 80 m from a traffic road. Each stage of MOUDI has 50% cut sizes ranging from 0.055 to 18.0 μm in an aerodynamic diameter at a sampling flow rate of 30 l min−1. The available cut-off diameters of the MOUDI are 0.055, 0.095, 0.17, 0.32, 0.55, 1.00, 1.8, 3.1, 6.2, 9.9, and 18.0 μm. The MOUDI samples were collected on pre-baked and pre-weighed 47 mm quartz-fiber filters. A 47 mm Teflon substrate was used as a back-up filter, but the WSOC and HULIS were not analyzed due to a very low mass deposited on the back-up filter. Aerosol sampling was conducted for about forty-eight hours to provide reliable results for the quantity of size-resolved HULIS. According to the time series of hourly PM10 mass concentration (Fig. 1), an Asian dust (AD) storm event occurred between 22nd and 24th February at the site (http://www.web.kma.go.kr/eng/weather/asiandust/timeseries), so 24 h aerosol samples were collected for AD. A total of 10 sets of MOUDI samples were made, with 2 sets out of 10 consisting of AD samples.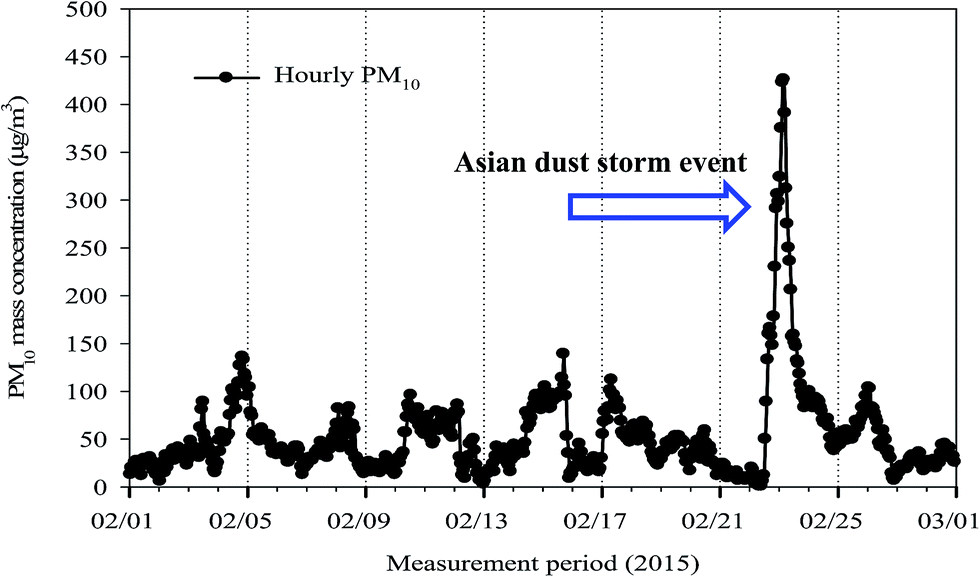 | ||
| Fig. 1 Time-series of hourly PM10 mass concentration over the study period. | ||
Ambient aerosol pollution in the city has been previously found to be a result of local emissions, regional pollution, and AD storms.15,54–58 Since the Korean peninsula is located downwind of China, anthropogenic and natural aerosols from China contribute to particulate matter pollution over the Korean peninsula. The details of the sampling site were described in our previous work,12,59 and the collected MOUDI samples were thus used to determine the mass, water-soluble ionic species, total water-soluble organic carbon (WSOC), and HULIS-C (μg C m−3) concentrations. Field blanks were also analyzed, and their background values were corrected to obtain the actual values of ambient aerosol particles at each impactor stage.
Chemical analysis of water-soluble components
Quartz-fibers and Teflon filters were weighed before and after collecting the samples by using a microbalance with a 1 μg sensitivity (Sartorius CP2P-F). The filters were conditioned for 24 h in a clean chamber that was maintained at a relative humidity of 40% at a temperature of 20 °C. The samples were weighed, and then size-segregated samples were extracted with 40 mL of ultrapure water (18.2 MΩ cm, Barnstead Nanopure ultrapure water system, Thermo Scientific, USA) in an ultrasonic bath at room temperature for 60 min. The extracts were filtered with a 0.45 μm membrane filter (Millipore) to remove insoluble materials before the analysis. The presence of eight ionic species (Na+, NH4+, K+, Ca2+, Mg2+, Cl−, NO3−, and SO42−) was determined using an ion chromatography (IC) system (Metrohm 861), and the amount of WSOC was also analyzed using a total organic carbon (TOC) analyzer (Sievers 5310C, USA). Detailed descriptions of the IC and TOC measurements were given in our previous studies.15,60,61In this study, a Hydrophilic-Lipophilic Balanced (HLB) solid phase extraction (SPE) method based on the one used by Lin et al.21 was applied to isolate the HULIS from the water extracts. HULIS was isolated in the water extracts by using a HLB SPE cartridge (Oasis HLB, 30 μm, 60 mg per cartridge, Waters, USA) by following three procedures: pretreatment of the SPE cartridge, loading the extracts on the cartridge, and concentrating the eluate. First, the cartridge was pre-treated with 1 mL of methanol (Honeywell Burdick & Jackson, HPLC grade > 99.9%) and 1 mL of ultrapure water, and this pretreatment cycle was repeated two times. It was then acidified with HCl (Daejung, Korea, purity 35.0%) to a pH ≈ 2. Second, the water extracts were acidified to a pH ≈ 2 by using HCl, and these were loaded onto the SPE cartridge. The cartridge sorbent was rinsed with 2 mL ultrapure water, and then the HULIS fraction was eluted from the SPE cartridge with 1.5 mL methanol containing 2% ammonia (w/w) (Daejung, Korea, purity 25–28%). Finally, the HULIS eluate was evaporated to dryness under a gentle stream of nitrogen and was re-dissolved in ultrapure water. The isolated HULIS fraction was then quantified using a TOC analyzer.
Recovery test of HULIS using the SPE method
Water-soluble inorganic ions, low molecular weight organic acids (e.g., oxalic acid), and sugars (e.g., levoglucosan) are hydrophilic substances that pass through the SPE resin cartridge with little retention, and those that are retained on the cartridge are strongly related to HULIS, including the hydrophobic fraction of WSOC.16,19,21,59,60 The performance of the HLB SPE isolation method used in this study was evaluated by testing the water-soluble inorganic ions in ambient aerosol particles taken from a PM2.5 low-volume sampler at our sampling site. Aqueous solutions were analyzed three times before and after passing through the SPE cartridge for eight inorganic ions by using an IC to check that they had been retained by the SPE cartridge. The concentrations of the inorganic ionic species (Cl−, NO3−, SO42−, Na+, K+, Ca2+, and Mg2+) were compared before and after the cartridge separation of water extracts for ambient aerosol samples, as shown in Fig. 2. More than 99.0% of these ionic species (NO3−, SO42−, Na+, K+, Ca2+, and Mg2+) had passed through the SPE cartridge, but a low penetration efficiency (∼85%) for Cl− was obtained. The low penetration efficiency of Cl− compared to those of the other ions is probably due to the acidification of both the SPE cartridge and the aqueous sample solution by HCl.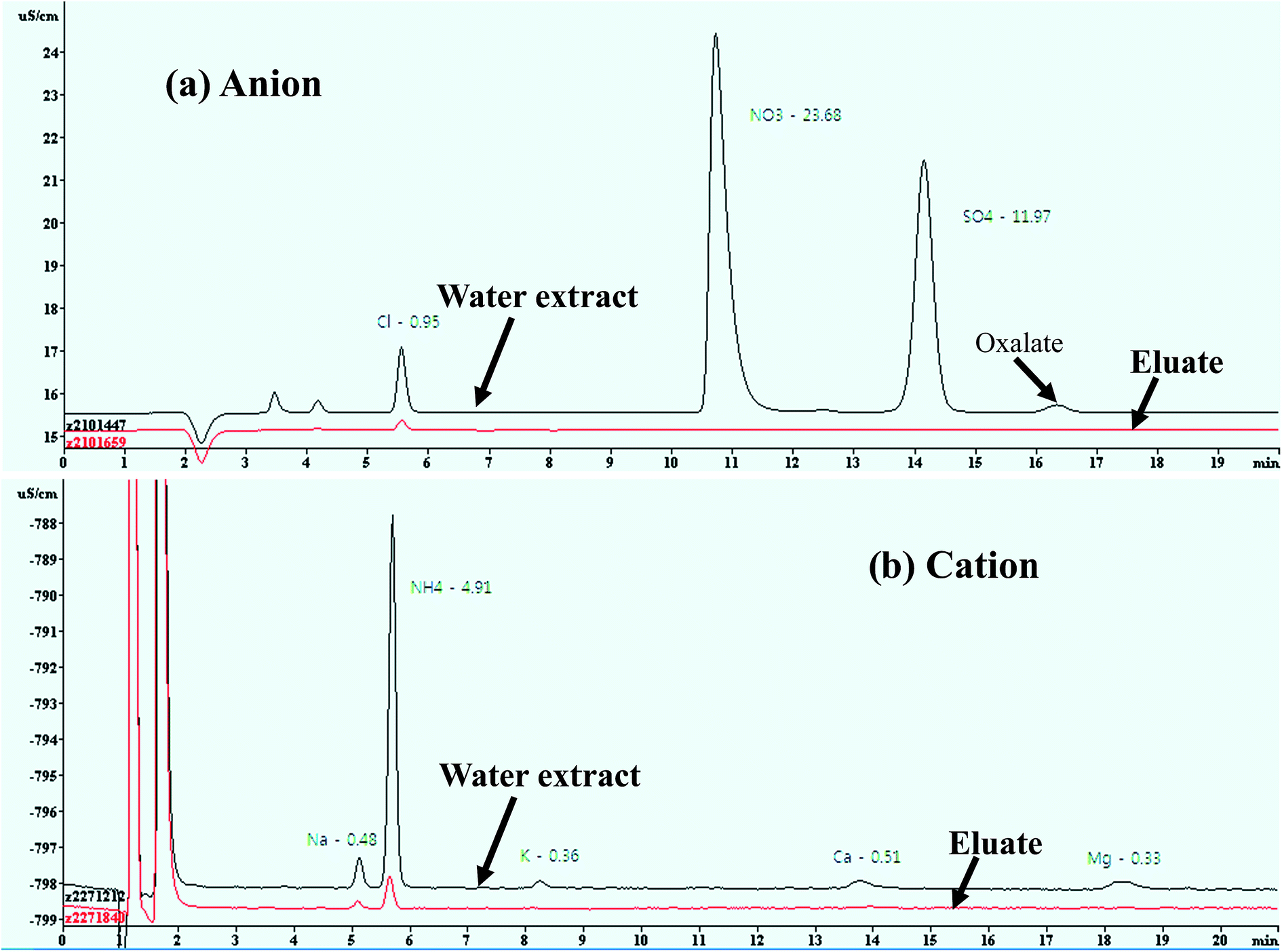 | ||
| Fig. 2 Interference of water-soluble ionic species to measured HULIS concentrations in ambient aerosol samples. | ||
In this study, the Suwannee River Fulvic Acid (SRFA) and Suwannee River Humic Acid (SRHA) HULIS standards were used to evaluate the recovery on the HLB SPE resin cartridge. The recovery experiments were carried out three times for each of the HULIS standard solution concentrations that were tested. The results of the recovery tests for the two HULIS standards are summarized in Table 1. The recovery efficiencies of the SRFA concentrations that were tested (620–5070 μg C L−1) from the HLB SPE cartridge ranged from 90.0 to 106.1%, depending on the initial concentrations. For SRHA, the recovery efficiency also varied with the initial concentration that was tested (530–3600 μg C L−1), ranging from 90.0 to 100.9%. Based on triplicate analyses of the same HULIS standard solution, the relative standard deviation for SRFA and SRHA was estimated to be <5.5% and <5.7%, respectively.
| HULIS type | Initial conc. (μg C L−1) | HULIS-C conc. (μg C L−1) | % Recovery |
|---|---|---|---|
| Suwannee River Fulvic Acid (SRFA) | 618 ± 8 | 587 ± 32 | 95.0 ± 4.2 |
| 1053 ± 40 | 1054 ± 17 | 100.1 ± 2.4 | |
| 2417 ± 38 | 2564 ± 79 | 106.1 ± 1.6 | |
| 2563 ± 6 | 2701 ± 235 | 101.3 ± 8.3 | |
| 5073 ± 131 | 4564 ± 89 | 90.0 ± 1.4 | |
| Suwannee River Humic Acid (SRHA) | 529 ± 10 | 507 ± 25 | 95.9 ± 3.1 |
| 1750 ± 20 | 1604 ± 92 | 95.1 ± 1.7 | |
| 1843 ± 6 | 1861 ± 55 | 100.9 ± 2.7 | |
| 3617 ± 50 | 3551 ± 60 | 90.0 ± 7.1 |
Results and discussion
General characteristics of water-soluble components in fine and coarse particles
As previously mentioned, Asian dust storms from China covered the entire Korean peninsula from February 22 to 24. Thus, the entire sampling period was divided into non-Asian dust (NAD) and Asian dust (AD) periods. Since MOUDI does not have a cut-off diameter of 2.5 μm, a diameter of 1.8 μm was defined as the cut-off point to distinguish fine from coarse particles. Therefore, PM1.8 and PM3.1–10, respectively, indicate fine and coarse particles. The mass fraction of organic aerosol (OA) is important when assessing the chemical mass balance closure of aerosol particles and the contribution of OA to the total PM mass. In this study, the size-resolved reconstructed PM mass from the MOUDI measurements was calculated as follows: reconstructed PM mass (μg m−3) = WSOM + Na+ + NH4+ + K+ + Ca2+ + Mg2+ + Cl− + SO42− + NO3−. Since composition data for elemental carbon (EC), organic carbon (OC), and elemental constituents are not available, the size-resolved PM mass was determined by using the concentration of water-soluble organic and inorganic species only. Based on the results reported in the literature,20,21,42 a conversion factor of ∼1.9 was applied to estimate the concentrations (μg m−3) of WSOM (water-soluble organic mass) and HULIS from the WSOC and HULIS-C concentrations in μg C m−3. With the exception of the MOUDI data collected during the AD period, a comparison of the measured and reconstructed size-resolved PM mass concentrations showed an excellent correlation with a slope of 0.74 and an R2 of 0.85 (not shown here). The low regression slope (0.74) was attributed to the absence of EC, OC, and crustal elements.The average PM10 concentrations for the NAD and AD periods were 34.2 ± 13.6 and 57.7 ± 24.4 μg m−3, respectively (Table 2). The average fine particle (PM1.8) concentration accounted for 69.2% (65.6–77.2%) of the PM10 for NAD and 53.9% (50.7–57.2%) for AD, respectively. The low fraction of the fine mode particle concentration during the AD period was a result of the higher concentration of crustal materials. The mass fractions of WSOC (as WSOM), HULIS-C (as HULIS), and secondary inorganic species (SIS = NO3− + SO42− + NH4+) in the PM1.8 are summarized in Table 2. The contributions of WSOM and HULIS to PM1.8 were approximately two times higher in the aerosol samples collected during NAD than those collected during AD, and their contributions were 23.2 and 8.0% for NAD and 12.8 and 4.2% for AD, respectively. The HULIS fraction during NAD was comparable to the results reported in other studies. The HULIS in PM2.5 from the Pearl River Delta (PRD) region in Southern China had been found to constitute 8.5–11.7% of the samples.16,21 A little difference was observed in the fraction of HULIS-C to WSOC in PM2.5 between the NAD (35.0 ± 6.9%) and AD (34.7 ± 5.2%) periods. The results from previous studies indicated that the high HULIS levels and the high fraction of HULIS-C in WSOC could be attributed to significant influences of the BB aerosol emissions.21,38,41 The HULIS-C fraction was observed to have a low value of ∼30% in fresh BB aerosols,21,41 and our results have values that are quite lower than those of the urban and suburban sites (48–60%) in the PRD region,16,21 where the influence of BB emissions is significant during the sampling period, and those in urban and rural sites (54–68%) in Europe.20,38,62 The low HULIS fractions during the NAD and AD periods in this study suggest a small contribution from BB emissions, and the concentration of SIS in fine particles was higher in the NAD (51.2 ± 3.7%) than in AD (34.4 ± 14.8%), suggesting that aerosol particles were more atmospherically processed during NAD than during AD.
| Conditions | PM10 (μg m−3) | PM1.8 (μg m−3) | PM1.8/PM10 (−) | WSOM/PM1.8 (−) | HULIS/PM1.8 (−) | SIC/PM1.8 (−) |
|---|---|---|---|---|---|---|
| a WSOM (=1.9 × WSOC): water-soluble organic mass, HULIS (=1.9 × HULIS-C), and SIC: sum of secondary inorganic components (=NO3− + SO42− + NH4+). | ||||||
| Non-Asian dust | 34.2 ± 13.6 | 24.0 ± 11.2 | 0.69 ± 0.04 | 0.23 ± 0.04 | 0.08 ± 0.02 | 0.51 ± 0.04 |
| Asian dust | 57.7 ± 24.4 | 30.5 ± 10.5 | 0.54 ± 0.05 | 0.13 ± 0.08 | 0.04 ± 0.02 | 0.34 ± 0.15 |
Size-resolved chemical composition of water-soluble components
Fig. 3 shows the average size-resolved chemical composition of water-soluble components from the NAD and AD samples. The “residue” in Fig. 3 indicates the sum of EC, water-insoluble OC, and mineral dust mass concentrations, which is the size-resolved total water-soluble matter concentration subtracted from the measured size-resolved PM concentration. The non-HULIS WSOM is calculated as the difference between WSOM and HULIS. In the case of the NAD period, the contribution of non-HULIS WSOM in the ultrafine mode (<0.1 μm) was dominant. The secondary NO3−, SO42−, and NH4+ components were dominant contributors in the fine mode (0.17–1.8 μm), followed by non-HULIS WSOM and HULIS. In the coarse mode (>3.1 μm), NO3−, SO42−, and non-HULIS WSOM were significant water-soluble components, with the exception of the unresolved chemical components. However, for the AD period, non-HULIS WSOM and SO42− were the main contributors to the ultrafine mode PM while SO42−, NO3−, NH4+, non-HULIS WSOM, and HULIS contributions were dominant in the fine mode (0.17–1.8 μm). In the coarse mode, NO3−, SO42−, non-HULIS WSOM, and Ca2+ were significant contributors. Fig. 4 shows the size-resolved relative contributions of HULIS-C to WSOC for the NAD and AD periods. The average contribution of HULIS-C to WSOC in the ambient particles was 33.5 ± 6.1% for NAD and 33.6 ± 7.3% for AD. However the highest contribution of HULIS-C to WSOC for the NAD and AD periods was observed for a particle size cut of 0.55–1.0 μm (44.9 ± 6.4%) and for a size cut of 1.8–3.1 μm (43.7 ± 7.4%), respectively. In the case of the NAD period, a high HULIS-C/WSOC existed mostly for fine particles, but the high contribution of HULIS-C for the AD period occurred for coarse mode particles as well as for fine particles due to the increased concentration of crustal materials. Further details of this issue are provided below.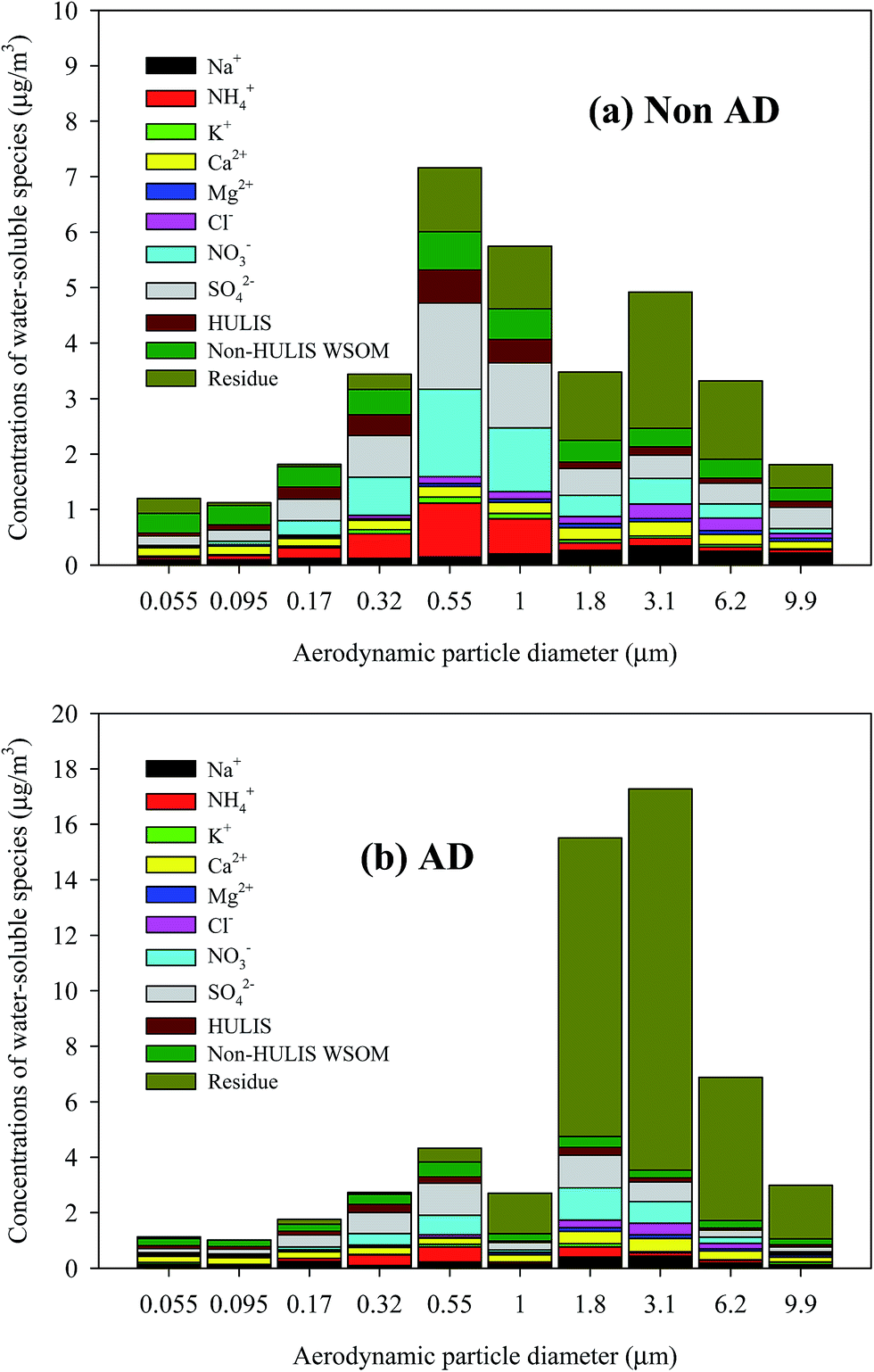 | ||
| Fig. 3 Average chemical composition of size-resolved PM for (a) NAD and (b) AD periods. | ||
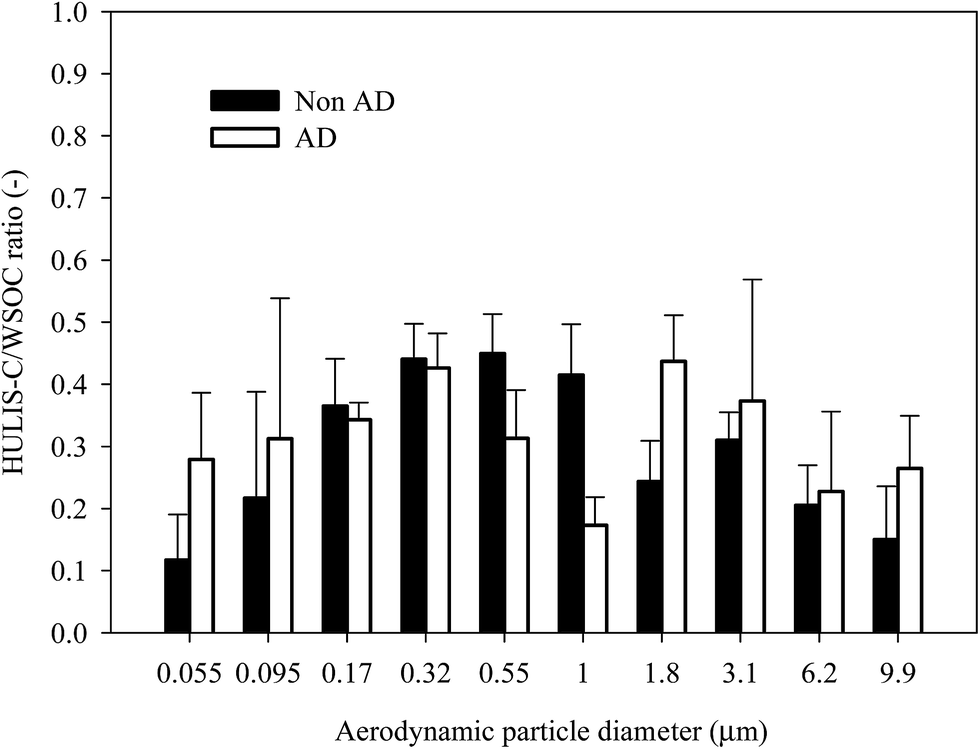 | ||
| Fig. 4 Size-resolved HULIS-C/WSOC ratio for NAD and AD periods. | ||
Size distribution of HULIS
Fig. 5 shows the size distributions of the PM, WSOC, HULIS-C, and major water-soluble inorganic species (NO3−, SO42−, NH4+, and K+) for both the NAD and AD periods. Similar size distributions in the PM and in other water-soluble chemical species were found for the NAD and AD samples in the fine particle mode, but in the coarse mode, different shapes of the size distribution were found. As shown in Fig. 5, the mass size distributions of the WSOC and HULIS-C were similar to those of NO3−, SO42−, NH4+, and K+, suggesting that they were likely produced through similar atmospheric transformation processes and/or primary emissions. For the NAD and AD periods, the WSOC and HULIS-C exhibited size distributions with a dominant droplet mode peaking at 0.55 μm or 0.31 μm. Also the NO3−, SO42−, NH4+, and K+ concentrations exhibited dominant droplet modes peaking with particle sizes of 0.55 μm. During the AD period, the PM and all chemical species peaked in the size ranging from 1.8 to 3.1 μm, as well as in the droplet mode, probably as a result of the long-range transport of anthropogenic aerosols in polluted and/or industrialized regions of China and crustal materials from the source regions of dust storms. Previous studies indicated that Asian dust storms carry significant quantities of anthropogenic aerosols during transport in addition to large amounts of crustal elements, resulting in the enrichment of anthropogenic pollution in downwind locations.55,58,63 | ||
| Fig. 5 Size distributions of PM, WSOC, HULIS-C, secondary ionic species, and K+ concentrations for NAD and AD periods. | ||
For NAD, the mass fractions of WSOM and HULIS in particle cut sizes of 0.55 and 0.32–1.0 μm accounted for 19.0 and 45.7% of the total WSOM, respectively, and 25.1 and 58.4% of the total HULIS, respectively. NO3−, SO42−, and NH4+ contributions were higher than WSOC and HULIS, accounting for 30.7, 25.1, and 34.0% of their total concentration at 0.55 μm, respectively, and for 66.6, 56.2, and 71.7% at 0.32–1.0 μm, respectively. When compared to the contribution of water-soluble species concentrations collected from NAD samples, the contributions to AD samples were observed to be rather low for fine mode particles. For the AD, the size resolved mass fraction of WSOM to the total WSOM contributed 15.6 and 35.4% at 0.55 and 0.32–1.0 μm, respectively. The HULIS at 0.55 and 0.32–1.0 μm contributed 14.2 and 35.8% of the total HULIS, respectively. NO3−, SO42−, and NH4+ contributions to their total concentrations were 19.3, 21.1, and 28.1% at 0.55 μm and 32.9, 39.9, and 51.3% at 0.32–1.0 μm, respectively.
Resolving the sources of fine and coarse mode HULIS
Fig. 6 shows the correlation of WSOC and HULIS-C with water-soluble inorganic species for all size-resolved measurements during the NAD and AD periods. HULIS-C exhibited a very high correlation (R2 = 0.73–0.94) with WSOC for two periods, indicating their similar chemical characteristics. However, a higher regression slope was obtained for HULIS-C/WSOC from the NAD samples (0.59) than from the AD samples (0.39), suggesting additional sources and/or further atmospheric processing of HULIS in size-segregated aerosol samples measured during NAD. The measurement data indicate that the HULIS for the NAD period was strongly associated with the biomass burning (BB) emission marker (K+) and the secondary inorganic species (NO3−, SO42−, and NH4+), suggesting that BB emissions and secondary formation processes could be important sources of HULIS during NAD. However the HULIS for the AD period was associated with secondary ionic species (NO3−, SO42−, and NH4+), suggesting its association with secondary formation processes.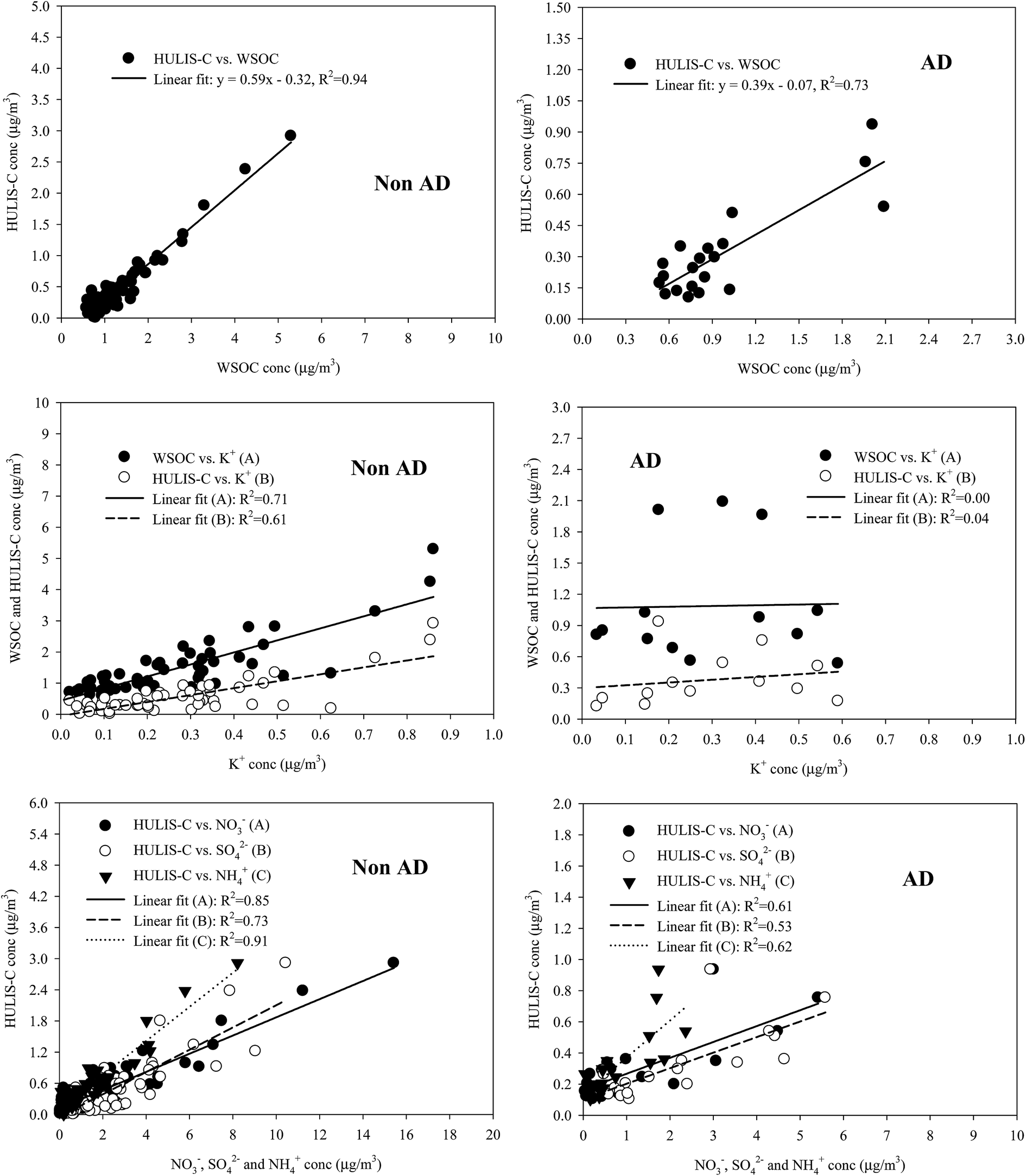 | ||
| Fig. 6 Correlations of HULIS-C and WSOC with K+, NO3−, SO42−, and NH4+ for NAD and AD periods. | ||
To further identify HULIS sources in fine and coarse modes, the relationships between HULIS-C and water-soluble inorganic ions (Na+, Cl−, K+, Ca2+, SO42−, NO3−, and NH4+) were examined, and the results are summarized in Table 3. For the NAD period, HULIS-C was strongly correlated with NH4+, NO3−, SO42−, and K+ in the fine particle mode at an R2 of 0.92, 0.88, 0.75 and 0.78, but was weakly correlated with water-soluble ions in the coarse mode. The droplet modes of SO42− and NO3− are known to be predominantly produced through aqueous-phase reactions of SO2 and NO2 in clouds or in aerosol surfaces.64 The strong correlations of HULIS-C with NO3−, SO42−, and K+ in the fine mode for the NAD indicates that the fine mode HULIS was likely produced from secondary transformation processes similar to those of NO3− and SO42− (i.e., aqueous-phase oxidation and/or cloud processing), as well as from biomass burning emissions.21,41,45,53 However, it is hard to identify the sources of low HULIS in the coarse mode during NAD due to poor correlations (R2 = 0.00–0.34) of HULIS-C with water-soluble ionic species in the coarse mode.
| Dependent variable | NAD period | AD period | |||
|---|---|---|---|---|---|
| PM1.8 | PM3.1–10 | PM1.8 | PM3.1–10 | ||
| a Superscripts * and ** are significant at p values of < 0.05 and < 0.01, respectively. | |||||
| HULIS-C | WSOC | 0.98 ** | 0.57 | 0.72 ** | 0.72 ** |
| Na+ | 0.01 | 0.00 | 0.14 | 0.84 ** | |
| Cl− | 0.28 | 0.05 | 0.24 | 0.83 ** | |
| K+ | 0.78 ** | 0.04 | 0.25 | 0.04 | |
| Ca2+ | 0.08 | 0.34 | 0.02 | 0.67 * | |
| NH4+ | 0.92 ** | 0.08 | 0.56 ** | 0.36 | |
| NO3− | 0.88 ** | 0.19 | 0.66 ** | 0.79 ** | |
| SO42− | 0.75 ** | 0.06 | 0.48 ** | 0.63 * | |
For the AD period, fine mode HULIS-C was also correlated with secondary NH4+, NO3−, and SO42− at an R2 of 0.56, 0.66, and 0.48, respectively, while the correlation between HULIS-C and K+ was not significant, suggesting that fine mode HULIS is mainly associated with secondary formation processes similar to those of NO3− and SO42−. However, unlike the HULIS-C observed during the NAD, coarse mode HULIS-C observed during AD was strongly correlated with Na+, Cl−, Ca2+, NO3−, and SO42− at an R2 of 0.84, 0.83, 0.67, 0.79, and 0.63, respectively. The coarse mode Na+ and Cl− are mostly from marine aerosols, and the coarse mode Ca2+ is derived from soil CaCO3 particles. Once they are emitted in the atmosphere, the reaction of NaCl and CaCO3 with HNO3 and H2SO4 may occur during transport.45,65 Previous studies indicated that the coarse mode WSOC is associated with either sea salt particles (Na+) or soil-related particles (Ca2+).46,59 In the coarse mode, strong correlations of Na+ (R2 = 0.88, p < 0.01) and Ca2+ (R2 = 0.76, p < 0.01) with NO3− and good correlations of Na+ (R2 = 0.73, p < 0.01) and Ca2+ (R2 = 0.60, p < 0.05) with SO42− suggest that the coarse mode NO3− and SO42− were likely associated with reactions of NaCl(s) and CaCO3(s) with HNO3(g) and H2SO4(g). As a consequence, the results from these correlations and from previous studies suggest that the coarse mode HULIS during AD was possibly condensed on the surface of the NaNO3 and Na2SO4 and/or the Ca(NO3)2 and CaSO4 particles.
Summary and conclusion
Forty-eight hour size-resolved measurements of ambient aerosol particles were made at an urban site during the period from February 3 through 28, 2015 in order to investigate the size distribution and sources of HULIS. The size-resolved HULIS from water extracts was isolated by HLB solid phase extraction and was quantified with a TOC analyzer. The HULIS-C concentration in fine particles (PM1.8) accounted for 35 ± 7% and 35 ± 5% of the WSOC concentration during the NAD and AD periods, respectively, with the highest HULIS-C/WSOC ratio in the particle cut sizes from 0.55 to 1.0 μm for the NAD (45 ± 6%) and 1.8 to 3.1 μm for AD (44 ± 7%). The size distribution of HULIS was similar to those of PM, WSOC, NO3−, SO42−, NH4+, and K+. HULIS exhibited a dominant droplet mode peaking at 0.55 μm during the NAD period and showed a bimodal size distribution peaking at both 0.32 and 1.8 μm during the AD period.The correlation of the size-resolved HULIS with other water-soluble ionic species strongly indicates that fine mode HULIS is produced by formation processes of SOA similar to those of secondary ionic species and primary BB emissions during NAD but is directly associated with SOA formation processes during the AD period. In the coarse mode, strong correlations of HULIS with Na+, Cl−, Ca2+, NO3−, and SO42− during the AD period suggest that the coarse mode HULIS during the AD period was likely to condense on the surface of sea-salt particles (NaNO3 and Na2SO4) and/or soil-related particles [Ca(NO3)2 and CaSO4]. However, the processes that could lead to the small presence of HULIS in the coarse mode during the NAD period are not known.
Acknowledgements
This research was supported by the Basic Science Research Programs through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2014R1A1A4A01003896).References
- S. S. Park and S. Y. Cho, Atmos. Environ., 2011, 45(1), 60–72 CrossRef CAS.
- P. Saxena, L. M. Hildemann, P. H. McMurry and J. H. Seinfeld, J. Geophys. Res., 1995, 100, 18755–18770 CrossRef.
- M. C. Facchini, et al. , Nature, 1999, 401, 257–259 CrossRef CAS.
- Y. Miyazaki, Y. Kondo, N. Takegawa, Y. Komazaki, K. Kawamura, M. Mochida, K. Okuzawa and R. J. Weber, J. Geophys. Res., 2006, 111, D23206, DOI:10.1029/2006jd007125.
- S. Zappoli, et al. , Atmos. Environ., 1999, 33, 2733–2743 CrossRef CAS.
- M. P. Fraser, G. R. Cass and B. R. T. Simoneit, Atmos. Environ., 1999, 33, 2715–2724 CrossRef CAS.
- J. H. Seinfeld and S. N. Pandis, Atmospheric Chemistry and Physics: from Air Pollution to Climate Change, John Wiley & Sons, Inc., New York, USA, 2nd edn, 2006 Search PubMed.
- A. Hecobian, X. Zhang, M. Zheng, N. Frank, E. S. Edgerton and R. J. Weber, Atmos. Chem. Phys., 2010, 10, 5965–5977 CAS.
- X. Zhang, A. Hecobian, M. Zheng, N. H. Frank and R. J. Weber, Atmos. Chem. Phys., 2010, 10, 6839–6853 CAS.
- Z. Du, et al. , Atmos. Environ., 2014, 92, 514–521 CrossRef CAS.
- G. H. Yu, S. Y. Cho, M. S. Bae and S. S. Park, Environ. Sci.: Processes Impacts, 2014, 16, 1726–1736 CAS.
- S. S. Park, S. Y. Cho and M. S. Bae, Sci. Total Environ., 2015, 533, 410–421 CrossRef CAS PubMed.
- K. F. Ho, et al. , Atmos. Chem. Phys., 2006, 6, 4569–4576 CrossRef CAS.
- S. Saarikoski, et al. , Atmos. Chem. Phys., 2008, 8, 6281–6295 CrossRef CAS.
- S. Y. Cho and S. S. Park, Environ. Sci.: Processes Impacts, 2013, 15(2), 524–534 CAS.
- B. Y. Kuang, P. Lin, X. H. H. Huang and J. Z. Yu, Atmos. Chem. Phys., 2015, 15, 1995–2008 CAS.
- R. D. B. Duarte and A. C. Duarte, J. Atmos. Chem., 2005, 51, 79–93 CrossRef CAS.
- E. R. Graber and Y. Rudich, Atmos. Chem. Phys., 2006, 6, 729–753 CrossRef CAS.
- A. P. Sullivan and R. J. Weber, J. Geophys. Res., 2006, 111, D05314, DOI:10.1029/2005jd006485.
- G. Kiss, B. Varga, I. Galambos and I. Ganszky, J. Geophys. Res., 2002, 107(D21), 8339, DOI:10.1029/2001jd000603.
- P. Lin, X. F. Huang, L. Y. He and J. Z. Yu, J. Aerosol Sci., 2010, 41, 74–87 CrossRef CAS.
- S. Decesari, M. C. Facchini, S. Fuzzi and E. Tagliavini, J. Geophys. Res., 2000, 105, 1481–1489, DOI:10.1029/1999jd900950.
- M. C. Facchini, et al. , Atmos. Environ., 2000, 34, 4853–4857 CrossRef CAS.
- G. Kiss, E. Tombacz and H. C. Hansson, J. Atmos. Chem., 2005, 50, 279–294 CrossRef CAS.
- P. Sannigrahi, A. P. Sullivan, R. J. Weber and E. D. Ingall, Environ. Sci. Technol., 2006, 40, 666–672 CrossRef CAS PubMed.
- R. Duarte, E. Santos, C. A. Pio and A. C. Duarte, Atmos. Environ., 2007, 41, 8100–8113 CrossRef CAS.
- T. Reemtsma, et al. , Anal. Chem., 2006, 78, 8299–8304 CrossRef CAS PubMed.
- K. E. Altieri, B. J. Turpin and S. P. Seitzinger, Atmos. Chem. Phys., 2009, 9, 2533–2542 CAS.
- L. R. Mazzoleni, et al. , Environ. Sci. Technol., 2010, 44, 3690–3697 CrossRef CAS PubMed.
- M. Gysel, et al. , Atmos. Chem. Phys., 2004, 4, 35–50 CrossRef CAS.
- E. Dinar, et al. , Atmos. Chem. Phys., 2006, 6, 2465–2481 CrossRef CAS.
- A. Hoffer, et al. , Atmos. Chem. Phys., 2006, 6, 3563–3570 CrossRef CAS.
- H. Lukács, et al. , J. Geophys. Res., 2007, 112, D23S18, DOI:10.1029/2006jd008151.
- M. Moonshine, Y. Rudich, S. Katsman and E. R. Graber, Geophys. Res. Lett., 2008, 35, L20807 CrossRef.
- V. Verma, et al. , Environ. Sci. Technol., 2012, 46, 11384–11392 CrossRef CAS PubMed.
- T. Feczko, et al. , J. Geophys. Res., 2007, 112, D23S10, DOI:10.1029/2006jd008331.
- P. Lin, G. Engling and J. Z. Yu, Atmos. Chem. Phys., 2010, 10, 6487–6500, DOI:10.5194/acp-10-6487-2010.
- I. Salma, T. Mészáros, W. Maenhaut, E. Vass and Z. Majer, Atmos. Chem. Phys., 2010, 10, 1315–1327 CrossRef CAS.
- S. C. Son, M. S. Bae and S. Park, J. Korean Soc. Atmos. Environ., 2015, 31(3), 239–254 CrossRef.
- F. Cavalli, et al. , J. Geophys. Res., 2004, 109, D24215 CrossRef.
- O. L. Mayol-Bracero, P. Guyon, B. Graham, G. Roberts, M. O. Andreae, S. Decesari, M. C. Facchini, S. Fuzzi and P. Artaxoet, J. Geophys. Res., 2002, 107(D20), 8091, DOI:10.1029/2001jd000522.
- Z. Krivácsy, et al. , J. Atmos. Chem., 2001, 39, 235–259 CrossRef.
- Z. Krivácsy, et al. , Atmos. Res., 2008, 87, 1–12 CrossRef.
- S. Decesari, M. C. Facchini, S. Fuzzi, G. B. McFiggans, H. Coe and K. N. Bower, Atmos. Environ., 2005, 39, 211–222 CrossRef CAS.
- X. F. Huang, J. Z. Yu, L. Y. He and Z. Yuan, J. Geophys. Res., 2006, 111, D22212 CrossRef.
- H. J. Timonen, et al. , Atmos. Chem. Phys., 2008, 8, 5635–5647 CrossRef CAS.
- Y. L. Zhao and Y. Gao, Atmos. Environ., 2008, 42, 4063–4078 CrossRef CAS.
- G. Wang, K. Kawamura, N. Umemoto, M. Xie, S. Hu and Z. Wang, J. Geophys. Res., 2009, 114, D19208, DOI:10.1029/2008jd011390.
- S. S. Park and D. M. Shin, Part. Aerosol Res., 2013, 9(1), 7–21 CrossRef.
- A. K. Frey, et al. , Boreal Environ. Res., 2009, 14, 255–271 CAS.
- M. S. Bae and S. S. Park, Asian J. Atmos. Environ., 2013, 7(2), 95–104 CrossRef CAS.
- S. S. Park, S. Y. Sim, M. S. Bae and J. J. Schauer, Atmos. Environ., 2013, 73, 62–72 CrossRef CAS.
- K. Saarnio, et al. , Sci. Total Environ., 2010, 408, 2527–2542 CrossRef CAS PubMed.
- S. S. Park, M. S. Bae, J. J. Schauer, Y. J. Kim, S. Y. Cho and S. J. Kim, Atmos. Environ., 2006, 40, 4182–4198 CrossRef CAS.
- S. S. Park, Y. J. Kim, S. Y. Cho and S. J. Kim, J. Air Waste Manage. Assoc., 2007, 57, 434–443 CAS.
- H. L. Lee, S. S. Park, K. W. Kim and Y. J. Kim, Atmos. Res., 2008, 88, 199–211 CrossRef CAS.
- J. Jung and Y. J. Kim, J. Geophys. Res., 2011, 116, D02206 CrossRef.
- S. S. Park and S. Y. Cho, Aerosol Air Qual. Res., 2013, 13, 1019–1033 CAS.
- S. S. Park and J. H. Kim, Atmos. Environ., 2014, 94, 134–143 CrossRef CAS.
- S. S. Park, J. U. Jeong and S. Y. Cho, Asian J. Atmos. Environ., 2012, 6, 67–72 CrossRef CAS.
- S. S. Park, J. H. Kim and J. U. Jeong, J. Environ. Monit., 2012, 14, 224–232 RSC.
- I. Salma, R. Ocskay, X. Chi and W. Maenhaut, Atmos. Environ., 2007, 41, 4106–4118 CrossRef CAS.
- S. C. Son and S. S. Park, Environ. Sci.: Processes Impacts, 2015, 17, 561–569 CAS.
- X. Wang, et al. , Atmos. Environ., 2012, 63, 68–76 CrossRef CAS.
- A. Eldering, et al. , Atmos. Environ., 1991, 25A, 2091–2102 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |