
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Phototransformation of pesticides in prairie potholes: effect of dissolved organic matter in triplet-induced oxidation†
M. Ekrem
Karpuzcu‡
*ab,
Andrew J.
McCabe‡
a and
William A.
Arnold
a
aUniversity of Minnesota, Department of Civil, Environmental, and Geo-Engineering, Minneapolis, MN 55455, USA. E-mail: karpuzcu@itu.edu.tr
bIstanbul Technical University, Department of Environmental Engineering, Maslak 34469, Istanbul, Turkey
First published on 8th January 2016
Abstract
Photochemical reactions involving a variety of photosensitizers contribute to the abiotic transformation of pesticides in prairie pothole lakes (PPLs). Despite the fact that triplet excited state dissolved organic matter (DOM) enhances phototransformation of pesticides by acting as a photosensitizer, it may also decrease the overall phototransformation rate through various mechanisms. In this study, the effect of DOM on the phototransformation of four commonly applied pesticides in four different PPL waters was investigated under simulated sunlight using photoexcited benzophenone-4-carboxylate as the oxidant with DOM serving as an anti-oxidant. For atrazine and mesotrione, a decrease in phototransformation rates was observed, while phototransformations of metolachlor and isoproturon were not affected by DOM inhibition. Phototransformation rates and the extent of inhibition/enhancement by DOM varied spatially and temporally across the wetlands studied. Characterization of DOM from the sites and different seasons suggested that the DOM type and variations in the DOM structure are important factors controlling phototransformation rates of pesticides in PPLs.
Environmental impactThe role of DOM as a photosensitizer in phototransformation of organic contaminants has been well known, but the potential of DOM as an antioxidant during photolysis has only recently been explored. By investigating whole prairie pothole waters and their seasonal variations, this study tries to directly address the environmental relevance of what so far has been a laboratory phenomenon. The inhibitory effect of DOM is especially important for the attenuation of pesticides that are recalcitrant to biotransformation and are inefficiently depleted by direct phototransformation. |
1. Introduction
Prairie pothole lakes (PPLs) are small depressional wetlands scattered across the northern Great Plains of the United States and Canada.1 They serve as wildlife habitat and a major component of regional hydrology.2,3 The Prairie Pothole Region (PPR) of North America is dominated by agricultural land uses4 and wetlands in the PPR have been severely degraded by agricultural practices, including drainage of potholes, sedimentation, and the application of agricultural pesticides and fertilizers.1 More than half of the original wetland area in the PPR has been drained for agricultural use.5,6 Remaining PPLs exhibit a scattered distribution within the agricultural landscape and are often artificially connected to rivers and streams through drainages or ditch networks.7–9 Thus, nonpoint source pollutants such as nutrients and pesticides from adjacent farmland that enter PPLs may reach downstream water bodies and negatively impact water quality as well as the overall biodiversity and productivity of the PPR.10–12 A number of pesticides, including but not limited to atrazine, metolachlor, bentazon, diuron and trifluralin, have been detected in the PPR aquatic ecosystems.13–15 There is a need for a better understanding of the fate and transformation mechanisms of pesticides in PPL systems to mitigate their environmental impacts.Previous studies reported that photochemical reactions play an important role in the abiotic transformation of a wide array of pesticides via direct and/or indirect pathways.16–19 Direct photolysis occurs when light energy is directly absorbed by a contaminant resulting in chemical transformation.20 Indirect photolysis occurs when another chemical species (photosensitizer) absorbs light and becomes electronically excited and consequently reacts directly with the contaminant of interest or produces “photochemically produced reactive intermediates” (PPRIs) which are capable of transforming the target compound. A variety of photosensitizers, including dissolved organic matter (DOM), nitrate/nitrite, and iron ions, as well as secondary species such as bicarbonate and halide ions are present in natural waters.20
DOM plays a critical role in aquatic health because it serves as an important component of the carbon cycle and plays a critical role in the transformation of aquatic contaminants, among several other roles. Inputs of water from agricultural fields,21 storm water,22,23 or wastewater effluent24 may affect the quantity and composition of the dissolved organic matter present. While DOM increases the phototransformation rates of pesticides by acting as a photosensitizer as mentioned above, it may also decrease the direct phototransformation rate by attenuating light as it proceeds through the water column (i.e., light screening), decrease indirect photolysis rates by quenching reactive species (PPRIs), or lead to reformation of the parent compound by acting as an antioxidant.25,26 The antioxidant pathways were first explored by Canonica and Laubscher.25 In their proposed model, the organic contaminant P reacts with an oxidizing radical denoted by Rad˙ or an excited triplet state denoted by 3Sens* to form a radical cation P˙+. Subsequently, P˙+ undergoes parallel reactions where it is either irreversibly oxidized to a product, Pox, or reduced back to its parent compound P by DOM (the antioxidant) forming an oxidized DOM radical, DOM˙+ (eqn (1)–(4)). The formed radical cation may further react with oxygen to form peroxy radicals.27
| Rad˙ + P → Rad− + P˙+ | (1) |
| 3Sens* + P → Sens˙− + P˙+ | (2) |
| P˙+ → Pox | (3) |
| P˙+ + DOM → P + DOM˙+ | (4) |
Thus, reduction of P˙+ by DOM would decrease the overall rate of contaminant transformation. It has been suggested that the antioxidant properties of DOM manifest through a variety of organic moieties that DOM contains, such as phenolic groups.26,28
Most PPLs have large surface areas with relatively shallow depths (typically < 1.5 m), which greatly increases the potential of sunlight to penetrate the water column.10,29 Another feature that makes the surface waters of PPLs suitable for photosensitized reactions is that they often contain high levels of DOM.17 It has been previously shown that Suwannee River fulvic acid, used as a reference DOM, exhibited an inhibitory effect on the photolytic reactions of some organic contaminants such as anilines, cyanophenol and the antibiotic drug trimethoprim, while it enhanced the overall phototransformation of some other compounds such as isoproturon and 4-methylphenol.25 The main purposes of this study were to understand the potential of DOM as an antioxidant during phototransformation of pesticides in PPL waters and to determine the effect of season and DOM properties on the extent of inhibition or enhancement of phototransformation.
2. Materials and methods
2.1 Study area and sampling locations
The 92 ha Cottonwood Lake study area is located in south-central North Dakota near Jamestown (Fig. 1) and has been the focus of biological and hydrological research since the U.S. Fish and Wildlife Service purchased the site in 1963.30 The study area contains approximately 17 ha of wetlands which are divided among 18 individual basins that have been numbered P1 through P9 and T1 through T9. The wetlands denoted by the letter “P” represent all semi-permanently flooded wetlands that only go dry during periods of drought, while the temporal “T” wetlands have a seasonal water regime. They have a wet–dry cycle each year unless there are extraordinary high precipitation events. The temporary wetlands do not have a deep marsh zone and typically consist of shallow marsh vegetation surrounded by a band of wet meadow and a band of low prairie.30 Depending on local precipitation, topography, and soil hydraulic conductivity, the wetlands may be groundwater recharge, flow-through, or discharge wetlands.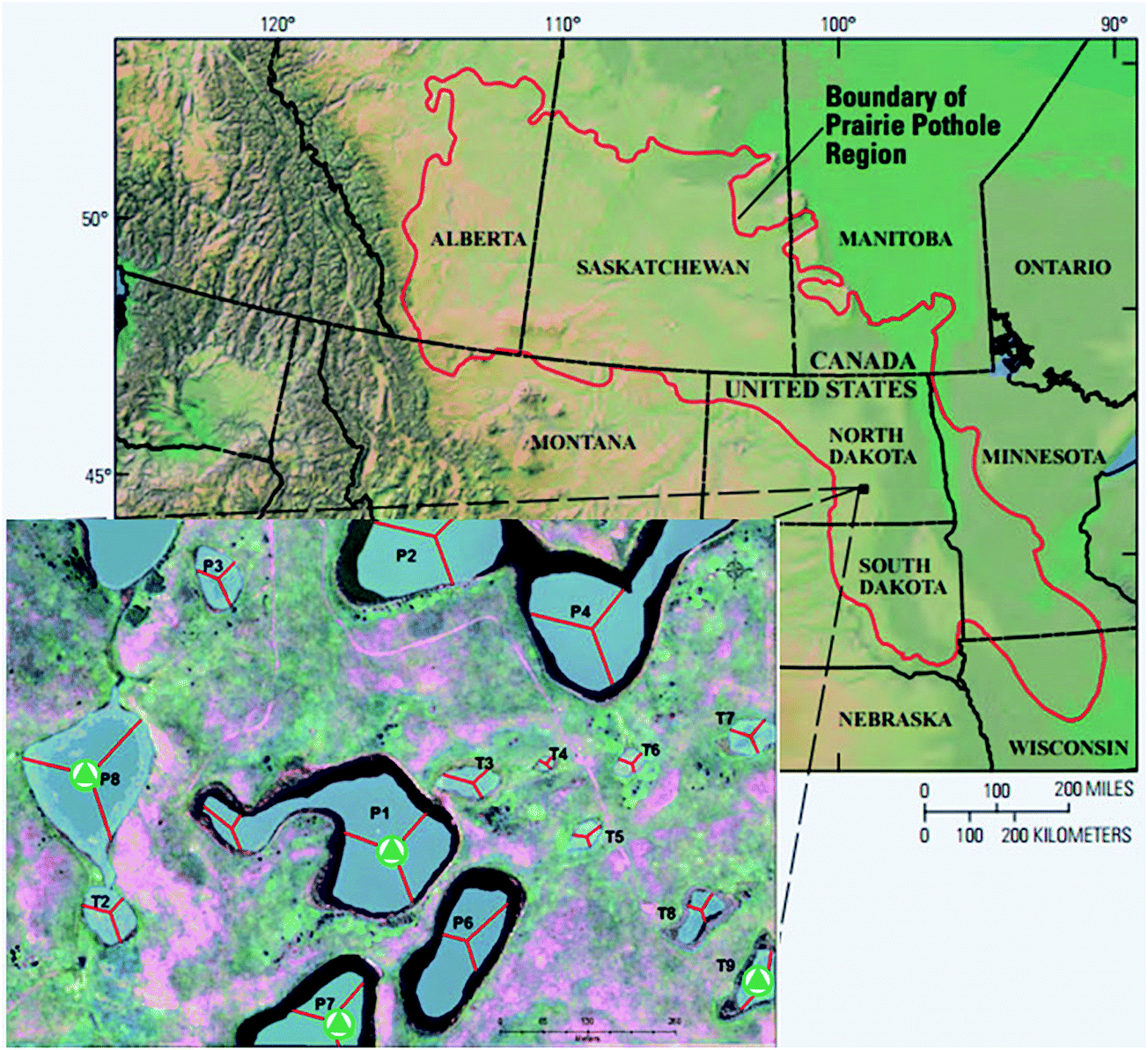 | ||
| Fig. 1 Map of the prairie pothole region (PPR) of North America (map adapted from USGS). The inset map shows the Cottonwood Lake study area. The sampling sites are marked with triangles. | ||
2.2 Sample collection and analysis
Prairie pothole surface waters were collected from four wetlands (Fig. 1) across the hydrologic gradient, T9 (recharge), P7 (flow-through), and P1 and P8 (discharge). Samples were collected in pre-combusted (550 °C for ≥5 hours) glass bottles, transported on ice to the University of Minnesota, pre-filtered through pre-combusted 0.7 μm glass-fiber filters, and subsequently filter-sterilized through 0.22 μm mixed cellulose membrane filters (GSWP, Millipore Corp.). The samples were stored in the dark at 4 °C until use. The water samples used were collected in the summer (July) and fall (November) of 2013.2.3 Experimental design
The four target pesticides were atrazine, metolachlor, isoproturon, and mesotrione, which are all used in the PPR and represent a range of pesticide classes. Additional information about the chemical reagents and their sources is given in the ESI.† All reactions were carried out in either filter sterilized prairie pothole wetland water (pH ranging from 8.10 to 9.70 (Table 1)) or in 10 mM borate buffer (pH 8.5). The excited triplet state of benzophenone-4-carboxylate (CBBP) was chosen as a model oxidant due to its high standard one-electron reduction potential (E0 = 1.83 V) and its capability of oxidizing a range of different compounds.25 Using CBBP as the primary oxidant allows DOM to serve as the antioxidant.| Site/season | pH | DOC (mg L−1) | SUVA254 (L mg−1 m−1) | S 250–500 (nm−1) | S 275–295 (nm−1) | S 350–400 (nm−1) | S R |
E
2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :E3 :E3 |
E
4:E6 |
Fluorescence index (FI) |
|---|---|---|---|---|---|---|---|---|---|---|
| T9 summer | 9.70 | 36.86 | 7.70 | 0.0181 | 0.0184 | 0.0192 | 0.96 | 6.87 | 15.67 | 1.57 |
| T9 fall | 8.10 | 20.70 | 6.53 | 0.0183 | 0.0187 | 0.0187 | 1.00 | 7.22 | 20.11 | 1.57 |
| P7 summer | 8.94 | 32.59 | 4.17 | 0.0201 | 0.0244 | 0.0208 | 1.18 | 11.46 | 8.00 | 1.63 |
| P7 fall | 8.72 | 29.78 | 4.31 | 0.0200 | 0.0241 | 0.0196 | 1.23 | 10.85 | 12.36 | 1.65 |
| P1 summer | 8.75 | 38.41 | 4.36 | 0.0236 | 0.0276 | 0.0212 | 1.30 | 14.66 | 8.14 | 1.62 |
| P1 fall | 8.99 | 35.26 | 4.29 | 0.0236 | 0.0284 | 0.0212 | 1.34 | 14.65 | 7.25 | 1.61 |
| P8 summer | 8.94 | 23.45 | 5.96 | 0.0197 | 0.0217 | 0.0196 | 1.11 | 8.60 | 11.63 | 1.59 |
| P8 fall | 8.24 | 21.85 | 5.39 | 0.0209 | 0.0227 | 0.0203 | 1.12 | 9.67 | 10.85 | 1.63 |
The photolytic reactions were conducted in an Atlas Suntest CPS+ solar simulator equipped with a 1500 W xenon arc lamp. The lamp was fitted with a UV-Suprax optical filter (passing wavelengths ranging from 290 to 800 nm), and the light intensity was set at 765 W m−2. The temperature of the reaction solutions was kept below 30 °C by circulating ambient air through the reaction chamber. The phototransformation experiments were carried out using 10 mL quartz test tubes. Test tubes were held at approximately a 30° angle from horizontal. All reactions were conducted in duplicate. Beside 10 μM of the target pesticide, aqueous solutions contained: treatment 1: borate buffer (blank); treatment 2: 40 μM CBBP and borate buffer; treatment 3: PPL water; treatment 4: PPL water and 40 μM CBBP; dark control: the same as treatment 4, but the test tubes were wrapped with aluminum foil to block light. These treatments follow the protocols outlined previously.25
Pesticide degradation was quantified with an Agilent 1100 HPLC equipped with a multiwavelength UV absorbance detector. A Discovery RP-amide C16 column (15 cm × 4.6 mm; 5 μm) was used for chromatographic separation. More technical details about the HPLC analysis are provided in the ESI.† Prior to irradiation experiments, UV-visible absorption spectra of the PPL water samples, target pesticides and CBBP were measured with a Shimadzu UV-1601PC spectrometer (Fig. S21, ESI†). Spectral slopes (S) were determined by fitting the spectra between 250 and 500 nm (corrected with absorbance at 700 nm) to an exponential trend line using Microsoft Excel®. Fluorescence spectra were collected using an Aqualog UV-800C with a 150 W ozone-free Xe source (Horiba, Jobin Yvon). Fluorescence indexes were computed from corrected spectra as the ratio of emission intensities at 470 nm to 520 nm at an excitation wavelength of 370 nm.31,32 Dissolved organic carbon (DOC) was measured on a Shimadzu TOC-L total organic carbon analyzer. Samples were acidified and purged with carbon-free air to remove any inorganic carbon prior to analysis. Iron(III) concentrations in the PPL samples were measured following a slightly modified version of the Ferrozine method of Viollier et al. (2000).33
2.4 Kinetics calculations
A first order decay model was used to estimate the observed phototransformation rate constants, kobs, (s−1) of the pesticides over the irradiation period (eqn (5)), where Ct is the pesticide concentration at a given time, C0 is the initial pesticide concentration and t is time (s).| Ct = C0e−kobst | (5) |
The rate constant for depletion of the target pesticide in the presence of both CBBP and DOM from the PPL water (treatment 4), kCB,PPL, was corrected by subtracting the rate constant for depletion in the presence of DOM from the PPL water only (treatment 3), kPPL, to compensate for pesticide loss due to DOM-induced phototransformation. When using kPPL in eqn (6), light screening by CBBP was taken into account by multiplying the kPPL value with a light screening factor, SDOM,CB (Table S2, ESI†). The rate constant for depletion of the target pesticide in the presence of CBBP (treatment 2), kCB, was corrected by subtracting the rate constant of the blank (treatment 1), kdirect, to compensate for the degradation caused by direct phototransformation rather than the presence of CBBP. When using kdirect in eqn (7), light screening by CBBP was taken into account and the kdirect value was corrected by multiplying it with a light screening factor, SCB (Table S2, ESI†). Screening factors were calculated as a ratio of the rates of light absorption in the presence and absence of the species responsible for screening. The details of light screening correction calculations are presented in the ESI.† Finally, the “Inhibition Factor”, IF,25 was defined as the ratio of the corrected rate constant in the presence of both CBBP and DOM, kcorr,CB,PPL, and the corrected rate constant in the presence of only CBBP, kcorr,CB, as described by eqn (6)–(8). When using kcorr,CB in eqn (8), light screening by DOM was taken into account by multiplying the kcorr,CB value with the light screening factor SCB,DOM (Table S2, ESI†). It should be noted that an IF value >1 indicates enhancement of phototransformation, while an IF value <1 indicates inhibition.
| kcorr,CB,PPL = kCB,PPL − kPPL | (6) |
| kcorr,CB = kCB − kdirect | (7) |
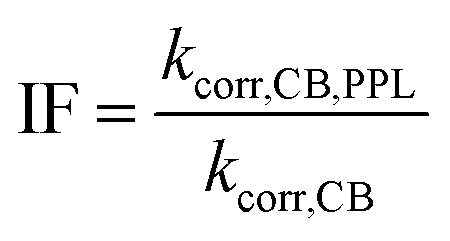 | (8) |
2.5 Statistical analysis
Nonlinear regression analysis of spectral slope calculations was performed using Matlab 8.0 and Statistics Toolbox 8.1 (The Mathworks, Inc., Natick, Massachusetts, U.S.). All other statistical analyses including Pearson correlation analyses, linear regression analyses, calculation of confidence intervals and standard errors of the mean, were performed using version 22 of the IBM SPSS software package (IBM, Armonk, New York, U.S.).3. Results and discussion
Representative results for atrazine in the four photolysis treatments in T9, P7, P1, and P8 waters from the summer season are shown in Fig. 2. The plots for the other compounds and seasons are in the ESI.†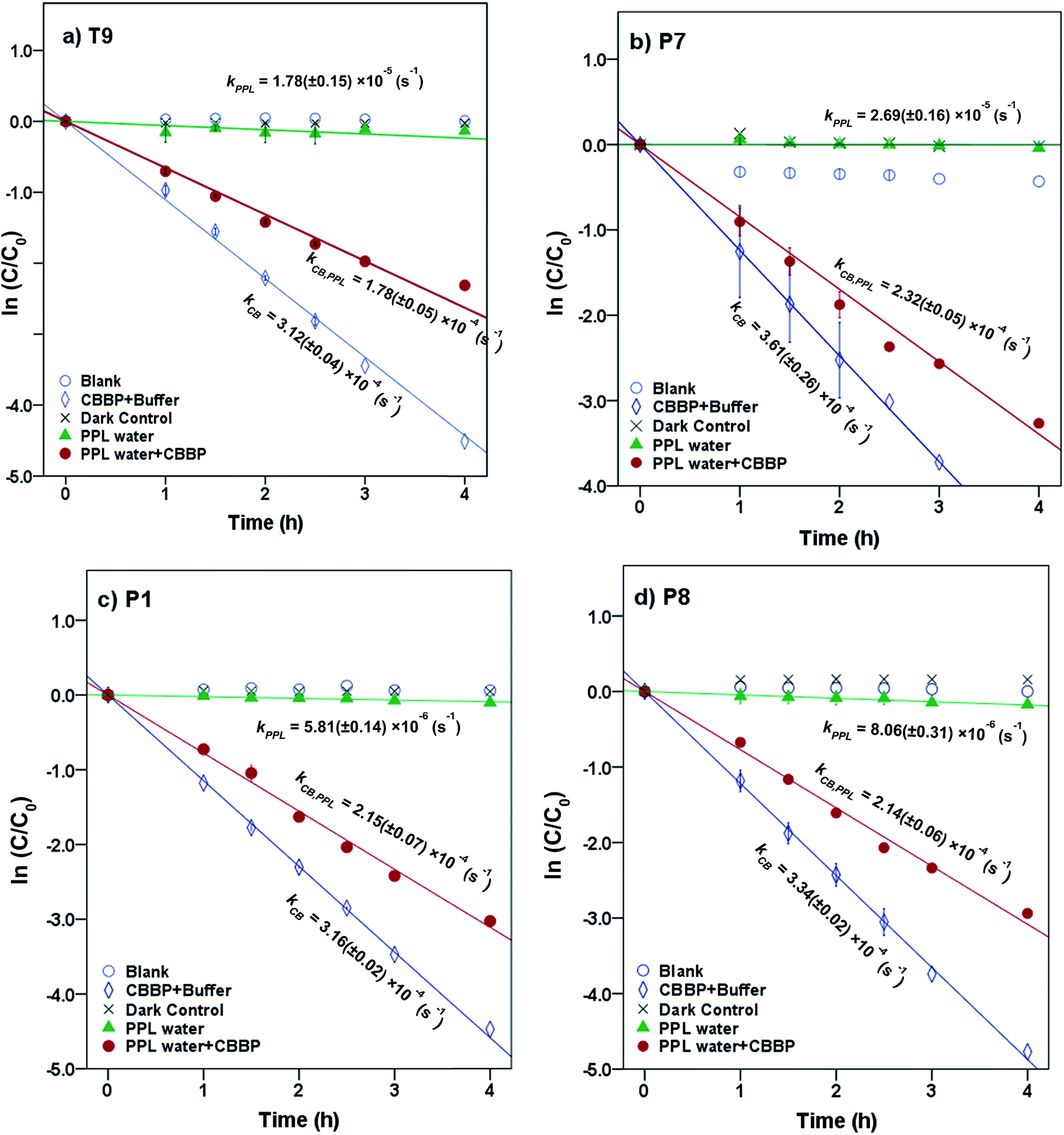 | ||
| Fig. 2 Phototransformation kinetics of atrazine in four photolysis treatments in (a) T9, (b) P7, (c) P1 and (d) P8 waters from the summer season. Error bars represent 95% confidence intervals. | ||
3.1 Direct phototransformation of pesticides
Hydrolysis rates (dark control) of pesticides were examined in PPL water with CBBP during the phototransformation experiments. All of the target pesticides exhibited negligible hydrolysis/sorption/volatilization rates that were several orders of magnitude lower than the observed direct and indirect phototransformation rates. Hence, no correction for losses in the dark controls was necessary within the irradiation time frame of the pesticides. The pseudo first-order rate constants for the direct photolysis in pH 8.5 borate buffer were calculated by eqn (5). Metolachlor exhibited the highest mean (±standard error of the mean) direct photolysis rate (kdirect, s−1 = 5.12(±0.23) × 10−6), followed by atrazine (kdirect, s−1 = 4.04(±2.11) × 10−6), isoproturon (kdirect, s−1 = 2.16(±0.31) × 10−6) and mesotrione (kdirect, s−1 = 9.69(±4.04) × 10−7).3.2 Phototransformation of pesticides in PPL waters
Indirect phototransformation of the target pesticides was investigated in PPL waters. The overall rate constants accounting for loss processes caused by all PPRIs present were calculated as follows:| kindirect = kPPL − kdirect | (9) |
When using kdirect in eqn (9), light screening by DOM was taken into account by multiplying the kdirect value with the light screening factor SF,DOM (Table S2; ESI†). Pesticides showed seasonal and spatial variation in observed overall indirect phototransformation rates across the field sites (Fig. 3). Isoproturon had the fastest mean (±standard error of the mean) indirect phototransformation rate overall (kindirect, s−1 = 9.08(±0.70) × 10−5), followed by mesotrione (kindirect, s−1 = 9.46(±0.62) × 10−6), atrazine (kindirect, s−1 = 7.79(±1.79) × 10−6) and metolachlor (kindirect, s−1 = 3.67(±0.96) × 10−6). Zeng et al. (2013)17 observed similar results in P8 water using the same solar simulator where kindirect (s−1) was reported to be 7.56(±0.12) × 10−5 for isoproturon, 7.38(±0.15) × 10−6 for atrazine, 5.26(±0.10) × 10−6 for mesotrione, and 7.66(±0.15) × 10−6 for metolachlor. A potential explanation for the difference in the magnitude of the indirect photolysis reaction rates between isoproturon and the other three compounds is the difference in one electron oxidation potential (E1 or the Gibbs free energy change of electron transfer) of these compounds which has been suggested as a potential predictor of reaction rate constants with 3DOM* in photoactive systems.34 Using computational chemistry methods, E1 values for 70 compounds including the pesticides used in this study have been calculated by Arnold (2014).34 Isoproturon has the highest E1 value (−1.36 V vs. NHE) among the pesticides included in this study and therefore expected to have the highest indirect phototransformation rate. Although isoproturon reacts with carbonate radical35 and model triplet chromophores,36 it has been shown that the single electron oxidation observed in the presence of DOM is due to the reaction with 3DOM* rather than reactions with 1O2, CO3−˙ or ˙OH.19,29,37 Atrazine and metolachlor have similar E1 values (−2.41 V vs. NHE and −2.40 V vs. NHE, respectively) and these pesticides exhibited indirect phototransformation rates of the same magnitude. Although mesotrione had the lowest E1 value (−2.96 V vs. NHE), it exhibited a higher indirect phototransformation rate compared to atrazine and metolachlor. This might be due to other possible mechanisms described in previous studies.38 It has been reported that while H-donors present in DOM have an inhibitory effect on the photolysis of mesotrione, this effect is counterbalanced by the reaction of mesotrione with singlet oxygen produced by 3DOM*.38
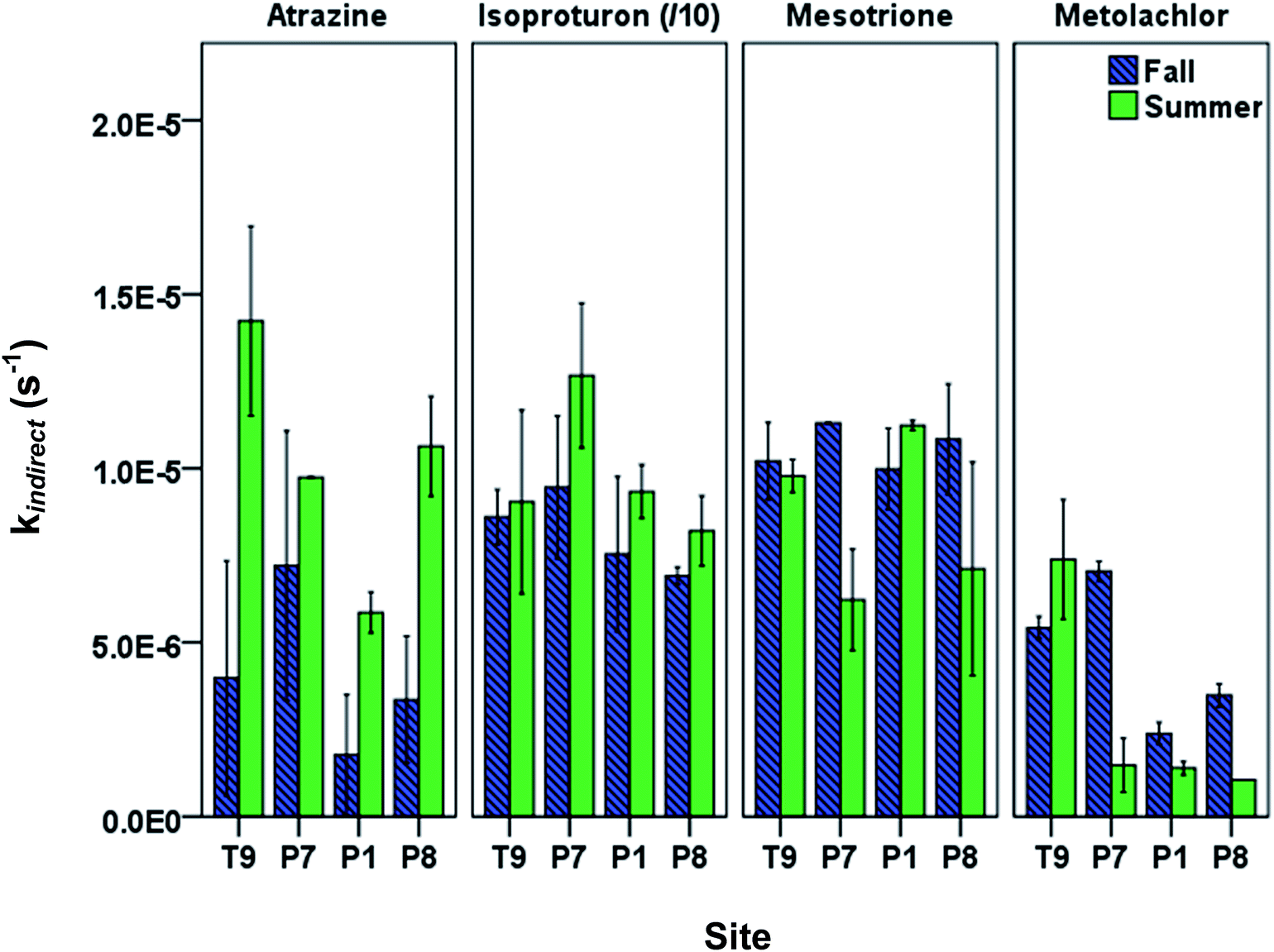 | ||
| Fig. 3 Measured overall indirect phototransformation rate constants of pesticides in PPL waters in fall and summer seasons. Data are arranged along the hydrologic gradient from recharge to discharge. The (/10) notation for isoproturon indicates that the displayed rate constant is ten times less than the actual rate constant. Error bars indicate 95% confidence intervals. | ||
A seasonal analysis of our results also indicates that indirect phototransformation rates were higher in summer compared to fall for atrazine and isoproturon at a significance level of 0.05, while there was no statistically significant seasonal difference for mesotrione and metolachlor (p = 0.40 and 0.22 for mesotrione and metolachlor, respectively). The seasonal differences may be explained by the loss of chromophores as a result of DOM photobleaching throughout the summer. Photobleaching depletes chromophoric dissolved organic matter (CDOM) which in turn may cause a reduction in indirect phototransformation rates of organic contaminants.39
3.3 DOM-induced inhibition of oxidation
Under the oxidative conditions generated by photoirradiated CBBP in PPL waters, atrazine and mesotrione displayed an overall decrease in phototransformation reaction rates caused by DOM originating from PPLs (Fig. 4). The most pronounced inhibition was observed with atrazine for both seasons (fall IF (mean ± standard error of the mean) = 0.75 ± 0.01, and summer IF = 0.76 ± 0.02) followed by mesotrione (fall IF = 0.95 ± 0.06, and summer IF = 0.96 ± 0.04).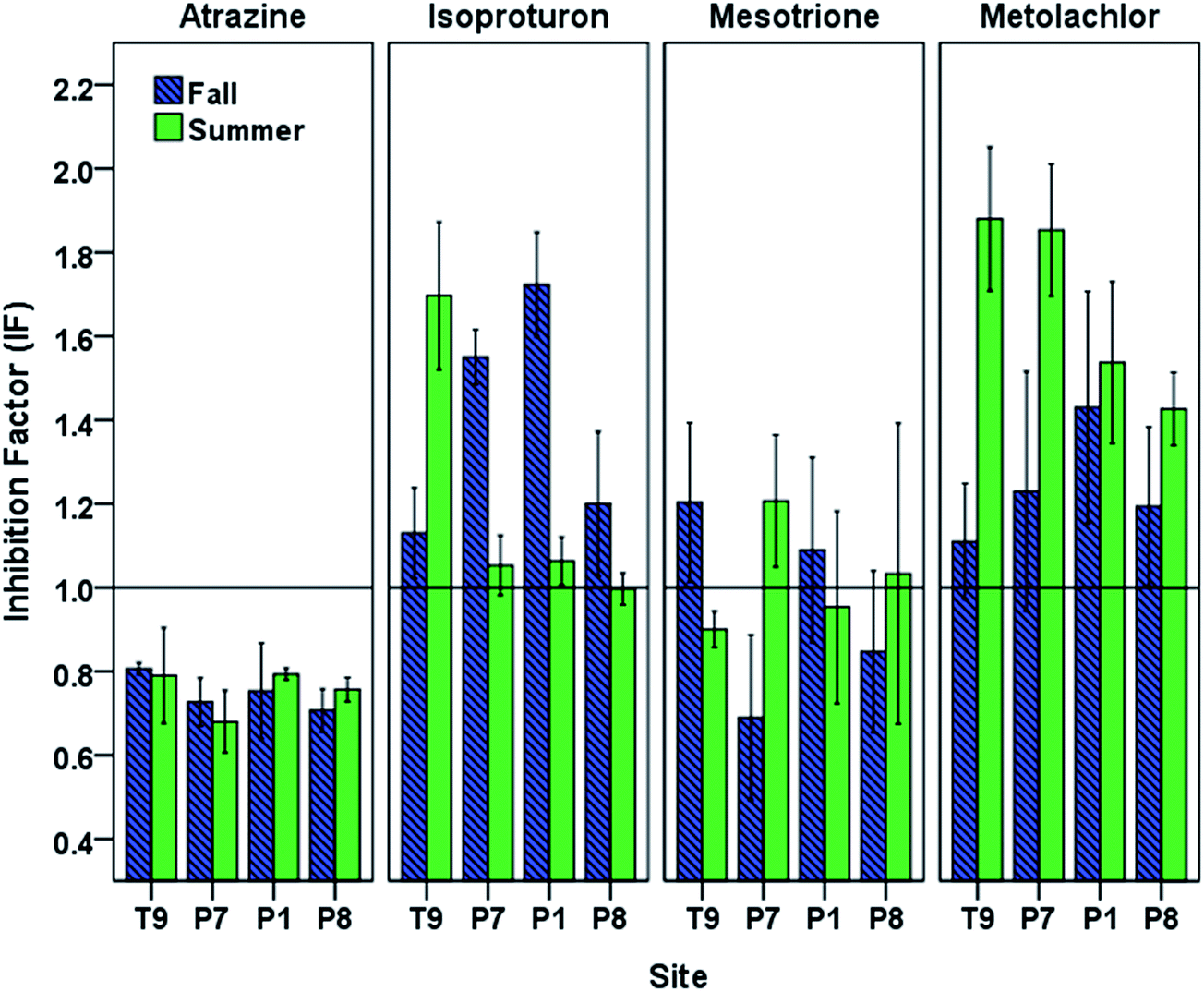 | ||
| Fig. 4 Inhibition factors for the phototransformation of target pesticides in PPL waters in fall and summer seasons. Data are arranged along the hydrologic gradient from recharge to discharge. Error bars indicate 95% confidence intervals. | ||
The isoproturon phototransformation rate was enhanced in the presence of DOM for both seasons (fall IF = 1.40 ± 0.08, and summer IF = 1.20 ± 0.09). This result is in agreement with the expectation that the inhibitory effect of DOM would not play a significant role for electron-rich phenylureas.25 The metolachlor phototransformation rate was also enhanced in the presence of DOM for both seasons (fall IF = 1.24 ± 0.04, and summer IF = 1.67 ± 0.06). The observed seasonal and spatial variation in the inhibition (or enhancement) of phototransformation caused by DOM (Fig. 4) indicated possible differences in DOM properties and suggested the need for DOM characterization of the PPL waters at different seasons.
Previous studies have also documented temporal and spatial changes in the PPL water chemistry in terms of composition and reactivity of DOM.40–43 Both internal processes and external sources such as introduction of wastewater effluent, agricultural runoff, or stormwater flows may alter the DOM composition in a system. The exact chemical structure of aquatic DOM is unknown because DOM is comprised of a collection of numerous molecules originating from microbial (autochthonous) and terrestrial (allochtonous) precursor materials.44–46 Allochtonous DOM is characterized by its generally higher molecular weight, more hydrophobic nature, and greater aromaticity relative to autochthonous DOM.46 The two major classes of DOM also have quite different spectroscopic properties.46–48 Several spectral parameters have been defined to extract information from these spectra about CDOM properties. To track changes in the average molecular weight of DOM, an E2:E3 ratio (the ratio of absorption at 250 to 365 nm) was defined by de Haan and de Boer.49 They showed that the E2:E3 ratio was inversely proportional to the molecular size of DOM because of stronger light absorption by high molecular weight CDOM.49 Another absorption ratio, E4:E6 (the ratio of absorption at 465 to 665 nm), was reported to be inversely related to the aromaticity of CDOM.50 In many natural waters, absorption at 254 nm or 280 nm has been used as an indicator of aromaticity instead of the E4:E6 ratio, because there was often very little or no measurable absorption at 665 nm.46 Weishaar et al.48 showed that a parameter called specific UV absorbance or SUVA254 (UV absorption at 254 nm divided by dissolved organic carbon (DOC) concentration), correlated strongly with DOM aromaticity for a large number of humic substance isolates.
In addition to the parameters mentioned above, spectral slopes (S, nm−1) have been used to gain further insights into CDOM characterization such as determining the ratio of fulvic acids to humic acids.46S values have also been shown to correlate with the molecular weight of isolates of fulvic acids, however, their usefulness is limited by the fact that their values depend on the wavelength interval over which they are calculated.46 A wide range in S values is reported in the literature even for similar sample types and the lack of standardization has made comparisons of published S values difficult.46 To overcome the potential artifacts mentioned above, Helms et al. (2008)46 defined a dimensionless parameter called “slope ratio” or SR by calculating the ratio of the slope of the shorter wavelength region (275–295 nm) to that of the longer wavelength region (350–400 nm). The advantage of this approach is that it avoids the use of the spectral data near the instrumental detection limits and focuses on absorbance values that change significantly with the photochemical alteration of CDOM.46 Fluorescence spectroscopy has also been extensively used to characterize the source and properties of DOM.31,47 McKnight et al.47 defined the fluorescence index (FI) (the ratio of the emission intensity at 450 nm to 500 nm obtained with an excitation wavelength of 370 nm) and suggested that it can be used as a simple index for DOM characterization.
Based on the current literature, there is no consensus on which spectral parameter is the best for the characterization of DOM. In this study, all of the aforementioned spectral parameters were calculated for the PPL waters (Table 1, and Fig. S17–20 in the ESI†). It has been previously reported that iron is a source of interference in the spectroscopic analysis of DOM.51 Iron concentrations ([Fe2+] + [Fe3+]) in the PPL samples were below 3 μM, hence no iron related correction for the spectral parameters was necessary. It was determined that SR and the E2:E3 ratio exhibited the most reliable and consistent results in correlation with the Inhibition Factor (lowest p-values and highest correlation coefficients in Pearson correlation analyses, Table 2).
| Compound | Season | Correlation with IF | S R | SUVA254 |
E
2:E3 |
E
4:E6 |
S 250−500 | FI |
|---|---|---|---|---|---|---|---|---|
| Atrazine | Summer | Pearson correlation coefficient, r | 0.93* | −0.727 | 0.950* | 0.745 | 0.938 | 0.621 |
| p | 0.049 | 0.273 | 0.050 | 0.255 | 0.062 | 0.379 | ||
| Fall | Pearson correlation coefficient, r | −0.227 | 0.483 | −0.091 | 0.426 | −0.196 | −0.935 | |
| p | 0.773 | 0.517 | 0.909 | 0.574 | 0.804 | 0.065 | ||
| Isoproturon | Summer | Pearson correlation coefficient, r | −0.805 | 0.829 | −0647 | 0.844 | −0.685 | −0.679 |
| p | 0.195 | 0.171 | 0.353 | 0.156 | 0.315 | 0.321 | ||
| Fall | Pearson correlation coefficient, r | 0.989* | −0.950* | 0.949* | −0.817 | 0.837 | 0.511 | |
| p | 0.011 | 0.050 | 0.050 | 0.183 | 0.163 | 0.489 | ||
| Mesotrione | Summer | Pearson correlation coefficient, r | 0.123 | −0.500 | 0.099 | 0.470 | 0.140 | 0.635 |
| p | 0.877 | 0.500 | 0.901 | 0.530 | 0.860 | 0.365 | ||
| Fall | Pearson correlation coefficient, r | −0.311 | 0.565 | −0.166 | 0.471 | −0.246 | −0.967* | |
| p | 0.689 | 0.435 | 0.834 | 0.529 | 0.754 | 0.033 | ||
| Metolachlor | Summer | Pearson correlation coefficient, r | −0.454 | 0.252 | −0.272 | 0.289 | −0.289 | 0.001 |
| p | 0.546 | 0.748 | 0.728 | 0.711 | 0.711 | 1.000 | ||
| Fall | Pearson correlation coefficient, r | 0.985* | −0.899 | 0.996* | −0.933 | 0.964* | 0.463 | |
| p | 0.015 | 0.101 | 0.004 | 0.067 | 0.036 | 0.537 |
There was a positive correlation between the inhibition factors and the SR and E2:E3 ratio values for atrazine in summer (Table 2). The inhibition factor for atrazine also had a relatively weak negative correlation (at a significance level of α = 0.10) with SUVA254 in summer samples (p = −0.73, r = −0.090). Inhibition factors for mesotrione did not exhibit any significant correlation with SR and E2:E3 ratio parameters, suggesting the role of other possible processes in its phototransformation as mentioned previously. Inhibition factors for both isoproturon and metolachlor were positively correlated with SR and the E2:E3 ratio in fall samples. Metolachlor also exhibited a positive correlation with the S250–500 parameter, while isoproturon exhibited a negative correlation with SUVA in fall (Table 2).
Previous studies reported evidence that photobleaching and shifts from high molecular weight (HMW) DOM to low molecular weight (LMW) DOM cause an increase in SR and S275–295 values.46 In this study, this phenomenon is supported by the fact that measured SR values increased in fall compared to summer at all PPL sites, suggesting the importance of photobleaching processes over the summer. The same relationship did not apply at all PPL sites for SUVA254 values, indicating that the observed seasonal differences in DOM behavior cannot be attributed to the changes in aromaticity alone. Together with the relationship of SUVA254 values to the aromaticity of DOM,48 our correlation analyses suggest that for the pesticide exhibiting the lowest IF values (atrazine), photobleaching and shift from HMW to LMW DOM were the dominant processes affecting the observed phototransformation inhibition by DOM followed by changes in aromaticity.
Thus, our results indicate that allochtonous, HMW and highly aromatic DOM (with higher SUVA and lower SR values) generally presents a relatively higher efficiency to inhibit excited triplet-induced oxidation than mainly autochthonous DOM. These results are in agreement with previous studies reporting that for the contaminants subject to inhibition of oxidation, their phototransformation induced by 3DOM* will vary greatly depending on the DOM type and will be inhibited more by highly aromatic DOM.26,44 These trends are largely consistent among the individual pesticides which are from different structural classes. More work is necessary to evaluate these correlations among more pesticides (across and within classes) as well as with additional water samples across a range of sites and seasons.
4. Conclusion
In this study, the effect of DOM on the phototransformation of pesticides in PPL waters was investigated. For atrazine and mesotrione, a decrease in phototransformation rates was observed; while phototransformation of metolachlor and isoproturon were not affected by DOM inhibition. Characterization of DOM from the sites and different seasons suggested that the DOM type and variations in the DOM structure are important factors controlling phototransformation rates of pesticides in PPLs. In general, the more aromatic, HMW DOM caused more inhibition to the phototransformation of pesticides in PPL waters. At some of the study sites, DOM in PPLs showed a more allochtonous, HMW and more aromatic character in summer which shifted towards less aromatic and LMW DOM in fall. Given that phototransformation processes are mostly important in summer when PPL waters are exposed to abundant sunlight and when they mostly receive irrigation runoff from adjacent agricultural fields, the retardation of indirect phototransformation by DOM may become critical for pesticide attenuation at the sites exhibiting these types of seasonal shifts. This is especially important for the attenuation of pesticides that are recalcitrant to biotransformation and are inefficiently depleted by direct phototransformation.The results of this study are part of an effort to understand factors controlling the phototransformation of pesticides in PPL waters which still serve as collection points for non-point source agricultural runoff. Given the interconnectedness of water resources in the PPR, a better understanding of pesticide phototransformation in PPLs may shed light on future water and land use management strategies to mitigate the impact of pesticides on downstream surface water resources.
Acknowledgements
This work was supported by the U.S. Geological Survey through the National Institutes for Water Resources (2012MN344 G) and the Joseph T. and Rose S. Ling Professorship. The authors thank Rose M. Cory and Katherine H. Harrold for their help in collecting excitation-emission spectra.References
- N. H. Euliss and D. M. Mushet, Wetlands, 1996, 16, 587–593 CrossRef.
- T. Ballard, R. Seager, J. E. Smerdon, B. I. Cook, A. J. Ray, B. Rajagopalan, Y. Kushnir, J. Nakamura and N. Henderson, Earth Interact., 2014, 18, 1–28 CrossRef.
- J. W. LaBaugh, T. C. Winter and D. O. Rosenberry, Great Plains Res., 1998, 8, 17–37 Search PubMed.
- R. R. Johnson, F. T. Oslund and D. R. Hertel, J. Soil Water Conserv., 2008, 63, 84A–87A CrossRef.
- J. N. H. Euliss, R. A. Gleason, A. Olness, R. L. McDougal, H. R. Murkin, R. D. Robarts, R. A. Bourbonniere and B. G. Warner, Sci. Total Environ., 2006, 361, 179 CrossRef PubMed.
- F. T. Oslund, R. R. Johnson and D. R. Hertel, Journal of Fish and Wildlife Management, 2010, 1, 131–135 Search PubMed.
- C. F. Lenhart, E. S. Verry, K. N. Brooks and J. A. Magner, River Res. Appl., 2012, 28, 1609–1619 CrossRef.
- M. J. Anteau, Wetlands, 2012, 32, 1–9 CrossRef.
- N. N. Brunet and C. J. Westbrook, Agric., Ecosyst. Environ., 2012, 146, 1–12 CrossRef CAS.
- L. G. Goldsborough and W. G. Crumpton, Great Plains Res., 1998, 8, 73–95 Search PubMed.
- N. E. Detenbeck, C. M. Elonen, D. L. Taylor, A. M. Cotter, F. A. Puglisi and W. D. Sanville, Wetlands Ecol. Manage., 2002, 10, 335–354 CrossRef CAS.
- S. Sura, M. Waiser, V. Tumber, J. R. Lawrence, A. J. Cessna and N. Glozier, J. Environ. Qual., 2012, 41, 732 CrossRef CAS PubMed.
- D. B. Donald, N. P. Gurprasad, L. Quinnett-Abbott and K. Cash, Environ. Toxicol. Chem., 2001, 20, 273–279 CrossRef CAS PubMed.
- D. B. Donald and N. E. Glozier, Environ. Health Perspect., 2007, 115, 1183–1191 CrossRef CAS PubMed.
- D. Degenhardt, A. J. Cessna, R. Raina, A. Farenhorst and D. J. Pennock, Environ. Toxicol. Chem., 2011, 30, 1982–1989 CrossRef CAS PubMed.
- H. D. Burrows, M. L. Canle, J. A. Santaballa and S. Steenken, J. Photochem. Photobiol., B, 2002, 67, 71–108 CrossRef CAS.
- T. Zeng and W. A. Arnold, Environ. Sci. Technol., 2013, 47, 6735–6745 CrossRef CAS PubMed.
- P. V. L. Reddy and K.-H. Kim, J. Hazard. Mater., 2015, 285, 325–335 CrossRef CAS PubMed.
- C. K. Remucal, Environ. Sci.: Processes Impacts, 2014, 16, 628–653 CAS.
- R. Schwarzenbach, P. Gschwend and M. Imboden, Environmental Organic Chemistry, Hoboken, NJ, 2003 Search PubMed.
- H. F. Wilson and M. A. Xenopoulos, Nat. Geosci., 2009, 2, 37–41 CrossRef CAS.
- S. Inamdar, S. Singh, S. Dutta, D. Levia, M. Mitchell, D. Scott, H. Bais and P. McHale, J. Geophys. Res.: Biogeosci., 2011, 116, G03043 Search PubMed.
- S. P. McElmurry, D. T. Long and T. C. Voice, Environ. Sci. Technol., 2014, 48, 45–53 CrossRef CAS PubMed.
- M. L. Quaranta, M. D. Mendes and A. A. MacKay, Water Res., 2012, 46, 284–294 CrossRef CAS PubMed.
- S. Canonica and H.-U. Laubscher, Photochem. Photobiol. Sci., 2008, 7, 547–551 CAS.
- J. Wenk, U. Von Gunten and S. Canonica, Environ. Sci. Technol., 2011, 45, 1334–1340 CrossRef CAS PubMed.
- W. Haag, J. Res. Natl. Bur. Stand., 1988, 93, 285–288 CrossRef CAS.
- J. Wenk and S. Canonica, Environ. Sci. Technol., 2012, 46, 5455–5462 CrossRef CAS PubMed.
- T. Zeng, Abiotic Transformations of Pesticides in Prairie Potholes, Doctoral Dissertation, University of Minnesota, 2012.
- D. M. Mushet, N. H. Euliss Jr, S. P. Lane and C. M. Goldade, The Flora of the Cottonwood Lake Study Area, USGS, Stutsman County, North Dakota, 2004 Search PubMed.
- R. M. Cory, M. P. Miller, D. M. McKnight, J. J. Guerard and P. L. Miller, Limnol. Oceanogr.: Methods, 2010, 8, 67–78 CrossRef CAS.
- K. R. Murphy, C. A. Stedmon, D. Graeber and R. Bro, Anal. Methods, 2013, 5, 6557–6566 RSC.
- E. Viollier, P. W. Inglett, K. Hunter, A. N. Roychoudhury and P. van Cappellen, Appl. Geochem., 2000, 15, 785–790 CrossRef CAS.
- W. A. Arnold, Environ. Sci.: Processes Impacts, 2014, 16, 832–838 CAS.
- S. Canonica, T. Kohn, M. Mac, F. J. Real, J. Wirz and U. von Gunten, Environ. Sci. Technol., 2005, 39, 9182–9188 CrossRef CAS PubMed.
- S. Canonica, B. Hellrung, P. Müller and J. Wirz, Environ. Sci. Technol., 2006, 40, 6636–6641 CrossRef CAS PubMed.
- A. C. Gerecke, S. Canonica, S. R. Müller, M. Schärer and R. P. Schwarzenbach, Environ. Sci. Technol., 2001, 35, 3915–3923 CrossRef CAS PubMed.
- A. ter Halle and C. Richard, Environ. Sci. Technol., 2006, 40, 3842–3847 CrossRef CAS PubMed.
- M. C. Langlois, L. K. Weavers and Y.-P. Chin, Environ. Sci.: Processes Impacts, 2014, 16, 2098–2107 Search PubMed.
- M. J. Waiser and R. D. Robarts, Limnol. Oceanogr., 2000, 45, 763–774 CrossRef CAS.
- M. J. Waiser, J. Geophys. Res.: Biogeosci., 2006, 111, G02024 Search PubMed.
- M. J. Waiser and R. D. Robarts, Biogeochemistry, 2004, 69, 263–284 CrossRef CAS.
- K. L. Ziegelgruber, T. Zeng, W. A. Arnold and Y. P. Chin, Limnol. Oceanogr., 2013, 58, 1136–1146 CrossRef CAS.
- J. J. Guerard, P. L. Miller, T. D. Trouts and Y.-P. Chin, Aquat. Sci., 2009, 71, 160–169 CrossRef CAS.
- D. M. McKnight, G. R. Aiken and R. L. Smith, Limnol. Oceanogr., 1991, 36, 998–1006 CrossRef CAS.
- J. R. Helms, A. Stubbins, J. D. Ritchie, E. C. Minor, D. J. Kieber and K. Mopper, Limnol. Oceanogr., 2008, 53, 955–969 CrossRef.
- D. M. McKnight, E. W. Boyer, P. K. Westerhoff, P. T. Doran, T. Kulbe and D. T. Andersen, Limnol. Oceanogr., 2001, 46, 38–48 CrossRef CAS.
- J. L. Weishaar, G. R. Aiken, B. A. Bergamaschi, M. S. Fram, R. Fujii and K. Mopper, Environ. Sci. Technol., 2003, 37, 4702–4708 CrossRef CAS PubMed.
- H. de Haan and T. de Boer, Water Res., 1987, 21, 731–734 CrossRef CAS.
- R. Summers, P. Cornel and P. Roberts, Sci. Total Environ., 1987, 62, 27–37 CrossRef CAS.
- B. A. Poulin, J. N. Ryan and G. R. Aiken, Environ. Sci. Technol., 2014, 48, 10098–10106 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5em00374a |
| ‡ Authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2016 |