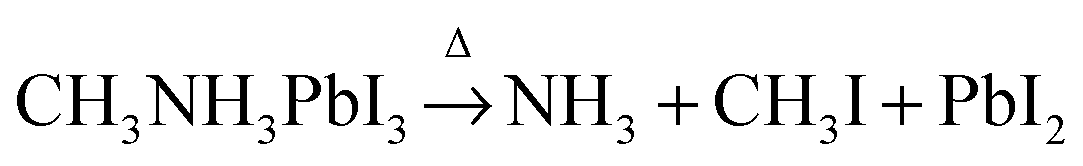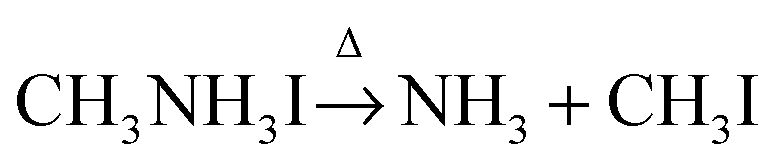DOI:
10.1039/C6EE02016J
(Communication)
Energy Environ. Sci., 2016,
9, 3406-3410
Thermal degradation of CH3NH3PbI3 perovskite into NH3 and CH3I gases observed by coupled thermogravimetry–mass spectrometry analysis†
Received
13th July 2016
, Accepted 1st September 2016
First published on 1st September 2016
Abstract
Thermal gravimetric and differential thermal analysis (TG-DTA) coupled with quadrupole mass spectrometry (MS) and first principles calculations were employed to elucidate the chemical nature of released gases during the thermal decomposition of CH3NH3PbI3. In contrast to the common wisdom that CH3NH3PbI3 is decomposed into CH3NH2 and HI, the major gases were methyliodide (CH3I) and ammonia (NH3). We anticipate that our findings will provide new insights into further formulations of the perovskite active material and device design that can prevent methylammonium decomposition and thus increase the long-term stability of perovskite-based optoelectronic devices.
Broader context
In the past few years, we have all witnessed the unprecedented zeal for the rapid growth of the organometal halide perovskite solar cell field. The tremendous efforts devoted to device fabrication and optimization have led to PCEs exceeding 20%, which gives perovskite solar cells a competitive advantage over many other well-known solar technologies. On the other hand, some challenges remain with the lifetime as the key. At present, even the most long-lasting perovskite solar cells can only retain stable PCEs up to a few thousands of hours, which is way behind, for example, silicon cells. To further improve perovskite solar cells, we must overcome/mitigate the degradation issue. For this reason, a thorough detailed understanding of perovskite solar cell degradation processes becomes an imperative first step.
|
Introduction
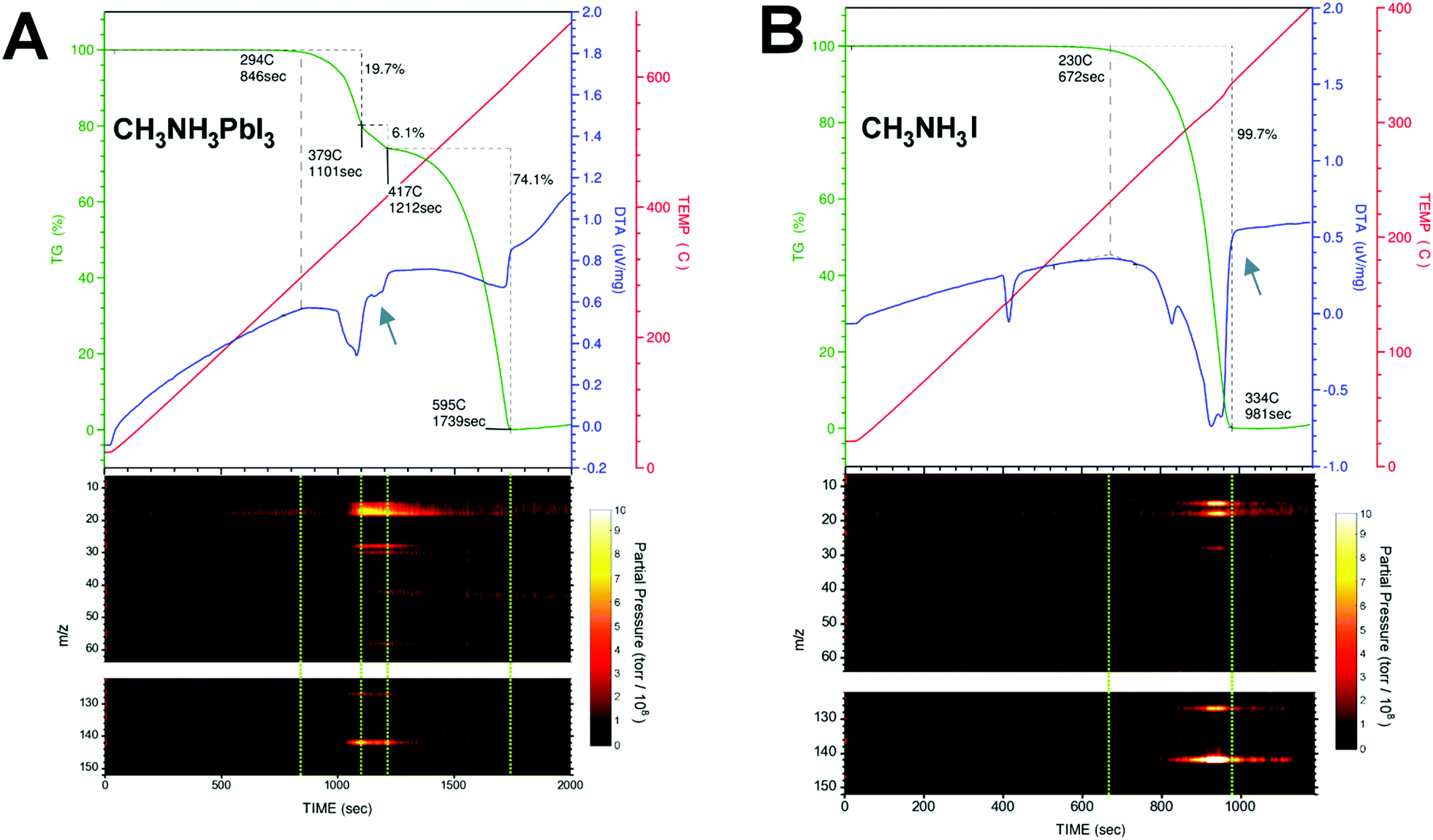
Photovoltaic technologies based on hybrid organometal halide perovskites have already achieved outstanding efficiencies.1 The main challenge now is to overcome their limited lifetime under operating conditions.2 The degradation mechanisms of perovskites have been a topic under intense investigation.3–6 The fundamental understanding of physicochemical processes that trigger degradation is needed to further engineer perovskite solar cells for enhanced stability. Generally speaking, extrinsic factors (e.g., light, temperature, humidity, and O2) can contribute to the degradation processes. Different degradation pathways have been proposed and the topic is still under debate. But it is clear that CH3NH3PbI3 perovskite loses its excellent light harvesting properties over time because it evolves into PbI2 after a nominal loss of CH3NH3I.7 The degradation from CH3NH3PbI3 to PbI2 is most likely accompanied by a release of gases via simple sublimation or assisted chemical reaction. Therefore, the determination of the composition of these released gases during controlled thermal degradation of CH3NH3PbI3 is expected to pinpoint the degradation pathways. A suitable method to identify the mass loss directly from perovskite degradation is the thermal gravimetric and differential thermal analysis (TG-DTA). Using TG-DTA, we can detect the endothermic/exothermic phase transitions and also quantify the percentage of mass loss during the heating of the material. However, this technique does not characterize the chemical nature of the released gases. For that reason, we connected a mass spectrometer (MS) to the gas exhaust line from the TG-DTA equipment in order to identify unambiguously the molecular mass of the released gases. Typically, the TG trace for CH3NH3PbI3 shows two consecutive mass loss steps, with the former ascribed to the nominal loss of methylammonium iodide CH3NH3I (ca. ∼25%, 300–420 °C) and the latter to the inorganic PbI2 component (∼75% above 420 °C).8–16 A careful comparison of the TG trace during the first mass loss step in CH3NH3PbI3 and the TG trace of the CH3NH3I salt sublimation shows that the former is at least composed of two intermediate mass loss steps evolving at different rates; see the portion of TG-DTA traces indicated by blue arrows in Fig. 1A and B.
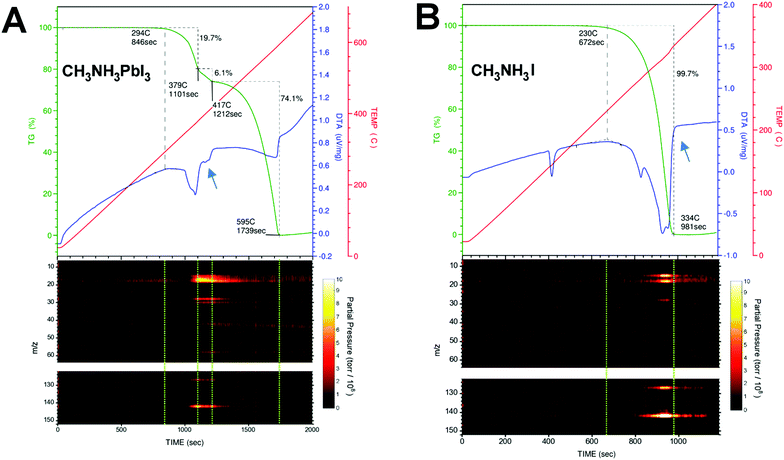 |
| | Fig. 1 TG-DTA traces (upper panel, dark green and blue color, respectively.) and the m/z peaks registered simultaneously during the thermal degradation (heating rate of 20 °C min−1) of (A) CH3NH3PbI3 and (B) CH3NH3I. Blue arrows indicate an endothermic process present in the DTA trace in CH3NH3PbI3 perovskite, but absent in the CH3NH3I salt. Dashed green lines in the m/z vs. time contour plots are guides to the eye delimiting the mass loss steps in the TG/DTA graph. | |
Different interpretations can be found in the literature regarding the chemical nature of these released gases during thermal degradation. Dualeh et al.11 and Nenon et al.13 proposed that CH3NH2 and HI are the gases released sequentially11 during the first mass loss step of CH3NH3PbI3 decomposition. On the other hand, Williams et al.15 found that the gases released are CH3I and NH3. In the study by Williams et al., the determination of CH3I and NH3 was clearly demonstrated for CH3NH3I, but it is ambiguous for the perovskite sample because of the limitation of the methodology employed. They performed the simultaneous thermal analysis-Fourier Transform infrared spectroscopy (STA-FTIR) measurements on samples of perovskite precursor solution. Therefore, the reported infrared spectra of the gases released during the perovskite formation/degradation processes are convoluted with the thermal degradation of the solvent, and the assignment of CH3I and NH3 is ambiguous in this case. The assignment of degradation gas species can get complicated by the use of perovskite solutions instead of solid samples (e.g., small perovskite crystals used in this work). For example, a close inspection of Fig. 4a in the report by Nenon et al.13 reveals that there is no 31 m/z peak for methylamine in the “Degraded” zone. Peak 31 m/z only appears during perovskite formation and drying steps. Also, their MS scan reached only up to 100 m/z leaving out of sight the 142 m/z CH3I signature peak.
It is noteworthy that the presence of CH3I molecules in the early stage of formation of CH3NH3PbI3 using vacuum deposition was proposed by Liu et al. in their X-ray photoelectron spectroscopy (XPS) study.17 Also, a recent study by Xu et al. provided further evidence using soft X-ray spectroscopy and density functional theory calculations.18
Results and discussion
Simultaneous thermogravimetry (TG) and differential thermal analysis (DTA) coupled with mass spectrometry (TG-DTA/MS) measurements were carried out under an inert atmosphere (He) to identify the chemical compositions of the released gases during the thermal decomposition process of CH3NH3PbI3 perovskite monocrystals (volume ∼ 1 mm3, weight of ∼7–15 mg) and CH3NH3I polycrystalline powder. The gases generated during the TG-DTA measurements are collected using a quadrupole mass filter, which is employed primarily for residual gas analysis (RGA). In this work, the starting and end points of the mass loss steps are delimited using the slope changes caused by thermal events in the DTA trace, which is recorded simultaneously during the TG measurements. Fig. 1A and B show the TG-DTA traces and the mass-to-charge ratio (m/z) peaks recorded simultaneously during the thermal degradation of CH3NH3PbI3 and CH3NH3I. For simplicity, only the relevant peaks in the m/z vs. time contour plots in Fig. 1 are shown for discussion. The complete MS results can be found in Fig. S1 (ESI†). Table 1 summarizes the m/z peaks observed during the thermal degradation of CH3NH3PbI3 and CH3NH3I together with their assignments. Dashed green lines in the m/z vs. time contour plots are guide to the eye, delimiting the mass loss steps in the TG-DTA graph. Contrary to common wisdom, CH3NH2 (methylamine, Tbp = −6 °C, m/z = 31) and HI (hydrogen iodide, Tbp = −34 °C, m/z = 128) gases were not detected during the thermal degradation of CH3NH3PbI3 or CH3NH3I. Although the m/z = 30 and 127 fragment peaks were observed, their corresponding parent molecules of m/z = 31 and 128 amu were not detected; see Fig. 1, Fig. S2 and S3 (ESI†). When performing measurements with MS systems care should be taken to consider the fragmentation of molecules during the ionization process (electron bombardment) within the MS system. As the control experiment, commercially available CH3NH2 and HI gases were fed directly into our MS system and m/z = 31 and 128 were indeed detected (Fig. S4a and b, ESI†) in addition to the m/z = 30 and 127 fragment peaks. In the thermal degradation process of CH3NH3PbI3 and CH3NH3I, because the parent molecular peaks corresponding to m/z = 31 and 128 peaks were missing, the amounts of CH3NH2 and HI gases can be concluded to be negligible. Instead, the m/z peaks observed in our TG-DTA/MS measurements can be unambiguously assigned to NH3 (ammonia gas, Tbp = −33 °C, m/z = 17) and CH3I (methyliodide, Tbp = 42 °C, m/z = 142) gases; see Fig. 1, Fig. S2, S3 (ESI†) and Table 1. It is noteworthy to mention that Nenon et al.13 performed MS in the range from m/z = 1 to 100, so they could not observe the m/z peaks at 127, 141, and 142, the latter two of which correspond to CH3I.
Table 1 Assignments of parent and fragmentation peaks in the TG-DTA/MS spectra
|
m/z |
Detected ion |
Parent/fragment molecule |
| 15 |
CH3+ |
CH3I fragment |
| 16 |
NH2+ |
NH3 fragment |
| 17 |
NH3+ |
NH3 parent peak |
| 18 |
NH4+ |
NH3 + H+ proton transfer |
| 18 |
H2O+ |
H2O parent peak21 |
| 28 |
N2+ |
N2 parent peak |
| 30 |
CH4N+ |
(CH3)3N fragment |
| 42 |
C2H4N+ |
(CH3)3N fragment |
| 44 |
C2H6N+ |
(CH3)3N fragment |
| 58 |
C3H8N+ |
(CH3)3N Hydrogen dissociation |
| 59 |
(CH3)3N+ |
(CH3)3N parent peak |
| 127 |
I+ |
CH3I fragment |
| 141 |
CH2I+ |
CH3I Hydrogen dissociation |
| 142 |
CH3I+ |
CH3I parent peak |
Based on the observations above, it is proposed that the first mass loss step during the thermal degradation of CH3NH3PbI3 and CH3NH3I under an inert atmosphere proceeds as:
| | 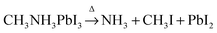 | (1a) |
| | 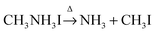 | (1b) |
Therefore, the decomposition reactions of the methylammonium ions (CH
3NH
3+) assisted by iodide proceed through the so-called reverse Menshutkin reaction (a bimolecular nucleophilic substitution S
N2 simultaneously breaking a C–N bond and forming a C–I bond) as it is well described for quaternary ammonium salts.
19,20 But, in contrast to the quaternary ammonium salts, the decomposition product of methylammonium
via reverse Menshutkin reaction forms ammonia instead of trimethylamine.
The high concentrations of NH4+ (m/z = 18), N2 (m/z = 28) and H2, (m/z = 2), which were also detected by our MS system, indicate that NH3 undergoes further decomposition and rearrangement reactions (Fig. S2 and S3, ESI†). Interestingly, during the CH3NH3PbI3 thermal degradation, unidentified traces can be detected only at the end of the first mass loss release (Fig. 1a and Fig. S2, ESI†). The m/z fragments at 30, 42, 44, 58, and 59 were tentatively identified as the fragmentation pattern of trimethylamine (CH3)3N (Tbp = 3 °C, m/z = 59, see Fig. S5, ESI†), which corroborates with the proposed pathway (eqn (2c)). On the other hand, we did not observe intermediates such as CH3NH2 (eqn (2a)) and (CH3)2NH (eqn (2b)), possibly due to the detection limit or the fact that the kinetics of the chain reaction is too fast to follow and we could only observe the final products (e.g., trimethylamine).
Therefore, the proposed mechanism for the formation of (CH3)3N is as follows:
| | | CH3NH3+ + NH3 → CH3NH2 + NH4+ | (2a) |
| | | CH3NH2 + CH3I → (CH3)2NH + H+ + I− | (2b) |
| | | (CH3)2NH + CH3I → (CH3)3N + H+ + I− | (2c) |
Here we propose that the previously formed NH
3 can react with the remaining methylammonium ions trapped in the PbI
3− octahedral network
via a simple acid–base reaction (
eqn (2a)), and the excess of CH
3I reacts with the as-formed primary amines (
eqn (2b)) and secondary amines (
eqn (2c))via the conventional Menshutkin reaction until the formation of the tertiary amine (CH
3)
3N that ends the chain reaction set. We hypothesize that the
(2a) reaction needs to proceed with trapped methylammonium ions inside the bulk perovskite, which would explain why these trace amounts of (CH
3)
3N are not observed during the thermal degradation of CH
3NH
3I.
Photovoltaic devices usually operate under light irradiation and, most likely, with moisture and oxygen invasion. Therefore, the chemical stability of perovskite under light irradiation and ambient conditions was studied using a modified TG-DTA/MS setup; see Fig. S9a (ESI†). Overall, the main difference observed when using an oxygen or water rich carrier gas during the TG-DTA/MS measurements was a different TG trace during the high temperature interval time. In all experiments performed with such oxidizing agents in the carrier gas, the PbI2 is partially converted to lead oxide (PbxOy) ashes,22 which remain intact after the measurement is finished; see Fig. S9, panels b, c, e and f (ESI†). On the other hand, the common feature is that the gases released during the thermal decomposition of CH3NH3PbI3 above 300 °C are still ammonia and methyliodide. Specifically, the 142 m/z signature peak for methyliodide release is clearly observed for all cases. The detection of the CH3I release at low temperature using our TG-DTA/MS setup equipment is nontrivial because such a low release rate of CH3I will be below the detection limit if using the same measurement conditions as before. Therefore, we have performed a specifically designed measurement to obtain the experimental results confirming that the proposed decomposition pattern also takes place at low temperatures such as 80 °C; see Section S3 in the ESI† for full details.
First-principles density functional theory (DFT) calculations using second order perturbation correction (Double-Hybrid Functional, see the ESI† files for full details) were carried out to gain further insight into the thermochemistry of the two chemical reaction pathways suggested for the perovskite degradation, represented by chemical eqn (3a) and (3b):
| | | CH3NH3+(g) + I−(g) → CH3I(g) + NH3(g) | (3a) |
| | | CH3NH3+(g) + I−(g) → CH3NH2(g) + HI(g) | (3b) |
Δ
G (Gibbs free enthalpy) evaluation in the temperature range of 25–600 °C reveals that both reactions are thermodynamically spontaneous and
reaction (3a) shows ∼13–14 kcal mol
−1 in Δ
G lower than
reaction (3b). Therefore, both degradation processes are favorable thermodynamically, but it does not explain why
eqn (3a) is kinetically favored compared to
(3b). A description of the reaction mechanism is therefore needed, in which the molecular structures of reactants, intermediates and products are correlated with their energy reaction pathways. In this case, the proposed thermodynamic cycle describing the degradation mechanism consists of 3 processes: (1) reactants infinitely separated are brought together to one specific interacting state; (2) chemical transformation of these reactants to molecular products by means of a transition state (TS); and (3) infinite distance separation of the products.
Fig. 2 depicts this hypothetical thermodynamic cycle for the (
(3a), blue) and (
(3b), red) reaction pathways. Based on our calculations, the interacting disposition of reactants as depicted in
Fig. 2,
reaction (3b) (state 6), is favored by ∼30 kcal mol
−1 compared to
reaction (3a) (state 2). However, a TS (state 3) that connects reactants (state 2) and products (state 4) can only be obtained for the
reaction (3a) pathway (three short movie files showing the relaxed scan calculations along the reaction coordinate for both chemical reactions and the imaginary frequency oscillation for the TS state 3 are given in the ESI
†). The activation energy (enthalpy difference between state 3 and state 2) is only 0.4–0.2 kcal mol
−1 in the temperature range studied. On the other hand, there are no stationary ground state geometrical dispositions for HI and CH
3NH
2 molecules where such molecules do not react evolving to state 6. Consequently, a TS for the
reaction (3b) pathway cannot be found. Therefore, the absolute lowest energy state for the molecular gas system is defined as CH
3I and NH
3 molecular products infinitely separated (state 5). State 6, however, remains as a local minimum in enthalpy energy but in the reactant side of the
(3b) chemical path. Only this state 6 could produce directly the degradation products (HI and CH
3NH
2) (state 7) going up the hill in enthalpy energy. In summary, these first-principles calculations support the experimental results,
i.e. the molecular ionic pair (CH
3NH
3+ and I
−) decomposes to energetically favored thermal degradation products (CH
3I and NH
3) in strong contrast to the previously suggested decomposition pathway to CH
3NH
2 and HI. However, it is not possible to completely rule out that under certain experimental conditions different from what was used in this experimental/theoretical study, the favored pathway would be decomposition to CH
3NH
2 and HI. Also, these first-principles DFT calculations are carried out using state 1 as the initial state of the system and studying its evolution until obtaining the degradation products (state 5 or 7). Deliberately, we have left out of this theoretical study state 0 of the system (the solid crystal phase for CH
3NH
3I or CH
3NH
3PbI
3) because it is out of the scope and it is unnecessary to explain the evolution of the molecular gas system. However, we have included in the ESI
† an estimation of such crystallization enthalpy from state 0 to state 1.
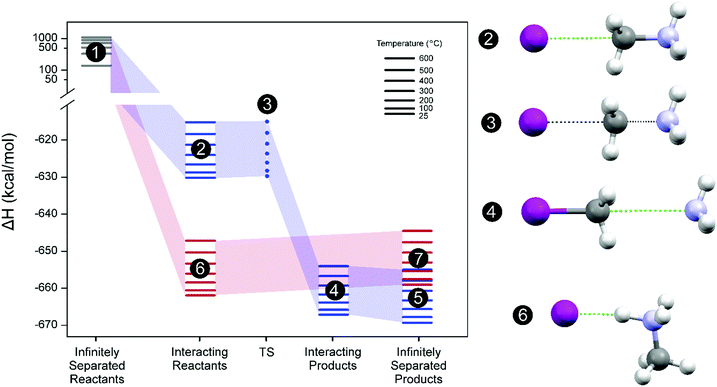 |
| | Fig. 2 Left panel: Thermodynamic cycle depicting the decomposition process of CH3NH3+ assisted by I− for the chemical reaction pathways producing CH3I and NH3 (eqn (3a), blue) and CH3NH2 and HI (eqn (3b), red). Enthalpy energy profiles with reference to the infinitely separated reactant system (CH3NH3+ + I−) at 25 °C, ΔH° = 0. Colored lines mean optimized ground state system and dot transition state (TS). Right panel: Optimized molecular geometries for states 2–4 and 6. White color sphere: H; purple: I; grey: C; cyan: N. Inset: Temperature range for the calculated thermochemical variables of the state. | |
Conclusions
In summary, by employing coupled TG-DTA and MS measurements, we demonstrate experimentally that the thermal degradation of CH3NH3PbI3 and CH3NH3I releases NH3 and CH3I as degradation products. Also, first-principles DFT calculations support the experimental results and show that only such degradation gas release can be allowed by a thermo- and kinetically favored chemical path of reaction in the temperature range of 20–600 °C and under atmospheric pressure. This result is in sharp contrast with the common wisdom. Our findings may serve as the first cornerstone in the way to obtaining long-term stabilized perovskite based photovoltaic devices.
Acknowledgements
This work was supported by funding from the Energy Materials and Surface Sciences Unit of the Okinawa Institute of Science and Technology Graduate University and JSPS KAKENHI Grant Number 15K17925. We thank Steven D. Aird, the Technical Editor at the Okinawa Institute of Science and Technology Graduate University, for valuable suggestions in revising the manuscript.
Notes and references
- N. R. E. L. (NREL), Best Research-Cell Efficiencies, http://www.nrel.gov/ncpv/images/efficiency_chart.jpg, Accessed 16 Mar, 2016.
- X. Li, M. Tschumi, H. Han, S. S. Babkair, R. A. Alzubaydi, A. A. Ansari, S. S. Habib, M. K. Nazeeruddin, S. M. Zakeeruddin and M. Gratzel, Energy Technol., 2015, 3, 551–555 CrossRef CAS.
- N. Aristidou, I. Sanchez-Molina, T. Chotchuangchutchaval, M. Brown, L. Martinez, T. Rath and S. A. Haque, Angew. Chem., Int. Ed., 2015, 54, 8208–8212 CrossRef CAS PubMed.
- D. Bryant, N. Aristidou, S. Pont, I. Sanchez-Molina, T. Chotchunangatchaval, S. Wheeler, J. R. Durrant and S. A. Haque, Energy Environ. Sci., 2016, 9, 1655–1660 CAS.
- A. D. Sheikh, A. Bera, M. A. Haque, R. B. Rakhi, S. D. Gobbo, H. N. Alshareef and T. Wu, Sol. Energy Mater. Sol. Cells, 2015, 137, 6–14 CrossRef CAS.
- R. K. Misra, S. Aharon, B. Li, D. Mogilyansky, I. Visoly-Fisher, L. Etgar and E. A. Katz, J. Phys. Chem. Lett., 2015, 6, 326–330 CrossRef CAS PubMed.
- T. A. Berhe, W.-N. Su, C.-H. Chen, C.-J. Pan, J.-H. Cheng, H.-M. Chen, M.-C. Tsai, L.-Y. Chen, A. A. Dubale and B.-J. Hwang, Energy Environ. Sci., 2016, 9, 323–356 CAS.
- T. Baikie, Y. Fang, J. M. Kadro, M. Schreyer, F. Wei, S. G. Mhaisalkar, M. Graetzel and T. J. White, J. Mater. Chem. A, 2013, 1, 5628–5641 CAS.
- Y. Dang, Y. Liu, Y. Sun, D. Yuan, X. Liu, W. Lu, G. Liu, H. Xia and X. Tao, CrystEngComm, 2015, 17, 665–670 RSC.
- L. Dimesso, M. Dimamay, M. Hamburger and W. Jaegermann, Chem. Mater., 2014, 26, 6762–6770 CrossRef CAS.
- A. Dualeh, P. Gao, S. I. Seok, M. K. Nazeeruddin and M. Gratzel, Chem. Mater., 2014, 26, 6160–6164 CrossRef CAS.
- J.-H. Im, C.-R. Lee, J.-W. Lee, S.-W. Park and N.-G. Park, Nanoscale, 2011, 3, 4088–4093 RSC.
- D. P. Nenon, J. A. Christians, L. M. Wheeler, J. L. Blackburn, E. M. Sanehira, B. Dou, M. L. Olsen, K. Zhu, J. J. Berry and J. M. Luther, Energy Environ. Sci., 2016, 9, 2072–2082 CAS.
- B. Wang, K. Young Wong, X. Xiao and T. Chen, Sci. Rep., 2015, 5, 10557 CrossRef PubMed.
- A. E. Williams, P. J. Holliman, M. J. Carnie, M. L. Davies, D. A. Worsley and T. M. Watson, J. Mater. Chem. A, 2014, 2, 19338–19346 CAS.
- W. Zhang, M. Saliba, D. T. Moore, S. K. Pathak, M. T. Horantner, T. Stergiopoulos, S. D. Stranks, G. E. Eperon, J. A. Alexander-Webber and A. Abate, et al., Nat. Commun., 2015, 6, 6142 CrossRef CAS PubMed.
- L. Liu, J. A. McLeod, R. Wang, P. Shen and S. Duhm, Appl. Phys. Lett., 2015, 107, 061904 CrossRef.
- W. Xu, L. Liu, L. Yang, P. Shen, B. Sun and J. A. McLeod, Nano Lett., 2016, 16, 4720–4725 CrossRef CAS PubMed.
- I. Yushina, B. Rudakov, I. Krivtsov and E. Bartashevich, J. Therm. Anal. Calorim., 2014, 118, 425–429 CrossRef CAS.
- M. Sawicka, P. Storoniak, P. Skurski, J. Błażejowski and J. Rak, Chem. Phys., 2006, 324, 425–437 CrossRef CAS.
- C. Muller, T. Glaser, M. Plogmeyer, M. Sendner, S. Doring, A. A. Bakulin, C. Brzuska, R. Scheer, M. S. Pshenichnikov and W. Kowalsky, et al., Chem. Mater., 2015, 27, 7835–7841 CrossRef.
- A. L. Roe, K. F. Hayes, C. Chisholm-Brause, G. E. Brown Jr, G. A. Parks, K. O. Hodgson and J. O. Leckie, Langmuir, 1991, 7, 367–373 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details for synthesis of CH3NH3PbI3 single crystals, TG/DTA-MS measurements, and DFT calculations, supplementary figures and table. See DOI: 10.1039/c6ee02016j |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ,
Zafer
Hawash
,
Sonia R.
Raga
,
Luis K.
Ono
and
Yabing
Qi
*
,
Zafer
Hawash
,
Sonia R.
Raga
,
Luis K.
Ono
and
Yabing
Qi
*