
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structurally stable Mg-doped P2-Na2/3Mn1−yMgyO2 sodium-ion battery cathodes with high rate performance: insights from electrochemical, NMR and diffraction studies†
Raphaële J.
Clément
a,
Juliette
Billaud
b,
A.
Robert Armstrong
b,
Gurpreet
Singh
c,
Teófilo
Rojo
c,
Peter G.
Bruce
bd and
Clare P.
Grey
*a
aDepartment of Chemistry, University of Cambridge, Cambridge CB2 1EW, UK. E-mail: cpg27@cam.ac.uk
bSchool of Chemistry, University of St. Andrews, St. Andrews, Fife KY16 9ST, UK
cCIC ENERGIGUNE, Parque Tecnológico de Álava, Albert Einstein 48, ED. CIC, 01510 Miñano, Spain
dDepartment of Materials, University of Oxford, Oxford OX1 3PH, UK
First published on 19th August 2016
Abstract
Sodium-ion batteries are a more sustainable alternative to the existing lithium-ion technology and could alleviate some of the stress on the global lithium market as a result of the growing electric car and portable electronics industries. Fundamental research focused on understanding the structural and electronic processes occurring on electrochemical cycling is key to devising rechargeable batteries with improved performance. We present an in-depth investigation of the effect of Mg doping on the electrochemical performance and structural stability of Na2/3MnO2 with a P2 layer stacking by comparing three compositions: Na2/3Mn1−yMgyO2 (y = 0.0, 0.05, 0.1). We show that Mg substitution leads to smoother electrochemistry, with fewer distinct electrochemical processes, improved rate performance and better capacity retention. These observations are attributed to the more gradual structural changes upon charge and discharge, as observed with synchrotron, powder X-ray, and neutron diffraction. Mg doping reduces the number of Mn3+ Jahn–Teller centers and delays the high voltage phase transition occurring in P2-Na2/3MnO2. The local structure is investigated using 23Na solid-state nuclear magnetic resonance (ssNMR) spectroscopy. The ssNMR data provide direct evidence for fewer oxygen layer shearing events, leading to a stabilized P2 phase, and an enhanced Na-ion mobility up to 3.8 V vs. Na+/Na upon Mg doping. The 5% Mg-doped phase exhibits one of the best rate performances reported to date for sodium-ion cathodes with a P2 structure, with a reversible capacity of 106 mA h g−1 at the very high discharge rate of 5000 mA g−1. In addition, its structure is highly reversible and stable cycling is obtained between 1.5 and 4.0 V vs. Na+/Na, with a capacity of approximately 140 mA h g−1 retained after 50 cycles at a rate of 1000 mA g−1.
Broader contextRechargeable lithium (Li)-ion batteries are currently the technology of choice for applications in portable electronics and in the electric vehicle industry. Yet, Li sources are scarce and discretely located on Earth, driving the search for novel, more sustainable chemistries for electrical energy storage. Sodium(Na)-based systems are one of the more promising emerging technologies, and much effort is currently put into finding high performance Na-ion electrodes and electrolytes. Sodium transition metal oxides (NaxTMO2) stand out as good cathode materials with a high volumetric energy density. Electrodes composed of a combination of low cost, abundant elements, e.g. Na, Mg, Fe, Mn, are ideally suited for large-scale applications such as grid storage. Here, we focus on a family of layered P2-Na2/3Mn1−yMgyO2 (y = 0.0, 0.05, 0.1) cathodes. Since a good understanding of the structure–composition–property relationship is key to devising better electrodes, the electrochemical performance and structural stability of the Na2/3Mn1−yMgyO2 series of compounds are carefully monitored as a function of Mg content (y), using a combination of diffraction and solid-state NMR techniques. The electrochemical performance of the y = 0.05 cathode is exceptional, the material exhibiting very stable electrochemistry upon extended cycling and one of the highest rate performance observed to date for this class of materials. |
1. Introduction
The growing demand for portable energy has driven the search for new electrode materials for rechargeable battery applications. Lithium-ion batteries (LIBs) have so far been preferred to their sodium-ion counterparts for their higher energy density and operating voltages,1 but concerns about Li supply and its rising cost have encouraged the scientific community to turn its attention to the more sustainable sodium-ion batteries (NIBs).2–8 The latter have until recently received little attention, leaving room for the exploration of new energy storage materials. Due to the abundance and low cost of Na, NIBs are considered for grid storage applications, and could be major players in next-generation low-carbon energy technologies and in the promotion of sustainable global economic growth.Sodium transition metal oxides (NaxTMO2, TM = transition metal) are well-suited for electrochemical energy storage applications as the Na+ ions can be reversibly removed (deintercalated) from or reinserted (intercalated) into the two dimensional layered structure when the electrochemical cell is charged and discharged, respectively.9–12 One of the major challenges faced by this particular class of materials are phase transformations occurring upon Na extraction and reinsertion.13 Such phenomena can result in poor reversibility upon extended cycling and are characterized by staircase-like voltage profiles.14
Mn has a high earth-abundance and low toxicity, making NaxMnO2 derivatives excellent candidates for safe, low cost, sustainable cathodes. Paulsen and Dahn investigated the structural properties and phase stability of sodium manganese bronzes based on P2-type Na2/3MnO2 (where P2 indicates that the Na+ ions are in trigonal prismatic sites (P), while the Mn ions are in octahedral sites, and that the O2− layer stacking is AB AB (2)15) and of their lithium analogs obtained by ion exchange. They showed that the substitution of Mn by Li, Ni or Co reduced and eventually suppressed the Jahn–Teller distortions present in the initial Na2/3MnO2 structure, and extended the stability range of the P2 phase.16 Since then, a large number of studies have looked at the substitution of the Jahn–Teller distorted Mn3+ ions by various transition metals in P2-type sodium transition metal oxides, as discussed in our recent review paper.17 For instance P2-Na2/3Fe1/2Mn1/2O2 exhibits good discharge capacities on cycling up to 4.0 V, but its capacity retention is drastically reduced upon cycling up to 4.3 V due to a phase transition at high voltage.18–20 Johnson and coworkers showed that Li substitution in the transition metal layer of P2-type NaxNiyMnzO2 compounds prevents the unfavorable phase transformation to the O2 phase induced by oxygen layer glides during electrochemical cycling.21 Smooth charge/discharge curves have been obtained for Na0.85[Li0.17Ni0.21Mn0.64]O2 and Na0.8[Li0.12Ni0.22Mn0.66]O2,21,22 in contrast to the staircase-like profile reported for the analogous P2-Na2/3Ni1/3Mn2/3O2 compound.23,24 A recent study showed that Mg-substituted P2-Na2/3Mn2/3Ni1/3−yMgyO2 cathodes exhibit sloping electrochemical profiles (particularly for y ≥ 0.1), no high voltage P2 to O2 phase transition, an improved cycling stability and a higher rate performance as compared with the undoped compound.25
The present study has been motivated by recent reports which suggest that Mg substitution for Mn in P2-type NaxMnO2 cathodes leads to smooth electrochemical profiles,26 enhanced Na-ion conduction,27 and high reversible capacity.28 In a preliminary electrochemical study on the P2-NaxMn1−yMgyO2 (y = 0.0, 0.05, 0.10) series, we observed high capacities and good capacity retention upon extended cycling and at high discharge rates for the Mg-doped compounds.29 Some of us recently reported on the influence of the applied discharge current on the structural evolution of Na0.67Mn0.8Mg0.2O2.30 Given the lack of a detailed investigation of the dependence of the structural and electrochemical properties of P2-NaxMn1−yMgyO2 cathodes on the Mg dopant (y) and Na (x) contents, we carry out here a comparative analysis on three different compositions: NaxMn1−yMgyO2 (y = 0.0, 0.05, 0.10). A combination of long-range (powder X-ray, neutron, and synchrotron diffraction) and short-range (solid-state nuclear magnetic resonance) structural techniques are employed ex situ to characterize the evolution of the layered framework upon electrochemical cycling at the macroscale and microscale. We show that low levels of Mg substitution in P2-Na2/3MnO2 lead to a significant enhancement of the electrochemical properties and delay the end-of-charge phase transformation. In particular, we find that the 5% Mg-doped compound exhibits exceptional electrochemical performance, with a very high rate capability comparable to the highest reported to date on P2-type Na-ion battery cathode materials,31 and excellent structural stability.
2. Experimental
2.1. Synthesis
NaxMnO2 samples (0.6 ≤ x ≤ 0.7) were prepared by solid-state synthesis. Appropriate amounts of Na2CO3 (Sigma Aldrich, anhydrous ≥99.5%) and Mn2O3 (Alfa Aesar, 99%) were ground under acetone using an agate mortar and pestle. The powder was then pelletized and calcined at 1000 °C for 15 hours in air, followed by quenching to room temperature. Various Na2/3Mn1−yMgyO2 samples (with y = 0.05 and 0.1) were prepared via a co-precipitation method. Stoichiometric amounts of Mn(CH3COO)2·4H2O and Mg(CH3COO)2·4H2O (both Aldrich) were dissolved in distilled water. In a separate container, a solution of Na2CO3 (Fisher) was dissolved in H2O. Co-precipitation was initiated by adding the acetate mixture dropwise to the Na2CO3 solution while stirring vigorously. H2O was removed by rotary evaporation and the resulting solid was dried at 300 °C for 15 hours in air. Pellets were pressed, calcined at 600 °C for 15 hours in air, quenched, ground, pelletized again and final calcination was performed at 800 °C for 15 hours in air, followed by a quench. After the heat treatment the samples were transferred to an Ar-filled glovebox for further handling.2.2. Electrochemistry
The electrochemical performance of Na2/3Mn1−yMgyO2 (y = 0.0, 0.05, 0.10) was evaluated at room temperature. Powder samples (AM) were mixed with carbon black (CB) and polyvinylidene fluoride (PVDF) in a wt% ratio AM![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CB:PVDF of 75:15:10. A slurry of the composite material was made using N-methyl-2-pyrrolidone (NMP). The components were pre-ground using a mortar and pestle and the mixture was stirred for 2–4 h prior to casting. The slurry was coated on a 50 μm thick aluminum foil using a doctor blade. The cast thickness of the film was approximately 30 μm (before pressing). The coated foil was dried at 120 °C for two hours under vacuum. Electrodes were punched and pressed under a load of ∼4 tons cm−2. The active material mass loading was approximately 5 mg cm−2. After compression, the electrodes were dried in vacuum overnight. They were then transferred to an Ar-filled glovebox in which O2 and H2O levels are maintained below 1 ppm. CR2032 coin cells were made inside the glovebox using 1 M NaClO4 in EC:PC as electrolyte, Na metal as counter electrode, and Whatman® glass-fiber filter paper as separator. The cells were charged/discharged galvanostatically at different current rates using a Maccor® battery tester. Samples for diffraction and NMR measurements were prepared as pellets of combined active material, super S carbon and Kynar Flex 2801 (a co-polymer based on PVDF) binder in a mass ratio of 75:18:7. After cycling at a rate of 10 mA g−1, the cells were transferred to an Ar-filled glovebox before opening and extracting the active material. The electrodes were then rinsed with a small amount of dry solvent to remove residual electrolyte and binder. They were left under dynamic vacuum overnight to ensure all solvent had evaporated. The sample powders were then transferred to capillaries/NMR rotors.
:CB:PVDF of 75:15:10. A slurry of the composite material was made using N-methyl-2-pyrrolidone (NMP). The components were pre-ground using a mortar and pestle and the mixture was stirred for 2–4 h prior to casting. The slurry was coated on a 50 μm thick aluminum foil using a doctor blade. The cast thickness of the film was approximately 30 μm (before pressing). The coated foil was dried at 120 °C for two hours under vacuum. Electrodes were punched and pressed under a load of ∼4 tons cm−2. The active material mass loading was approximately 5 mg cm−2. After compression, the electrodes were dried in vacuum overnight. They were then transferred to an Ar-filled glovebox in which O2 and H2O levels are maintained below 1 ppm. CR2032 coin cells were made inside the glovebox using 1 M NaClO4 in EC:PC as electrolyte, Na metal as counter electrode, and Whatman® glass-fiber filter paper as separator. The cells were charged/discharged galvanostatically at different current rates using a Maccor® battery tester. Samples for diffraction and NMR measurements were prepared as pellets of combined active material, super S carbon and Kynar Flex 2801 (a co-polymer based on PVDF) binder in a mass ratio of 75:18:7. After cycling at a rate of 10 mA g−1, the cells were transferred to an Ar-filled glovebox before opening and extracting the active material. The electrodes were then rinsed with a small amount of dry solvent to remove residual electrolyte and binder. They were left under dynamic vacuum overnight to ensure all solvent had evaporated. The sample powders were then transferred to capillaries/NMR rotors.
2.3. Diffraction
Powder X-ray diffraction measurements were performed on a Stoe STADI/P diffractometer operating in capillary mode, with FeKα1 radiation (λ = 1.936 Å) to eliminate Mn fluorescence. Samples were loaded in 0.5 mm glass capillaries and data were collected overnight. Synchrotron powder X-ray diffraction data were obtained on the same capillaries using the I11 diffractometer at the Diamond Light Source, UK. Time-of-flight powder neutron diffraction data for selected compositions were obtained using 2 mm quartz capillaries on the Polaris instrument at ISIS at the Rutherford Appleton Laboratory, UK. Rietveld refinements of the structures were carried out using the program Topas Academic.322.4. 23Na solid-state nuclear magnetic resonance (NMR)
All NMR experiments were performed under 60 kHz magic angle spinning (MAS), using a 1.3 mm double-resonance HX probe.23Na 1D spin echo spectra were recorded at room temperature on a Bruker Avance III 200 wide-bore spectrometer, at a Larmor frequency of −53.0 MHz. 23Na NMR chemical shifts were referenced against solid NaCl at 7.21 ppm. 23Na spin echo NMR spectra were acquired using a 90° RF (radio frequency) pulse of 1 μs at 25.04 W, a 180° RF pulse of 2 μs at 25.04 W, and a recycle delay of 20 ms. The NMR sample temperature is estimated to be in the range 320–330 K due to frictional heating introduced by fast MAS (60 kHz). All NMR spectra were processed with a 1 kHz applied line broadening. Lineshape analysis was carried out using the SOLA lineshape simulation package within the Bruker Topspin software. Transverse (T2′) relaxation times were obtained from an exponential fit of the decay of the signal intensity obtained as the echo delay was increased in an NMR spin echo pulse sequence. The fits and statistical analyses of the relaxation data were performed in MATLAB using an in-house program written by Prof. Andrew J. Pell. The error bars depicted in Fig. 10 represent the 95% confidence intervals for the plotted T2′ values. Longitudinal (T1) relaxation times were determined with an inversion-recovery NMR experiment. The T1 measurements confirmed that a recycle delay of 20 ms used in the spin echo experiments was long enough for the bulk magnetization to return to equilibrium between the RF pulses.3. Results and discussion
Electrochemical data obtained on the Na2/3Mn1−yMgyO2 (y = 0.0, 0.05, 0.10) phases, complementary to that reported in our earlier work,29 are presented below. In order to understand the effect of Mg doping on the structural and electronic behavior of P2-NaxMnO2 upon sodium extraction and reinsertion, various compositions on the first electrochemical cycle of the undoped, and 5 and 10% Mg-doped phases were investigated using a range of structural techniques.3.1. Electrochemical performance of Na2/3Mn1−yMgyO2 (y = 0.0, 0.05, 0.10)
Plots of the differential capacity vs. voltage are shown in Fig. 1a for the various samples, and their charge/discharge curves are shown in Fig. 1b, for the second cycle, since this allows the electrochemistry for the full range of sodium stoichiometries to be explored (Fig. 1). The first cycle electrochemical data are shown in Fig. S1 in the ESI.†
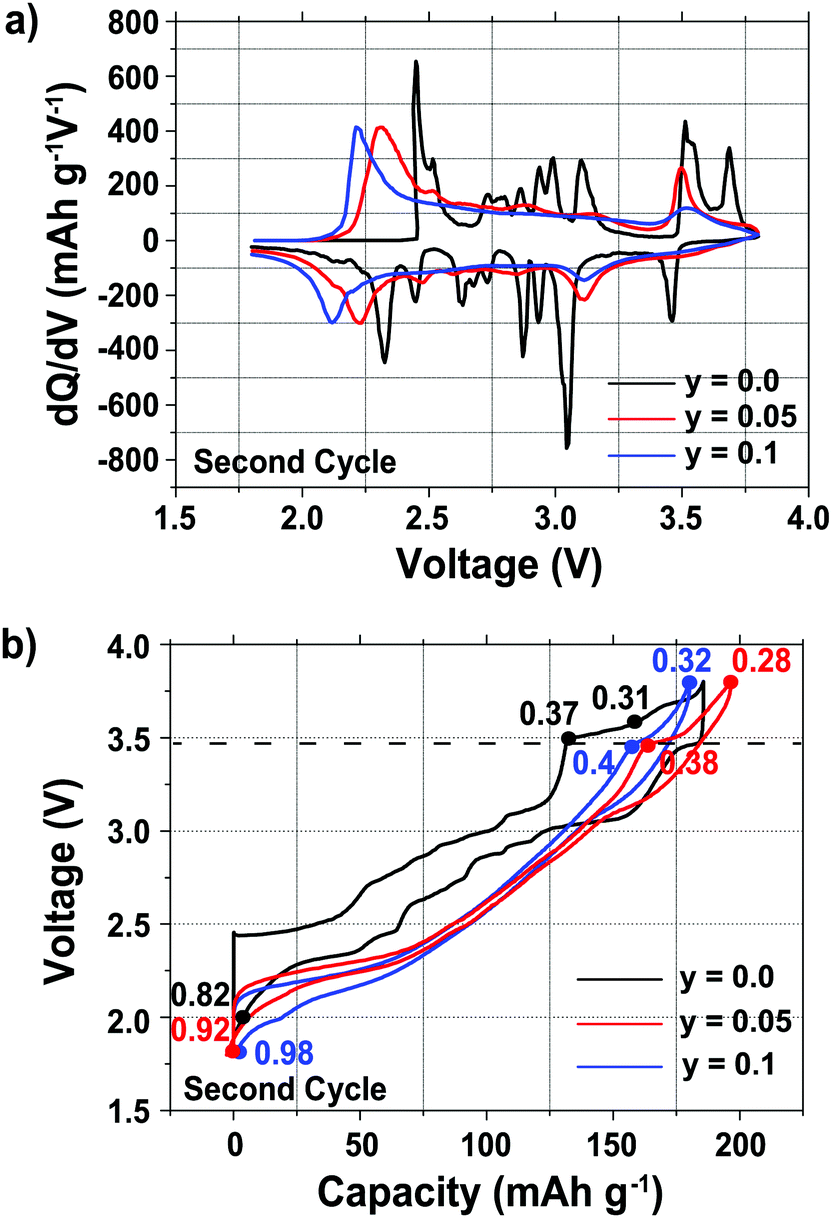 | ||
| Fig. 1 (a) Differential capacity vs. voltage plots, and (b) electrochemical profiles of the Na2/3Mn1−yMgyO2 (y = 0.0, 0.05 and 0.1) compositions. The compounds were cycled at 10 mA g−1 between 1.8 and 3.8 V vs. Na+/Na. The electrochemical cells were stopped at different points indicated on the charge/discharge curves to collect the ex situ diffraction data (the numbers on the curves correspond to the Na content, x, at which the cells were stopped). The dotted line at ca. 3.5 V indicates the voltage above which a phase transformation occurs in all compositions. The second cycles are shown in order to observe the electrochemical processes occurring below the open circuit voltage (OCV) of the as-synthesized materials, at ca. 2.5 V vs. Na+/Na. | ||
A number of sharp, low intensity peaks are observed in the differential capacity vs. voltage curve (Fig. 1a) of the undoped (y = 0.0) phase, but do not appear after Mg substitution for Mn. These peaks correspond to charge ordering and structural transitions resulting from the Jahn–Teller activity of Mn3+ ions in the as-synthesized compounds, as previously reported for related NaxMnO2 phases.11,14,33,34 For all three compositions, the oxidation and reduction peaks present at ca. 3.5 V and 3.0 V, respectively, are assigned to a reversible phase transition discussed in more detail in the later sections. Additional oxidation and reduction peaks for the undoped sample at ca. 3.7 V and 3.5 V, respectively, are assigned to a Na+ ion/vacancy ordering phenomenon. A shift is observed in the end-of-discharge peak position, presumably due to different reaction mechanisms occurring at the end of discharge upon Mg doping.
As reported previously,29 the electrochemical profiles of the Mg-doped samples, shown in Fig. 1b, are smoother than that of the undoped sample. Below 3.5 V, the Mg-doped compounds exhibit a largely sloping electrochemical profile when Na is inserted and extracted from the material. Upon galvanostatic cycling, the Mg-doped samples show a smaller hysteresis and end-of-charge polarization, reflected in the decrease in the potential difference between the oxidation and reduction peaks in the differential capacity plots (Fig. 1a), than the undoped sample.
At a low cycling rate of 12 mA g−1 the initial capacity of the y = 0.1 phase is lower than that of the y = 0.0 and 0.05 compounds (see Fig. S2 in the ESI†), yet Mg doping improves the cycling stability after 25 cycles, as discussed in our earlier work.29 The rate capability of the Na2/3Mn1−yMgyO2 (y = 0.0, 0.05, 0.10) samples was further investigated in SCFD (slow charge fast discharge) mode, by testing cells at discharge currents of 100, 200, and 500 mA g−1, while keeping the charging current constant at 12 mA g−1, as shown in Fig. 2. The y = 0.0 compound exhibits fairly stable cycling behavior at all rates investigated. At high rates of discharge, the discharge capacity of the Mg-substituted samples is noticeably higher than for the undoped compound. At a discharge rate of 500 mA g−1, for example, discharge capacities of 110, 140 and 135 mA h g−1 are observed after 50 cycles for the y = 0.0, 0.05 and 0.1 compositions, respectively. At this high Na insertion rate (500 mA g−1), the 5% Mg-doped composition exhibits remarkable capacity retention (almost 100%) with a high discharge capacity of 140 mA h g−1 (62% of the theoretical capacity). By comparison, only 45% of the theoretical capacity is retained after 50 cycles for the y = 0.0 composition.
 | ||
| Fig. 2 Evolution of the discharge capacity as a function of the cycle number for Na2/3Mn1−yMgyO2: (a) y = 0.0; (b) y = 0.05; and (c) y = 0.1. The cells were cycled between 1.5 and 4.0 V vs. Na+/Na, charged at 12 mA g−1 and discharged at various rates, as indicated. | ||
The rate capability and cycling ability reported here for the y = 0.05 and 0.1 compounds cycled between 1.5 and 4.0 V are higher than those reported by Yabuuchi et al. for the y = 0.22 material cycled between 1.5 and 4.4 V.28 The poorer rate and cycling performance for the y = 0.22 phase may result from the kinetically slow phase transformation in the high voltage region (>4.0 V) not explored in the present work.
000 mA g−1.
The rate performance of the 5% Mg-doped compound, shown in Fig. 3, is exceptional. When discharge rates of 100 mA g−1 (∼0.6C), 1000 mA g−1 (∼6C), and 2000 mA g−1 (∼11.5C) are applied, initial discharge capacities of ca. 170, 146, and 135 mA h g−1, respectively, are observed. A 106 mA h g−1 reversible capacity is observed when a very high discharge rate of 5000 mA g−1 (28.6C) is applied.31 Note that at these high rates, further electrode optimization is generally required in order to obtain improved rate performance. The rate capability of P2-Na2/3Mn0.95Mg0.05O2 is close to the highest rate performance ever reported for a P2-type cathode, namely, P2-Na2/3Mn1/2Fe1/4Co1/4O2 with a 128 mA h g−1 reversible capacity at a 30C discharge rate.31 Unlike Co-containing compounds (which generally exhibit high rate capabilities31,35,36), the materials in the present study are composed of cheap, abundant and non-toxic elements, a significant advantage for large-scale applications.
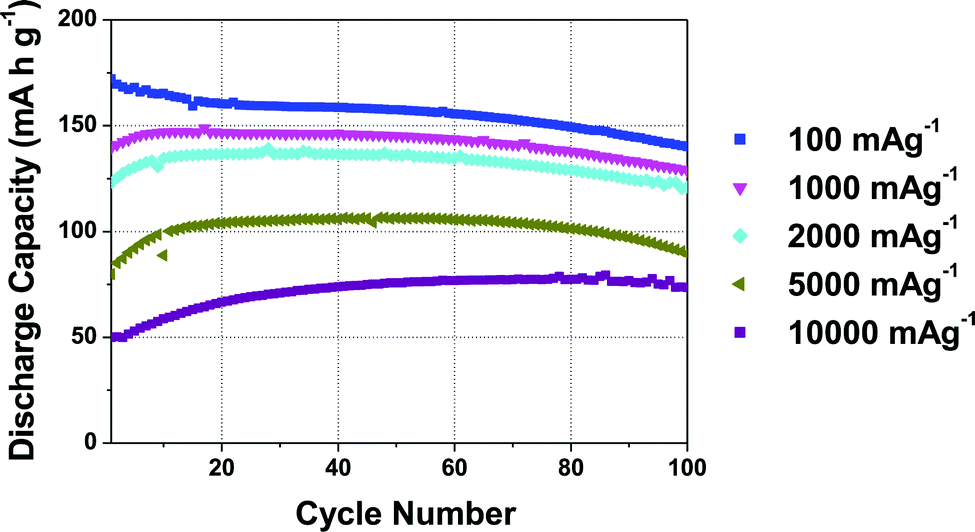 | ||
| Fig. 3 Cycling performance of the 5% Mg-doped Na2/3Mn0.95Mg0.05O2 cathode, evaluated at various discharge rates. The cells were cycled between 1.5 and 4.0 V vs. Na+/Na, charged at 13 mA g−1 and discharged at various rates, as indicated. | ||
As suggested by Sharma et al. for the 20% Mg-doped phase, the decrease in capacity observed at very high rates of 5000 and 10000 mA g−1 may be related to a lower utilization of the cathode material at the end of discharge.30 For the 20% Mg-doped compound, a drop in reversible capacity occurs as the discharge rate is increased from 100 to 400 mA g−1. A lower Mg content y = 0.05 results in a higher rate capability, which demonstrates that rate performance does not increase linearly with Mg content. We return to discuss the reasons for the good performance of the 5% Mg doped compound at the end of the paper, once a better understanding of the structural changes occurring in the various materials upon electrochemical cycling has been obtained.
3.2. Structural evolution of the Na2/3Mn1−yMgyO2 (y = 0.0, 0.05, 0.10) compounds upon cycling
The long-range structural evolution of NaxMn1−yMgyO2 (y = 0.0, 0.05, 0.10) upon cycling was monitored by ex situ powder X-ray, synchrotron X-ray, and neutron diffraction. The Na local environments in all three compositions were probed using ex situ ssNMR spectroscopy.3.2.1.i. Powder XRD and Neutron diffraction results. As-prepared Na2/3MnO2 lies on a plateau at 2.4 V (Fig. 1b) and is composed of a mixture of two phases with lower and higher sodium contents. Rietveld refinement of the diffraction pattern indicates the presence of an orthorhombic phase (space group Cmcm) and of a monoclinic phase (space group C2/n) in a 70
:30 ratio, as shown in Fig. S3 and Table S1 in the ESI.† These two very similar phases are distorted forms of the ideal hexagonal P2 structure (space group P63/mmc). The distortion is induced by the presence of Jahn–Teller active Mn3+ ions and denoted by a prime (i.e. P2′).15 In view of the two-phase behavior noted for x = 2/3, a range of different compositions were synthesized in an attempt to prepare a single-phase material. A single monoclinic (C2/n) phase was obtained when using a Na stoichiometry corresponding to x = 0.62 (Fig. S4 and Table S2 in the ESI†).
At the beginning of charge, the monoclinic distortion disappears and is replaced by an orthorhombic phase, as illustrated by the Na0.37MnO2 sample obtained after charging the material to 3.5 V vs. Na+/Na (Fig. S5a and Table S3a in the ESI†). The expansion of the c lattice parameter (corresponding to the interlayer spacing) upon charge, observed for all Na2/3Mn1−yMgyO2 (y = 0.0, 0.05, 0.10) phases, is due to the increase in the repulsive electrostatic interactions between adjacent MnO2 layers as Na+ ions are removed. Upon further Na extraction (for x ≤ 0.31 and at potentials above 3.6 V), shearing of every other MnO2 layer in the P2 structure (AB AB stacking) leads to the formation of an OP4 phase (AB BA CB BC stacking)37,38 – the OP4 structure is best described in the P63/mmc space group,39 which converts every other prismatic layer into an octahedral layer. The Na0.31MnO2 compound is composed of 66% of an orthorhombic phase and 34% of an OP4 phase with a smaller c lattice parameter (Fig. S5b and Table S3b in the ESI†). At the end of charge (x < 0.23), only the OP4 phase is observed and most of the peaks in the X-ray diffraction pattern show considerable hkl-dependent line broadening, presumably due to deviations from the ideal OP4 stacking sequence.29
As Na is reinserted, the orthorhombic phase (space group Cmcm) gradually reappears and the OP4 phase vanishes. The sample obtained at the end of discharge, Na0.82MnO2, is composed of two P2′ orthorhombic phases with different Na contents: the major phase (56%) with x = 2/3, and the minor phase (44%) with full sodium occupancy (Fig. S5c and Table S3c in the ESI†). The latter shows a highly distorted orthorhombic structure (space group Cmcm). The short distance between edge- and face-centered Na sites, shown in Fig. 4, means that only the former are occupied in the stoichiometric Na1.0MnO2 phase.
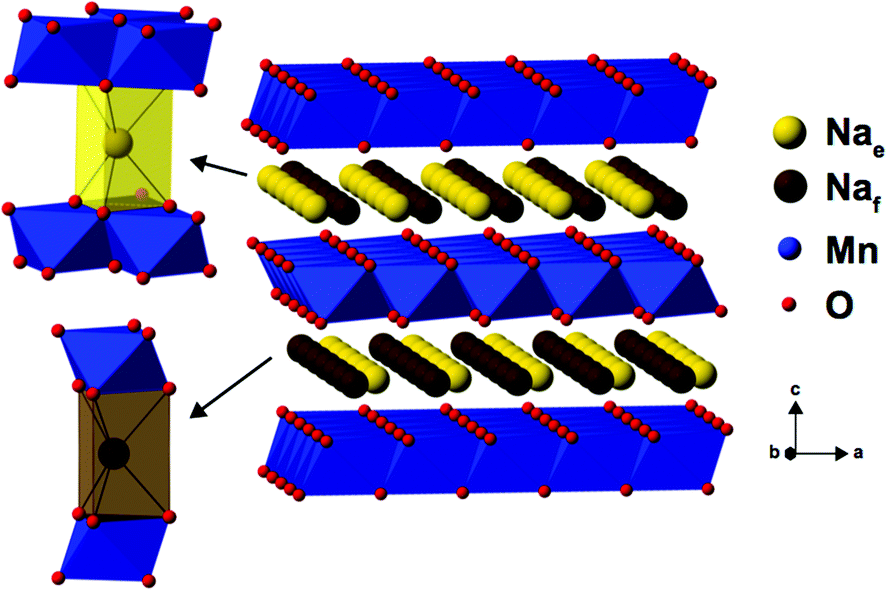 | ||
| Fig. 4 The edge-centered (Nae) and face-centered (Naf) Na sites present in P2-type NaxMn1−yMgyO2 structures. | ||
3.2.1.ii. NMR results. The spectra obtained for the undoped NaxMnO2 phase are presented in Fig. 5. The NMR spectrum of biphasic Na2/3MnO2 exhibits a number of overlapping peaks. The similarity between this and the spectrum obtained for Na0.74MnO2 collected on the end-of-discharge electrochemical plateau (at 2.3 V) confirms that the two phases present in the pristine sample are formed again at the end of the first electrochemical cycle. An increase in the Fermi contact shift (the main component of the overall 23Na shift in paramagnetic NaxMn1−yMgyO2 compounds) upon Mn3+ to Mn4+ oxidation has been reported in many Li/Na NMR studies of manganese-containing compounds.40–44 We therefore assign the high frequency (1500 to 1850 ppm range) and low frequency (650 to 1150 ppm range) set of resonances to Na sites with predominantly Mn4+ around and with predominantly Mn3+ around, respectively. The low frequency, relatively sharp 23Na resonance appearing at about 700 ppm when Na2/3MnO2 is initially discharged (see the Na0.9MnO2 spectrum), and after one charge/discharge cycle (see the Na0.82MnO2 spectrum), is in good agreement with a low average Mn oxidation state, close to +III.
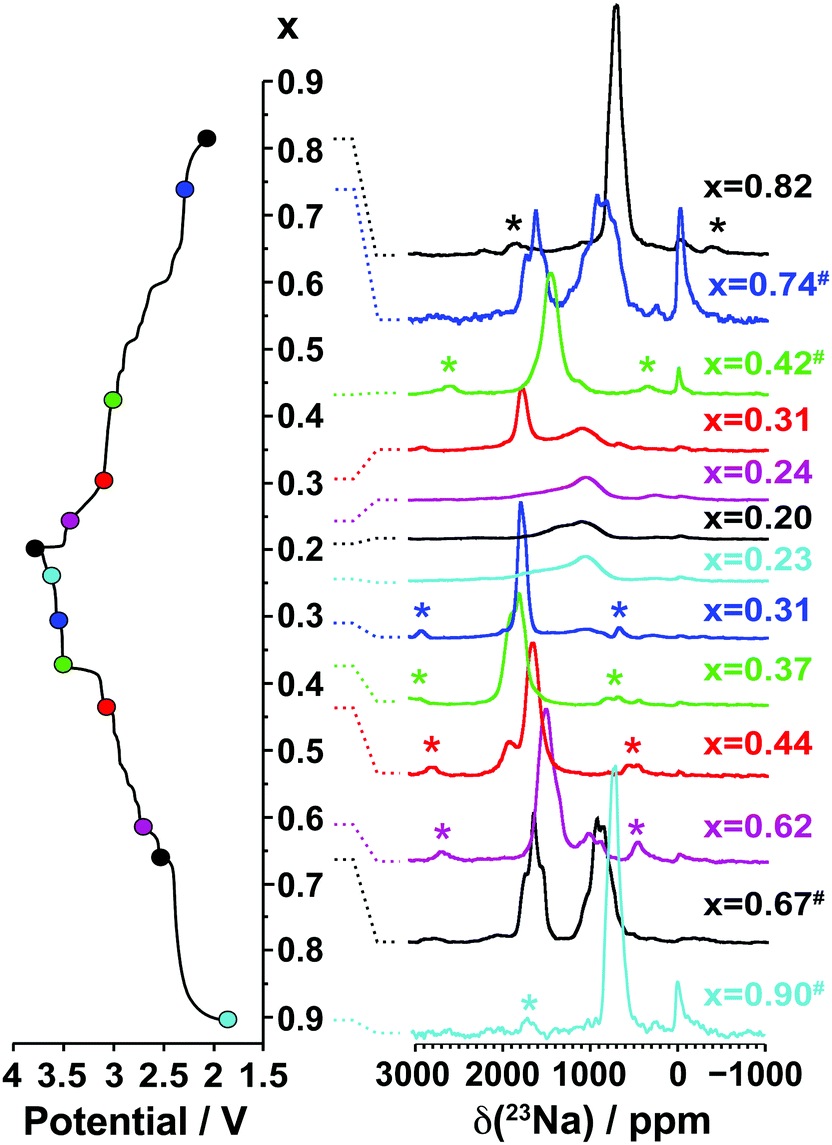 | ||
| Fig. 5 Ex situ 23Na solid-state MAS NMR spectra collected on cells stopped at different points along the first electrochemical charge/discharge cycle of Na2/3MnO2, as indicated by the colored dots on the electrochemical curve on the left hand side. These and the spectra in Fig. 8a and b have been scaled according to the number of scans collected during the NMR experiment, the amount of sample in the NMR rotor, and the NMR signal decay obtained from transverse relaxation time measurements on each sample. The hash (#) indicates samples for which a lack of experimental data prevented proper scaling of the spectrum. The peak near 0 ppm is due to Na+ ions in a diamagnetic environment, most probably from residual electrolyte or its decomposition products formed during cycling. Here and in all the 23Na spectra, the asterisks (*) indicate the spinning sidebands of the main 23Na NMR resonances introduced by fast rotation of the sample (MAS). | ||
As expected, the 23Na NMR shift increases upon charge. The numerous electrochemical plateaus observed between x = 2/3 and x = 0.31 (i.e. between 2.5 and 3.5 V on charge, see Fig. 1b) are indicative of Jahn–Teller induced structural transitions, Mn3+/Mn4+ orderings on the transition metal lattice, and/or Na+ ion/vacancy ordering transitions in the Na layers,11,14,33,34 and account for the complex changes observed in the 23Na spectra. As the Na content drops from 0.31 to 0.23, a broad peak centered around 1100 ppm (assigned to the OP4 phase observed in the Rietveld refinements) grows at the expense of the sharper peak at higher frequencies (corresponding to the orthorhombic P2′ phase). The broadening of the NMR spectrum results from Mn3+/Mn4+ oxidation and from partial layer shearing at low Na contents giving rise to a range of Na local environments and hence to a distribution of 23Na resonant frequencies. In addition, the contraction of the structure at high voltage is likely to hamper Na-ion hopping between sites, leading to low Na-ion mobility and further broadening of the NMR peaks. The origin of the decrease in the 23Na resonant frequency at the end of charge (from ca. 1800 to 1100 ppm) is discussed in the ESI.† The evolution of the NMR spectra upon discharge demonstrates the reversibility of the processes occurring upon charge.
3.2.2.i. Synchrotron XRD and neutron diffraction results. The as-prepared P2′ Na0.67Mn0.95Mg0.05O2 and Na0.67Mn0.9Mg0.1O2 phases adopt an orthorhombic symmetry (Cmcm space group), as reported by Billaud et al.29 Superlattice peaks are absent from the XRD data collected on the 5 and 10% Mg doped phases, suggesting no long-range ordered pattern of the Mg and Mn ions on the transition metal lattice, unlike that reported for the P2-type NaxMn0.89Mg0.11O2 and Na0.67Mn0.72Mg0.28O2 compounds.26,28
Upon charge, the structure of the 5% Mg-doped material remains orthorhombic down to x = 0.38 (Fig. 6a and Table S4a in the ESI†). The 10% Mg-doped compound crystallizes in the ideal P2 structure with hexagonal symmetry (P63/mmc space group) when x = 0.40 (Fig. 7a and Table S7a in the ESI†), due to the smaller proportion of Mn3+ ions in this material (22% of all Mn). At the end of charge, the Mg-doped materials do not fully transform to the OP4 phase, unlike NaxMnO2. They are composed of a major P2 phase and of a minor disordered OP4 phase (also in the P63/mmc space group), in a 60:40, and a 65:35 ratio, respectively (Fig. 6b, 7b and Tables S4b, S7b in the ESI†).
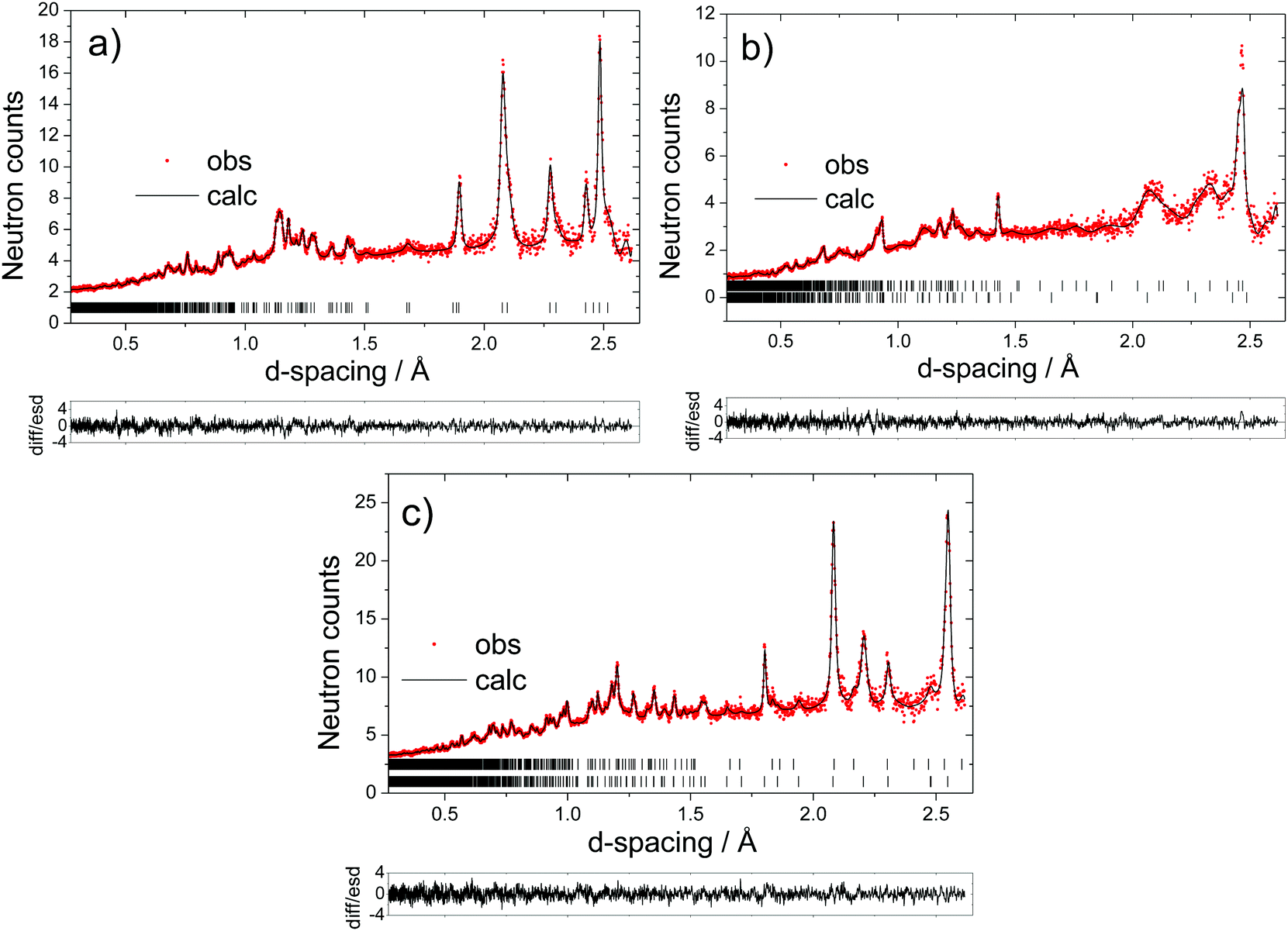 | ||
| Fig. 6 Fitted powder neutron diffraction patterns for 5% Mg-doped: (a) Na0.38Mn0.95Mg0.05O2 formed before the end-of-charge voltage plateau; (b) Na0.28Mn0.95Mg0.05O2 formed after the end-of-charge voltage plateau (upper tick marks correspond to the OP4 phase and lower tick marks to the P2 phase); and (c) Na0.92Mn0.95Mg0.05O2 obtained at the end of discharge (lower tick marks correspond to the Na1Mn0.95Mg0.05O2 reflections and upper tick marks to the Na0.7Mn0.95Mg0.05O2 reflections). In these and all subsequent diffraction patterns, the red dots represent the observed data and the solid line the calculated pattern; the lower line is the difference/esd; the tick marks represent the allowed reflections. | ||
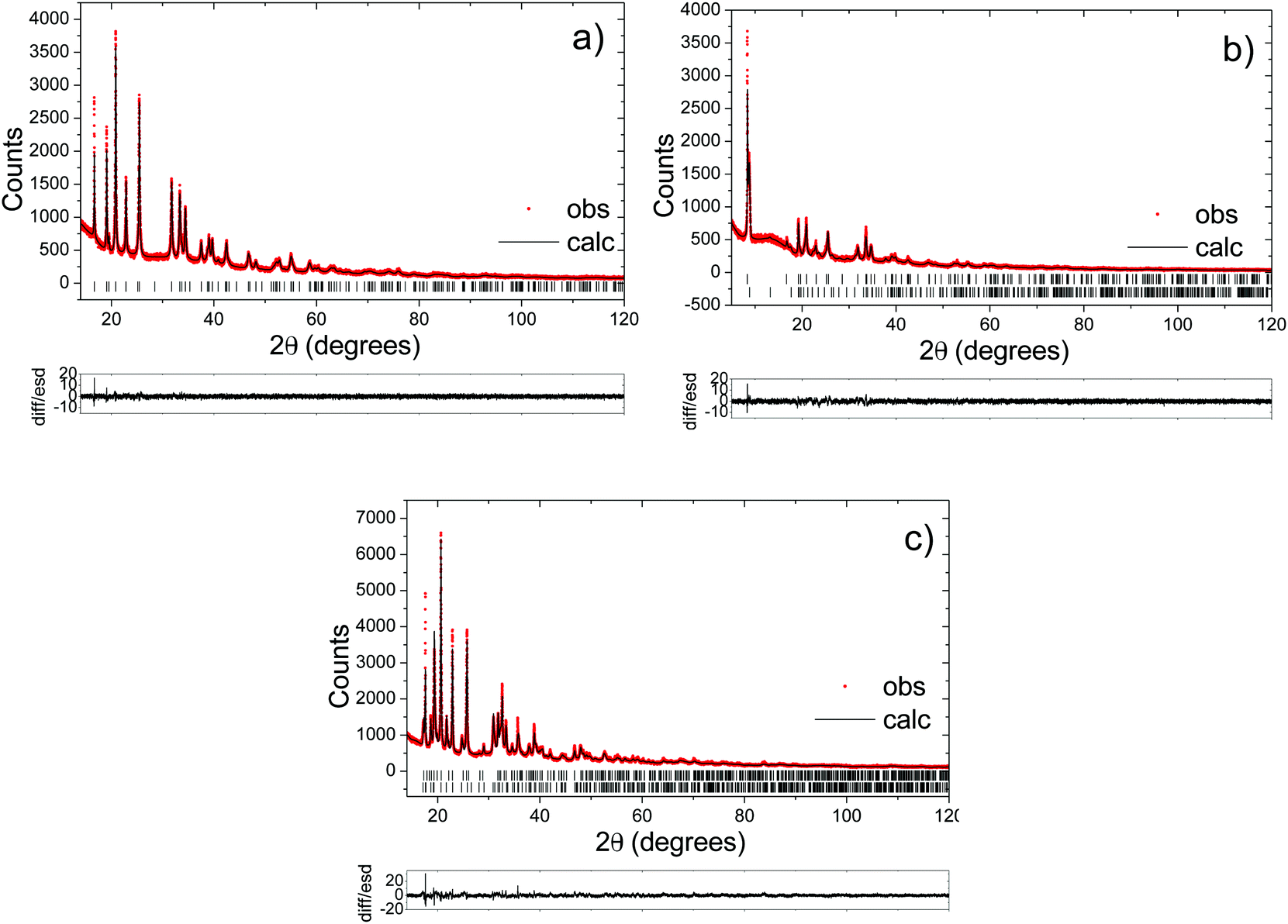 | ||
| Fig. 7 Fitted synchrotron diffraction patterns for 10% Mg-doped: (a) Na0.40Mn0.9Mg0.1O2 formed before the end-of-charge voltage plateau; (b) Na0.32Mn0.9Mg0.1O2 formed after the end-of-charge voltage plateau (upper tick marks correspond to the P2 phase and lower tick marks to the OP4 phase); and (c) Na0.98Mn0.9Mg0.1O2 obtained at the end of discharge (lower tick marks correspond to the Na1Mn0.9Mg0.1O2 reflections and upper tick marks to the Na0.7Mn0.9Mg0.1O2 reflections). | ||
As Na is reinserted, the OP4 phase disappears. For the 5% doped compound, the x = 0.40 composition crystallizes in the ideal P2 structure (Fig. S6 and Table S5 in the ESI†), and a P2′ orthorhombic phase is formed upon further Na reinsertion (Fig. S7 and Table S6 in the ESI†). As the 10% doped cathode material is discharged and x ≥ 0.43, the structure becomes and remains orthorhombic until the final stage of discharge (Fig. S8 and Table S8 in the ESI†). The Mg-doped materials exhibit a two-phase region at high Na contents (x > 0.9). Na0.92Mn0.95Mg0.05O2, obtained when the 5% Mg-doped material has been fully discharged, consists of a major phase (78%) with a Na stoichiometry of 1, and of a minor phase (22%) with a lower Na content (Fig. 6c and Table S4c in the ESI†). The 10% Mg-doped Na0.98Mn0.9Mg0.1O2 sample is composed of a major phase (88%) with x = 1, and of a minor phase (12%) with a lower Na content (Fig. 7c and Table S7c in the ESI†).
3.2.2.ii. NMR results. The 23Na NMR spectra obtained for the Mg-doped (y = 0.05, 0.1) phases are presented in Fig. 8a and b.
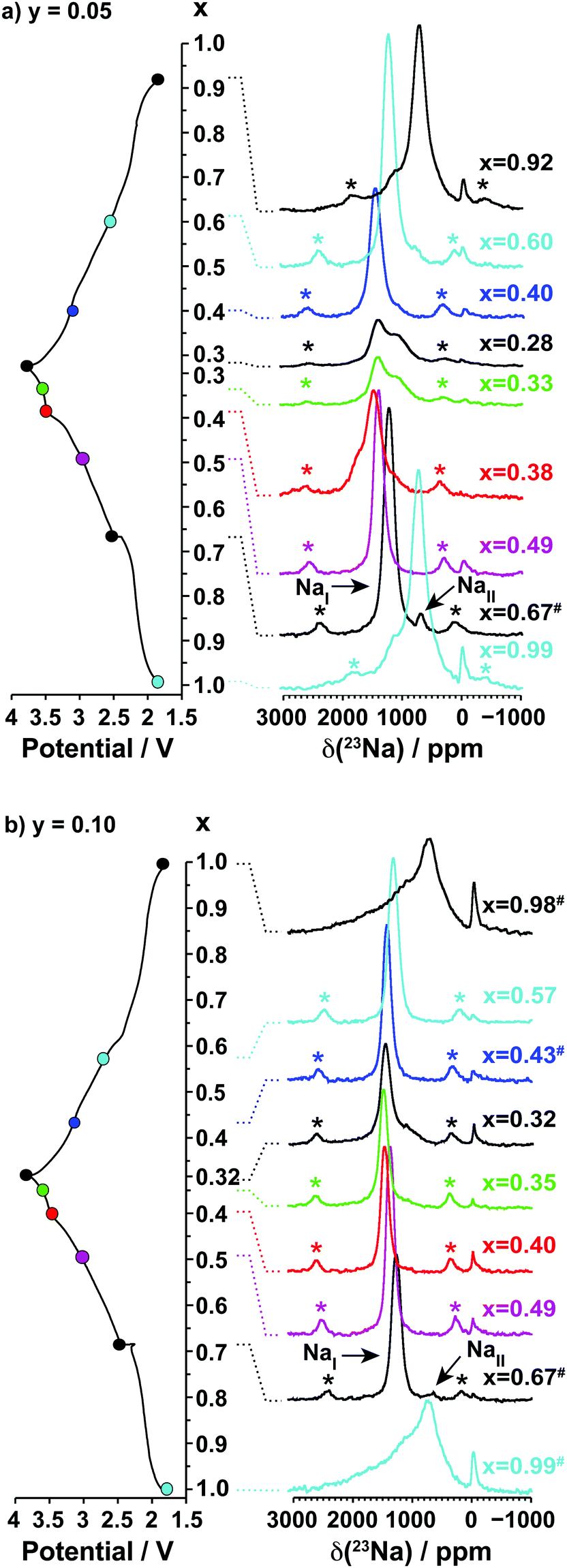 | ||
| Fig. 8 Ex situ 23Na solid-state MAS NMR spectra collected on cells stopped at different points along the first electrochemical charge/discharge cycle of Na2/3Mn1−yMgyO2: (a) y = 0.05, and (b) y = 0.10, as indicated by the colored dots on the electrochemical curve on the left hand side of the spectra. The peak near 0 ppm is due to Na+ ions in a diamagnetic environment, most probably from residual electrolyte or its decomposition products formed during cycling. | ||
Two 23Na resonances, denoted as NaI and NaII, are present in the spectra of the as-synthesized Na2/3Mn1−yMgyO2 (y = 0.05, 0.1) samples. Fits of the spectra indicate a NaI:NaII occupation ratio of 20:1 for Na0.67Mn0.95Mg0.05O2, and of 50:1 for Na0.67Mn0.9Mg0.1O2. The large population difference of the two environments rules out an assignment of NaI and NaII to the two crystallographic edge- and face-centered sites in the P2′ orthorhombic structure. Instead, the NaI resonance is assigned to an average signal resulting from rapid exchange on the NMR timescale of Na+ ions between edge- and face-centered interlayer sites in the P2′ phase (fast Na-ion motion results in coalescence of the resonances from these sites). This single, relatively sharp NaI resonance is in stark contrast to the broad overlapping peaks observed for the Na2/3MnO2 phase, the latter suggesting multiple phases and slower Na-ion motion within the Na layers in the undoped material. The low frequency NaII signal indicates the presence of a Mn3+-rich phase. It is assigned to an O3′ α-NaMnO2 impurity present in small amounts and not detected in the XRD data, as shown in Fig. S10 and discussed in more detail in the ESI.† The NaII resonance disappears at early stages of charge (as soon as x = 0.49), suggesting Na-ion removal from the α-NaMnO2 phase or domains at low voltage.
Upon Na deintercalation, the 23Na resonances shift towards higher frequencies, as noted previously for the undoped material (see Fig. 5). Mg doping, however, leads to more continuous changes in the Na resonances, and to fewer, sharper peaks throughout the first electrochemical cycle (when x ≤ 2/3). These observations, together with the smoother electrochemical curves obtained upon Mg doping and the XRD data presented earlier, suggest more gradual electronic and structural changes upon Na (de)intercalation. Unlike in NaxMnO2, there is no evidence from NMR and XRD data for Na+ ion/vacancy and/or Mn3+/Mn4+ ordering transitions upon charge of the 10% Mg-doped compound. The spectra obtained on the Na2/3Mn1−yMgyO2 (y = 0.05, 0.1) phases charged to 3.8 V indicate that the proportion of Na+ ions in OP4 distorted prismatic edge-centered sites, with a shift around 1100 ppm, decreases as the Mg doping level is increased from 5 to 10%. In principle, the 1100 ppm resonance could result from O2 stacking faults within the P2 phase, however, the OP4 phase is observed by XRD for all Mg substitution levels, consistent with the assignment of the 1100 ppm resonance to this phase rather than to an environment within a disordered P2 phase.
The evolution upon electrochemical cycling of the 23Na transverse (T2′) NMR relaxation time is shown in Fig. 9 for Na2/3Mn1−yMgyO2 (y = 0.0, 0.05, 0.1). The two T2′ values recorded at certain Na compositions correspond to two different phases: the P2 and OP4 phases in the end-of-charge samples, and the two P2′ phases with different Na contents in the end-of-discharge samples. For 2/3 ≥ x > 0.3, very short transverse relaxation times, in the range of 33 to 112 μs, are obtained for the Mg-doped phases. Li-ion motion on the timescale of μs to ms has recently been shown to be a major source of fast T2′ relaxation (short relaxation times) in lithium-containing paramagnetic cathodes.45 By analogy, we ascribe the short 23Na T2′ times to rapid Na-ion hopping between edge- and face-centered prismatic sites in the P-type layers. The range of Na compositions over which transverse relaxation is fast is reduced for NaxMnO2, in agreement with the various ordering and structural transitions occurring upon cycling and affecting fast Na-ion conduction.
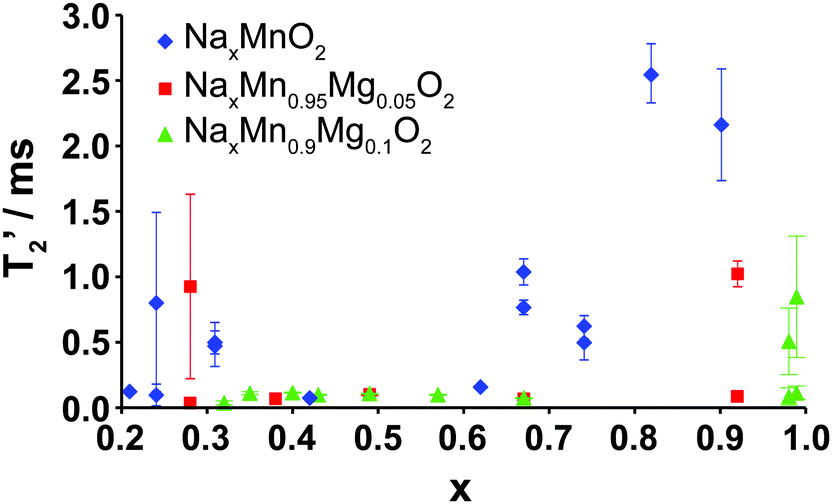 | ||
| Fig. 9 Plot of the transverse NMR relaxation time (T2′) obtained on samples at different points along the first electrochemical charge/discharge cycle of Na2/3Mn1−yMgyO2 (y = 0.0, 0.05, 0.1) as a function of Na content (x). Two relaxation times are plotted on the graph when two phases or local environments were identified in the sample. Error bars for the determination of the T2′ values represent the 95% confidence intervals. | ||
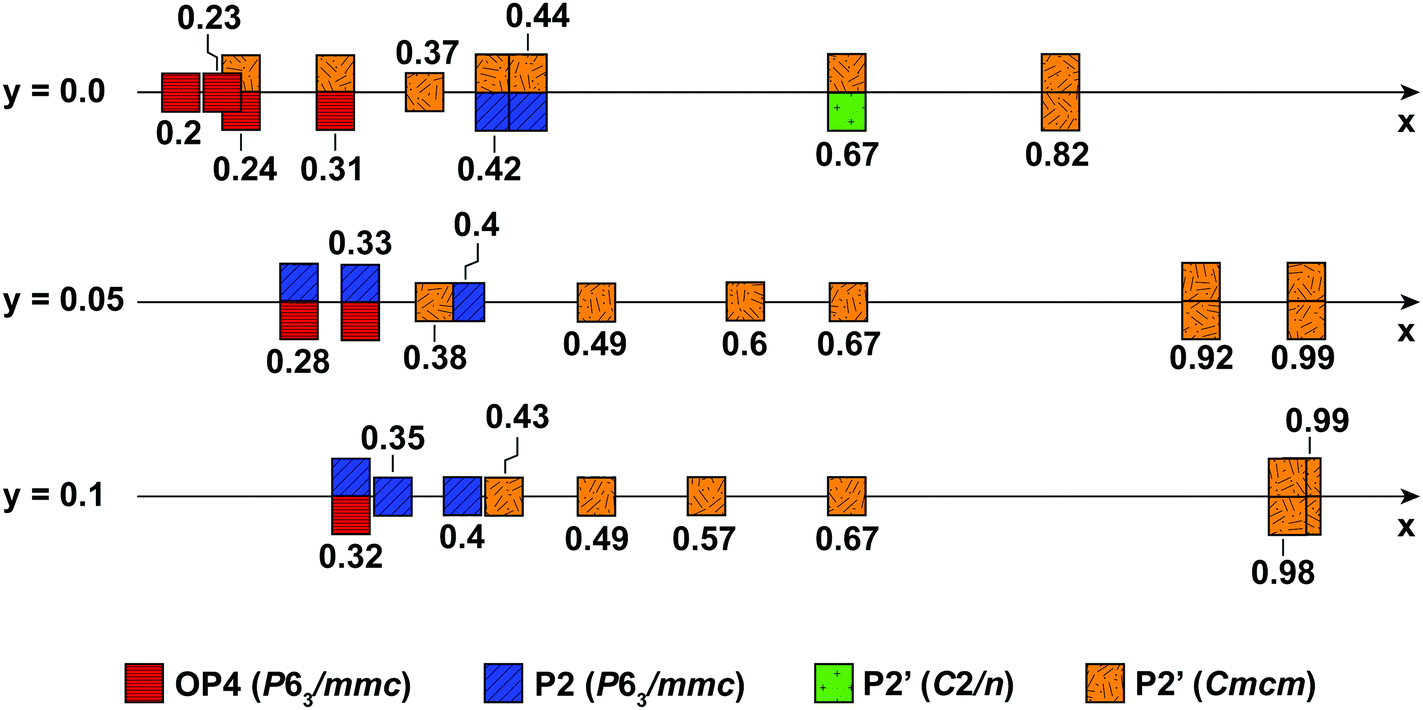 | ||
| Fig. 10 Ranges of P2, P2′ and OP4 phase stability for NaxMn1−yMgyO2 (y = 0.0, 0.05, 0.1) upon electrochemical Na removal and reinsertion, obtained from Rietveld refinements of diffraction data. The ideal hexagonal P2 phase is depicted in blue, the OP4 phase in red, and the Jahn–Teller distorted P2′ phases are depicted in green (monoclinic distortion) and orange (orthorhombic distortion). When more than one phase is observed, e.g. for the 10% Mg-doped material at x = 0.98 composed of two orthorhombic phases, two squares are shown above and below the x axis. | ||
Long T2′ relaxation times (>2.0 ms) are recorded for the discharged NaxMnO2 samples. The broad resonances observed in the spectra collected on the biphasic 5 and 10% Mg-doped end-of-discharge (x ≥ 0.92) samples (see Fig. 8a and b) result from a combination of frequency overlap between the signals assigned to the two phases and a spread in the 23Na resonant frequencies due to a range of TM–TM (and TM–O) bond distances in the severely Jahn–Teller distorted phases.18 The two phases have very different T2′ relaxation behaviors. The slow relaxing component (T2′ > 0.5 ms) is assigned to rigid Na+ ions in the orthorhombically-distorted x = 1.0 phase, leading to further broadening of the spectra. The faster relaxing component is assigned to the more mobile Na+ ions in the second phase with a lower Na content.
At the end of charge, broad resonances and increased T2′ relaxation times (0.8–1.0 ms) are recorded for the 0 and 5% Mg-doped materials. The broad spectra are due to partial layer shearing and increased structural disorder at short length scales, as well as overlap between the residual P2 resonance around 1450 ppm, and the OP4 resonance around 1100 ppm (see Fig. 5 and 8a). 10% Mg substitution leads to significant improvements in Na-ion mobility up to 3.8 V charge, demonstrated by narrow 23Na NMR peaks and transverse relaxation times below 112 μs throughout cycling (for x ≤ 2/3).
3.3. How does Mg doping improve the structural and electrochemical properties of P2-Na2/3MnO2?
Fig. 10 summarizes the structural evolution of NaxMn1−yMgyO2 (y = 0.0, 0.05, 0.1) as a function of Na content (x), as inferred from Rietveld refinements on different samples obtained by electrochemical Na extraction/insertion presented in this paper, in the ESI† or in our previous study.29 While structures with an orthorhombic and/or monoclinic P2′ distortion (shown as orange and green squares, respectively) are generally observed at higher Na contents, hexagonal P2 structures (blue squares) and OP4 phases (red squares) are obtained for x ≤ 0.44.By increasing the average Mn oxidation state, i.e. by reducing the number of Jahn–Teller active ions in P2 NaxMnO2, Mg doping leads to more gradual and more reversible structural and electronic changes upon cycling, as indicated by diffraction and NMR results. Sloping profiles are obtained at the beginning of charge and up to 3.5 V for the Mg-doped compounds (see Fig. 1).
The extent of layer shearing at high voltage depends on the number of remaining Na+ ions holding the MnO2 layers together. As shown in Fig. 10, the P2 to OP4 phase transformation occurs around x = 0.31–0.33, irrespective of Mg content. In the undoped material, complete phase transformation to the OP4 phase occurs by 3.6 V, when x < 0.24. In the 5 and 10% Mg-doped compounds, with an end-of-charge Na content of 0.28 and 0.32, respectively, the frequency of layer shearing events is reduced and partial P2 to OP4 phase transition takes place at a higher potential.
The P2 to OP4 phase transformation leads to oxidation peaks above 3.5 V in the electrochemical dQ/dV curves (Fig. 1a) and to a rise in the overpotential of the cell, because of the activation barrier associated with the phase transition process. The decrease in the end-of-charge overpotential (Fig. 1b) is in agreement with the smaller fraction of OP4 phase formed as more Mg is doped into P2-NaxMnO2.
The formation of an OP4 phase, intermediate between the P2 and O2 layer stackings, appears to be less structurally damaging, hence more reversible, than the P2–O2 phase transition observed upon Na extraction from e.g. P2-Na2/3Ni1/3Mn2/3O2.24 By increasing the average Mn oxidation state for a given Na content and allowing Na to be extracted at higher voltage, Mg substitution delays, rather than completely prevents, O layer glides. As reported for P2-Na2/3Mn2/3Ni1/3−yMgyO2 compounds,25 the stabilization of the P2-NaxMnO2 structure upon Mg doping leads to good cycling stability.
The high voltage plateau is observed at all rates explored in this work, suggesting that the partial P2 to OP4 phase transformation, which induces minimal structural changes as compared with the P2 to Cmcm phase transition, is not rate limiting in the 5% Mg-doped material. The exact cause for poor cycling stability in P2-type cathodes is still not clearly understood, yet it has been related to the high voltage phase transition in P2-NaxMn1/2Fe1/2O2,18,20,50 and in P2-NaxMn1/2Fe1/4Co1/4O2.31 Here, the large polarization and hysteresis observed at high voltage (see Fig. 1b) presumably leads to capacity fade upon extended cycling (see Fig. 2), which is exacerbated at high discharge rates (see Fig. 3). As suggested for P2-NaxMn1/2Fe1/2O2,50 the large volume changes associated with the high voltage transition from the P2 to the OP4 phase likely contribute to structural irreversibility. Rapid expansion of the y = 0.05 structure when the OP4 component converts back to the P2 phase leads to poorer structural stability, hence poorer capacity retention, at very high discharge rates.
4. Conclusions
An in-depth comparative study of the electrochemical properties and structural changes occurring upon cycling of P2-type NaxMn1−yMgyO2 (y = 0.0, 0.05, 0.1) materials showed that low levels of Mg doping (5 and 10%) improve the electrochemical performance and structural stability of the material. While long-range structural changes were monitored using powder X-ray, neutron, and synchrotron diffraction, local changes were characterized with 23Na solid-state NMR to obtain a full picture of the structural evolution of the cathode materials at both lengthscales. The presence of Mg on the transition metal lattice leads to fewer Jahn–Teller distorted Mn3+ ions and disrupts potential Mn3+/Mn4+ ordering. As a result, fewer structural and electronic processes take place as the cell is cycled and smoother load curves are obtained. In addition, the greater number of Na+ ions present at the end of charge, when Mg is substituted in the compound, increases the voltage range over which the P2 phase is stable and delays the occurrence of oxygen layer glides leading to the formation of an OP4 phase. Although the theoretical capacity decreases as more electrochemically-inactive Mg is introduced into the material, the presence of Mg leads to higher capacity retention after 50 cycles at a 500 mA g−1 discharge rate (110 mA h g−1 for the undoped phase, vs. 140 and 135 mA h g−1 for the Mg doped phases). The 5% Mg-doped compound exhibits exceptional rate performance, with ca. 106 mA h g−1 initial reversible capacity observed at a 5000 mA g−1 discharge rate, and excellent capacity retention. Such rate performance is among the highest observed for P2-type cathode materials.Acknowledgements
This work was partially supported by the Assistant Secretary for Energy Efficiency and Renewable Energy, Office of Vehicle Technologies of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231, under the Batteries for Advanced Transportation Technologies (BATT) Program subcontract #7057154 (R. J. C.). C. P. G. and R. J. C. thank the EU ERC for an Advanced Fellowship for CPG. This work was partially supported by the LINABATT project from the Ministerio de Economía Competitividad under Contract No. ENE2013-44330-R (G. S. and T. R.). P. G. B. is grateful to the EPSRC, including the SUPREGEN programme, for financial support. Please find Oxford University Research Archive (ORA) deposit at DOI: 10.5287/bodleian:r1mEe405n.References
- M. Armand and J.-M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed
.
- S.-W. Kim, D.-H. Seo, X. Ma, G. Ceder and K. Kang, Adv. Energy Mater., 2012, 2, 710–721 CrossRef CAS
- H. Pan, Y.-S. Hu and L. Chen, Energy Environ. Sci., 2013, 6, 2338 CAS
- M. D. Slater, D. Kim, E. Lee and C. S. Johnson, Adv. Funct. Mater., 2013, 23, 947–958 CrossRef CAS
- N. Yabuuchi, K. Kubota, M. Dahbi and S. Komaba, Chem. Rev., 2014, 114, 11636–11682 CrossRef CAS PubMed
- V. Palomares, M. Casas-Cabanas, E. Castillo-Martínez, M. H. Han and T. Rojo, Energy Environ. Sci., 2013, 6, 2312 CAS
- V. Palomares, P. Serras, I. Villaluenga, K. B. Hueso, J. Carretero-González and T. Rojo, Energy Environ. Sci., 2012, 5, 5884–5901 CAS
- M. Dahbi, N. Yabuuchi, K. Kubota, K. Tokiwa and S. Komaba, Phys. Chem. Chem. Phys., 2014, 16, 15007–15028 RSC
- Y. Takeda, K. Nakahara, M. Nishijima and N. Imanishi, Mater. Res. Bull., 1994, 29, 659–666 CrossRef CAS
- S. Komaba, C. Takei, T. Nakayama, A. Ogata and N. Yabuuchi, Electrochem. Commun., 2010, 12, 355–358 CrossRef CAS
- A. Mendiboure, C. Delmas and P. Hagenmuller, J. Solid State Chem., 1985, 57, 323–331 CrossRef CAS
- M. H. Han, E. Gonzalo, G. Singh and T. Rojo, Energy Environ. Sci., 2015, 8, 81–102 CAS
- M. Galceran, V. Roddatis, F. J. Zúñiga, J. M. Pérez-Mato, B. Acebedo, R. Arenal, I. Peral, T. Rojo and M. Casas-Cabanas, Chem. Mater., 2014, 26, 3289–3294 CrossRef CAS
- A. Caballero, L. Hernán, J. Morales, L. Sánchez, J. S. Peña and M. A. G. Aranda, J. Mater. Chem., 2002, 12, 1142–1147 RSC
- C. Delmas, C. Fouassier and P. Hagenmuller, Physica B+C, 1980, 99, 81–85 CrossRef CAS
- J. M. Paulsen and J. R. Dahn, Solid State Ionics, 1999, 126, 3–24 CrossRef CAS
- R. J. Clément, P. G. Bruce and C. P. Grey, J. Electrochem. Soc., 2015, 162, A2589–A2604 CrossRef
- B. Mortemard de Boisse, D. Carlier, M. Guignard, L. Bourgeois and C. Delmas, Inorg. Chem., 2014, 53, 11197–11205 CrossRef CAS PubMed
- G. Singh, J. M. L. del Amo, M. Galceran, S. Pérez-Villar and T. Rojo, J. Mater. Chem. A, 2015, 3, 6954–6961 CAS
- E. Talaie, V. Duffort, H. L. Smith, B. Fultz and L. F. Nazar, Energy Environ. Sci., 2015, 8, 2512–2523 CAS
- D. Kim, S.-H. Kang, M. Slater, S. Rood, J. T. Vaughey, N. Karan, M. Balasubramanian and C. S. Johnson, Adv. Energy Mater., 2011, 1, 333–336 CrossRef CAS
- J. Xu, D. H. Lee, R. J. Clément, X. Yu, M. Leskes, A. J. Pell, G. Pintacuda, X.-Q. Yang, C. P. Grey and Y. S. Meng, Chem. Mater., 2014, 26, 1260–1269 CrossRef CAS
- Z. Lu and J. R. Dahn, J. Electrochem. Soc., 2001, 148, A1225–A1229 CrossRef CAS
- D. H. Lee, J. Xu and Y. S. Meng, Phys. Chem. Chem. Phys., 2013, 15, 3304–3312 RSC
- P. F. Wang, Y. You, Y.-X. Yin, Y. S. Wang, L.-J. Wan, L. Gu and Y.-G. Guo, Angew. Chem., 2016, 128, 7571–7575 CrossRef
- D. Buchholz, C. Vaalma, L. G. Chagas and S. Passerini, J. Power Sources, 2015, 282, 581–585 CrossRef CAS
- Z.-Y. Li, R. Gao, J. Zhang, X. Zhang, Z. Hu and X. Liu, J. Mater. Chem. A, 2016, 4, 3453–3461 CAS
- N. Yabuuchi, R. Hara, K. Kubota, J. Paulsen, S. Kumakura and S. Komaba, J. Mater. Chem. A, 2014, 2, 16851–16855 CAS
- J. Billaud, G. Singh, A. R. Armstrong, E. Gonzalo, V. Roddatis, M. Armand, T. Rojo and P. G. Bruce, Energy Environ. Sci., 2014, 7, 1387–1391 CAS
- N. Sharma, N. Tapia-Ruiz, G. Singh, A. R. Armstrong, J. C. Pramudita, H. E. A. Brand, J. Billaud, P. G. Bruce and T. Rojo, Chem. Mater., 2015, 27, 6976–6986 CrossRef CAS
- L. Liu, X. Li, S.-H. Bo, Y. Wang, H. Chen, N. Twu, D. Wu and G. Ceder, Adv. Energy Mater., 2015, 5, 1500944 CrossRef
- A. A. Coelho, J. Appl. Crystallogr., 2000, 33, 899–908 CrossRef CAS
- X. Li, X. Ma, D. Su, L. Liu, R. Chisnell, S. P. Ong, H. Chen, A. Toumar, J.-C. Idrobo, Y. Lei, J. Bai, F. Wang, J. W. Lynn, Y. S. Lee and G. Ceder, Nat. Mater., 2014, 13, 586–592 CrossRef CAS PubMed
- D. Su, C. Wang, H. J. Ahn and G. Wang, Chem. – Eur. J., 2013, 19, 10884–10889 CrossRef CAS PubMed
- Z.-Y. Li, R. Gao, L. Sun, Z. Hu and X. Liu, J. Mater. Chem. A, 2015, 3, 16272–16278 CAS
- T. Shibata, Y. Fukuzumi, W. Kobayashi and Y. Moritomo, Sci. Rep., 2015, 5, 9006 CrossRef CAS PubMed
- Z. Ren, J. Shen, S. Jiang, X. Chen, C. Feng, Z. Xu and G. Cao, J. Phys.: Condens. Matter, 2006, 18, L379 CrossRef CAS
- R. Balsys, Solid State Ionics, 1997, 93, 279–282 CrossRef CAS
- R. Berthelot, M. Pollet, D. Carlier and C. Delmas, Inorg. Chem., 2011, 50, 2420–2430 CrossRef CAS PubMed
- J. Billaud, R. J. Clément, A. R. Armstrong, J. Canales-Vázquez, P. Rozier, C. P. Grey and P. G. Bruce, J. Am. Chem. Soc., 2014, 136, 17243–17248 CrossRef CAS PubMed
- Y. J. Lee and C. P. Grey, Chem. Mater., 2000, 12, 3871–3878 CrossRef CAS
- K. A. Aldi, J. Cabana, P. J. Sideris and C. P. Grey, Am. Mineral., 2012, 97, 883–889 CrossRef CAS
- H. Duncan, B. Hai, M. Leskes, C. P. Grey and G. Chen, Chem. Mater., 2014, 26, 5374–5382 CrossRef CAS
- Y. J. Lee, F. Wang and C. P. Grey, J. Am. Chem. Soc., 1998, 120, 12601–12613 CrossRef CAS
- L. Zhou, M. Leskes, T. Liu and C. P. Grey, Angew. Chem., Int. Ed., 2015, 54, 14782–14786 CrossRef CAS PubMed
- G. J. Shu and F. C. Chou, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 052101 CrossRef
- A. J. Toumar, S. P. Ong, W. D. Richards, S. Dacek and G. Ceder, Phys. Rev. Appl., 2015, 4, 064002 CrossRef
- Y. Wang, R. Xiao, Y.-S. Hu, M. Avdeev and L. Chen, Nat. Commun., 2015, 6, 6954 CrossRef CAS PubMed
- N. Bucher, S. Hartung, J. B. Franklin, A. M. Wise, L. Y. Lim, H.-Y. Chen, J. N. Weker, M. F. Toney and M. Srinivasan, Chem. Mater., 2016, 28, 2041–2051 CrossRef CAS
- W. K. Pang, S. Kalluri, V. K. Peterson, N. Sharma, J. Kimpton, B. Johannessen, H. K. Liu, S. X. Dou and Z. Guo, Chem. Mater., 2015, 27, 3150–3158 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ee01750a |
| This journal is © The Royal Society of Chemistry 2016 |