
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNitrogenase bioelectrocatalysis: heterogeneous ammonia and hydrogen production by MoFe protein†
Ross D.
Milton
ab,
Sofiene
Abdellaoui
a,
Nimesh
Khadka
c,
Dennis R.
Dean
d,
Dónal
Leech
b,
Lance C.
Seefeldt
c and
Shelley D.
Minteer
*a
aDepartment of Chemistry, University of Utah, 315 S 1400 E Room 2020, Salt Lake City, Utah 84112, USA. E-mail: minteer@chem.utah.edu
bSchool of Chemistry, National University of Ireland Galway, University Road, Galway, Ireland
cDepartment of Chemistry and Biochemistry, Utah State University, Logan, Utah 84322, USA
dDepartment of Biochemistry, Virginia Tech University, Blacksburg, Virginia 24061, USA
First published on 20th June 2016
Abstract
Nitrogenase is the only enzyme known to catalyze the reduction of N2 to 2NH3. In vivo, the MoFe protein component of nitrogenase is exclusively reduced by the ATP-hydrolyzing Fe protein in a series of transient association/dissociation steps that are linked to the hydrolysis of two ATP for each electron transferred. We report MoFe protein immobilized at an electrode surface, where cobaltocene (as an electron mediator that can be observed in real time at a carbon electrode) is used to reduce the MoFe protein (independent of the Fe protein and of ATP hydrolysis) and support the bioelectrocatalytic reduction of protons to dihydrogen, azide to ammonia, and nitrite to ammonia. Bulk bioelectrosynthetic N3− or NO2− reduction (50 mM) for 30 minutes yielded 70 ± 9 nmol NH3 and 234 ± 62 nmol NH3, with NO2− reduction operating at high faradaic efficiency.
Broader contextConsuming over 1% of the world's energy resources in the estimated production of 150 million metric tons per year, ammonia (NH3) is an important chemical commodity to modern civilization. Currently the majority of NH3 (approximately 66%) is produced by the Haber–Bosch process from N2 and H2, although high temperatures and high pressures (400 °C and 20 MPa) are required, which are estimated to be responsible for approximately 3% of CO2 emissions. Nitrogenase is the only enzyme known to reduce N2 to NH3 (along with concomitant H2 production) and it is able to do so at room temperature, neutral pH and ambient pressure although a large input of chemical energy is also required (16 ATP per N2). We have bypassed the reducing- and ATP-hydrolyzing properties of the Fe protein of nitrogenase by immobilizing the catalytic protein of nitrogenase (MoFe protein) at a carbon electrode surface whereby a suitable electron mediator (cobaltocene) is able to support the bioelectrocatalytic reduction of 2H+ to H2, N3− to NH3 and NO2− to NH3 under mild conditions (room temperature, neutral pH and ambient pressure). |
The reduction of effectively-inert dinitrogen (N2), the major constituent of the Earth's atmosphere (79%), to more biologically and industrially useful forms of nitrogen (i.e. NH3) is a key step in the global biogeochemical N cycle.1 From a biological standpoint, a select group of diazotrophic microorganisms (limited to bacteria and archaea) are able to reduce atmospheric N2 to NH3 under both aerobic and anaerobic growth conditions by way of a single enzyme, nitrogenase.2 Industrially, the renowned Haber–Bosch process highlights the importance of N2 fixation at the expense of high pressures, high temperatures and consumes approximately 1% of the world's energy resources (as of 2002).3,4 The ability to electrochemically produce ammonia at room temperature under ambient pressure would present a significant alternative technology to the Haber–Bosch process.
Nitrogenase is an enzyme that is able to reduce N2 to NH3 at the expense of ATP hydrolysis and a reductant (typically ferredoxin or flavodoxin in vivo), of which three major classes are characterized by their Mo-, V- or Fe-dependent catalytic cofactors (FeMo-co, VFe-co, FeFe-co respectively);5–8 this study focuses on Mo-dependent nitrogenase. In addition to the ability of nitrogenase to reduce N2, other interesting substrates include H+, C2H2, N3−, HCN, NO2−, N2H4, CO2, CO, R-CN and R-NC.1,4,9,10
Mo-dependent nitrogenase consists of two protein components that are highly sensitive to O2; a Fe protein and a MoFe protein.1 The Fe protein (a ∼66 kDa homodimer) is responsible for reducing the MoFe protein (a ∼240 kDa dimer of dimers), in a series of individual electron transfer events that are coupled to the hydrolysis of two MgATP molecules to MgADP (Fig. 1).2 The Fe protein bound to two MgATP molecules transiently associates with the MoFe protein, where a single electron is transferred from the Fe4S4 cluster of the Fe protein to the MoFe protein in a process that is coupled to the hydrolysis of two MgATP to two MgADP. These events are followed by the dissociation of the Fe protein from the MoFe protein. Within the MoFe protein, electrons are shuttled from the P cluster to the active site FeMo-cofactor where N2 is reduced to 2NH3 with the concomitant reduction of 2H+ to H2 (eqn (1)).2
| 16MgATP + 8e− + 8H+ + N2 → 16MgADP + 2NH3 + H2 + 16Pi | (1) |
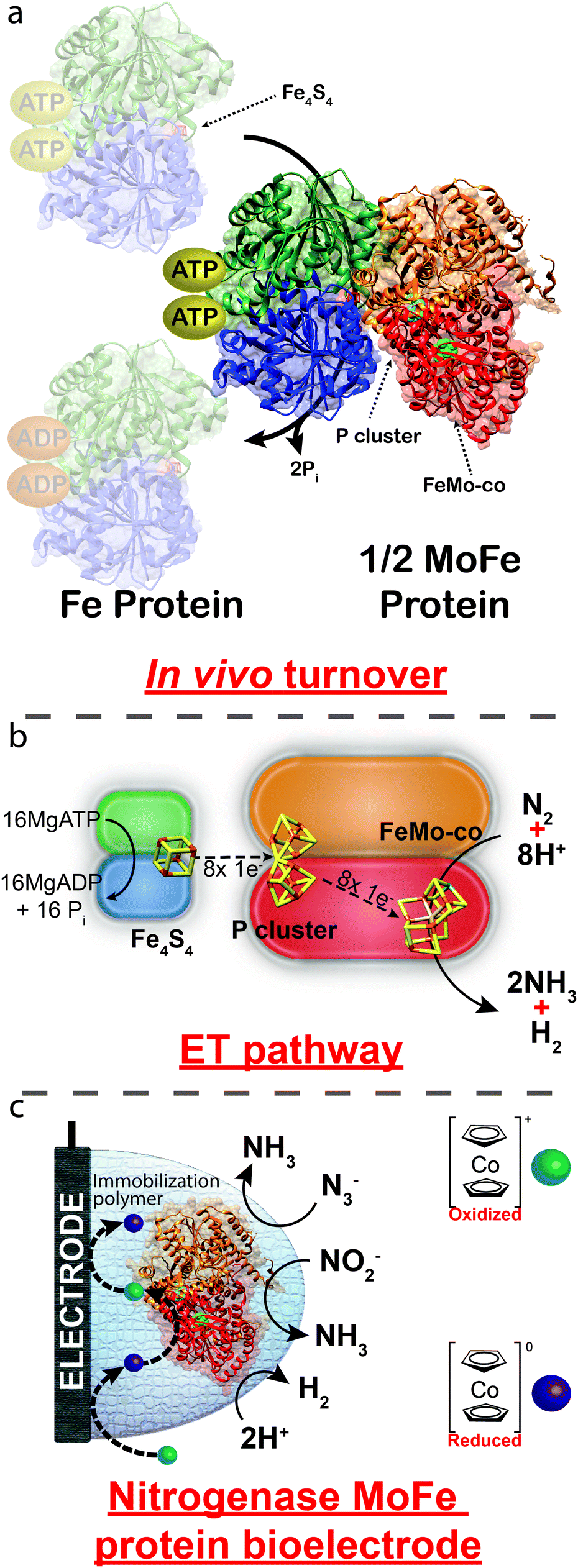 | ||
| Fig. 1 (a) Crystal structure of nitrogenase (PDB: 4WZA) from Azotobacter vinelandii illustrating the transient association of the Fe protein to the MoFe protein. (b) ET between the Fe4S4 cluster of the Fe protein to the P cluster and the FeMo-co of the MoFe protein. (c) Proposed route of heterogeneous ET using cobaltocene as an electron mediator. Reactions depicted in (c) are unbalanced. | ||
To achieve sustainable substrate reduction by nitrogenase without the need for ATP, there is considerable interest in delivering electrons directly to the MoFe protein. Toward this end, alternative in vitro reducing agents have been demonstrated to be able to reduce the MoFe protein and support substrate reduction; however, success is commonly limited to the reduction of substrates other than N2, such as N2H4, H+, HCN and N3− and at very low rates.13–15 Recently, light-dependent reduction of the MoFe protein by CdS nanorods has been shown to be able to support N2 reduction to 2NH3 with a high turnover number and quantum efficiency.16
To date multiple enzymes (such as glucose oxidase, bilirubin oxidase, hydrogenase and carbon monoxide dehydrogenase, to name a few) have been immobilized at electrode surfaces where heterogeneous ET to/from the enzyme supports enzymatic substrate reduction or oxidation, respectively.17–22 ET takes place either directly (DET) or through the use of an electron mediator (MET), whereby the ability to directly observe electron transfer in real time to/from an enzyme provides a powerful tool for the evaluation of enzymatic mechanisms and kinetics, as well as the ability to create devices that are able to produce electrical energy from alternative energy dense fuels (enzymatic fuel cells, EFCs).23,24 Additionally, the ability to bioelectrosynthetically produce an important chemical at the expense of electrical energy could circumvent the necessity of harsh reaction conditions or expensive catalysts.
Here we report the immobilization of the MoFe protein of wild-type nitrogenase (purified from Azotobacter vinelandii, an aerobic diazotroph) at an electrode surface, where the use of an unnatural electron mediator facilitates the real-time electrochemically-observable reduction of H+ to H2 and azide (N3−) and nitrite (NO2−) to NH3. Importantly, observed bioelectrocatalysis is obtained in the absence of the Fe protein for wild-type MoFe protein, negating the requirement of all mechanistic steps associated with the Fe protein cycle, including the rate-limiting step for overall catalysis. We therefore anticipate that this approach will be valuable in revealing mechanistic details of nitrogenase as well as provide a technological basis to establish a bioelectrosynthetic technique for the production of hydrogen, ammonia and hydrocarbons (using CO or CO2 as substrate) at room temperature and neutral pH.
Initially, nitrogenase MoFe protein was immobilized within a polymer at the surface of a glassy carbon electrode and preliminary cyclic voltammetry studies were performed against a series of metal-containing inorganic/organometallic redox active complexes in an attempt to screen for compatible electron mediators (data not shown). MoFe protein was immobilized at the electrode surface under a chemically-crosslinked poly(vinylamine) support to effectively increase the concentration of the MoFe protein at the electrode surface in an attempt to achieve greater sensitivities for the detection of apparent MoFe protein activity. In addition, a nitrogenase MoFe protein with a single amino acid substitution, β-98Tyr→His, was utilized that has previously been demonstrated to improve the ability of the MoFe protein to accept electrons from unnatural reducing agents.13,25
While many metallocene complexes exist, ferrocene is well established in the bioelectrochemical field following the discovery that the ferrocene/ferrocenium couple can efficiently mediate electron transfer between glucose oxidase and an electrode surface, resulting in a system that is able to bioelectrocatalytically oxidize glucose.26 In order to drive a bioelectrocatalytic reductive reaction with nitrogenase, however, the formal potential of ferrocene is expected to be too positive. Thus, its structural analogue cobaltocene (Cc+, bis(cyclopentadienyl)cobalt(III)) was investigated as a more suitable electron mediator (E° = −1.16 V vs. Ag/AgCl sat'd KCl).27
Fig. 2 presents cyclic voltammograms for the resulting MoFe protein bioelectrodes in a solution containing Cc+, where a single electron redox couple can be observed between the oxidized cobaltocenium cationic complex and its reduced cobaltocene complex (Cc+/Cc). In the presence of enzymatically active MoFe protein and Cc+ (Fig. 2a), a reductive catalytic wave is observed that is attributed to the reduction of 2H+ to H2 by the MoFe protein bioelectrode, with an onset potential of approximately −1.04 V (vs. SCE). Following the addition of 50 mM N3− (Fig. 2b) the reduction current increases, which is attributed to the reduction of N3− to NH3 and N2; there are three major proposed pathways for the reduction of N3− or HN3 (eqn (2) and (3)).28 The addition of 50 mM NO2− results in a catalytic reductive wave assigned to the 6e− reduction of NO2− to NH3 and 2H2O (eqn (4)).10,28
| N3− + 3H+ + 2e− → N2 + NH3 | (2) |
| N3− + 9H+ + 8e− → 3NH3 | (3) |
| NO2− + 7H+ + 6e− → NH3 + 2H2O | (4) |
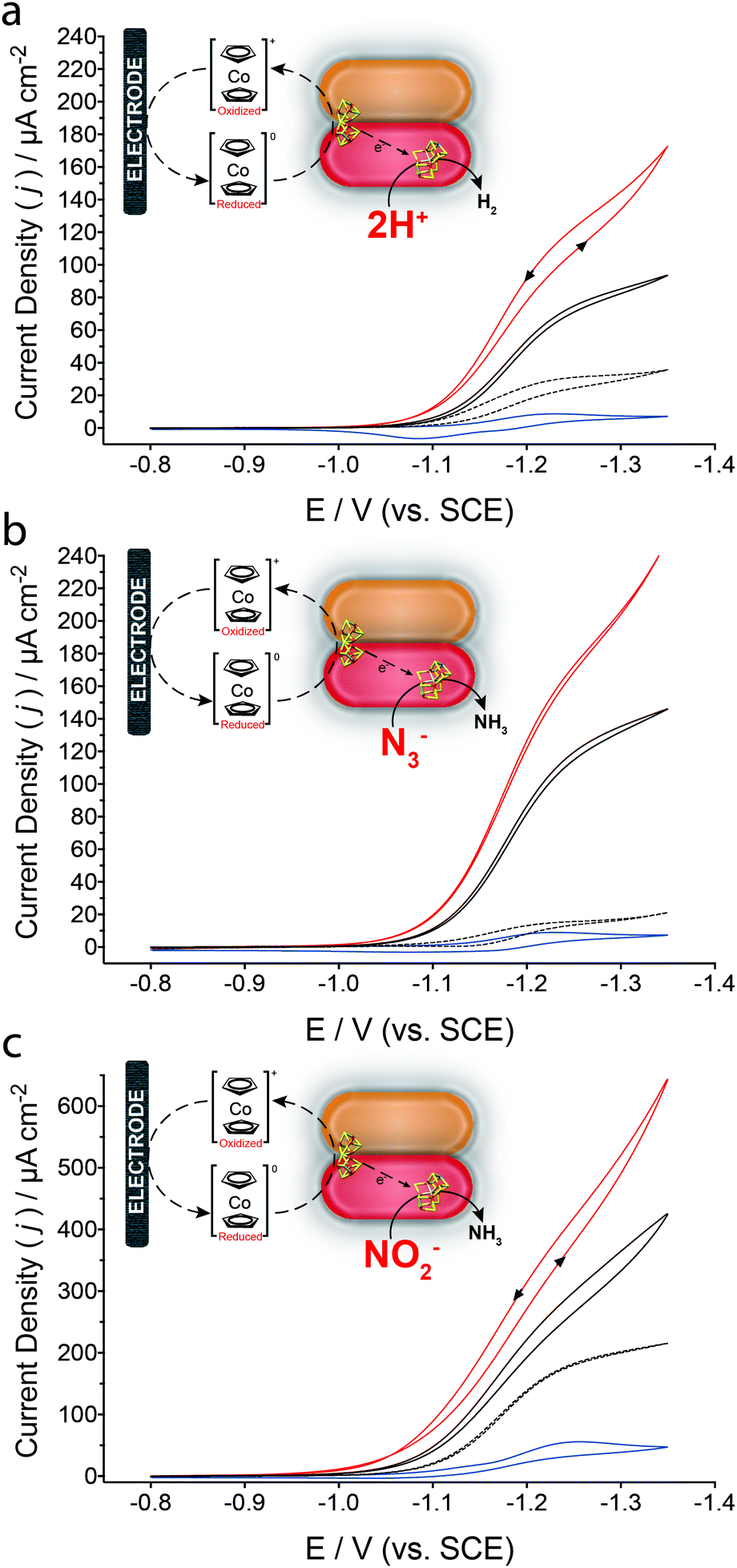 | ||
| Fig. 2 Cyclic voltammograms of wild-type MoFe protein bioelectrode in HEPES buffer (pH 7.4, 250 mM) containing 200 μM Cc+ at a scan rate of 2 mV s−1 (solid black lines) in the (a) absence of any additional substrates, or in the presence of (b) 50 mM N3− or (c) 50 mM NO2−. MoFe protein bioelectrodes were also prepared with the β-98Tyr→His MoFe protein (red solid lines). Equivalent control bioelectrodes were either prepared with BSA (blue line) or apo-MoFe protein (black dashed line) in the presence of the respective substrates. All voltammetric experiments were performed in an anaerobic tent (Ar) where the O2 concentration was continuously <1 ppm, at room temperature (∼21 °C). Reactions depicted in (b) and (c) are unbalanced. | ||
In all cases, MoFe protein bioelectrodes prepared with the β-98Tyr→His modified MoFe protein result in improved catalytic currents; this is in agreement with previous reports whereby this single amino acid substitution improves the ability of the MoFe protein to accept electrons from unnatural electron donors.13 Control bioelectrodes consisted of either an equivalent loading of BSA (bovine serum albumin, by mass) in place of the MoFe protein, or apo-MoFe protein (obtained from a nifB-deficient strain of A. vinelandii yielding a MoFe protein lacking FeMo-co). Under non-steady state conditions, minimal contributions are observed for bioelectrodes prepared with apo-MoFe protein for H+ and N3− reduction. An increased background contribution is observed for apo-MoFe protein bioelectrodes and BSA control bioelectrodes in the presence of NO2−; increased apparent catalytic reductive current for NO2− at apo-MoFe protein bioelectrodes suggests the P cluster may be able to catalytically reduce NO2−. NO2− reduction by the carbon electrode surface and Cc+ is evaluated within the ESI† (Fig. S1).29 Further controls were prepared by systematically eliminating individual MoFe protein bioelectrode components (ESI,† Fig. S2). Additionally, it is important to note that FeMo-co extracted from the MoFe protein is extremely unstable in aqueous solutions.30,31 Table S1 (ESI†) reports the catalytic current densities obtained for each substrate, corrected to apo-MoFe protein bioelectrodes.
In addition to cyclic voltammetry, steady-state amperometric analyses were also performed to validate the catalytic turnover of N3− by the wild-type and β-98Tyr→His MoFe protein bioelectrodes (Fig. 3). To facilitate bioelectrosynthetic reduction of the MoFe protein and thus N3−, a potential of −1.25 V (vs. SCE) was applied and substrate injections resulted in an increasing catalytic current. Prior to N3− injections, the stead-state current is due to the reduction of 2H+ to H2. Following additions of N3− into the bulk solution, the reduction current increases as a function of the bioelectrocatalytic reduction of N3− by the MoFe protein, whereby electrons are delivered to the protein by the Cc/Cc+ electron mediator (as in Fig. 1c).
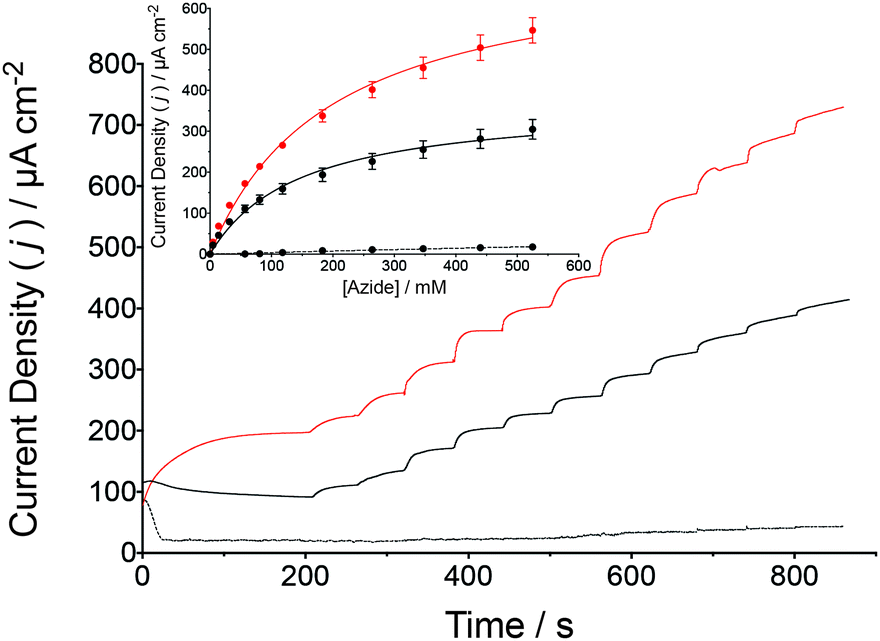 | ||
| Fig. 3 Amperometric i–t curve for the reduction of N3− by a wild-type MoFe protein bioelectrode (black solid line) and the β-98Tyr→His MoFe protein (red solid line) in stirred HEPES buffer (pH 7.4, 250 mM) containing 200 μM Cc+ (Eapplied = −1.25 V vs. SCE). Successive injections of N3− were made from a stock solution also containing 200 μM Cc+. Control experiments were performed under the same conditions although apo-MoFe protein was used (as described for Fig. 2, black dashed line). The inset presents the normalized catalytic current response to N3− injections for wild-type (black fit) and β-98Tyr→His (red fit) MoFe protein bioelectrodes compared to control bioelectrodes prepared with apo-MoFe protein (black dashed line fit, n = 3). The Michaelis–Menten kinetic model was applied by nonlinear regression. The N3− stock solution was prepared with 200 μM Cc+ and 250 mM HEPES buffer, to prevent dilution due to N3− injections. | ||
The change in the catalytic current following each addition of N3− was analyzed against the Michaelis–Menten kinetic model by plotting the corrected current density against the corresponding N3− concentration. Apparent Michaelis constants (KM) for N3− were calculated by nonlinear regression to be 146 ± 15 mM N3− for the wild-type MoFe protein bioelectrodes, with an increase to 196 ± 14 mM N3− observed for the β-98Tyr→His MoFe protein bioelectrode. Interestingly, the apparent maximum current density (Jmax) for the wild-type MoFe protein bioelectrodes was calculated to be 370 ± 14 μA cm−2, which almost doubled for the β-98Tyr→His MoFe protein bioelectrodes to 725 ± 22 μA cm−2. An increased Jmax as well as a similar KM provide further support for bioelectrocatalysis by intact MoFe protein at the electrode surface (and not by unfolded protein or dissociated cofactor), whereby substrate affinity remains largely unchanged although improved ET to FeMo-co result in enhanced catalytic currents (since the single β-98Tyr→His substitution has previously been demonstrated to improve substrate turnover by the MoFe protein in the absence of the Fe protein).13,25
Control experiments were performed using apo-MoFe protein bioelectrodes; under these steady-state conditions, negligible bioelectrocatalytic currents were observed. Additionally, apparent kinetics were not determined for NO2− reduction under steady-state conditions at this stage; the background reductive wave for NO2− at control bioelectrodes in the absence and presence of Cc+ results in a low signal to noise ratio. Apparent kinetics for NO2− reduction will be evaluated using an alternative bioelectrode architecture and/or MET platform.
Finally, bulk bioelectrosynthesis (using β-98Tyr→His MoFe protein bioelectrodes) was performed at −1.25 V (vs. SCE) following an injection of either N3− or NO2− (50 mM, in the presence of Cc+) to confirm NH3 production; NH3 was quantified using a fluorometric assay whereby NH3 forms a fluorescent complex with ortho-phthalaldehyde (ESI,† Fig. S3 and S4).32,33 Bioelectrosynthetic reduction of N3− for 30 min yielded 70 ± 9 nmol of NH3 where the theoretical quantity of NH3 expected to be produced (as a function of the charge passed during the experiment) varied depending on the pathway of N3− reduction (Table 1, eqn (2) and (3)). Bulk bioelectrosynthetic NO2− reduction (performed under the same conditions) yielded 234 ± 62 nmol NH3 where the theoretical quantity of NH3 expected was 231 ± 65 nmol NH3, corresponding to a faradaic efficiency of 101 ± 39% (ESI,† Fig. S4). The high faradaic efficiency suggests that the reduced MoFe protein (reduced by Cc) largely favors the reduction of NO2 as opposed to 2H+, when in the presence of NO2−.
| Reduction reaction | NH3 calculated (nmol) | NH3 detected (nmol) | Faradaic efficiency (%) |
|---|---|---|---|
| N3− + 3H+ + 2e− → N2 + NH3 | 203 ± 35 | 70 ± 9 | 35 ± 18 |
| N3− + 9H+ + 8e− → 3NH3 | 152 ± 26 | 46 ± 18 | |
| NO2− + 7H+ + 6e− → NH3 + 2H2O | 231 ± 65 | 234 ± 62 | 101 ± 39 |
Conclusions
In conclusion, we report the heterogeneous reduction of immobilized nitrogenase wild-type and β-98Tyr→His MoFe protein, where the resulting reduced MoFe protein bioelectrode is able to support the reduction of 2H+ to H2, N3− to NH3, and NO2− to NH3 using Cc/Cc+ as an electron mediator. Since reduced Cc efficiently reduces MoFe protein at the electrode surface a mediated reductive catalytic response is observed, providing an alternative methodology to investigate kinetics, substrate interactions, inhibitory effects and ET pathways of the MoFe protein. Additionally, this methodology presents a novel bioelectrosynthetic system for the production of NH3 and H2 under mild conditions (such as room temperature and neutral pH). Future work will investigate the possibility of immobilizing the MoFe protein within a suitable redox polymer (controlling the concentration of localized electron mediator) with an aim to increase the electron flux through MoFe protein and achieve N2 reduction. In addition to immobilizing the protein within an appropriately-designed redox polymer, improved understanding of the role of ATP in vivo would help achieve N2 reduction at this bioelectrode surface.Abbreviations
| SCE | Saturated calomel electrode |
| MoFe protein | Molybdenum iron protein of nitrogenase |
| Fe protein | Iron protein of nitrogenase |
| Cc/Cc+ | Cobaltocene/cobaltocenium |
| FeMo-co | Molybdenum iron cofactor of nitrogenase |
Acknowledgements
R. D. M., D. L. and S. D. M. acknowledge funding from a Marie Curie-Skłodowska Individual Fellowship (Global) under the EUR Commission's Horizon 2020 Framework (project 654836, “Bioelectroammonia”). S. A. and S. D. M. thank the Army Research Office for funding. N. K., D. R. D. and L. C. S. thank the US Department of Energy, Office of Science, Office of Basic Energy Sciences.Notes and references
- B. K. Burgess and D. J. Lowe, Chem. Rev., 1996, 96, 2983–3012 CrossRef CAS PubMed.
- J. Christiansen, D. R. Dean and L. C. Seefeldt, Annu. Rev. Plant Physiol. Plant Mol. Biol., 2001, 52, 269–295 CrossRef CAS PubMed.
- B. E. Smith, Science, 2002, 297, 1654–1655 CrossRef CAS PubMed.
- Z.-Y. Yang, K. Danyal and L. C. Seefeldt, in Nitrogen Fixation, Methods in Molecular Biology 766, ed. M. W. Ribbe, Springer, New York, 2011, ch. 2 Search PubMed.
- Y. Hu, C. C. Lee and M. W. Ribbe, Science, 2011, 333, 753–755 CrossRef CAS PubMed.
- J. W. Peters, a. K. Fisher and D. R. Dean, Annu. Rev. Microbiol., 1995, 49, 335–366 CrossRef CAS PubMed.
- I. Dance, Chem. Commun., 2013, 49, 10893–10907 RSC.
- B. M. Hoffman, D. Lukoyanov, Z.-Y. Yang, D. R. Dean and L. C. Seefeldt, Chem. Rev., 2014, 114, 4041–4062 CrossRef CAS PubMed.
- L. C. Seefeldt, B. M. Hoffman and D. R. Dean, Annu. Rev. Biochem., 2009, 78, 701–722 CrossRef CAS PubMed.
- S. Shaw, D. Lukoyanov, K. Danyal, D. R. Dean, B. M. Hoffman and L. C. Seefeldt, J. Am. Chem. Soc., 2014, 136, 12776–12783 CrossRef CAS PubMed.
- S. Duval, K. Danyal, S. Shaw, A. K. Lytle, D. R. Dean, B. M. Hoffman, E. Antony and L. C. Seefeldt, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 16414–16419 CrossRef CAS PubMed.
- K. Danyal, D. R. Dean, B. M. Hoffman and L. C. Seefeldt, Biochemistry, 2011, 50, 9255–9263 CrossRef CAS PubMed.
- K. Danyal, B. S. Inglet, K. A. Vincent, B. M. Barney, B. M. Hoffman, F. A. Armstrong, D. R. Dean and L. C. Seefeldt, J. Am. Chem. Soc., 2010, 132, 13197–13199 CrossRef CAS PubMed.
- L. E. Roth and F. A. Tezcan, J. Am. Chem. Soc., 2012, 134, 8416–8419 CrossRef CAS PubMed.
- K. Danyal, A. J. Rasmussen, S. M. Keable, B. S. Inglet, S. Shaw, O. A. Zadvornyy, S. Duval, D. R. Dean, S. Raugei, J. W. Peters and L. C. Seefeldt, Biochemistry, 2015, 54, 2456–2462 CrossRef CAS PubMed.
- K. A. Brown, D. F. Harris, M. B. Wilker, A. Rasmussen, N. Khadka, H. Hamby, S. Keable, G. Dukovic, J. W. Peters, L. C. Seefeldt and P. W. King, Science, 2016, 352, 448–450 CrossRef CAS PubMed.
- M. Holzinger, A. Le Goff and S. Cosnier, Front. Chem., 2014, 2, 63 Search PubMed.
- D. Leech, P. Kavanagh and W. Schuhmann, Electrochim. Acta, 2012, 84, 223–234 CrossRef CAS.
- N. Mano, Appl. Microbiol. Biotechnol., 2012, 96, 301–307 CrossRef CAS PubMed.
- M. T. Meredith and S. D. Minteer, Annu. Rev. Anal. Chem., 2012, 5, 157–179 CrossRef CAS PubMed.
- A. Szczupak, J. Halamek, L. Halamkova, V. Bocharova, L. Alfonta and E. Katz, Energy Environ. Sci., 2012, 5, 8891–8895 Search PubMed.
- T. W. Woolerton, S. Sheard, E. Reisner, E. Pierce, S. W. Ragsdale and F. A. Armstrong, J. Am. Chem. Soc., 2010, 132, 2132–2133 CrossRef CAS PubMed.
- V. Fourmond, S. Stapf, H. Li, D. Buesen, J. Birrell, O. Rüdiger, W. Lubitz, W. Schuhmann, N. Plumeré and C. Léger, J. Am. Chem. Soc., 2015, 137, 5494–5505 CrossRef CAS PubMed.
- R. D. Milton, D. P. Hickey, S. Abdellaoui, K. Lim, F. Wu, B. Tan and S. D. Minteer, Chem. Sci., 2015, 6, 4867–4875 RSC.
- J. W. Peters, K. Fisher, W. E. Newton and D. R. Dean, J. Biol. Chem., 1995, 270, 27007–27013 CrossRef CAS PubMed.
- A. E. G. Cass, G. Davis, G. D. Francis, H. A. O. Hill, W. J. Aston, I. J. Higgins, E. V. Plotkin, L. D. L. Scott and A. P. F. Turner, Anal. Chem., 1984, 56, 667–671 CrossRef CAS PubMed.
- L. A. Khanova, V. V. Topolev and L. I. Krishtalik, Chem. Phys., 2006, 326, 33–42 CrossRef CAS.
- W. E. Newton and M. J. Dilworth, in Nitrogen Fixation, Methods in Molecular Biology 766, ed. M. W. Ribbe, Springer, New York, 2011, ch. 8 Search PubMed.
- N. Chebotareva and T. Nyokong, J. Appl. Electrochem., 1997, 27, 975–981 CrossRef CAS.
- V. K. Shah and W. J. Brill, Proc. Natl. Acad. Sci. U. S. A., 1977, 74, 3249–3253 CrossRef CAS.
- A. W. Fay, C. C. Lee, J. A. Wiig, Y. Hu and M. W. Ribbe, in Nitrogen Fixation, Methods in Molecular Biology 766, ed. M. W. Ribbe, Springer, New York, 2011, ch. 16 Search PubMed.
- M. Duca, J. R. Weeks, J. G. Fedor, J. H. Weiner and K. A. Vincent, ChemElectroChem, 2015, 2, 1086–1089 CrossRef CAS.
- R. M. Holmes, A. Aminot, R. Kerouel, B. A. Hooker and B. J. Peterson, Can. J. Fish. Aquat. Sci., 1999, 56, 1801–1808 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ee01432a |
| This journal is © The Royal Society of Chemistry 2016 |