
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rational wiring of photosystem II to hierarchical indium tin oxide electrodes using redox polymers†
Katarzyna P.
Sokol
a,
Dirk
Mersch
a,
Volker
Hartmann
b,
Jenny Z.
Zhang
a,
Marc M.
Nowaczyk
b,
Matthias
Rögner
b,
Adrian
Ruff
c,
Wolfgang
Schuhmann
c,
Nicolas
Plumeré
*d and
Erwin
Reisner
*a
aDepartment of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, UK. E-mail: reisner@ch.cam.ac.uk
bPlant Biochemistry, Faculty of Biology & Biotechnology, Ruhr-Universität Bochum, Universitätsstr. 150, 44780 Bochum, Germany
cAnalytical Chemistry – Center for Electrochemical Sciences (CES), Faculty of Chemistry and Biochemistry, Ruhr-Universität Bochum, Universitätsstr. 150, 44780 Bochum, Germany
dCenter for Electrochemical Sciences (CES) – Molecular Nanostructures, Ruhr-Universität Bochum, Universitätsstr. 150, 44780 Bochum, Germany. E-mail: nicolas.plumere@rub.de
First published on 5th July 2016
Abstract
Photosystem II (PSII) is a multi-subunit enzyme responsible for solar-driven water oxidation to release O2 and highly reducing electrons during photosynthesis. The study of PSII in protein film photoelectrochemistry sheds light into its biological function and provides a blueprint for artificial water-splitting systems. However, the integration of macromolecules, such as PSII, into hybrid bio-electrodes is often plagued by poor electrical wiring between the protein guest and the material host. Here, we report a new benchmark PSII–electrode system that combines the efficient wiring afforded by redox-active polymers with the high loading provided by hierarchically-structured inverse opal indium tin oxide (IO-ITO) electrodes. Compared to flat electrodes, the hierarchical IO-ITO electrodes enabled up to an approximately 50-fold increase in the immobilisation of an Os complex-modified and a phenothiazine-modified polymer. When the Os complex-modified polymer is co-adsorbed with PSII on the hierarchical electrodes, photocurrent densities of up to ∼410 μA cm−2 at 0.5 V vs. SHE were observed in the absence of diffusional mediators, demonstrating a substantially improved wiring of PSII to the IO-ITO electrode with the redox polymer. The high photocurrent density allowed for the quantification of O2 evolution, and a Faradaic efficiency of 85 ± 9% was measured. As such, we have demonstrated a high performing and fully integrated host–guest system with excellent electronic wiring and loading capacity. This assembly strategy may form the basis of all-integrated electrode designs for a wide range of biological and synthetic catalysts.
Broader contextIn natural photosynthesis, solar energy drives the conversion of CO2 and H2O into chemical energy carriers and building blocks, releasing O2 as a by-product. Artificial photosynthesis attempts to mimic this process to produce a renewable and storable fuel. Photosystem II (PSII), the first protein complex in oxygenic photosynthesis, is capable of harnessing solar energy required to perform photocatalytic water oxidation, a bottleneck in artificial photosynthesis. As such, there is substantial interest in integrating PSII onto electrode scaffolds to gain better insight into fundamental protein function and also in protein film photoelectrochemical (PF-PEC) platforms for proof-of-principle solar electricity and solar fuel generation. Here, we describe a rational approach for a PSII-based electrode assembly, where we electrically wired PSII using a redox polymer matrix to a high surface area electrode scaffold. The PSII–polymer mixture integrated in a hierarchical indium tin oxide electrode provides the basis for high performance PSII-photoelectrochemistry and will be relevant for future enzyme-driven semi-artificial photosynthetic systems. |
Introduction
The immobilisation of photosynthetic proteins onto electrodes is of importance to a range of current and future innovations, including biosensors,1–3 biophotovoltaic systems4–7 and photoelectrochemical (PEC) platforms.8,9 Photosystem II (PSII) is a photosynthetic enzyme with the ability to photocatalyse water oxidation, a bottleneck reaction in artificial photosynthesis, at theoretical rates of up to 250 mol O2 (mol PSII monomer)−1 s−1.10,11 As such, there is considerable interest in the integration of PSII as a guest into electrode scaffolds,12,13 in particular to improve our fundamental understanding of the protein function and also in PEC cells for proof-of-principle solar electricity/fuel generation.14–17Several strategies for the integration of PSII into electrodes are currently employed, each with unique advantages. Before these approaches are discussed, some knowledge of the mechanism behind PSII water oxidation is required. Briefly, light is absorbed by pigments within PSII, and funnelled into the reaction centre complex where charge formation and separation at the P680 primary electron donor site occurs. The photogenerated electrons are then transferred via pheophytin and plastoquinone A (QA) to the terminal electron acceptor plastoquinone B (QB), which is located on the stromal side of the enzyme. Holes generated at the P680 are transferred in the lumenal direction, via a redox-active tyrosine side chain (TyrZ) to the oxygen-evolving complex (OEC), where water is oxidised to liberate H+ and O2.18,19 If the PSII is adsorbed in the correct orientation on an electrode, direct electron transfer from the QA/QB to the electrode can take place.9,20 However, a QB mimic, such as 2,6-dichloro-1,4-benzoquinone (DCBQ), is typically required as a diffusional mediator between the insufficiently wired PSII and the electrode to substantially enhance photocurrent generation.19
A traditional approach for the immobilisation of photosynthetic reaction centres is to align the proteins on chemically-modified electrodes functionalised with linkers such as quinonoid,21N-hydroxy-succinimidyl ester,22 nickel nitrilotriacetic acid,23,24 cytochrome c25,26 and carboxylic acid/amino groups.27 However, the magnitude of the photocurrent is limited by the attachment of a single monolayer of photosynthetic reactions centres that can be assembled on the electrode.
A strategy to enhance the loading of electrically wired PSII onto electrodes is to entrap PSII in a redox-active polymer matrix on an electrode surface.28,29 In this approach, PSII of any orientation can in principle be efficiently wired to the electrode by the redox-active moieties that are homogeneously distributed in the matrix, which can mediate charge transfer via an electron hopping mechanism.30 The benchmark system using this approach consists of a flat gold electrode on which PSII is embedded in an Os complex-modified polymer (E1/2 = 0.395–0.505 V versus the standard hydrogen electrode; vs. SHE).31 Photocurrents of up to 45 μA cm−2 at an applied potential (Eapp) of 0.5 V vs. SHE were reported for this biophotoanode. Despite its advantages, the performance of this system was limited by the intrinsic properties of the polymeric matrix. Independently of the total loading at the electrode surface, the amount of electroactive enzyme is defined by the rate of charge transfer via electron hopping, which limits the maximum (photo-)electrocatalytic response that can be detected.32 On modified flat electrodes where enzymes are entrapped in redox polymers, the current generation typically arises from catalysts present within a thin layer (a few micrometer thick) at the electrode/hydrogel interface; the remaining catalysts in the outer layers of the film are electro-inactive and do not contribute to current generation.33
An emerging and effective enzyme immobilisation strategy involves the adoption of highly structured electrode morphologies34–36 to increase the available surface area for enzyme adsorption.9,27,37 In a recent benchmark system, PSII was adsorbed on a hierarchically-structured indium tin oxide (ITO) electrode that incorporated macroporosity (for enhanced enzyme and substrate penetration) and mesoporosity (to enhance the effective surface area and enzyme anchoring) with high thickness.9 As a result, a 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000-fold increase in PSII loading was observed compared to conventional flat electrodes.6,31 However, insufficient wiring at the PSII–electrode interface was still apparent, with non-mediated photocurrents of 20 μA cm−2 being observed in contrast to 1 mA cm−2 in the presence of a freely diffusing mediator. A further limitation of the electrode is poor PSII photostability, with the electrode exhibiting a photocurrent half-life time of only a few minutes.
000-fold increase in PSII loading was observed compared to conventional flat electrodes.6,31 However, insufficient wiring at the PSII–electrode interface was still apparent, with non-mediated photocurrents of 20 μA cm−2 being observed in contrast to 1 mA cm−2 in the presence of a freely diffusing mediator. A further limitation of the electrode is poor PSII photostability, with the electrode exhibiting a photocurrent half-life time of only a few minutes.
Here, we report a high performing PSII–electrode system that combines the best aspects of two advanced enzyme immobilisation strategies: the use of a redox polymer matrix to enable efficient PSII wiring, and the use of high surface area hierarchically-structured ITO electrodes to enable high loading of both the polymer and the PSII (Fig. 1a). The highly structured electrode scaffold increases the polymer–electrode interface and reduces the average charge transfer distance between the PSII and the electrode surface via the polymeric matrix. This enables the wiring of a large population of PSII to the electrode, which translates to high effective loading. We first compared the performance of two promising redox polymers differing in chemical and redox properties as electron conducting matrices for PSII in inverse opal mesoporous ITO (IO-ITO) electrodes (Fig. 1b). We then focused on the optimisation of the lead ITO–polymer–PSII system to ultimately deliver high photocurrents in the absence of diffusional mediators, at an extended operating lifetime.
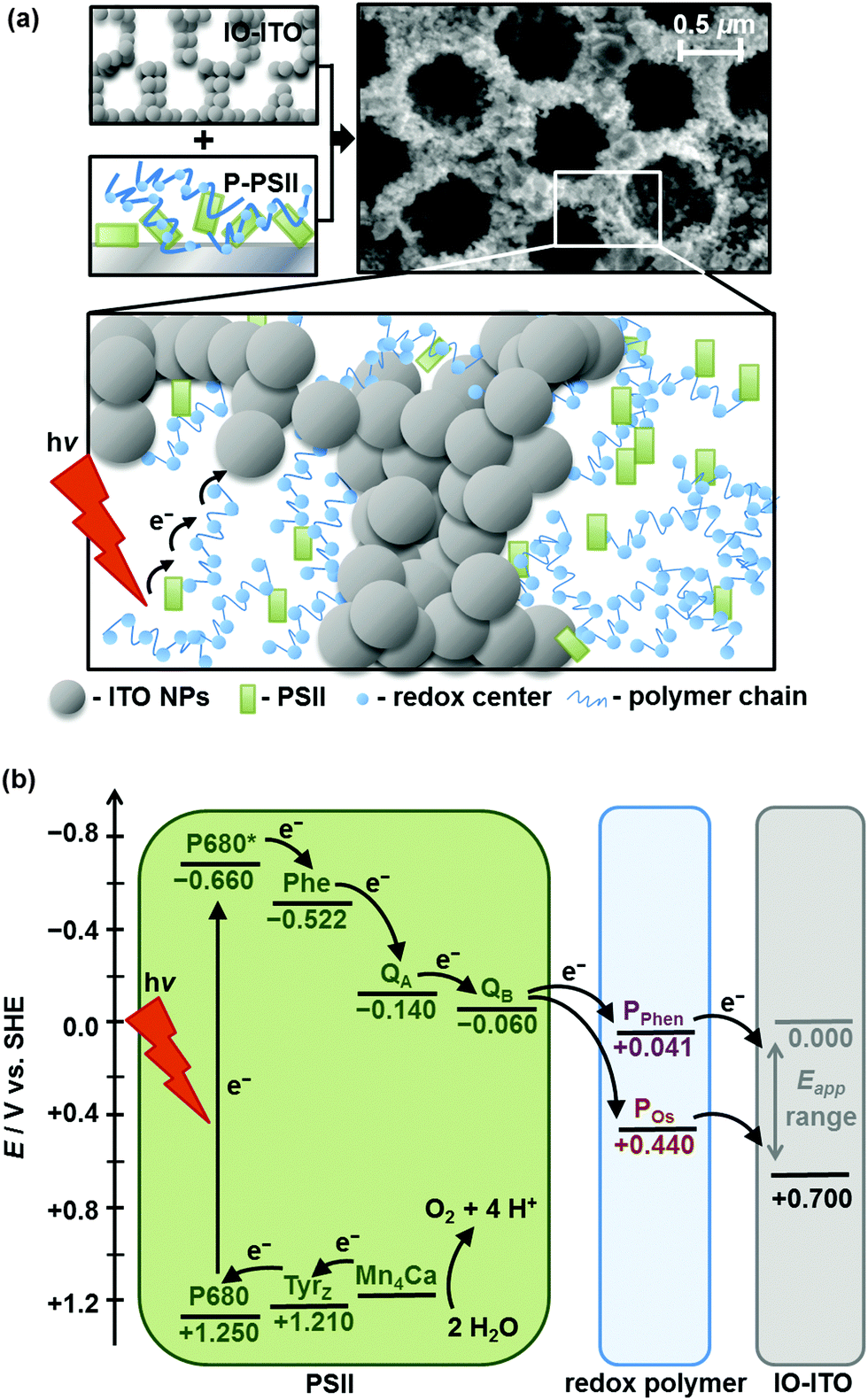 | ||
| Fig. 1 (a) Schematic representation of PSII wired via a redox polymer network to a hierarchically-structured IO-ITO electrode (species size not drawn to scale), indicating the electron transfer from photoexcited PSII to the electrode via the redox-active centres. The SEM image of IO-ITO is also shown. (b) Energy level diagram showing electron transfer pathways between PSII, the redox polymer (PPhen or POs, at pH 6.5) and the IO-ITO electrode (Eapp refers to the applied electrochemical potential, which determines the Fermi level at the ITO electrode). Abbreviations: P680 – primary electron donor site, Phe – pheophytin, QA/QB – electron acceptor plastoquinones, TyrZ – tyrosine, Mn4Ca – oxygen-evolving complex. | ||
Experimental section
Materials
All chemicals 1-vinylimidazole, 2,2′-bipyridine, allyl amine, K2OsCl6, butyl acrylate, allyl methacrylate, poly(ethylene glycol) methacrylate (Mn = 500 g mol−1), 2,2′-azobis(2-methylpropionitrile), toluidine blue (all Sigma Aldrich), DCBQ (Sigma Aldrich), 2-(N-morpholino)ethane sulfonic acid (MES, Alfa Aesar), CaCl2 (Breckland Scientific), MgCl2 (Fisher Scientific), KCl (Alfa Aesar), KOH (Breckland Scientific), NH4OH (30%) solution (Fisher Scientific), H2O2 (30%) solution (Fisher Scientific), polystyrene beads (Polysciences, Inc., 750 nm diameter, 2.6% w/v suspension in H2O), ITO nanoparticles (NPs) (Sigma Aldrich; <50 nm diameter) and fluorine-doped tin oxide (FTO) coated glass slides (8 Ω sq−1; Sigma Aldrich) were purchased from commercial suppliers and used without further purification unless otherwise noted. Methanol, absolute ethanol, 2-propanol, dimethyl sulfoxide (HPLC grade) were purchased from Sigma Aldrich. Poly(ethylene glycol) diglycidyl ether (PEGDGE) (Polyscience, USA) and 2,2′-(ethylenedioxy)(diethanethiol) (Sigma Aldrich) were purchased from commercial suppliers. PSII was isolated from the thermophilic cyanobacterium Thermosynechococcus elongatus and purified according to a previously reported procedure,38 resulting in purified PSII with an average oxygen-evolving activity of approximately 5300 μmol O2 h−1 mg−1 of chlorophyll a (Chl a). A stock PSII solution containing 2.6 mg Chl a mL−1 (83 μM PSII monomer) was stored in a liquid N2 Dewar.Polymer synthesis
The synthetic approaches to obtain the poly(1-vinylimidazole-co-allylamine) backbone, the Os precursor cis-[OsIICl2(bipy)2] (bipy = 2,2′-bipyridine) and the Os complex cis-[OsIICl(1-(n-butyl)-imidazole)(bipy)2](PF6) were described previously.31,39 The redox polymer poly(1-vinylimidazole)-Os(bipy)2Cl-polymer (POs),31 and phenothiazine-modified polymer (PPhen, phenothiazine moiety = toluidine blue)6 were synthesised according to previously reported procedures (for characterisation see ESI†), with POs prepared with slight modifications. In brief, after stirring a mixture of cis-[OsCl2(bipy)2] and the poly-(1-vinylimidazole-co-allylamine) backbone (1:1.65 weight ratio) dissolved in ethanol for 5 days at 90 °C, the product (POs) was precipitated upon addition of diethyl ether. The precipitate was separated by centrifugation, thoroughly washed with diethyl ether and dried under vacuum to obtain a reddish powder. Aqueous stock solutions of POs (10 mg mL−1) and PPhen (10 mg mL−1) were used.
Physical characterisation
The surface morphology of the electrodes was analysed by scanning electron microscopy (SEM; Philips SFEG XL30). A 5804 Eppendorf centrifuge and Carbolite furnace (ELF 11/14B/301) were used for electrode preparation. UV-vis absorption spectra were recorded on a Varian Cary 50 or Agilent Cary 60 UV-vis spectrophotometer, using cuvettes with an optical path length of 1 cm. Nuclear magnetic resonance (NMR) experiments were conducted with a Bruker 200 DPX spectrometer with a proton resonance frequency of 200.13 MHz (the residual solvent peak was used as internal standard). All dynamic light scattering (DLS) measurements were carried out with a Malvern Zetasizer Nano ZS (laser wavelength: 633 nm, measurement angle: 173° backscatter). The buffers were filtered through 450 nm membrane filters (polypropylene membranes bearing a borosilicate prefilter, Alltech) before dissolution of the polymers for DLS measurements; cuvettes were rinsed with buffer solution prior to measurements. For the filtration of polymer suspensions, non-pyrogenic 200 nm polyethersulfone membranes (Filtropur S, Sarstedt) were used.Preparation of IO-ITO|PSII electrodes
The IO-ITO electrodes were fabricated according to a previously reported co-assembly procedure.9 A standard IO-ITO electrode macropore diameter of 750 nm, film thickness of 20 μm and geometrical surface area of 0.25 cm2 were used, unless stated otherwise. An amount of 4.2 μL of the described polystyrene–ITO dispersion on a 0.25 cm2 geometrical surface area corresponds to a 10 μm thick IO-ITO structure. To obtain higher film thicknesses, the polystyrene–ITO mixture (4.2 μL) was deposited several times with a 4 h drying period in between. All current densities (μA cm−2) are reported with respect to the geometrical surface area. The IO-ITO|PSII modified electrodes were prepared as follows: a PSII stock solution (1 μL, 2.6 mg Chl a mL−1) was drop-cast onto the IO-ITO electrode and incubated in the dark for 15 min at room temperature. It was determined that 1 μL of PSII stock solution provided an excess of PSII for 20 μm thick IO-ITO and ensured full enzyme coverage on the electrode surface. Prior to electrochemical studies, the IO-ITO|PSII electrode was rinsed (3 × 500 μL) with the electrolyte solution to remove all unbound enzyme from the electrode surface.Preparation of IO-ITO|polymer–PSII electrodes
A PSII to polymer ratio of 1:1 (v/v) was defined based on 1 μL PSII stock solution (2.6 mg Chl a mL−1) and 1 μL polymer solution (10 mg mL−1). The PSII to polymer ratio was optimised on a 20 μm thick IO-ITO electrode by keeping the PSII solution volume (1 μL) and concentration (2.6 mg Chl a mL−1) constant, and varying the polymer solution concentration at constant volume (1 μL). The optimal PSII to polymer ratio was found to be 1 μL of PSII solution (2.6 mg Chl a mL−1) to 1 μL of the polymer solution (10 mg mL−1) per 20 μm thickness of IO-ITO. The IO-ITO|polymer–PSII electrodes were prepared as follows: the PSII stock solution (1 μL) was mixed with a redox polymer solution (1 μL) and the polymer–PSII mixture was drop-cast (2 μL) onto the IO-ITO electrode (20 μm thick) and incubated in the dark for 15 min at room temperature. Prior to electrochemical studies, the IO-ITO|polymer–PSII electrode was rinsed (3 × 500 μL) with the electrolyte solution.
Determination of PSII and polymer loading on IO-ITO
The amount of PSII on the IO-ITO surface was quantified by scratching off the IO-ITO from the glass substrate and washing with MeOH (500 μL) to extract Chl a from the electrode surface into a centrifuge vial. The vial was centrifuged (10000 rpm, 1 min), and the UV-vis spectrum of the supernatant was recorded (Fig. S6a, ESI†). The band with an absorption maximum of λmax = 665 nm assigned to Chl a (extinction coefficient ε = 79.95 (Chl a mg)−1 mL cm−1)40 was used to calculate the amount of PSII monomers assuming 35 Chl a molecules per PSII monomer.41 The Os complex loading in the POs polymer was determined by UV-vis spectroscopy in DMSO, using the freely diffusing Os complex analogue, cis-[OsIICl(1-(n-butyl)-imidazole)(bipy)2](PF6) for calibration, and confirmed by inductively-coupled plasma atomic emission spectroscopy (ICP-AES), obtained by washing off the POs from the IO-ITO electrode with aq. conc. HNO3 solution and measuring the concentration of the Os2+ metal ions relative to Osmium ICP standard (1 mg Os mL−1 in 20% HCl, Ricca Chemical).
Protein film photoelectrochemistry (PF-PEC) measurements
All electrochemical experiments (with the exceptions of O2 quantification and action spectra measurements) were performed with an Ivium Compactstat potentiostat with a purpose-built monochromatic red-light LED lamp (λ = 685 nm), collimated by two plano-convex lenses (THORLABS N-BK7 A Coated, ∅ = 7.5 cm, f = 5.0 cm). A light intensity flux (irradiance) (Ee) of 10 mW cm−2 was used, unless stated otherwise. Chronoamperometry and cyclic voltammetry (CV) measurements were carried out in a water-jacketed glass one-compartment cell at 25 °C with a three-electrode setup using an IO-ITO working, a Pt wire counter and a Ag/AgCl (3 M KCl) reference electrode. Measurements of the IO-ITO|polymer–PSII system were carried out in 4 mL aqueous pH 6.5 electrolyte solution containing CaCl2 (20 mM), MgCl2 (15 mM), KCl (50 mM) and MES (40 mM). For the mediated photocurrent measurements, a DCBQ solution in DMSO (40 μL, 100 mM) was added to give a final concentration of 1 mM in the electrolyte solution. The following correction factor was used to convert the reduction potential to SHE: ESHE = EAg/AgCl + 0.209 V (25 °C). IO-ITO|polymer–PSII electrodes were typically exposed to cycles of 30 s dark and 30 s light irradiation in the PF-PEC measurements. The photocurrent response was defined as the baseline-corrected photocurrent peak after the third light exposure, accounting for charging effects and to avoid overestimation.19 The action spectra were recorded using a Xenon lamp Solar Light Simulator (300 W) coupled to a monochromator (MSH300; both from LOT Quantum design). The light intensity was measured as a function of wavelength with a photodiode detector (SEL033/F/QNDS1/W) and power meter (ILT1400). For the O2 evolution measurements, an Ivium Modulight LED module (λ = 660 nm; Ee = 10 mW cm−2) and a gas-tight two-compartment glass cell with the IO-ITO|polymer–PSII working electrode separated from the counter electrode by a glass frit were employed in an anaerobic (O2 level <1 ppm) MBraun glovebox. The error analysis was based on the standard deviations resulting from at least three experiments.Product analysis
Quantification of O2 was performed with a calibrated fluorescence O2 sensor (Neofox; Ocean Optics FOSPHOR probe) inside an MBraun glovebox to avoid leakage of atmospheric O2. The probe was placed inside the cell headspace, protected from direct irradiation and the background signal was subtracted from all measurements using the OriginPro 9.0 software. The reported O2 values were corrected for dissolved O2 using Henry's Law.Equations (1)–(5):
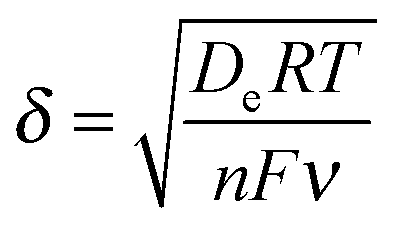
| (1) |
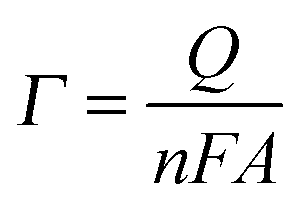 | (2) |
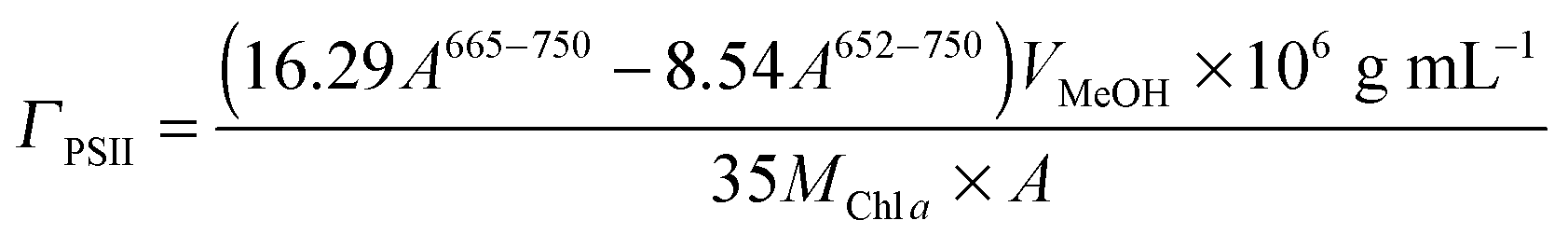 | (3) |
a – molecular mass of Chl a (893.5 g mol−1).40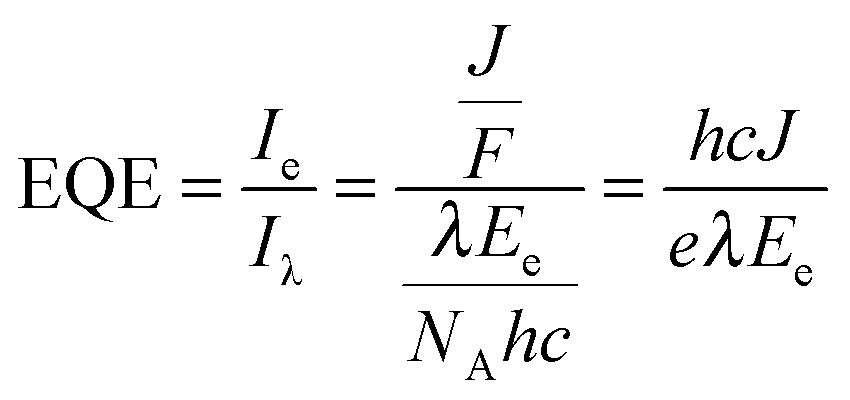 | (4) |
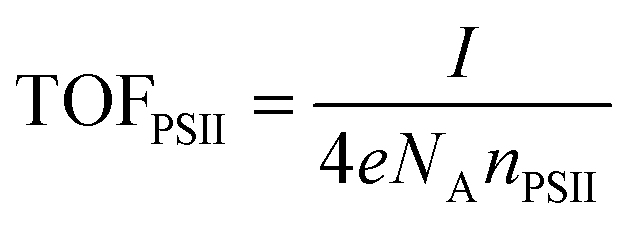 | (5) |
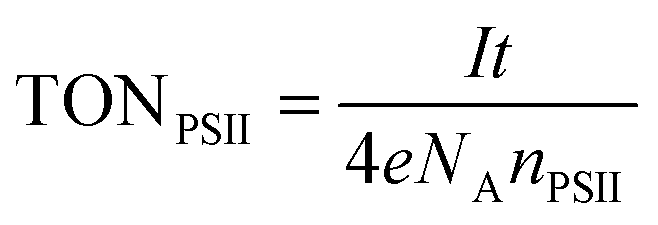 | (6) |
Results and discussion
Synthesis and characterisation of IO-ITO and polymers
This study uses a hierarchical IO-ITO electrode, which has previously demonstrated a high loading capacity for the large enzymes, PSII and hydrogenase (Fig. S1a, ESI†).9 The macropores with diameter of 750 nm and channels of 150 nm are also suitable for the penetration of macromolecular polymers; the mesopores with a diameter of approximately 50 nm provide a high effective surface area of ∼115 × 106 m2 m−3 for polymer/enzyme adsorption.9 The tunability of the film thickness (up to 80 μm, Fig. S1d, ESI†) provides extra flexibility in the optimisation of guest loading. The PSII used was isolated from the thermophilic cyanobacterium Thermosynechococcus elongatus given that cyanobacterial PSII is relatively well characterised,41,43,44 and it exhibits high activity and relative robustness.38,45The polymers chosen for this study include the Os complex-modified polymer POs (Fig. 2a), which has demonstrated excellent integration of PSII on flat electrodes;31 and the purely organic PPhen (Fig. 2b), which has a better matched redox potential with the QA/QB cofactors and has also demonstrated favourable wiring of PSII to flat electrodes.6 Both polymers are compatible with PSII and are stable under the acidic pH conditions for photocurrent measurements.6,39 The chemical structure, purity and size of the polymers were confirmed by 1H NMR (Fig. S2, ESI†), UV-vis spectroscopy (Fig. S3, ESI†) and DLS (Fig. S4, ESI†), respectively. The 1H NMR spectra of the polymer backbones correspond to the expected structure (Fig. S2, ESI†). Based on the integral ratio between methyl groups of terminal isopropyl units and the intra-chain imidazole unit, as well as the two signals assigned to the polymer chain, a molecular weight of ∼26 ± 3 kDa was estimated for the POs backbone. For the backbone of the PPhen polymer, analysis of the molecular weight via NMR spectroscopy was not possible due to overlapping signals in the spectrum of the backbone.
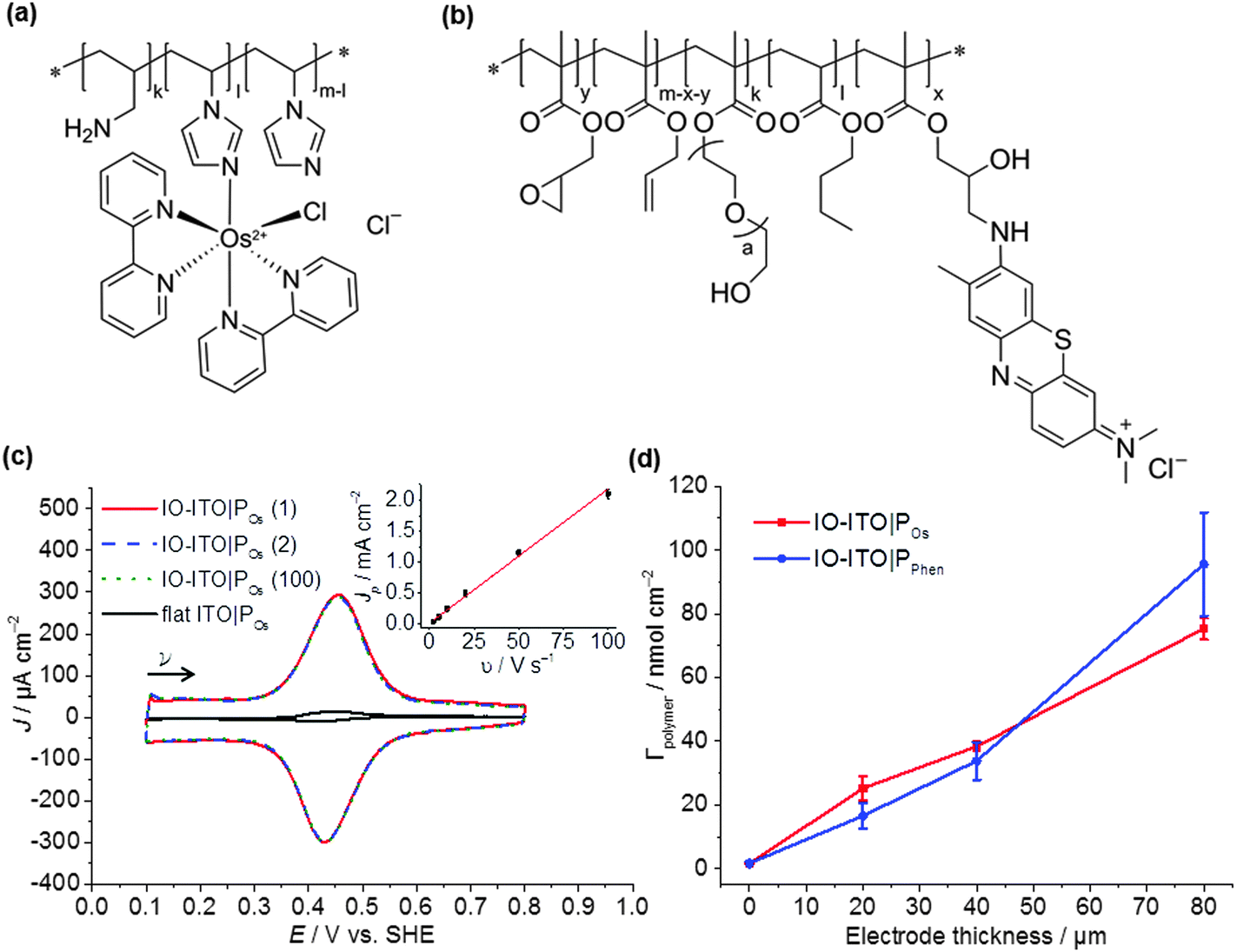 | ||
| Fig. 2 (a) Chemical structures of the POs and (b) PPhen polymers. (c) CV scans of POs adsorbed on 20 μm thick IO-ITO (adsorbed 25 ± 4 nmol Os cm−2) showing excellent stability on the electrode surface. The first scan and the 100th scan (dark, 10 mV s−1) are shown by the red solid trace and the dark green dotted trace, respectively. The second scan was the only scan measured during irradiation (λ = 685 nm; Ee = 10 mW cm−2), and is shown by the blue dashed line. A CV scan with POs-modified flat ITO (dark, 10 mV s−1) is shown for comparison (adsorbed 1.7 ± 0.2 nmol Os cm−2). The inset shows a linear dependence of the peak current density Jp with the scan rate ν, confirming electron transfer of a surface-confined redox species. (d) Linear dependence of the redox-active centres loading for both polymers POs and PPhen (up to 75 ± 3 and 96 ± 16 nmol cm−2, respectively) with the electrode thickness confirms effective infiltration of the polymers into the electrode matrix. The error bars correspond to the standard deviation (N = 4). All experiments were carried out in a MES electrolyte solution (pH = 6.5, T = 25 °C). | ||
The total number of Os complexes in POs was quantified using UV-vis spectroscopy in DMSO (0.74 ± 0.04 mmol g−1 polymer, Fig. S3, ESI†), which is consistent with ICP-AES measurements (0.67 ± 0.05 mmol Os g−1 polymer). Cis-[OsIICl(1-(n-butyl)-imidazole)(bipy)2](PF6) (Fig. S3c, ESI†), which can be regarded as the freely diffusing analogue to the Os complex moiety in the POs, was used as a reference for characterisation by UV-vis spectroscopy. The spectrum of the freely diffusing complex and the polymer exhibit the same spectral features (Fig. S3a, ESI†). Thus, for the calculation of the total number of Os complexes within the polymer, we assume that both species exhibit the same extinction coefficients. From the UV-vis studies, the ratio of non-complexed imidazole units to Os complex moieties was calculated to be ≈7:1, which corresponds to a molecular weight of ∼44 ± 5 kDa for POs. The same analysis was performed with the freely diffusing toluidine blue (Fig. S3c, ESI†) and PPhen. The spectral shapes of both species are again similar (Fig. S3d, ESI†), but the extinction coefficient of the toluidine blue moiety is increased upon covalent attachment to the polymer backbone (the primary amine in the toluidine blue monomer is converted to a secondary or even to a tertiary amine upon reaction with the epoxide functionality of the polymer backbone of PPhen). Thus, the estimation of the exact number of toluidine blue species was not possible (calculated values exceed the theoretical values).
The hydrodynamic particle diameter of POs and PPhen was determined using DLS (Fig. S4, ESI†) to be 16 ± 1 nm and ∼500 nm (broad distribution), respectively, which indicate the agglomeration of smaller polymer chains. Since both polymer solutions were filtered through a membrane with 200 nm pore size, it was concluded that the PPhen polymer chains form weak agglomerates that can be easily disassembled. The estimated sizes and agglomeration properties of POs and PPhen are expected to allow them to enter into the IO-ITO structure either by diffusional transport or by convection due to the capillary forces induced by pore filling and H2O evaporation.
Integration of PSII and polymer into IO-ITO electrodes
The polymers (1 μL, 10 mg mL−1) were drop-cast onto the IO-ITO and allowed to adsorb for 15 min at room temperature. The redox properties of the adsorbed polymers on the IO-ITO electrode (IO-ITO|polymer) were characterised using cyclic voltammetry (CV; Fig. 2c and Fig. S5, ESI†). The redox waves of POs and PPhen were attributed to the Os3+/2+ (1e− transfer process) and Phen+/PhenH (2e−/H+ transfer) redox couples, respectively.6 The positive reduction potential of the POs polymer (E1/2 = 0.44 V vs. SHE) is expected to provide a large driving force for electron transfer from the QA and QB (E1/2 = −0.14 V and −0.06 V vs. SHE, respectively)19 to the redox centres of the polymer (Fig. 1b). However, electron transfer between QA/QB and the Os complexes results also in a substantial potential loss (>0.4 V).5 The PPhen hydrogel exhibits a less positive reduction potential (E1/2 = 0.04 V vs. SHE), which matches the QB more closely (Fig. 1b). The reversibility of the electron transfer process for the surface-adsorbed redox polymers is evident in the almost symmetrical shape in the CV scans of POs and PPhen, which show minimal peak separation between the oxidation and reduction potentials (ΔEp = 0.02 ± 0.01 and 0.01 ± 0.005 V for POs and PPhen, respectively). Furthermore, an anodic to cathodic peak current ratio close to unity (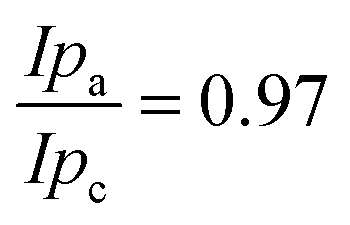 and 0.83 for POs and PPhen, respectively) can be observed, and the current density is linearly proportional to the scan rate up to 100 mV s−1 (Fig. 2c inset and Fig. S5, ESI†).42 The observed slight increase in the ΔEp at scan rates >10 mV s−1 (Fig. S5, ESI†) was attributed to the rate limiting charge transfer between the polymer and the electrode surface.46 In particular, PPhen showed small shoulder waves at high scan rates that could arise from the slow 2e−/H+ transfer rate at the iminium cation site. The voltammetric features of POs, even at the relatively fast scan rates used here, are characteristic for surface-confined species. The corresponding diffusion layer thicknesses of the electron (δ, calculated from eqn (1)) give an estimate of the film thickness that is accessible to the electrochemical process assuming planar semi-infinite diffusion. Based on the previously reported apparent electron diffusion coefficient of the electron for POs (De of 4.00 ± 0.47 × 10−9 cm2 s−1),39 the δ value corresponding to the scan rate of 100 mV s−1 is 320 nm. Hence, the diffusional range of the electrons within POs is in the range of the IO macropore radius (375 nm; Fig. S1a, ESI†) even at fast scan rates. As such, the IO structure should increase the total polymer loading that can participate in electron transfer in a given geometric surface area by taking advantage of the thick 3-D architecture.
and 0.83 for POs and PPhen, respectively) can be observed, and the current density is linearly proportional to the scan rate up to 100 mV s−1 (Fig. 2c inset and Fig. S5, ESI†).42 The observed slight increase in the ΔEp at scan rates >10 mV s−1 (Fig. S5, ESI†) was attributed to the rate limiting charge transfer between the polymer and the electrode surface.46 In particular, PPhen showed small shoulder waves at high scan rates that could arise from the slow 2e−/H+ transfer rate at the iminium cation site. The voltammetric features of POs, even at the relatively fast scan rates used here, are characteristic for surface-confined species. The corresponding diffusion layer thicknesses of the electron (δ, calculated from eqn (1)) give an estimate of the film thickness that is accessible to the electrochemical process assuming planar semi-infinite diffusion. Based on the previously reported apparent electron diffusion coefficient of the electron for POs (De of 4.00 ± 0.47 × 10−9 cm2 s−1),39 the δ value corresponding to the scan rate of 100 mV s−1 is 320 nm. Hence, the diffusional range of the electrons within POs is in the range of the IO macropore radius (375 nm; Fig. S1a, ESI†) even at fast scan rates. As such, the IO structure should increase the total polymer loading that can participate in electron transfer in a given geometric surface area by taking advantage of the thick 3-D architecture.
No photocurrent originating from the IO-ITO|polymer electrodes during irradiation (λ = 685 nm, Ee = 10 mW cm−2) was observed (Fig. 2c). The surface coverage (Γ) of the electrochemically-active redox centres connected to the electrode surface was calculated for each polymer using eqn (2); the total charge was calculated by integrating the area under the CV curve minus the background. A substantial enhancement in polymer loading (Fig. S5, ESI†) was observed for IO-ITO compared to flat electrodes. The polymer loading increased approximately linearly with the electrode thickness (Fig. 2d). Loadings of 1.7 ± 0.2 nmol cm−2 and 1.7 ± 0.5 nmol cm−2 were observed for flat ITO electrodes with the adsorbed polymers, POs and PPhen, respectively, which is comparable to previously reported values (1.8 ± 0.1 nmol cm−2) on flat glassy carbon electrodes.39 IO-ITO electrodes with a thickness of 20, 40 and 80 μm gave rise to a 15-, 23- and 45-fold increase in ΓOs (25 ± 4, 38 ± 1 and 75 ± 3 nmol cm−2) and 10-, 19- and 55-fold increase in ΓPhen (17 ± 4, 34 ± 5 and 96 ± 16 nmol cm−2) compared to a flat ITO electrode, respectively (Table S1, ESI†). The number of electrochemically-active Os complexes on 20 μm thick IO-ITO was found to be ∼85 ± 10% of the total number of immobilised Os atoms, quantified by ICP-AES and UV-vis spectroscopy. The IO-ITO|POs electrode exhibited excellent stability, showing no significant desorption or decomposition after 100 CV cycles at 10 mV s−1 scan rate (Fig. 2c). The IO-ITO|PPhen electrode exhibited lower stability (63% and 38% ΓPhen remaining after the second and third CV cycle at 10 mV s−1 scan rate, respectively, Fig. S5e, ESI†). The imidazole functionality in the POs is also likely to have a strong affinity for the ITO surface and act as an anchoring group, analogous to histidine-tagged enzymes.47 The toluidine blue centres of the PPhen (heterocyclic N and S atoms pKa < 7) are most likely deprotonated and the polymer backbone groups (amine functions pKa > 7) are protonated at pH 6.5. The hydrogel nature of the polymers allows the diffusion of small molecules throughout the network, although the lack of anchoring groups in PPhen prevents stable loading.
Following the assembly and characterisation of the IO-ITO|POs and IO-ITO|PPhen electrode systems, PSII was introduced into the electrode system. PSII (1 μL, 2.6 mg Chl a mL−1) and the redox polymer (1 μL, 10 mg mL−1) were mixed together and immediately drop-cast on the IO-ITO electrode (20 μm thick) as a uniform blend, then allowed to adsorb in the dark for 15 min at room temperature. The amount of PSII entrapped in the polymer matrix inside the electrode (ΓPSII) was quantified based on the absorption amplitude of Chl a (λmax = 665 nm, eqn (3)), extracted from PSII using MeOH (Fig. S6a, ESI†). UV-vis spectra of polymer solutions (0.02 mg mL−1) in the electrolyte solution and MeOH (Fig. S6b, ESI†) showed a negligible absorption at the irradiation wavelength used in PF-PEC (λ = 685 nm). Exceptionally high PSII loadings were observed for IO-ITO|POs–PSII (144 ± 21 pmol cm−2), IO-ITO|PPhen–PSII (149 ± 7 pmol cm−2) and IO-ITO|PSII (162 ± 17 pmol cm−2) (Fig. S6c, ESI†). The slightly higher PSII loading in the PSII-only system could be explained by more space being available (that could be filled by the enzymes) in the absence of polymers. The SEM images of the IO-ITO electrodes taken before and after POs–PSII and PPhen–PSII deposition (Fig. S1, ESI†) indicate no evident channel or pore blockages.
The effective assembly of PSII with the polymers can be attributed to favourable non-covalent interactions between the protein shell and the polymers. The hydrophilic nature of the polymers is bestowed primarily by the cationic Os complex/phenothiazine dye, with some contributions by the multiple polar functional side groups (POs: imidazole and amine groups; PPhen: polyethylene glycol side chains and OH-functions). At pH 6.5, POs is expected to behave as a cationic polyelectrolyte since the primary amine (pKa 10) and imidazole groups (pKa 7) are protonated. This also contributes to the close to optimal polymer solvation and swelling, supported by high De value previously observed.39
PF-PEC with IO-ITO|polymer–PSII electrodes
PF-PEC measurements were performed at 25 °C using an IO-ITO|polymer–PSII working, a Pt wire counter and a Ag/AgCl (3 M KCl) reference electrode. The electrolyte solution was adjusted to pH 6.5 and contained CaCl2 (20 mM), MgCl2 (15 mM), KCl (50 mM) and MES (40 mM). The action spectra of the IO-ITO|PSII, IO-ITO|POs–PSII and IO-ITO|PPhen–PSII photoelectrodes were recorded to determine appropriate wavelengths of light for photocurrent generation (Fig. 3 and Fig. S7, ESI†). In a typical experiment, the wavelength was decreased in steps of 20 nm starting from 760 nm at an applied potential of 0.5 V vs. SHE and the photoresponse was measured at each wavelength. The maximum photocurrent was observed at 680 nm, which matches the UV-vis absorption spectrum of PSII and supports the integrity of PSII in its native state during the immobilisation on the IO-ITO electrode.21,48 The action spectra of the control samples IO-ITO, IO-ITO|POs and IO-ITO|PPhen corresponded to the UV-vis absorption spectra of the respective polymers (Fig. S6b, ESI†) and confirmed no significant contribution to the photocurrent generation from the polymers (Fig. S7, ESI†).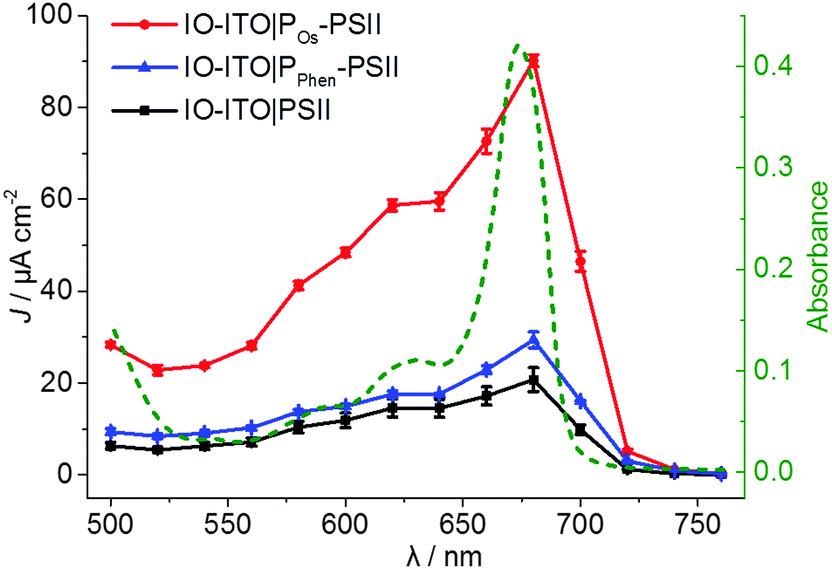 | ||
| Fig. 3 Action spectra (solid traces) showing the photocurrent density (left Y axis) vs. irradiation wavelength of the IO-ITO|PSII (black), IO-ITO|POs–PSII (red) and IO-ITO|PPhen–PSII (blue) photoelectrodes (20 μm thickness) recorded with monochromatic light measured in 20 nm steps (Ee = 3.25 to 6.26 mW cm−2) at Eapp = 0.5 V vs. SHE (pH = 6.5, T = 25 °C) in MES electrolyte solution (see Fig. S7 for raw data and more detailed information, ESI†). The error bars correspond to the standard deviation (N = 3). The UV-vis absorption spectrum of the PSII (1 μL, 2.6 mg Chl a mL−1) in MES electrolyte solution (0.5 mL) (dashed green line, right Y axis) matches the photocurrent response of PSII on the electrodes. | ||
Stepped chronoamperometry with chopped red-light irradiation (λ = 685 nm, Ee = 10 mW cm−2) was performed to characterise the onset potential (Eonset) of photocurrents in each IO-ITO|polymer–PSII system (Fig. S8, ESI†). In a typical experiment, the applied potential was gradually increased in steps of 0.1 V in the anodic direction. A summary of the photoresponse as a function of the Eapp is shown in Fig. 4. The IO-ITO|PPhen–PSII system showed an Eonset value of ∼0.1 V vs. SHE, which is slightly more positive than expected, possibly due to other minor interference charge transfer pathways. However, the Eonset of IO-ITO|PPhen–PSII is still clearly more negative than that of the IO-ITO|POs–PSII electrode (Eonset = ∼0.3 V vs. SHE; Fig. 4 inset), which is consistent with the lower E1/2 of PPhen (E1/2 = 0.04 V vs. SHE) compared to the POs (E1/2 = 0.44 V vs. SHE). The photocurrents for both the IO-ITO|POs–PSII and IO-ITO|PPhen–PSII electrodes reach a plateau at ∼0.5 V vs. SHE. No photoactivity and negligible dark current were observed for the IO-ITO|POs and IO-ITO|PPhen electrodes (Fig. S8d, ESI†). Upon prolonged irradiation at more positive potentials (Eapp > 0.6–0.7 V vs. SHE), a drop in photocurrent was observed. This drop in photocurrent is irreversible, as shown by the low photoresponse given by a backward scan in the negative direction (at 0.5 V vs. SHE, Fig. S9a, ESI†). CV scans of the IO-ITO|POs–PSII electrode (Fig. S9b, ESI†) confirmed the stability and homogeneity of the integrated PSII–polymer film on the electrode surface in the dark. However, CV scans performed with red light irradiation (Fig. S9c, ESI†) show a significant decrease in photocurrents after 3 potential sweep cycles over the range 0.1–0.8 V vs. SHE, which is indicative of POs–PSII film photodegradation (PSII-limiting system).49
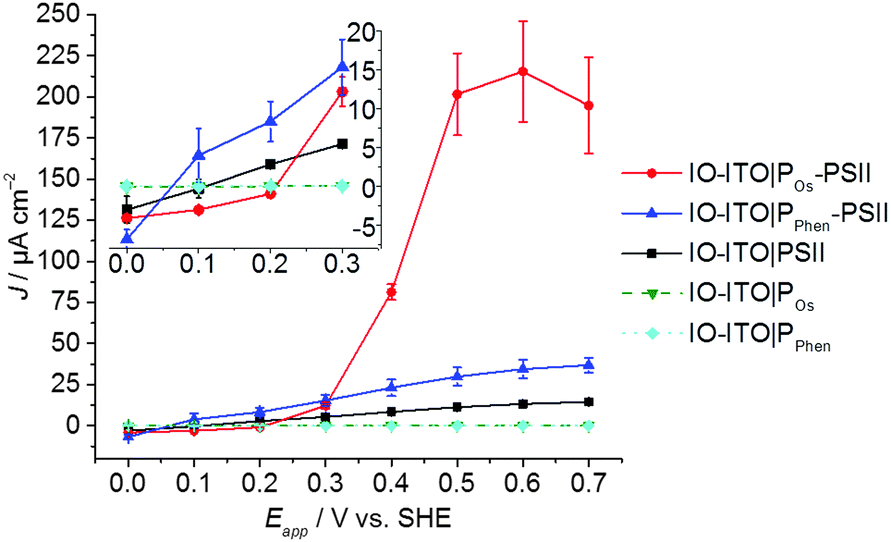 | ||
| Fig. 4 Photocurrent density as a function of the applied potential (Eapp) for the IO-ITO|polymer–PSII photoanodes determined by stepped potential chronoamperometry (pH = 6.5, T = 25 °C) (see Fig. S8 for raw data, ESI†). The inset shows a magnified region of the plot close to the onset potentials of the polymers. The photoresponse for PSII-free IO-ITO|polymer electrodes are shown for comparison. The error bars correspond to the standard deviation (N = 4). | ||
To investigate the quality of the wiring between the PSII and the ITO electrode in the IO-ITO|polymer–PSII systems, chronoamperometry at an applied potential of 0.5 V vs. SHE was performed in the presence and absence of the diffusional mediator, DCBQ, with chopped light irradiation (Fig. 5). Typical photocurrent densities for optimised 20 μm thick IO-ITO|PSII, IO-ITO|POs–PSII, and IO-ITO|PPhen–PSII electrodes in the absence of a diffusional mediator (Fig. 5a) were approximately 15, 230 and 45 μA cm−2, respectively, which compares favourably with PF-PEC of previously reported PSII–electrodes.9,19 Bare IO-ITO and IO-ITO|polymer electrodes exhibited photocurrent densities below 100 nA cm−2.
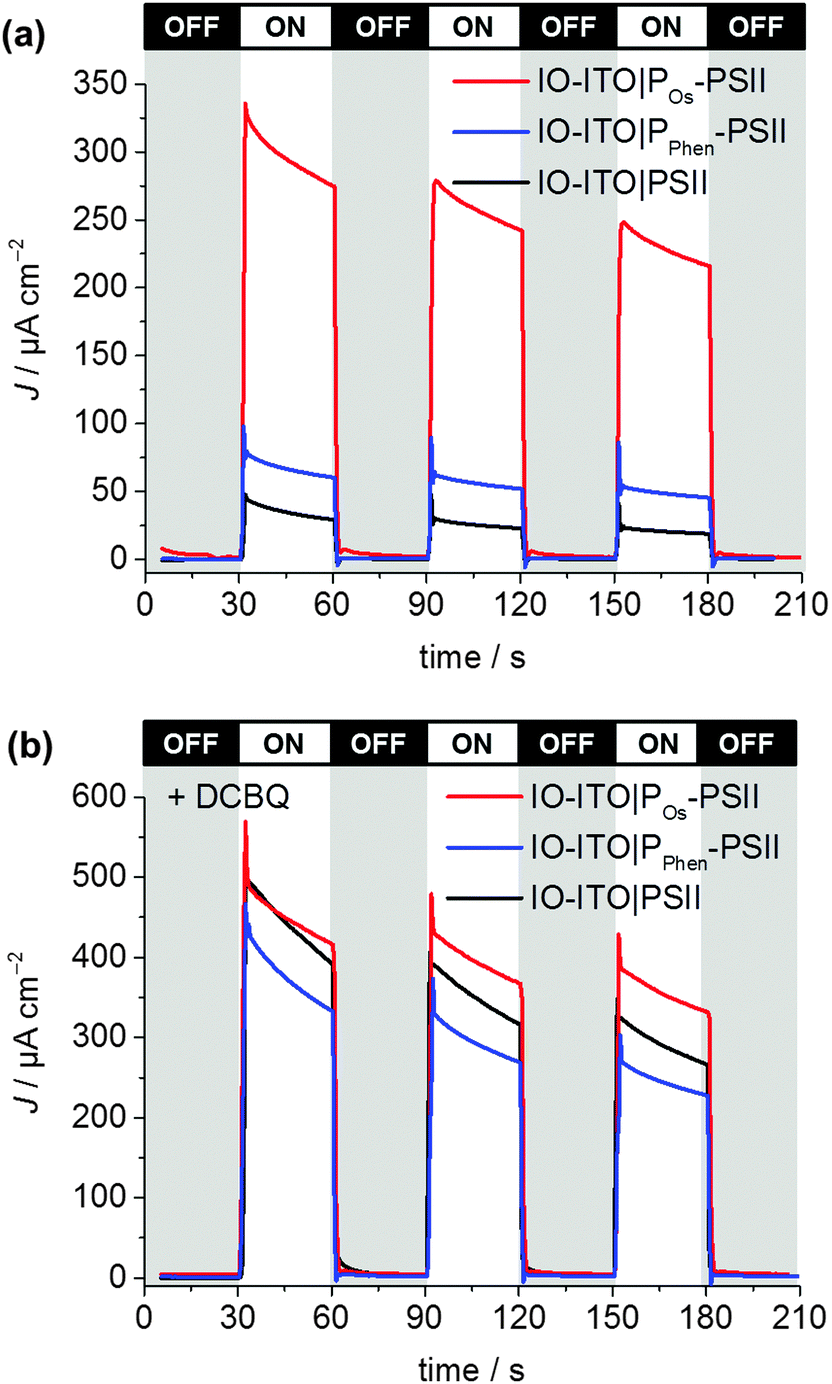 | ||
| Fig. 5 Photocurrent density of the IO-ITO|polymer–PSII and IO-ITO|PSII electrodes (20 μm thickness) measured with chopped illumination (λ = 685 nm; Ee = 10 mW cm−2) at Eapp = 0.5 V vs. SHE (pH = 6.5, T = 25 °C). No diffusional mediator is present in (a) and DCBQ (1 mM) was present in the electrolyte solution in (b). The reported photocurrent densities were defined as the right shoulder of the third peak. The PSII loading for each modified electrode (see Fig. S6, ESI†) was comparable: 162 ± 17 pmol cm−2 (IO-ITO|PSII), 144 ± 21 pmol cm−2 (IO-ITO|POs–PSII) and 149 ± 7 pmol cm−2 (IO-ITO|PPhen–PSII). | ||
The relatively large photoresponse observed for the IO-ITO|Pos–PSII system is indicative of efficient electronic communication between PSII and the electrode. An external quantum efficiency (EQE) of 4.4% (derived using eqn (4)) was obtained for the IO-ITO|POs–PSII system, which is 15-fold higher than for IO-ITO|PSII (EQE = 0.3%) and the highest reported so far for a diffusional mediator-free PSII-electrode.9,19 The photoresponse in the IO-ITO|PPhen–PSII system (EQE = 0.8%) is improved compared to IO-ITO|PSII, however the enhancement is not as great as the IO-ITO|POs–PSII system, which indicates that the PPhen is less efficient at wiring PSII to the electrode, possibly because of its significantly lower driving force for electron transfer.
The addition of DCBQ (0.36 V vs. SHE)9 to the IO-ITO|POs–PSII system gave rise to a further 1.5-fold photocurrent density increase (375 μA cm−2, EQE = 7.7%, Fig. 5b). Similarly, the addition of DCBQ to the IO-ITO|PPhen–PSII system gave rise to a 6-fold photocurrent density increase (236 μA cm−2, EQE = 4.6%). The addition of DCBQ to the IO-ITO|PSII system gave rise to an 18-fold increase in photoresponse (265 μA cm−2, EQE = 5.1%). This observation demonstrates that a significantly higher proportion of PSII was electrically connected to the electrode in the IO-ITO|POs–PSII system compared to IO-ITO|PPhen–PSII, and that the IO-ITO|polymer–PSII electrodes were better connected than the IO-ITO|PSII system. Addition of bifunctional cross-linkers (PEGDGE for POs31 and 2,2′-(ethylenedioxy)diethanethiol for PPhen6), to the IO-ITO|polymer–PSII systems resulted in no further photocurrent increase. This may be attributed to the stabilisation of the PSII–polymer matrix inside the 3-D-interconnected porous electrode framework.9
These results indicate favourable interactions between the POs and PSII, most likely between the side groups of the polymer (positively-charged Os3+ complex, primary amine and imidazolium units) and the polar residues of PSII,41,50 in particular the negatively charged region at the stromal side of PSII and near the QA site.37,51 In addition, a high number of electrochemically-active Os centres is estimated to be in close proximity to each PSII unit (based on the Os centre to PSII ratios (ΓOs/ΓPSII ∼ 175) co-adsorbed on the electrodes), which explains the favourable photoelectrochemical response of the system discussed earlier. The PPhen can also interact with PSII via its hydrophilic side chains and residual epoxide groups to give rise to possible cross-linking.41,50 However, the PPhen is expected to have weaker interactions with the ITO electrode surface (Fig. S5e, ESI†), and is more likely to undergo polymer aggregation, as indicated by DLS, to result in significantly lower polymer entrapment and retention of PSII. The estimated number of toluidine blue units per PSII unit is 108, which is significantly lower than in the IO-ITO|POs–PSII system.
Comparison of POs and PPhen
In the preceding experiments, PF-PEC was used to systematically compare the performance of two benchmark polymers for PSII entrapment when they are integrated into high surface area electrodes. The POs exhibited the most stable integration in 20 μm thick IO-ITO electrodes. When embedded with PSII, the IO-ITO|POs–PSII electrodes delivered high photocurrent densities that were at least 5-fold higher than systems connected by PPhen (Fig. 5a). Despite the fact that PPhen is free of noble metals and has a better matched E1/2 to the QA and QB (giving rise to earlier onset potentials for water oxidation), it exhibits lower adsorption stability on 20 μm thick IO-ITO electrodes. The IO-ITO|PPhen–PSII systems showed lower overall photoresponses compared to IO-ITO|POs–PSII, which can also be rationalised by their more negative redox potential values (providing less driving force) and slower (H+ diffusion-dependent) electron hopping process (2e−/H+vs. 1e− transfer, respectively). Overall, IO-ITO|POs–PSII electrodes demonstrated higher performance and more efficient wiring between the PSII and the ITO electrode.IO-ITO|POs–PSII performance
To determine the enhancement of the photoresponse with film thickness in IO-ITO|polymer–PSII, IO-ITO electrodes with varying thickness (from 20 to 80 μm) were prepared and studied by PF-PEC. The focus was placed on the optimisation of the top performing IO-ITO|POs–PSII systems.The maximum loading of PSII and POs on IO-ITO electrodes of different thicknesses are shown in Fig. 6a. POs and PSII loadings increase linearly as the thickness rises from 0 to 80 μm. In comparison, an adsorbent saturation point was reached for IO-ITO|POs–PSII electrodes beyond 40 μm. This was attributed to the accumulation of moderately viscous POs–PSII aggregates over deposition time, which limits the penetration depth of the POs due to the formation of channel blockages. No significant losses due to desorption upon long-term (60 min) immersion in the electrolyte solution with constant light irradiation were observed.
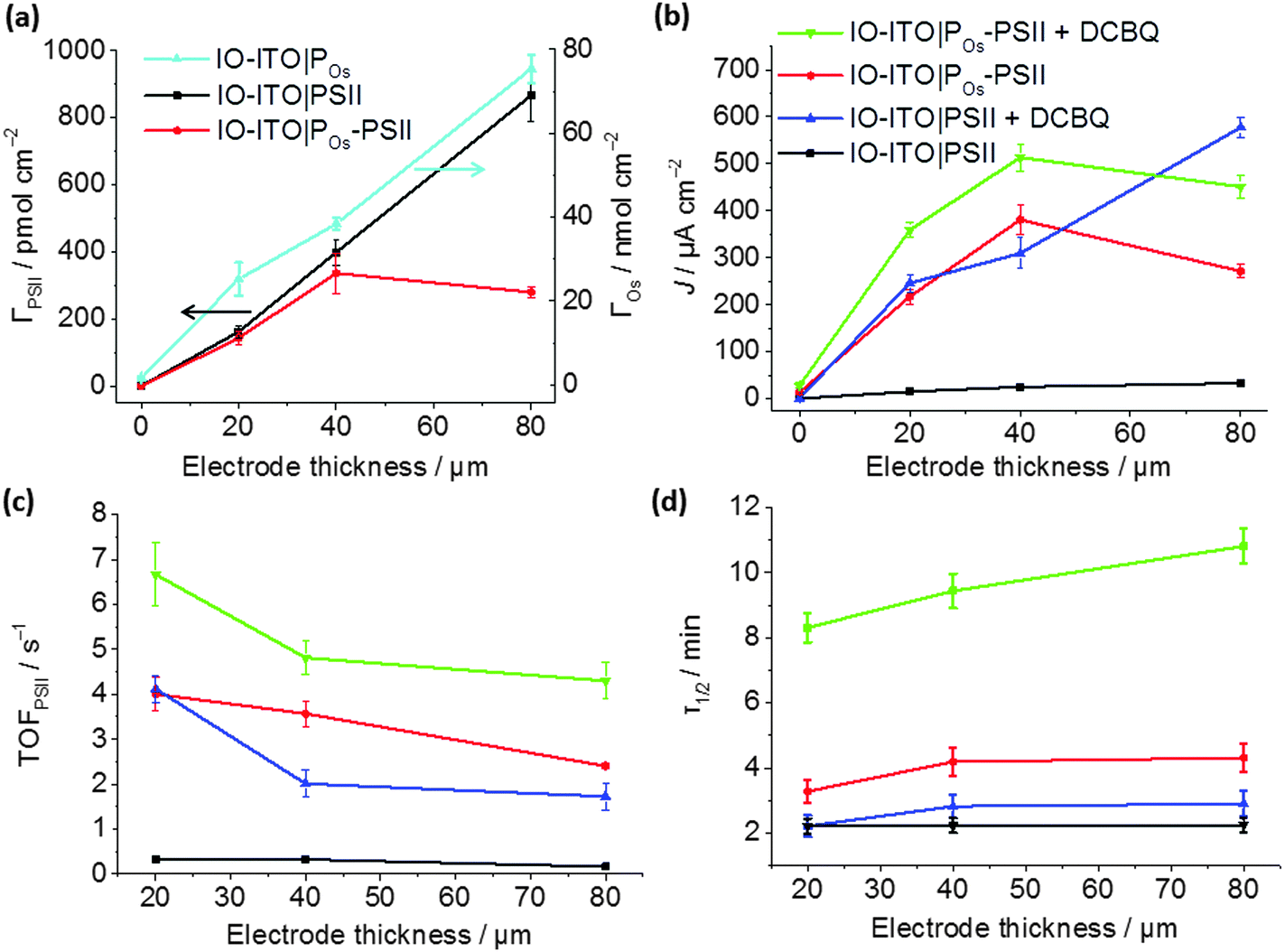 | ||
| Fig. 6 Characterisation of the IO-ITO|POs–PSII photoanode as a function of the electrode thickness: (a) PSII loading quantified by the amplitude of absorption at λ = 665 nm, and Os3/2+ redox centres loading determined by CV (Fig. S5b, ESI†); (b) photocurrent densities, (c) corresponding TOFPSII values and (d) photocurrent half-life times (τ1/2) measured upon light illumination (λ = 685 nm; Ee = 10 mW cm−2) at a fixed potential of 0.5 V vs. SHE without any additional diffusional mediator and upon addition of 1 mM of freely diffusing DCBQ mediator (pH = 6.5, T = 25 °C). The error bars correspond to the standard deviation (N = 4). | ||
The dependence of photocurrent density on the IO-ITO|POs–PSII electrode thickness is shown in Fig. 6b. A saturation photocurrent density of 381 ± 31 μA cm−2 (EQE = 6.9 ± 0.9%) for 40 μm thick electrodes was observed, which correlates with the maximum PSII loading reached at this thickness. Upon DCBQ addition, a further 1.35-fold photocurrent density increase was detected (513 ± 29 μA cm−2, EQE = 9.3 ± 1.2%). The IO-ITO|PSII electrode exhibited almost ideal linear increase in photocurrent densities with the ITO film thickness, which is also consistent with the trend of PSII loading in IO-ITO|PSII electrodes. Maximum photocurrent values of 33 ± 5 and 577 ± 21 μA cm−2 from 80 μm thick electrodes were observed in the absence and presence of DCBQ, respectively. The comparable maximum photocurrent densities reached by the IO-ITO|POs–PSII electrode in the absence of DCBQ and the IO-ITO|PSII electrode in the presence of DCBQ indicate efficient wiring of the PSII to the ITO surface by the POs matrix.
The theoretical TOFPSII of water oxidation was estimated (assuming 100% Faradaic efficiency) according to eqn (5) for the IO-ITO|POs–PSII electrodes of different thicknesses as shown in Fig. 6c. The maximum TOFPSII of 4.0 ± 0.4 s−1 was achieved using 20 μm thick IO-ITO|POs–PSII electrodes, which could be increased to 6.7 ± 0.7 s−1 by the addition of DCBQ. This is a 1.7-fold increase compared to the IO-ITO|PSII system in the presence of DCBQ, and indicates that the mediated IO-ITO|POs–PSII system is overall more efficiently wired than the mediated IO-ITO|PSII system due to the presence of the POs matrix.
The long-term photostability of the IO-ITO|POs–PSII system was evaluated at a relatively mild Eapp = 0.5 V vs. SHE and the results are presented in Fig. 6d. To determine the photocurrent half-life time (τ1/2), the photocurrent generated by IO-ITO|POs–PSII electrode under continuous light irradiation for 60 min was recorded starting at the third photoresponse peak (Fig. S10, ESI†). Across the entire thickness range, the IO-ITO|POs–PSII systems exhibited a 2-fold τ1/2 increase (maximum of 4.3 ± 0.4 min) compared to the IO-ITO|PSII systems (2.2 ± 0.2 min) in the absence of DCBQ. In the presence of DCBQ, further enhancement of the τ1/2 can be seen to reach ∼10 min in 80 μm thick IO-ITO|POs–PSII electrodes. After 60 min of constant light irradiation, ∼7% and 11% of the initial photocurrent was detected from the IO-ITO|POs–PSII electrode, without and with DCBQ addition, respectively. In contrast, less than 2% of the initial photocurrent was detected from the IO-ITO|PSII electrode. This can in part be attributed to the physical stabilisation of the PSII by the polymer matrix and the IO-ITO electrode architecture. The increased τ1/2 in the IO-ITO|POs–PSII system can also be partly attributed to reduced accumulation of pigments in the excited state due to more efficient electron transfer between PSII and the Os centres in POs.49 The higher efficiency in charge transfer would result in dampened formation of reactive oxygen species and deterioration of the D1 subunit in PSII.52
Finally, the photocurrent generated by the IO-ITO|POs–PSII electrode is high enough to enable the quantification of O2 evolution (Fig. 7). Controlled potential electrolysis at Eapp = 0.5 V vs. SHE was carried out in a two-compartment cell in the glovebox employing an optimised 40 μm thick IO-ITO|POs–PSII electrode upon light irradiation for 60 min (λ = 660 nm, Ee = 10 mW cm−2). The passage of 0.12 ± 0.03 C cm−2 charge was measured and the evolution of 0.24 ± 0.03 μmol O2 cm−2 was detected by a fluorescence O2 sensor, which corresponded to 85 ± 9% Faradaic efficiency. A turnover number TONPSII of 946 ± 96 mol O2 (mol PSII)−1, and an initial PSII-based TOFPSII of 3.6 ± 0.3 mol O2 (mol PSII)−1 s−1 was calculated based on quantified O2 and PSII using eqn (6) and (5), respectively. Previously, the generation of 0.23 ± 0.01 C cm−2 charge and the evolution of 0.45 ± 0.01 μmol O2 cm−2 (75 ± 4% Faradaic efficiency), corresponding to TONPSII of 4200 ± 200 mol O2 (mol PSII)−1 and TOFPSII of 12.9 ± 0.4 mol O2 (mol PSII)−1 s−1 were reported for the IO-ITO|PSII system in the presence of DCBQ.9 The absence of diffusion-limited mediators enables an all-integrated electrode design and eliminates problems such as those associated with concentration-dependent electron transfer. It also overcomes the issue of diffusional mass transport that may interfere with processes at the counter electrode and limit the performance of PSII-based PEC assemblies. Lastly, this electrode prototype allows all catalytic/electroactive material to be confined inside the porous electrode architecture, minimising the presence of high concentration catalytic/electroactive material in the bulk solution.
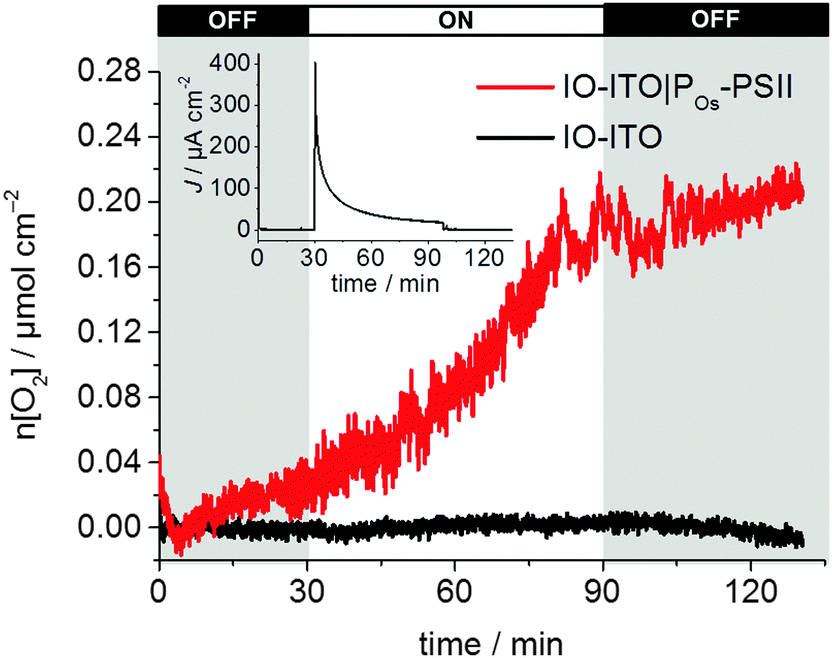 | ||
| Fig. 7 Quantification of O2 evolution and determination of Faradaic yield (85 ± 9%) for the IO-ITO|POs–PSII electrode (40 μm thickness) during continuous light illumination (λ = 660 nm; Ee = 10 mW cm−2) between 30 and 90 min with continuous stirring at Eapp = 0.5 V vs. SHE (pH = 6.5, T = 25 °C, red line). The chronoamperogram is shown in the inset. A control experiment in the absence of PSII is also shown (black curve). | ||
Conclusions
The present study has introduced a new benchmark PSII-based electrode, which was developed as a result of a rational design process that incorporated the best aspects of two leading enzyme immobilisation strategies. We integrated the stabilisation and efficient electronic wiring of enzymes within redox polymer matrices with the high loading capacity of hierarchically-structured electrodes. This enabled the demonstration of high photocurrent densities, TOFs and levels of evolved O2 that could be obtained for a PSII-driven PF-PEC system without the requirement for diffusional additives in the bulk solution. The photocurrents arising from PSII reported here also compare favourably with those reported for other wired photosynthetic proteins such as bacterial reaction centres53 or photosystem I.4,39,54The development of this IO-ITO|polymer–PSII system provides the basic concepts needed for the future design of enzyme-driven semi-artificial photosynthetic systems, including PEC tandem systems that incorporate other reaction centre or pigment-based proteins. We anticipate that this approach will also serve as an inspiration in the design of synthetic PEC water-splitting architectures. In the future, we expect that improvements in polymer design will lead to favourable changes to the electrode stability, electron hopping efficiency and formal redox potentials to better match the energy levels of the protein terminal electron acceptors. Lastly, hierarchical IO electrodes have demonstrated the potential to be highly versatile as a host system and may be used in various applications outside of PF-PEC, including batteries, fuel cells and solar cells.
Acknowledgements
This work was supported by the U.K. Engineering and Physical Sciences Research Council (EP/L015978/1 and EP/G037221/1, nanoDTC, and a DTA studentship to K. P. S.), the U.K. Biology and Biotechnological Sciences Research Council (BB/J000124/1), the Deutsch-Israelische Projektkooperation in the framework of the project “Nanoengineered optoelectronics with biomaterials and bioinspired assemblies”, the Cluster of Excellence RESOLV (EXC 1069) funded by the Deutsche Forschungsgemeinschaft (DFG), the COST Action TD1102 PHOTOTECH and a Marie Curie International Incoming Fellowship (PIIF-GA-2012-328085 RPSII to J. J. Z.). We would also like to thank Dr Sascha Pöller and Ms Sabine Alsaoub for their contributions to polymer synthesis, Dr Julien Warnan, Mr William Robinson, Dr Bertrand Reuillard and Dr Demetra Achilleos for valuable discussions and Mr Wayne Bailey and Mr Manuel Wentscher for building the monochromatic red-light LED lamp.References
- M. T. Giardi, M. Koblížek and J. Masojídek, Biosens. Bioelectron., 2001, 16, 1027–1033 CrossRef CAS PubMed.
- M. Koblížek, J. Malý, J. Masojídek, J. Komenda, T. Kučera, M. T. Giardi, A. K. Mattoo and R. Pilloton, Biotechnol. Bioeng., 2002, 78, 110–116 CrossRef PubMed.
- D. J. K. Swainsbury, V. M. Friebe, R. N. Frese and M. R. Jones, Biosens. Bioelectron., 2014, 58, 172–178 CrossRef CAS PubMed.
- A. Mershin, K. Matsumoto, L. Kaiser, D. Yu, M. Vaughn, M. K. Nazeeruddin, B. D. Bruce, M. Grätzel and S. Zhang, Sci. Rep., 2012, 2, 1–7 Search PubMed.
- T. Kothe, N. Plumeré, A. Badura, M. M. Nowaczyk, D. A. Guschin, M. Rögner and W. Schuhmann, Angew. Chem., Int. Ed., 2013, 52, 14233–14236 CrossRef CAS PubMed.
- V. Hartmann, T. Kothe, S. Pöller, E. El-Mohsnawy, M. M. Nowaczyk, N. Plumeré, W. Schuhmann and M. Rögner, Phys. Chem. Chem. Phys., 2014, 16, 11936–11941 RSC.
- K. Nguyen and B. D. Bruce, Biochim. Biophys. Acta, Bioenerg., 2014, 1837, 1553–1566 CrossRef CAS PubMed.
- O. Yehezkeli, R. Tel-Vered, D. Michaeli, R. Nechushtai and I. Willner, Small, 2013, 9, 2970–2978 CrossRef CAS PubMed.
- D. Mersch, C.-Y. Lee, J. Z. Zhang, K. Brinkert, J. C. Fontecilla-Camps, A. W. Rutherford and E. Reisner, J. Am. Chem. Soc., 2015, 137, 8541–8549 CrossRef CAS PubMed.
- G. Ananyev and G. C. Dismukes, Photosynth. Res., 2005, 84, 355–365 CrossRef CAS PubMed.
- D. J. Vinyard, G. M. Ananyev and G. C. Dismukes, Annu. Rev. Biochem., 2013, 82, 577–606 CrossRef CAS PubMed.
- R. Tel-Vered and I. Willner, ChemElectroChem, 2014, 1, 1778–1797 CrossRef CAS.
- O. Yehezkeli, R. Tel-Vered, D. Michaeli, I. Willner and R. Nechushtai, Photosynth. Res., 2014, 120, 71–85 CrossRef CAS PubMed.
- K. K. Rao, D. O. Hall, N. Vlachopoulos, M. Grätzel, M. C. W. Evans and M. Seibert, J. Photochem. Photobiol., B, 1990, 5, 379–389 CrossRef CAS.
- A. Badura, B. Esper, K. Ataka, C. Grunwald, C. Wöll, J. Kuhlmann, J. Heberle and M. Rögner, Photochem. Photobiol., 2006, 82, 1385–1390 CrossRef CAS PubMed.
- N. Terasaki, M. Iwai, N. Yamamoto, T. Hiraga, S. Yamada and Y. Inoue, Thin Solid Films, 2008, 516, 2553–2557 CrossRef CAS.
- O. Yehezkeli, R. Tel-Vered, J. Wasserman, A. Trifonov, D. Michaeli, R. Nechushtai and I. Willner, Nat. Commun., 2012, 3, 742–748 CrossRef PubMed.
- J. Barber and P. D. Tran, J. R. Soc., Interface, 2013, 10, 20120984 CrossRef PubMed.
- M. Kato, J. Z. Zhang, N. Paul and E. Reisner, Chem. Soc. Rev., 2014, 43, 6485–6497 RSC.
- S. A. Trammell, A. Spano, R. Price and N. Lebedev, Biosens. Bioelectron., 2006, 21, 1023–1028 CrossRef CAS PubMed.
- E. Y. Katz, A. Y. Shkuropatov, O. I. Vagabova and V. A. Shuvalov, Biochim. Biophys. Acta, Bioenerg., 1989, 976, 121–128 CrossRef.
- E. Katz, J. Electroanal. Chem., 1994, 365, 157–164 CrossRef CAS.
- C. Nakamura, M. Hasegawa, Y. Yasuda and J. Miyake, Appl. Biochem. Biotechnol., 2000, 84–86, 401–408 CrossRef CAS PubMed.
- S. A. Trammell, L. Wang, J. M. Zullo, R. Shashidhar and N. Lebedev, Biosens. Bioelectron., 2004, 19, 1649–1655 CrossRef CAS PubMed.
- N. Lebedev, S. A. Trammell, A. Spano, E. Lukashev, I. Griva and J. Schnur, J. Am. Chem. Soc., 2006, 128, 12044–12045 CrossRef CAS PubMed.
- H. Yaghoubi, Z. Li, D. Jun, E. Lafalce, X. Jiang, R. Schlaf, J. T. Beatty and A. Takshi, J. Phys. Chem. C, 2014, 118, 23509–23518 CAS.
- M. Kato, T. Cardona, A. W. Rutherford and E. Reisner, J. Am. Chem. Soc., 2013, 135, 10610–10613 CrossRef CAS PubMed.
- A. Heller, Curr. Opin. Chem. Biol., 2006, 10, 664–672 CrossRef CAS PubMed.
- R. Gracia and D. Mecerreyes, Polym. Chem., 2013, 4, 2206–2214 RSC.
- A. Badura, T. Kothe, W. Schuhmann and M. Rögner, Energy Environ. Sci., 2011, 4, 3263–3274 CAS.
- A. Badura, D. Guschin, B. Esper, T. Kothe, S. Neugebauer, W. Schuhmann and M. Rögner, Electroanalysis, 2008, 20, 1043–1047 CrossRef CAS.
- P. N. Bartlett and K. F. E. Pratt, J. Electroanal. Chem., 1995, 397, 61–78 CrossRef.
- V. Fourmond, S. Stapf, H. Li, D. Buesen, J. Birrell, O. Rüdiger, W. Lubitz, W. Schuhmann, N. Plumeré and C. Léger, J. Am. Chem. Soc., 2015, 137, 5494–5505 CrossRef CAS PubMed.
- Y. Li, Z.-Y. Fu and B.-L. Su, Adv. Funct. Mater., 2012, 22, 4634–4667 CrossRef CAS.
- P. Trogadas, V. Ramani, P. Strasser, T. F. Fuller and M.-O. Coppens, Angew. Chem., Int. Ed., 2015, 54, 2–29 CrossRef.
- K. R. Phillips, G. T. England, S. Sunny, E. Shirman, T. Shirman, N. Vogel and J. Aizenberg, Chem. Soc. Rev., 2016, 45, 281–322 RSC.
- M. Kato, T. Cardona, A. W. Rutherford and E. Reisner, J. Am. Chem. Soc., 2012, 134, 8332–8335 CrossRef CAS PubMed.
- H. Kuhl, J. Kruip, A. Seidler, A. Krieger-Liszkay, M. Bünker, D. Bald, A. J. Scheidig and M. Rögner, J. Biol. Chem., 2000, 275, 20652–20659 CrossRef CAS PubMed.
- T. Kothe, S. Pöller, F. Zhao, P. Fortgang, M. Rögner, W. Schuhmann and N. Plumeré, Chem. – Eur. J., 2014, 20, 11029–11034 CrossRef CAS PubMed.
- R. J. Porra, W. A. Thompson and P. E. Kriedemann, Biochim. Biophys. Acta, Bioenerg., 1989, 975, 384–394 CrossRef CAS.
- Y. Umena, K. Kawakami, J.-R. Shen and N. Kamiya, Nature, 2011, 473, 55–60 CrossRef CAS PubMed.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, Wiley, New York, 2nd edn, 2001 Search PubMed.
- A. R. Holzwarth, M. G. Müller, M. Reus, M. Nowaczyk, J. Sander and M. Rögner, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 6895–6900 CrossRef CAS PubMed.
- L. Rapatskiy, N. Cox, A. Savitsky, W. M. Ames, J. Sander, M. M. Nowaczyk, M. Rögner, A. Boussac, F. Neese, J. Messinger and W. Lubitz, J. Am. Chem. Soc., 2012, 134, 16619–16634 CrossRef CAS PubMed.
- J. Kern, B. Loll, C. Lüneberg, D. DiFiore, J. Biesiadka, K.-D. Irrgang and A. Zouni, Biochim. Biophys. Acta, Bioenerg., 2005, 1706, 147–157 CrossRef CAS PubMed.
- E. Laviron, J. Electroanal. Chem. Interfacial Electrochem., 1979, 101, 19–28 CrossRef CAS.
- N. Lebedev, A. Spano, S. Trammell, I. Griva, S. Tsoi and J. M. Schnur, Org. Photovoltaics, 2006, 6370, 63700T Search PubMed.
- Y.-H. Lai, M. Kato, D. Mersch and E. Reisner, Faraday Discuss., 2014, 176, 199–211 RSC.
- P. Cai, X. Feng, J. Fei, G. Li, J. Li, J. Huang and J. Li, Nanoscale, 2015, 7, 10908–10911 RSC.
- M. Suga, F. Akita, K. Hirata, G. Ueno, H. Murakami, Y. Nakajima, T. Shimizu, K. Yamashita, M. Yamamoto, H. Ago and J.-R. Shen, Nature, 2015, 517, 99–103 CrossRef CAS PubMed.
- S. Khan, J. S. Sun and G. W. Brudvig, J. Phys. Chem. B, 2015, 119, 7722–7728 CrossRef CAS PubMed.
- E.-M. Aro, I. Virgin and B. Andersson, Biochim. Biophys. Acta, Bioenerg., 1993, 1143, 113–134 CrossRef CAS.
- V. M. Friebe, J. D. Delgado, D. J. K. Swainsbury, J. M. Gruber, A. Chanaewa, R. Van Grondelle, E. Von Hauff, D. Millo, M. R. Jones and R. N. Frese, Adv. Funct. Mater., 2016, 26, 285–292 CrossRef CAS.
- A. Badura, D. Guschin, T. Kothe, M. J. Kopczak, W. Schuhmann and M. Rögner, Energy Environ. Sci., 2011, 4, 2435–2440 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. Additional data related to this publication are available at the University of Cambridge data repository (http://dx.doi.org/10.17863/CAM.671). See DOI: 10.1039/c6ee01363e |
| This journal is © The Royal Society of Chemistry 2016 |