
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tin/vanadium redox electrolyte for battery-like energy storage capacity combined with supercapacitor-like power handling†
Juhan
Lee
ab,
Benjamin
Krüner
ab,
Aura
Tolosa
ab,
Sethuraman
Sathyamoorthi
ac,
Daekyu
Kim
ad,
Soumyadip
Choudhury
a,
Kum-Hee
Seo
d and
Volker
Presser
*ab
aINM – Leibniz Institute for New Materials, Campus D2 2, 66123 Saarbrücken, Germany. E-mail: volker.presser@leibniz-inm.de
bDepartment of Materials Science and Engineering, Saarland University, Campus D2 2, 66123 Saarbrücken, Germany
cElectrochemical Process Engineering Division, CSIR-Central Electrochemical Research Institute, Karaikudi, 630 006, India
dSchool of Energy, Materials and Chemical Engineering, Korea University of Technology and Education, Chungjeol-ro 1600, 31253 Cheonan, Republic of Korea
First published on 9th June 2016
Abstract
We introduce a high performance hybrid electrochemical energy storage system based on an aqueous electrolyte containing tin sulfate (SnSO4) and vanadyl sulfate (VOSO4) with nanoporous activated carbon. The energy storage mechanism of this system benefits from the unique synergy of concurrent electric double-layer formation, reversible tin redox reactions, and three-step redox reactions of vanadium. The hybrid system showed excellent electrochemical properties such as a promising energy capacity (ca. 75 W h kg−1, 30 W h L−1) and a maximum power of up to 1.5 kW kg−1 (600 W L−1, 250 W m−2), exhibiting capacitor-like galvanostatic cycling stability and a low level of self-discharging rate.
Broader contextRedox electrolytes have received particular attention in supercapacitor research studies in the last decade as they could replace the conventional ‘electrochemically inert’ electrolyte, still contribute to the double layer formation, and significantly enhance the intrinsically low energy density of the supercapacitors. Via the redox reactions in a liquid state, there is no intrinsic stress/strain degradation as known from intercalation-type battery materials. So far, redox electrolyte systems have been investigated mostly using redox couples which do not have solid phase transition throughout the reactions since the solid phase transition is believed to bring negative effects such as low power performance and short cyclic lifetime. In this communication, we report that the redox activities of tin sulfate can be effectively coupled with the redox activities of vanadyl sulfate. Even with the partial solid phase transition of the tin redox ions, the tin/vanadium hybrid system can show capacitor-like high power performance and cycling stability when such complex redox phase transitions occur in the well-confined nanopores of the carbon based electrode material. Such advanced hybrid energy storage systems are envisioned to play an important role to exceed the current performance range of both supercapacitors and batteries. |
Electrochemical energy storage (EES) systems such as batteries and supercapacitors are key enabling technologies for a sustainable and efficient use of energy.1–3 With a growth in the demand for a reliable supply for renewable energy sources, EES systems have drawn more and more attention since the grid scale application of EES systems could resolve the intermittent power generation features of the renewable energy sources.4 In general, common batteries can provide high energy storage capacity (30–300 W h kg−1) while suffering from low power performance (<1 kW kg−1) and limited cycle lifetime (<5000 cycles). Supercapacitors are characterized by a high power performance (5–10 kW kg−1) and extremely high stability (>100
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles), but exhibit low energy storage capacity (<8 W h kg−1).5 These differences originate from different charge storage mechanisms.6 In principle, the charge storage mechanisms for EES devices can be classified into four categories (Fig. 1A): (1) electrical double-layer formation at the electrode/electrolyte interface (electrical double-layer capacitors), (2) redox intercalation through a layered structure via bulk reaction of the electrode (typical batteries), (3) solid phase redox reaction at the near surface of the electrode but not through the bulk material which can be pseudocapacitive,7 and (4) electrochemical reactions of soluble redox couples (redox electrolytes).8 Electric double-layer formation is accomplished by fast ion electrosorption and this purely physical charge separation process at the electrode/electrolyte interface enables rapid charge storage and recovery.9 Redox intercalation provides much higher charge storage capacity through the intercalation of ions (such as lithium, sodium, or potassium) into the layered structure of the electrode via a charge transfer process.10–12 The intercalation process, however, is severely limited by the diffusion kinetics of the ions and may degrade the layered structure of the electrode with increasing cycle numbers, particularly at a high rate.13 Nanoscale design and optimization of the electrode materials,14 or the use of novel nanotextured materials, like MXene,15 may provide unique possibilities to improve on this limitation, but alternative concepts are still in high demand. In particular, the non-ideal reversibility of chemical and/or structural changes in redox-active electrode materials complicates the achievement of longevity and stable performance.16
000 cycles), but exhibit low energy storage capacity (<8 W h kg−1).5 These differences originate from different charge storage mechanisms.6 In principle, the charge storage mechanisms for EES devices can be classified into four categories (Fig. 1A): (1) electrical double-layer formation at the electrode/electrolyte interface (electrical double-layer capacitors), (2) redox intercalation through a layered structure via bulk reaction of the electrode (typical batteries), (3) solid phase redox reaction at the near surface of the electrode but not through the bulk material which can be pseudocapacitive,7 and (4) electrochemical reactions of soluble redox couples (redox electrolytes).8 Electric double-layer formation is accomplished by fast ion electrosorption and this purely physical charge separation process at the electrode/electrolyte interface enables rapid charge storage and recovery.9 Redox intercalation provides much higher charge storage capacity through the intercalation of ions (such as lithium, sodium, or potassium) into the layered structure of the electrode via a charge transfer process.10–12 The intercalation process, however, is severely limited by the diffusion kinetics of the ions and may degrade the layered structure of the electrode with increasing cycle numbers, particularly at a high rate.13 Nanoscale design and optimization of the electrode materials,14 or the use of novel nanotextured materials, like MXene,15 may provide unique possibilities to improve on this limitation, but alternative concepts are still in high demand. In particular, the non-ideal reversibility of chemical and/or structural changes in redox-active electrode materials complicates the achievement of longevity and stable performance.16
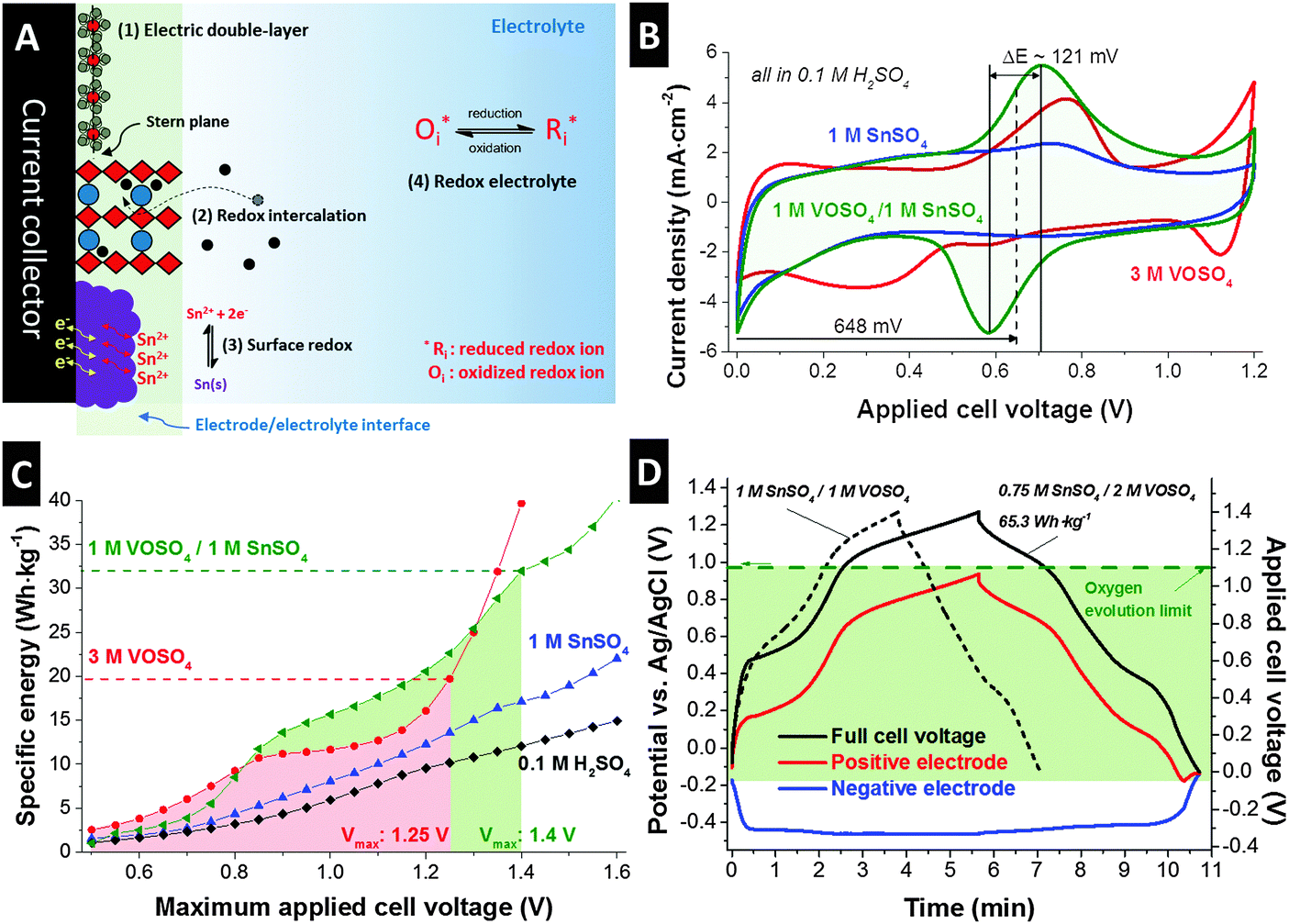 | ||
| Fig. 1 Schematic illustration of various electrochemical energy storage mechanisms (A). Electrochemical analysis with a full-cell configuration without a reference electrode (B and C) and with the Ag/AgCl reference electrode (D). (B) Cyclic voltammograms were obtained for various redox electrolytes at 1 mV s−1. (C) Specific energies were calculated from the cyclic voltammograms at 1 mV s−1via voltage range extension with the interval of 50 mV. The colored area indicates the maximum stability windows of each system, which are determined by a thorough investigation of the irreversible portion of the reaction or known as S-value analysis. The highest specific energy was obtained for the 1 M VOSO4/1 M SnSO4 system in a stable cell potential window. (D) A full-cell is charged up to 1.4 V while observing the potential of the negative electrode through an Ag/AgCl reference electrode. The 0.75 M SnSO4/2 M VOSO4 system shows the best optimized performance in terms of specific energy. | ||
While the use of redox-active electrode materials has attracted tremendous research interest, the field of redox-active electrolytes remains largely uncharted at present.17,18 Unlike the other mentioned charge storage mechanisms, energy storage via redox electrolytes is not exclusively confined to the electrode/electrolyte interface and encompasses the entire fluid volume. For instance, vanadyl sulfate (VOSO4) will not only provide charge storage via double-layer formation at the electrode/electrolyte interface, but additional charge transfer is accomplished through a series of vanadium redox states.19,20 This dual (hybrid) energy storage mechanism brings along the need for the implementation of ion exchange membranes to avoid ion shuttling and rapid self-discharge.21 When no solid phase transition occurs and there are no electrolyte cross-contamination issues,22 a redox electrolyte based energy storage system can offer a cycling lifetime of over 10000 cycles.23,24
To maximize the volumetric and gravimetric energy storage capacity, an intriguing approach has been used to employ a synergistic combination of ultrafast ion electrosorption, fast redox-electrolyte charge transfer, and high-energy interfacial redox reactions. For that purpose, we introduce in this work, for the first time, the effective and stable coupling of tin and vanadium redox activities. Our work employs conventional nanoporous carbon to illustrate the facile implementation of the approach to already existing electrode technologies. A large energy capacity is expected based on the large standard potential difference between V3+/V2+ and VO2+/VO2+ reactions (ESI,† Fig. S1) in combination with the reversible Sn2+/Sn phase transition inside carbon nanopores. The latter provides a large charge storage capacity involving the transfer of two electrons and the possibility of further oxidation to Sn[IV].25
We prepared free-standing carbon film electrodes made from YP80F activated carbon (Kuraray Chemicals) with a specific surface area of 1756 m2 g−1 and an average pore size of 1.6 nm as characterized by nitrogen gas adsorption analysis (ESI†).26 The final hot-rolled films were produced with 200 ± 6 μm thickness containing 5 mass% polytetrafluoroethylene (PTFE) as the binder (see ref. 27). For the redox electrolyte, vanadyl sulfate (VOSO4) with 97% purity, tin sulfate (SnSO4) with 95% purity, sodium sulfate (Na2SO4) with 99% purity, and H2SO4 with 99.99% purity were purchased from Sigma Aldrich and mixed with deionized water. For the stability of the electrolyte and enhanced redox activity, all redox electrolytes were prepared by addition of 0.1 M H2SO4. We used a type FAS15 anion exchange membrane (FuMa-Tech) to effectively prevent redox shuttling of vanadium and vanadium oxide ions between the two electrodes. As the reference electrode, we applied an Ag/AgCl electrode (Bioanalytical Systems, saturated, 3 M NaCl). For more information on electrochemical testing using a VMP300 potentiostat/galvanostat (Bio-Logic), see the ESI† (esp. Fig. S2). For post-mortem analysis, electrode samples were collected after charging the working electrode to −0.2 V and −0.57 V vs. Ag/AgCl in chronoamperometry mode for 30 min. After cell disassembly, the samples were soaked in 0.1 M H2SO4 aqueous solution for a day, then subsequently soaked in deionized water for a day, and dried at room temperature in a desiccator in order to remove the dissolved ions. The possible contribution of the graphite current collector to the energy storage capacity (e.g., via intercalation28,29) was excluded by comparative experiments with platinum current collectors (Fig. S4D, ESI†).
As seen from the cyclic voltammogram for a full cell presented in Fig. 1B, the aqueous VOSO4/SnSO4 redox electrolyte system with added 0.1 M H2SO4 is ideally suited to maximize the energy storage capacity over the entire range of accessible cell voltage (exemplified for up to 1.2 V). A large area under the discharging curve is indicative of a high-energy storage (recovery) capacity which can be quantified by integrating the discharging current. We obtained 42 mA h g−1 (16.8 mA h mL−1) for a mixture of 1 M VOSO4/1 M SnSO4, 38 mA h g−1 (15.2 mA h mL−1) for 3 M VOSO4, and 29 mA h g−1 (11.6 mA h mL−1) for 1 M SnSO4 at a slow rate of 1 mV s−1. For a comparison, only 9.1 mA h g−1 (3.6 mA h mL−1) was achieved when using the same carbon material in 1 M Na2SO4 or 17 mA h g−1 (6.8 mA h mL−1) for 1 H2SO4 in the same voltage range. A clearly visible and broad redox peak pair at around 648 mV with small ΔE for 1 M VOSO4/1 M SnSO4 is additionally beneficial to achieve a high specific energy. The maximum specific energy (W h kg−1) of a hybrid energy storage system greatly depends on the charge/voltage profile (voltage plateau) and the voltage stability window. Accordingly, a thorough assessment of the voltage stability window needs to predate further performance evaluation. Quantitative data analysis of cyclic voltammograms via S-value testing (Fig. S4A, ESI†) is a powerful tool providing (i) the onset potential window of the chemical decomposition potential by thorough investigation of the irreversible portion of the charge,30,31 (ii) specific energies at the applied voltage window, and (iii) cyclic voltammograms containing information about the redox reactions. The comprehensive data from the S-value testing can directly be converted to a plot of specific energy vs. maximum applied cell voltage, as can be seen from Fig. 1C. The limit for the stable cell voltage (marked green and red) is determined by an S-value of 0.1, which corresponds to a maximum deviation between the transferred charge during charging and the recovered charge during discharging of max. 10%. For the 1 M VOSO4/1 M SnSO4 system, we see that a stable cell performance is maintained (by definition of S < 0.1 and its second derivative <0.05) up to 1.4 V applied cell voltage (Fig. S4A, ESI†). These values for the cell voltage in acidic media are much higher than the theoretical thermodynamic limit of 1.23 V. Voltage floating confirmed good electrochemical performance stability for pure 0.1 M H2SO4 aqueous solution at 1.3 V and 1.4 V cell voltage (Fig. S4E, ESI†). This behavior may be associated with the cathodic limit extension via hydrogen adsorption and/or overpotential influence depending on the type and pore structure of the carbon.32–36
A high amount of Faradaic charge transfer associated with redox reactions contributes to an enhanced specific energy and occurs at a fixed cell voltage. For instance, 3 M VOSO4 exhibits two increases in specific energy at 0.5–0.8 V and >1.1 V (Fig. 1C), which correspond to the two large redox peaks observed in the cyclic voltammogram (Fig. 1B). As a result, a maximum specific energy rating of 19.8 W h kg−1 can be reached. For 3 M VOSO4, however, no further increase in specific energy is possible because of the voltage stability window being limited to ∼1.25 V, most probably due to water splitting. An even higher specific energy of 32 W h kg−1 was achieved by the 1 M VOSO4/1 M SnSO4 system, having a much larger voltage stability window of up to 1.4 V. This clearly implies a synergetic influence of the VOSO4/SnSO4 combination in terms of high specific energy. The individual application of VOSO4 and SnSO4 could not provide such high specific energy (Fig. 1C) due to their solubility limits (SnSO4: 1.6 M in water at 20 °C in absence of vanadyl sulfate, VOSO4: 3.3 M in water at 20 °C).37,38 Initially, we see an S-value (Fig. S4A, ESI†) above 0.1 from 0.5 V to 0.8 V. However, this value drops below 0.1 as the tin sulfate system becomes conditioned. This indicates that S-value analysis is to be carefully employed when adapting to redox systems.
The data so far relate to two-electrode cells. To gain a more detailed insight into the electrochemistry of the VOSO4/SnSO4 system, a three-electrode configuration was applied with an Ag/AgCl reference electrode (Fig. S2B, ESI†) to study the voltage development at each individual electrode. This particular configuration allows us to not only operate the cell voltage in a full-cell-like manner, but also access the potential information of the positive and negative electrode at the same time. Fig. S4B (ESI†) shows the potential profile of the positive and negative electrode while the voltage applied between the negative and positive electrodes was ranged from 0 V to 1.4 V. In the case of the 1 M VOSO4/1 M SnSO4 system, the potential at the positive electrode increases much faster than the potential at the negative electrode due to the strong redox activities at the negative potential at around −0.45 V vs. Ag/AgCl.
Since the asymmetric potential development of the negative and positive electrode implies an inefficient utilization of the total charge storage capacity of a system, charge balancing is required.39 The performance can be severely improved by balancing the charge storage capacity of both electrodes while keeping the potential of each electrode below their respective stability limits. One unique advantage of the redox electrolyte system is the highly facile charge balancing through concentration adjustment. In this way, charge balancing is much simpler as compared to the mass balancing commonly employed for supercapacitor devices, which brings complexity through imbalance in the geometrical shape of the electrodes.40 In order to maximize the specific energy of the system, the voltage imbalance of the negative and positive electrode can be adjusted by enhancing the charge storage capacity at the positive electrode. The 1 M VOSO4/1 M SnSO4 system already represented its maximum possible electrolytic concentration due to the low solubility of SnSO4 (ca. 1.6 M) as compared to that of VOSO4 (ca. 3.3 M). Further optimization of the electrochemical performance can be obtained by carefully increasing the VOSO4-to-SnSO4 ratio in order to balance the strong redox activity at the negative electrode via the enhancement of vanadium redox activities at the positive electrode. An array of different molar ratios is presented in the ESI,† Fig. S4C. The best performance was obtained for 0.75 M SnSO4/2 M VOSO4 in terms of high energy density as the potential increase at the positive electrode was suppressed by the redox activity of VOSO4 at around +0.75 V and +0.14 V vs. Ag/AgCl and remained below the oxygen evolution potential (Fig. 1D). The specific energy of this system was 65.3 W h kg−1 as calculated from the discharging potential profile shown in Fig. 1D, as compared to 37 W h kg−1 for the 1 M SnSO4/1 M VOSO4 cell.
For a comprehensive understanding of the redox activities of the negative and positive electrode, we employed a three-electrode configuration with an oversized counter electrode (oversized by ca. 35 times compared to the working electrode; Fig. S2B, ESI†). In the case of the 0.75 M SnSO4/2 M VOSO4 system, the VO2+/VO2+ redox reaction was investigated at the working electrode while keeping the potential of the counter electrode outside the range of the other redox activities (Fig. S3, ESI†). As a first step, 1 M SnSO4 and 1 M VOSO4 redox electrolytes were individually studied at the potential range from around −0.6 V to +0.8 V (Fig. 2A). The vanadium system shows three distinguishable redox peaks, which can be identified as V3+/V2+, VO2+/V3+, and VO2+/VO2+ at around −0.5 V, +0.1 V, and +0.75 V vs. Ag/AgCl.4
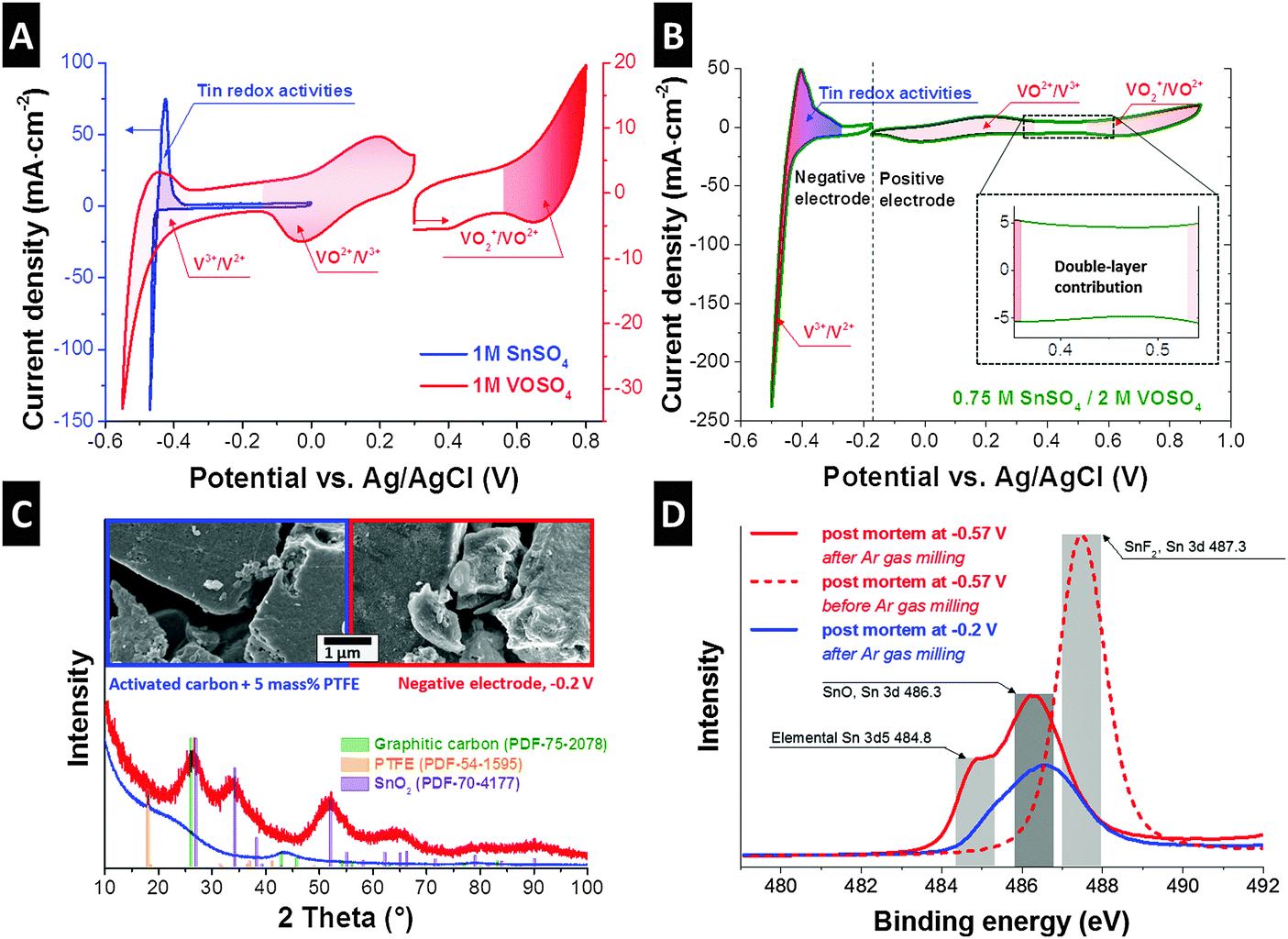 | ||
| Fig. 2 Cyclic voltammograms of various redox electrolyte systems with three-electrode configuration with an overbalanced counter electrode (A and B). (A) Cyclic voltammograms from individual 1 M SnSO4 system and 1 M VOSO4 system. (B) Cyclic voltammograms from the best optimized redox electrolytes containing 0.75 M SnSO4 and 2 M VOSO4. The inset shows a close-up on the part of the current response indicating double-layer formation. (C) SEM images and XRD data are obtained from the negative electrode. XRD peak positions indicate the formation of SnO2 with the domain size of around 2 nm. (D) XPS results, providing evidence for the formation of tin oxides, metallic tin, and superficial SnF2 at the negative electrode. | ||
In the case of the tin system, a very sharp and large peak appears at around −0.47 V, which aligns with the reduction of Sn2+ to metallic tin (tin electroplating). As the scanning direction changes to the anodic scan, the metallic tin is expected to be oxidized to Sn2+. A slightly broader anodic peak (−0.43 V) might indicate a series of tin oxidation reactions towards Sn[IV] (Fig. S1, ESI†). As a next step, two cells were prepared with 0.75 M SnSO4/0.2 M VOSO4 in order to study the electrochemical behavior of the positive and negative electrodes (Fig. 2B). Reversible reactions of the VO2+/V3+ and VO2+/VO2+ couples can be seen at the positive electrode. During the reduction of the negative electrode, a steep increase in cathodic current was observed at −0.43 V, which aligns with the reduction of Sn2+ to tin metal. The results of the post-mortem nitrogen gas adsorption test (Table S2, ESI†) are in excellent agreement with the results of the formation of metallic tin, possibly in the carbon nanopores with pore sizes below 3 nm (Fig. S5, ESI†). The formation of the metal is believed to block the carbon nanopores as supported by the lower specific surface area (minus 41%) and the total pore volume (minus 48%) of the reduced negative electrode as compared to the pristine electrode. A high cathodic peak at −0.5 V (Fig. 2B) indicates the contribution of the V3+ to V2+ reaction. During reverse polarization (Fig. 2B), a broad anodic peak arises at around −0.41 V, which aligns with the oxidation of V2+ to V3+ and tin metal to Sn2+. The dissolution of the tin metal to Sn2+ is also evidenced by the higher specific surface area and the total pore volume of the oxidized negative electrode (Table S2, ESI†) from the post-mortem nitrogen gas adsorption analysis (Fig. S5, ESI†). During further oxidation from −0.36 V to −0.27 V, we still see a high anodic current, perhaps implying further oxidization of tin to Sn[IV], which aligns with the lower specific surface area and total pore volume from the post-mortem analysis of the oxidized negative electrode. We see a capacitive behavior of the electrode in the range where no specific redox reactions are expected (Fig. 2B, inset) as characterized by a rectangular shape in the cyclic voltammogram. This capacitive contribution aligns with the formation of the electric double-layer by vanadium (V3+, V2+), vanadium oxides (VO2+, VO2+), and tin cations (Sn2+) as well as sulfates (SO42−), or by the pseudocapacitive mechanism of the redox species; the capacitive contribution can be also seen in the cyclic voltammogram obtained from a full-cell configuration (Fig. S4D, ESI†).
To confirm the oxidation states of the tin system, we carried out post-mortem characterization of the negative electrode. For that, the electrodes were charged to −0.2 V and −0.57 V vs. Ag/AgCl, washed with 0.1 M H2SO4 aqueous solution and deionized water, and dried in vacuum. The electron micrographs (Fig. 2C) show that the surface of the carbon particles became roughened after the electrode was exposed to −0.2 V vs. Ag/AgCl as compared to the pristine electrode (activated carbon with 5 mass% PTFE). The Raman spectra support the fact that no change in the carbon structure occurred, maintaining the same ID/IG ratio (1.08 ± 0.05) before and after electrochemical testing (Fig. S6A, ESI†). The XRD pattern of the pristine electrode shows the presence of amorphous carbon and PTFE binder (Fig. 2C). The negative electrode exhibits amorphous SnO2 peaks, meaning that the metallic tin could have been further oxidized to Sn[IV] through electrochemical operation. The formation of tin oxides is also supported by the increase in oxygen and tin content measured from EDX (ESI†). The calculated domain size (coherence length; measured via XRD) of SnO2 is approximately 2 nm, which is in the range of pore sizes of the activated carbon and supports the assumption that the Sn[IV] transition took place within the pores. Post-mortem nitrogen gas sorption analyses (Fig. S5, ESI†) support the formation of tin metal in nanopores with the possible size of below 3 nm. In order to confirm the oxidation state of tin present in the electrodes, XPS analysis was carried out (Fig. 2D). The samples were measured before and after ion milling with argon gas with the depth of approximately 1–2 μm to differentiate the signal information coming from the outer activated carbon particle surface and from inside. Samples before ion milling showed the peak with the binding energy of around 487.3 eV, which can be identified as the Sn 3d signal of tin fluoride (SnF2). The existence of SnF2 can also be supported by the F 1s electron binding energy (Fig. S6B, ESI†). After ion milling with argon gas (Fig. 2D), two distinctive peaks arise at around 484.8 eV and 486.3 eV, which are considered to stem from the electron binding energy of SnO or SnO2 (Sn 3d) and tin metal (Sn 3d5). Therefore, SnF2 formation as a result of the reaction between tin and fluoride from the PTFE-binder is possibly limited to the outer surface of the activated carbon. Inside the large volume of carbon nanopores, however, exclusively tin oxides and metallic tin are formed. We assume that the PTFE was partially decomposed to fluorine and carbon.
As investigated so far, the SnSO4/VOSO4 electrolyte system provides battery-like specific energy (>60 W h kg−1) through a complex charge storage mechanism of tin and vanadium redox activities and electric double-layer formation. For broader applications of this promising energy storage system, a high power performance is required. For the characterization of rate handling, galvanostatic charge/discharge was applied to the 0.75 M SnSO4/2 M VOSO4 cells with a full-cell configuration (Fig. S2A, ESI†) up to 1.4 V (Fig. 3A). For comparison, we added the data for the performance of a double-layer capacitor in 1 M Na2SO4 aqueous solution with the same type of activated carbon electrode. At a low specific current of around 55 mA g−1, the 0.75 M SnSO4/2 M VOSO4 system exhibited a battery-like energy specific energy of up to 75 W h kg−1, which can be converted to ca. 30 W h L−1 when normalized to the volume of the two electrodes, the membrane, and the electrolyte. At a low specific current, the double-layer capacitor employing 1 M Na2SO4 only provides a maximum of 5 W h kg−1 (2 W h L−1). At a specific current of 4 A g−1, the 0.75 M SnSO4/2 M VOSO4 system shows much higher specific energy (8.2 W h kg−1) than the 1 M Na2SO4 supercapacitor (3.1 W h kg−1). The poor energy rating at higher specific currents (>6 A g−1) is possibly due to the solid phase transition of Sn2+ ions to solid tin as also supported by the increase in ohmic resistance (see the ESI† for the calculation) at the specific current regime over 1 A g−1, which indicates the overpotential due to mass transfer. When the specific current is converted to specific power (Fig. S7, ESI†), the 0.75 M SnSO4/2 M VOSO4 system provides a maximum performance of up to ca. 1.5 kW kg−1, which can be converted to ca. 600 W L−1 or 250 W m−2 when normalized to the volume of the two electrodes, the membrane, and the electrolyte, or to the membrane area, respectively.
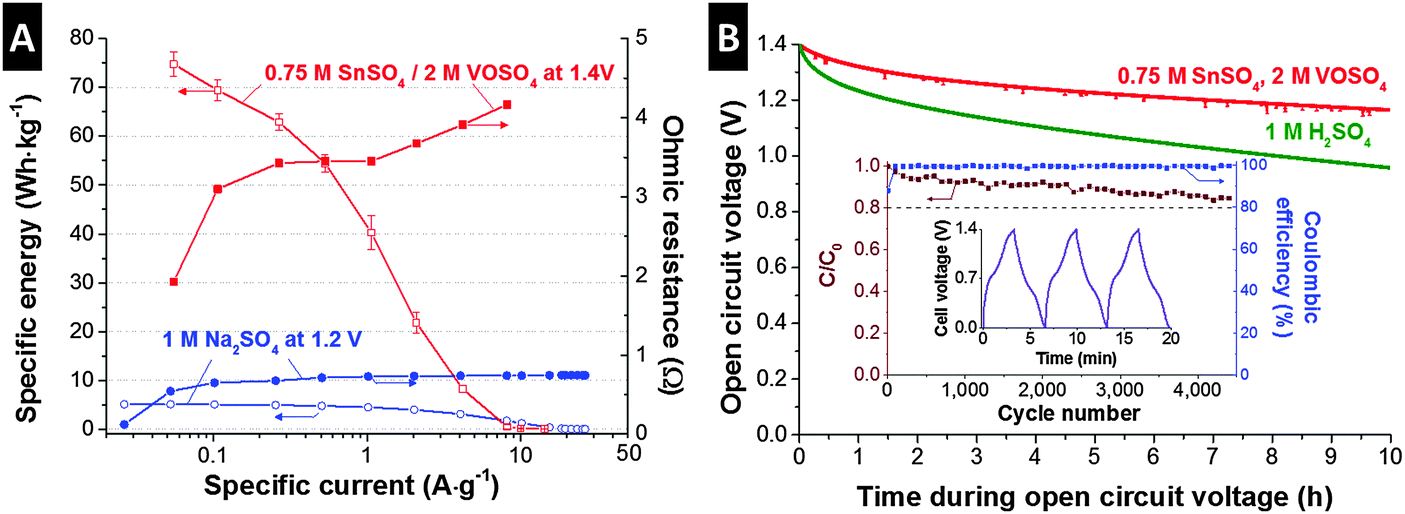 | ||
| Fig. 3 (A) The rate performance of the SnSO4/VOSO4 system is plotted, as well as the ohmic resistance of the cell measured at each applied specific current in comparison to the 1 M Na2SO4 system. (B) Open circuit voltage for 10 h after charging the cells up to 1.4 V. The SnSO4/VOSO4 system shows also a stable cyclic performance over 4000 cycles with a Coulombic efficiency close to 100%. The inset shows a profile of the galvanostatic charge/discharge for several cycles. | ||
An important but often not discussed property of energy storage systems is the self-discharge, or more specifically, the time-dependent loss of the open circuit voltage after charging up to a specific cell voltage. As reported in several studies,21,41,42 redox electrolyte based systems are expected to suffer from a large self-discharging rate due to redox shuttling when ions are not selectively separated, for instance, by application of an ion exchange membrane. To prevent the ion shuttling of the vanadium ions, we employed a type FAS15 ion exchange membrane and achieved a very stable behavior (Fig. 3B). After 10 h of OCV measurement, the initial voltage dropped to about 17%, which is only half the self-discharge of an aqueous supercapacitor operated in 1 M H2SO4. Interestingly, the redox electrolyte system yields a self-discharge superior to supercapacitors, possibly due to less effect of ion-redistribution on the potential loss as the system has a hybrid charge storage principle.43–45
The high performance stability (Fig. 3B, inset) is particularly remarkable when we consider that battery-like electrochemical energy storage systems commonly feature poor cycling stability. However, our redox electrolyte system presents very stable performance and after 4500 cycles (ca. 400 h of continuous testing), the specific capacity of the 0.75 M SnSO4/2 M VOSO4 cell degraded only by ca. 15% compared to the initial capacity of the system. The comparably high cycling stability is complemented by a high Coulombic efficiency of almost 100%, which is in excellent agreement with the electrochemical stability determined by the S-value analysis (Fig. S4C, ESI†).
In summary, we demonstrated a new type of hybrid energy storage system by applying an aqueous redox electrolyte with SnSO4 and VOSO4 into a nanoporous carbon based electrode. Through a simple charge balancing by adjusting the relative molar concentration of SnSO4 and VOSO4, the best optimized cell exhibited a broad voltage stability window of up to 1.4 V in a full-cell configuration, a promising specific energy of 75.4 ± 2.3 W h kg−1, a maximum specific power of ca. 1.5 kW kg−1 as well as a high cycling stability over 4500 cycles, and a low level of self-discharging rate. The origin of the high specific energy is the three-step redox activities of vanadium by changing the oxidation state from 2+ to 5+ and the large redox activities of tin at the negative electrode. From the XPS analysis, these tin redox activities are believed to be due to the formation of the metallic tin in the carbon micropores, further oxidation of metallic tin to Sn[IV], or a further reduction of metallic tin to tin fluoride on the outside of the carbon particles, where PTFE is accessible. As the solid phase transition through the redox activities of tin can narrow or block carbon micropores, studies regarding the detailed solid phase formation mechanism in the micropores are necessary which will allow the estimation of the best pore structure of the electrode material, thus, further improvement of the power performance is expected.
Acknowledgements
We acknowledge the funding from the German Federal Ministry for Research and Education (BMBF) in support of the nanoEES3D project (award number 03EK3013) as part of the strategic funding initiative energy storage framework. The authors thank Prof. Eduard Arzt (INM) for his continuing support, FuMA-Tech (esp. Dr Bauer and Dr Klicpera) for kindly providing membranes and helpful discussions, Dr Ingrid Grobelsek for the EDX and SEM measurements, Dr Mesut Aslan, Srimuk Pattarachai, Nicolas Jäckel, Simon Fleischmann, Friedrich Kaasik, and Anna Schreiber (all at INM) for useful discussions and technical support.References
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS PubMed.
- F. Béguin, V. Presser, A. Balducci and E. Frackowiak, Adv. Mater., 2014, 26, 2219–2251 CrossRef PubMed.
- M. Barghamadi, A. S. Best, A. I. Bhatt, A. F. Hollenkamp, M. Musameh, R. J. Rees and T. Ruther, Energy Environ. Sci., 2014, 7, 3902–3920 Search PubMed.
- Z. Yang, J. Zhang, M. C. Kintner-Meyer, X. Lu, D. Choi, J. P. Lemmon and J. Liu, Chem. Rev., 2011, 111, 3577–3613 CrossRef CAS PubMed.
- J. R. Miller and A. F. Burke, Electrochem. Soc. Interface, 2008, 17, 53 CAS.
- P. Simon, Y. Gogotsi and B. Dunn, Science, 2014, 343, 1210–1211 CrossRef CAS PubMed.
- V. Augustyn, P. Simon and B. Dunn, Energy Environ. Sci., 2014, 7, 1597–1614 Search PubMed.
- P. Bandaru, H. Yamada, R. Narayanan and M. Hoefer, Mater. Sci. Eng., R, 2015, 96, 1–69 CrossRef.
- D. Pech, M. Brunet, H. Durou, P. Huang, V. Mochalin, Y. Gogotsi, P.-L. Taberna and P. Simon, Nat. Nanotechnol., 2010, 5, 651–654 CrossRef CAS PubMed.
- V. Etacheri, R. Marom, R. Elazari, G. Salitra and D. Aurbach, Energy Environ. Sci., 2011, 4, 3243–3262 Search PubMed.
- W. Luo, J. Wan, B. Ozdemir, W. Bao, Y. Chen, J. Dai, H. Lin, Y. Xu, F. Gu, V. Barone and L. Hu, Nano Lett., 2015, 15, 7671–7677 CrossRef CAS PubMed.
- M. Shirpour, J. Cabana and M. Doeff, Energy Environ. Sci., 2013, 6, 2538–2547 Search PubMed.
- A. Mukhopadhyay and B. W. Sheldon, Prog. Mater. Sci., 2014, 63, 58–116 CrossRef CAS.
- V. Augustyn, J. Come, M. A. Lowe, J. W. Kim, P.-L. Taberna, S. H. Tolbert, H. D. Abruña, P. Simon and B. Dunn, Nat. Mater., 2013, 12, 518–522 CrossRef CAS PubMed.
- M. D. Levi, M. R. Lukatskaya, S. Sigalov, M. Beidaghi, N. Shpigel, L. Daikhin, D. Aurbach, M. W. Barsoum and Y. Gogotsi, Adv. Energy Mater, 2015, 5, 1400815 CrossRef.
- S. T. Senthilkumar, R. K. Selvan and J. S. Melo, J. Mater. Chem. A, 2013, 1, 12386–12394 RSC.
- B. Akinwolemiwa, C. Peng and G. Z. Chen, J. Electrochem. Soc., 2015, 162, A5054–A5059 CrossRef CAS.
- L. Guan, L. Yu and G. Z. Chen, Electrochim. Acta, 2016, 206, 464–478 CrossRef CAS.
- E. Frackowiak, K. Fic, M. Meller and G. Lota, ChemSusChem, 2012, 5, 1181–1185 CrossRef CAS PubMed.
- S. Senthilkumar, R. K. Selvan, N. Ponpandian, J. Melo and Y. Lee, J. Mater. Chem. A, 2013, 1, 7913–7919 RSC.
- L. Chen, H. Bai, Z. Huang and L. Li, Energy Environ. Sci., 2014, 7, 1750–1759 Search PubMed.
- D. Reed, E. Thomsen, W. Wang, Z. Nie, B. Li, X. Wei, B. Koeppel and V. Sprenkle, J. Power Sources, 2015, 285, 425–430 CrossRef CAS.
- K. Fic, M. Meller and E. Frackowiak, J. Electrochem. Soc., 2015, 162, A5140–A5147 CrossRef CAS.
- B. Krüner, J. Lee, N. Jäckel, A. Tolosa and V. Presser, ACS Appl. Mater. Interfaces, 2016, 8, 9104–9115 Search PubMed.
- R. Narayanan and P. R. Bandaru, J. Electrochem. Soc., 2014, 162, A86–A91 CrossRef.
- G. Y. Gor, M. Thommes, K. A. Cychosz and A. V. Neimark, Carbon, 2012, 50, 1583–1590 CrossRef CAS.
- N. Jäckel, D. Weingarth, M. Zeiger, M. Aslan, I. Grobelsek and V. Presser, J. Power Sources, 2014, 272, 1122–1133 CrossRef.
- N. E. Sorokina, M. A. Khaskov, V. V. Avdeev and I. V. Nikol'skaya, Russ. J. Gen. Chem., 2005, 75, 162–168 CrossRef CAS.
- M. D. Levi, E. Levi, Y. Gofer, D. Aurbach, E. Vieil and J. Serose, J. Phys. Chem. B, 1999, 103, 1499–1508 CrossRef CAS.
- D. Weingarth, H. Noh, A. Foelske-Schmitz, A. Wokaun and R. Kötz, Electrochim. Acta, 2013, 103, 119–124 CrossRef CAS.
- J. Lee, D. Weingarth, I. Grobelsek and V. Presser, Energy Technol., 2016, 4, 75–84 CrossRef CAS.
- C. Vix-Guterl, E. Frackowiak, K. Jurewicz, M. Friebe, J. Parmentier and F. Béguin, Carbon, 2005, 43, 1293–1302 CrossRef CAS.
- E. Frackowiak and F. Béguin, Carbon, 2002, 40, 1775–1787 CrossRef CAS.
- Q. Gao, L. Demarconnay, E. Raymundo-Pinero and F. Beguin, Energy Environ. Sci., 2012, 5, 9611–9617 Search PubMed.
- K. Fic, M. Meller, J. Menzel and E. Frackowiak, Electrochim. Acta, 2016, 206, 496–503 CrossRef CAS.
- K. Fic, G. Lota, M. Meller and E. Frackowiak, Energy Environ. Sci., 2012, 5, 5842–5850 Search PubMed.
- F. Rahman and M. Skyllas-Kazacos, J. Power Sources, 1998, 72, 105–110 CrossRef CAS.
- J. Donaldson and W. Moser, J. Chem. Soc., 1960, 4000–4003 RSC.
- M. Lazzari, F. Soavi and M. Mastragostino, J. Power Sources, 2008, 178, 490–496 CrossRef CAS.
- A. Brandt, P. Isken, A. Lex-Balducci and A. Balducci, J. Power Sources, 2012, 204, 213–219 CrossRef CAS.
- M. Tachibana, Y. Tsukada, T. Ohishi, H. Yamagishi and M. Murakami, Kobunshi Ronbunshu, 2009, 66, 259–265 CrossRef CAS.
- M. Tachibana, T. Ohishi, Y. Tsukada, A. Kitajima, H. Yamagishi and M. Murakami, Electrochemistry, 2011, 79, 882–886 CrossRef CAS.
- J. Kowal, E. Avaroglu, F. Chamekh, A. Šenfelds, T. Thien, D. Wijaya and D. U. Sauer, J. Power Sources, 2011, 196, 573–579 CrossRef CAS.
- M. Kaus, J. Kowal and D. U. Sauer, Electrochim. Acta, 2010, 55, 7516–7523 CrossRef CAS.
- H. A. Andreas, J. Electrochem. Soc., 2015, 162, A5047–A5053 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ee00712k |
| This journal is © The Royal Society of Chemistry 2016 |