
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence570 mV photovoltage, stabilized n-Si/CoOx heterojunction photoanodes fabricated using atomic layer deposition†
Xinghao
Zhou
ab,
Rui
Liu
a,
Ke
Sun
ac,
Kimberly M.
Papadantonakis
ac,
Bruce S.
Brunschwig
ad and
Nathan S.
Lewis
*acde
aJoint Center for Artificial Photosynthesis, California Institute of Technology, Pasadena, CA 91125, USA. E-mail: nslewis@caltech.edu
bDivision of Engineering and Applied Science, Department of Applied Physics and Materials Science, California Institute of Technology, Pasadena, CA 91125, USA
cDivision of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, CA 91125, USA
dBeckman Institute and Molecular Materials Research Center, California Institute of Technology, Pasadena, CA 91125, USA
eKavli Nanoscience Institute, California Institute of Technology, Pasadena, CA 91125, USA
First published on 8th January 2016
Abstract
Heterojunction photoanodes, consisting of n-type crystalline Si(100) substrates coated with a thin ∼50 nm film of cobalt oxide fabricated using atomic-layer deposition (ALD), exhibited photocurrent-onset potentials of −205 ± 20 mV relative to the formal potential for the oxygen-evolution reaction (OER), ideal regenerative solar-to-O2(g) conversion efficiencies of 1.42 ± 0.20%, and operated continuously for over 100 days (∼2500 h) in 1.0 M KOH(aq) under simulated solar illumination. The ALD CoOx thin film: (i) formed a heterojunction with the n-Si(100) that provided a photovoltage of 575 mV under 1 Sun of simulated solar illumination; (ii) stabilized Si photoanodes that are otherwise unstable when operated in aqueous alkaline electrolytes; and, (iii) catalyzed the oxidation of water, thereby reducing the kinetic overpotential required for the reaction and increasing the overall efficiency relative to electrodes that do not have an inherently electrocatalytic coating. The process provides a simple, effective method for enabling the use of planar n-Si(100) substrates as efficient and durable photoanodes in fully integrated, photovoltaic-biased solar fuels generators.
Broader contextPhotoelectrochemical water splitting is an attractive method for converting solar energy directly into hydrogen fuel. However, small-band-gap semiconductors undergo corrosion or passivation reactions in most aqueous electrolytes, especially while oxidizing water to produce O2(g). Various protection schemes have been developed that allow the use of such semiconductors as photoanodes, but these approaches generally rely on formation of an underlying buried p–n junction to obtain a high photovoltage. High quality emitters or heterojunctions can not however be formed on many semiconductors, and formation of an emitter introduces additional complexity into the fabrication process. We demonstrate herein the use of a thin layer of CoOx formed by atomic-layer deposition to form a high photovoltage, stabilized photoanode for water oxidation on planar, crystalline n-Si electrode surfaces, demonstrating the possibility of utilizing interfacial engineering to obtain high performance stabilized electrodes without formation of a buried diffused junction. The work provides a demonstration of a simple, scalable process for formation of scalable photoanodes for use in integrated solar fuels generators. |
The sustainable electrochemical production of fuels from aqueous electrolytes, accomplished either by reducing water to generate H2(g) or by reducing CO2(g) and water to generate hydrocarbons, requires the concomitant oxidation of water to O2(g), to liberate the electrons needed for the fuel-forming reactions.1–4 Efficient, intrinsically safe solar-driven water-splitting systems can be constructed in strongly alkaline or strongly acidic electrolytes.5–7 Such electrolytes also allow the use of commercially available, gas-impermeable, ion-exchange membranes, which have been developed for fuel cells and electrolyzers that use acidic or alkaline electrolytes.8
Protective coatings can stabilize technologically important small-band-gap semiconductors for use as oxygen-evolving photoanodes, even in strongly alkaline electrolytes. At least four types of protective coatings have been developed:9,10 TiO2, either in an insulating form as a thin (few nm thick) insulating tunnel barrier or in a “leaky” form as a thick (multiple tens of nm) anodically conductive barrier;11–16 thin metallic coatings;3 and p-type transparent, conductive transition-metal oxides.13,17–21 In addition to serving as a physical barrier between the semiconductor and electrolyte, the protective coating should provide high interfacial charge-transfer rates and either be inherently catalytic for the oxygen-evolution reaction (OER) or should support an active OER electrocatalyst, to reduce the kinetic overpotential required for the reaction as well as to assist in the removal of reactive photogenerated holes from the surface of the semiconductor.22,23 Protective coatings have been demonstrated to extend the lifetime of photoanodes fabricated from semiconductors such as Si, InP, and CdTe, which can form surface layers of insoluble, passivating oxides during operation.13,18
Integration of protection layers into efficient photoanodes generally has required deposition of the protection layer on top of a homogeneous np+ buried junction.13,18,24 However, high quality emitters or heterojunctions can not be formed on many semiconductors. Furthermore, formation of diffused junctions on inexpensive, small-grain-size, polycrystalline or thin-film semiconductors generally results in preferential migration of dopants down the grain boundaries, and thus produces deleterious minority-carrier recombination and majority-carrier shunts in the resulting device.25
CoOx is an electrocatalyst for the evolution of O2(g) from aqueous alkaline solutions,26,27 and has been explored as a protective coating for photoanodes.10,19,28 CoOx protective layers have been reported to provide only limited stability (a few hours or less) against corrosion for photoanodes in strongly alkaline solutions,10,28 or require substrates with homogeneous np+ buried junctions to produce high photovoltages (see ESI† for definition of photovoltage).19 Recently, deposition of a thin (∼2 nm) film of cobalt oxide (CoOx) prior to the sputter deposition of a multifunctional and protective NiOx coating onto n-Si surfaces has yielded photoanodes with an electrochemical performance that approaches the Shockley diode limit for the substrate. The interfacially engineered n-Si/SiOx,RCA/CoOx/NiOx structure demonstrates the fabrication of a stable and efficient device that utilizes a direct heterojunction contact between an n-Si substrate and a protective coating, thereby obviating the need for homogeneous np+ buried junctions in this system.29
We demonstrate herein that stable, high-photovoltage anodes for the oxidation of water from strongly alkaline electrolytes can be fabricated from n-Si substrates protected by a uniform layer of CoOx formed using atomic-layer deposition (ALD). The ability to fabricate a stable photoanode by deposition of a protective and electrocatalytic coating directly onto a planar substrate without sacrificing device efficiency simplifies the fabrication processes needed to obtain efficient and stable photoanodes for solar-driven water oxidation on Si surfaces. Although the interfacially engineered n-Si/SiOx,RCA/CoOx/NiOx device provides increased solar-to-O2(g) conversion efficiency relative to an n-Si photoanode protected by a multi-functional layer of ALD CoOx, the n-Si/SiOx,RCA/CoOx photoanodes exhibited a lower decay rate of their solar-to-O2(g) conversion efficiency than n-Si/SiOx,RCA/CoOx/NiOx photoanodes due to a compact and uniform ALD CoOx protection layer, along with a simplified fabrication process including lower temperature and fewer processing steps.
Detailed experimental procedures are provided in the ESI.† Briefly, planar Si(100) substrates (0.1–1 ohm cm resistivity, 525 μm thick) were immersed in a Radio Corporation of America Standard Clean-2 (RCA SC-2) etchant for 10 min at 75 °C. This procedure resulted in a thin (∼2 nm) SiOx layer on the surface of the Si (Si/SiOx,RCA). CoOx films were deposited via ALD onto Si/SiOx,RCA substrates at 150 °C, with each ALD cycle consisting of a 2 s pulse of a cobaltocene precursor, a 10 s N2(g) purge at a flow rate of 20 cm3 min−1, a 5 s pulse of ozone, and another 10 s N2(g) purge. 1000 ALD cycles (∼50 nm) of CoOx deposition produced optimal photocurrent-onset potentials relative to the formal potential for the oxygen-evolution reaction (OER) (E0′(O2/H2O) = 1.23 V versus a reversible hydrogen electrode, RHE, at pH = 14) for n-Si/SiOx,RCA/CoOx photoanodes (Fig. S1, ESI†).
Fig. 1A shows a cross-sectional transmission-electron micrograph of the n-Si/SiOx,RCA/CoOx interface collected using bright-field mode. The black region at the bottom of the image corresponds to the Si wafer, the thin bright region atop the Si corresponds to SiOx, and the polycrystalline layer atop the Si corresponds to the compact CoOx film, which was uniformly smooth and ∼50 nm thick, consistent with the low surface roughness (0.74 nm) determined using atomic-force microscopy (AFM, Fig. S2A, ESI†). Fig. 1B and C show high-resolution transmission-electron microscope (HRTEM) images of the interface. An amorphous SiOx layer ∼2 nm thick was present between the Si substrate and the CoOx layer, and crystalline grain boundaries were evident in the CoOx regions of the image. The grain boundaries and in/out-of-plane orientation of the diffraction patterns suggest that the ALD CoOx films deposited at 150 °C using these methods were polycrystalline rather than amorphous, consistent with results obtained previously using grazing incidence X-ray diffractometry (GIXRD).29
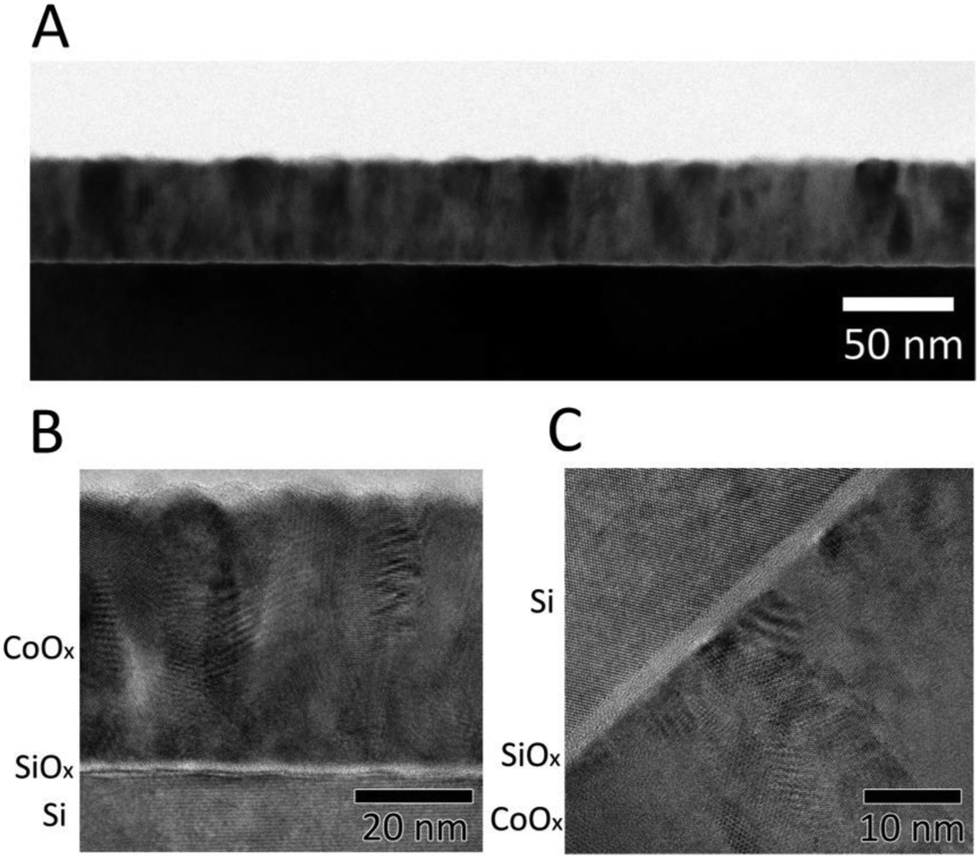 | ||
| Fig. 1 (A) Low-magnification bright-field transmission-electron micrograph (TEM) of a cross-section of an n-Si/SiOx,RCA/CoOx electrode. (B and C) High-resolution TEM cross-sectional images of an n-Si/SiOx,RCA/CoOx sample. The CoOx regions show the polycrystalline structure of the CoOx film. | ||
Fig. 2A shows typical current-density versus potential (J–E) behavior for n-Si/SiOx,RCA/CoOx photoanodes in contact with 1.0 M KOH(aq), in the dark or under 1.1 Sun of simulated solar illumination provided by a Xe lamp with AM 1.5G filters. The behavior of a nonphotoactive p+-Si/SiOx,RCA/CoOx electrode is also shown under the same conditions. The light-limited photocurrent density was 30.2 ± 1.1 mA cm−2 under these conditions (1.1 Sun illumination), and corresponds to a photocurrent density of ∼27 mA cm−2 under 1 Sun illumination. Fig. 2B shows the spectral response data (external quantum yield as a function of wavelength) for an n-Si/SiOx,RCA/CoOx photoanode in contact with 1.0 M KOH(aq) while the sample was maintained at 1.93 V versus RHE. When integrated with respect to the spectral irradiance distribution of the AM 1.5G 1.1 Sun solar spectrum, the spectral response of the n-Si/SiOx,RCA/CoOx would be expected to yield a photocurrent density of 30.6 mA cm−2, in good agreement with the light-limited photocurrent observed under 1.1 Sun illumination (see ESI† for detailed calculations).
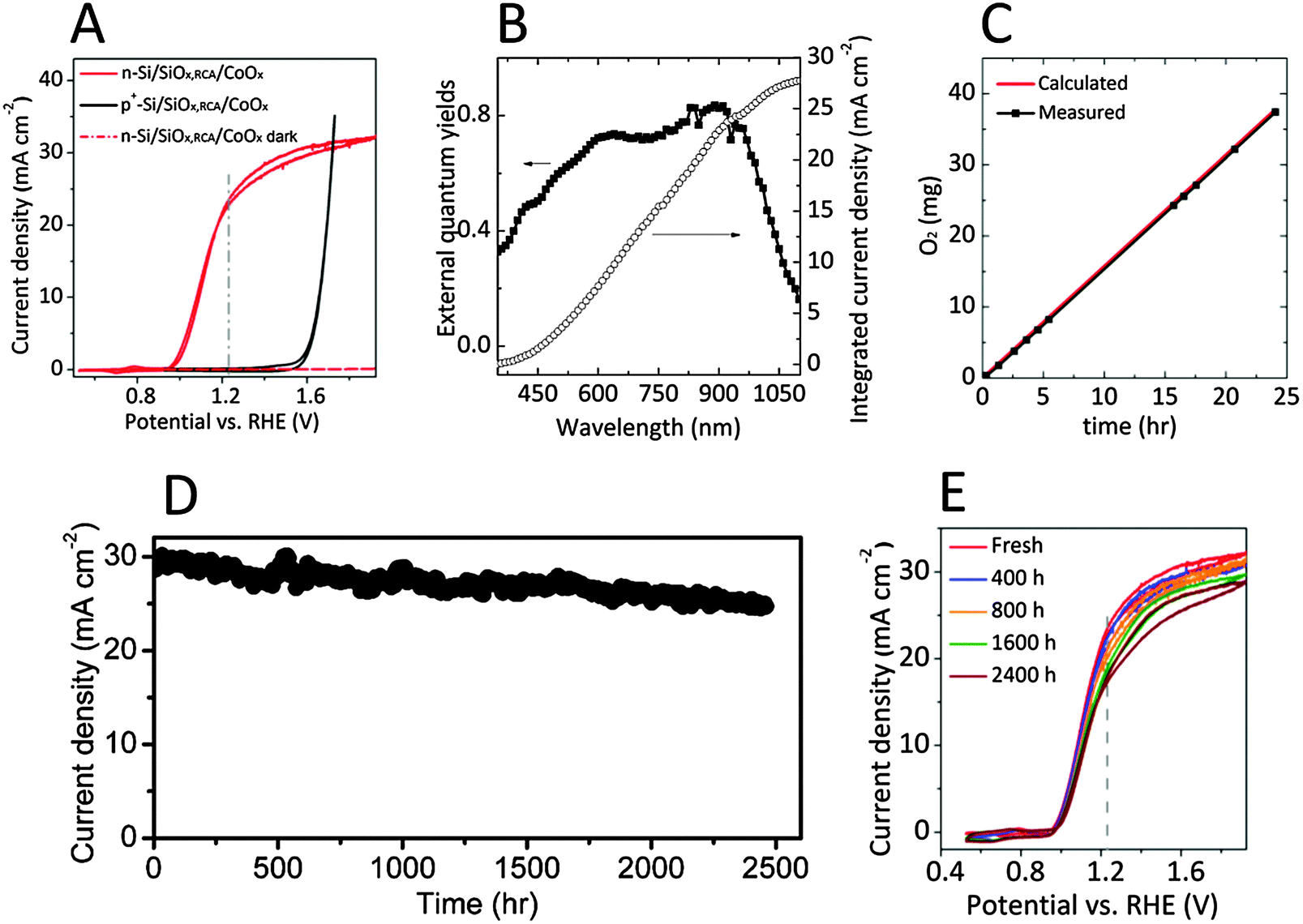 | ||
| Fig. 2 (A) Representative current-density versus potential (J–E) behavior for n-Si/SiOx,RCA/CoOx photoanodes in contact with a 1.0 M KOH(aq) solution in the dark or under 110 mW cm−2 of simulated AM 1.5G solar illumination. The J–E behavior of a non-photoactive p+-Si/SiOx,RCA/CoOx electrode is also shown. (B) External quantum yield for an n-Si/SiOx,RCA/CoOx photoanode biased at 1.93 V versus a reversible hydrogen electrode (RHE) and in contact with 1.0 M KOH(aq) while illuminated by light that had been passed through a monochromator (left ordinate), and the current density derived by integrating the external quantum yield with respect to the AM 1.5G spectrum (right ordinate). (C) O2(g) production measured for an n-Si/SiOx,RCA/CoOx photoanode held at a constant current of 6.7 mA cm−2 for 24 h while under AM 1.5G simulated illumination and in contact with 1.0 M KOH(aq), and O2(g) production calculated based on the charge passed assuming 100% Faradaic efficiency (red line). (D) Chronoamperometry of an n-Si/SiOx,RCA/CoOx photoanode held at 1.63 V vs. RHE while under 1.1 Sun of simulated solar illumination and in contact with 1.0 M KOH(aq). (E) J–E behavior of the n-Si/SiOx,RCA/CoOx photoanode as measured periodically during the chronoamperometric stability test shown in (D). | ||
A representative n-Si/SiOx,RCA/CoOx photoanode exhibited a photocurrent-onset potential of −212 mV relative to E0′(O2/H2O), 23.2 mA cm−2 of photocurrent density at E0′(O2/H2O), and a solar-to-O2(g) ideal-regenerative-cell conversion efficiency, ηIRC, of 1.5%.30 With three n-Si/SiOx,RCA/CoOx electrodes tested, the photocurrent-onset potentials were −205 ± 20 mV relative to E0′(O2/H2O), the photocurrent densities were 22.9 ± 1.6 mA cm−2 at E0′(O2/H2O), and the solar-to-O2(g) ηIRC was 1.42 ± 0.20%. The J–E behavior of the p+-Si/SiOx,RCA/CoOx dark electrode provides a measurement of the performance characteristics of the CoOx layer for the electrocatalysis of O2(g) production in 1.0 M KOH(aq). A load-line analysis31 indicated that a photodiode with an open-circuit voltage (Voc) of 575 ± 20 mV, a short-circuit photocurrent density (Jsc) of 30.2 ± 1.1 mA cm−2, a fill factor of 0.60 ± 0.01, and a photovoltaic efficiency of η = 11.1 ± 0.5% would be required for a photodiode connected in series with a dark p+-Si/SiOx,RCA/CoOx anode to exhibit the electrochemical characteristics observed for the n-Si/SiOx,RCA/CoOx photoanode.
Fig. 2C shows the mass of O2(g) generated as a function of time, as determined using an eudiometer, for an n-Si/SiOx,RCA/CoOx photoanode in contact with 1.0 M KOH(aq). The photoanode was biased to maintain a constant current density of 6.7 mA cm−2 (see ESI†), and was illuminated by 100 mW cm−2 of AM 1.5G simulated solar illumination. The required bias was 1.6 V and did not change during the 24 h experiment (Fig. S3, ESI†). The mass of O2(g) that would be produced as a function of time assuming 100% Faradaic efficiency for the oxidation of water to O2(g) is included as a red curve in Fig. 2C. The measured mass of O2(g) was within experimental error of the calculated value, indicating that the n-Si/SiOx,RCA/CoOx photoanodes exhibited essentially 100% Faradaic efficiency for the production of O2(g) under these conditions.
Fig. 2D shows the photocurrent density as a function of time for an n-Si/SiOx,RCA/CoOx photoanode in contact 1.0 M KOH(aq) while biased at 1.63 V versus RHE and under 1.1 Sun of ENH-type tungsten-halogen simulated solar illumination. The photocurrent density at 1.63 V versus RHE decreased by ∼14% after more than 100 days (∼2500 h) of continuous operation in 1.0 M KOH(aq), at which point the experiment was terminated but failure of the electrode was not apparent (see ESI,† discussions about stability test). For comparison, the n-Si/SiOx,RCA interface without the CoOx overlayer showed rapid surface oxidation (Fig. S4A, ESI†). Fig. 2E shows the J–E behavior of the photoelectrode as measured periodically during a 100-day stability test. The photocurrent-onset potential relative to E0′(O2/H2O) increased from −220 mV to −215 mV, −209 mV, −205 mV, and −202 mV, the photocurrent density at E0′(O2/H2O) decreased from 23.2 mA cm−2 to 22.1 mA cm−2, 20.9 mA cm−2, 18.7 mA cm−2, and 17.7 mA cm−2, the photocurrent density at 1.63 V versus RHE decreased from 30.4 mA cm−2 to 29.7 mA cm−2, 29.2 mA cm−2, 27.8 mA cm−2, and 26.8 mA cm−2 and the solar-to-O2(g) ηIRC decreased from 1.47% to 1.40%, 1.27%, 1.15% and 1.05% after 400 h, 800 h, 1600 h and 2400 h, respectively. The decay of the performance might be due to the dissolution of the CoOx layer, formation of pinholes at the electrode surface (Fig. S2B, ESI†), and the consequent generation of SiOx islands at the pinholes. The average decay rates of the photocurrent-onset potential relative to E0′(O2/H2O), the photocurrent density at E0′(O2/H2O), and the solar-to-O2(g) ηIRC for the n-Si/SiOx,RCA/CoOx photoanode were 7.5 mV, 2.3 mA cm−2 and 0.17% per 1000 h, respectively, thus significantly lower than those for the n-Si/SiOx,RCA/CoOx/NiOx photoanode, which were 36 mV, 7.8 mA cm−2 and 0.97% per 1000 h, respectively.29
The uniform and compact nature of the ALD CoOx film may explain the extended stability (∼2500 h) and reduced rate of performance decay for these devices in 1.0 M KOH(aq) because the compactness of the film may inhibit the formation of porous CoOOH at the grain boundaries, allowing effective isolation of the Si substrate from contact with the corrosive electrolyte. Also, XPS data (Fig. S5, ESI†) showed the formation of CoOOH at the electrode surface, which could function as an OER catalyst.32
Fig. 3A shows the differential capacitance vs. potential (Mott–Schottky) data for an n-Si/SiOx,RCA/CoOx photoanode in contact with an aqueous solution that contained the electrochemically reversible, one-electron Fe(CN)63−/4− redox couple. The flat-band potential, Efb, calculated from the x intercept of the linear region of the data, was −0.85 ± 0.02 V versus E(Fe(CN)63−/4−) (see ESI†), similar to the −0.83 ± 0.02 V versus E(Fe(CN)63−/4−) flat-band potential reported previously for n-Si/SiOx,RCA substrates coated with a thin layer of ALD CoOx as well as with a 100 nm layer of sputtered nickel oxide.29 The flat-band potential of the n-Si/SiOx,RCA/CoOx photoanodes is consistent with the presence of significant band bending and therefore with the presence of a large electric field in the space-charge region, which enables efficient separation of photogenerated charge carriers.
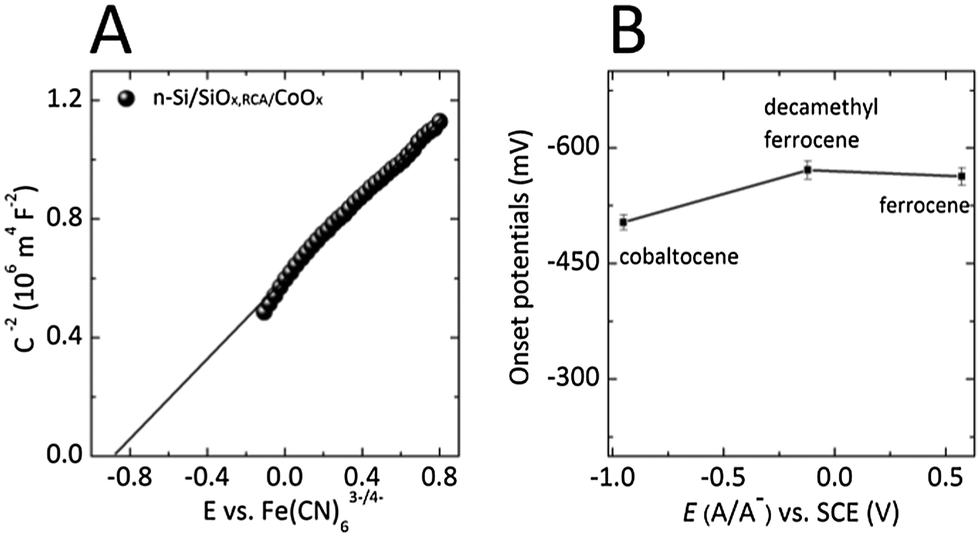 | ||
| Fig. 3 (A) Mott–Schottky plots of the inverse of the differential capacitance, C, of the electrode versus potential for an n-Si/SiOx,RCA/CoOx photoanode in contact with a solution of 50 mM K3Fe(CN)6, 350 mM K4Fe(CN)6 and 1.0 M KCl aqueous Fe(CN)63−/4− (B) Photocurrent-onset potentials versus the Nernstian potential of the solution for n-Si/SiOx,RCA/CoOx photoanodes under 100 mW cm−2 illumination and in contact with CH3CN solutions containing the cobaltocenium/cobaltocene, decamethylferrocenium/decamethylferrocene, and ferrocenium/ferrocene redox couples, respectively. The line only connects the experimentally observed values; no functional form is assumed. | ||
The properties of the n-Si/SiOx,RCA/CoOx junction were evaluated further using nonaqueous electrochemical measurements. Fig. 3B compares the photocurrent-onset potentials measured versus the Nernstian potential of the solution for n-Si/SiOx,RCA/CoOx photoanodes under 100 mW cm−2 of simulated solar illumination and in contact with CH3CN solutions that contained the one-electron redox couples cobaltocenium/cobaltocene (Co(Cp)2+/0), decamethylferrocenium/ferrocene (Me10Cp2Fe+/0), and ferrocenium/ferrocene (Fe(Cp)2+/0), respectively. The n-Si/SiOx,RCA/CoOx photoelectrodes exhibited photocurrent-onset potentials of −503 mV, −571 mV, and −563 mV relative to the Nernstian potential of the solution when in contact with Co(Cp)2+/0, Me10Cp2Fe+/0, and Fe(Cp)2+/0, respectively. The variation in photocurrent-onset potentials observed for n-Si/SiOx,RCA/CoOx photoelectrodes was negligible in comparison to the 1.524 V range over which the potential of the contacting solution was varied (−0.951 V for Co(Cp)2+/0, −0.121 V for Me10Cp2Fe+/0 and 0.573 V for Fe(Cp)2+/0versus the saturated calomel electrode (SCE)), indicating that the junction is almost completely buried and therefore unaffected by the Nernstian potential of the solution. These results are consistent with the presence of a compact CoOx film with few defects that minimize contact between the underlying Si and the solution. In contrast, n-Si/SiOx,RCA substrates coated with 2–3 nm of ALD CoOx exhibited photocurrent-onset potentials that varied by 510 mV in contact with these same redox species. Additionally, n-Si/SiOx,RCA substrates coated with ∼100 nm columnar films of sputtered nickel oxide exhibited photocurrent-onset potentials that varied by 225 mV in contact with the same set of redox couples (Fig. S6, ESI†). These results suggest that 50 nm thick ALD CoOx films exhibit fewer pinhole-type defects, which allow contact between the underlying Si and the electrolyte, than are exhibited by sputtered NiOx films, which in turn exhibit fewer through-film defects than thin ALD CoOx films.
The compact ALD CoOx layer required an overpotential of 433 mV to drive the OER on p+-Si at a rate corresponding to a current density of 10 mA cm−2 under these conditions. For comparison, under the same conditions, sputtered columnar NiOx films on p+-Si require a 330 mV overpotential to produce a current density of 10 mA cm−2.17 Compared to the n-Si/sputtered-catalyst interface, the n-Si/ALD-catalyst interface was free of detrimental effects produced by the oxygen plasma during the sputtering process, and was conformal and compact. Assuming that the interface characteristics do not depend on the doping of the substrate, the electrochemical behavior of the p+-Si anodes can be compared to the photoelectrochemical behavior of the n-Si photoanodes, indicating that the n-Si/SiOx,RCA/CoOx electrodes exhibited both a higher photovoltage and a higher overpotential, by 165 and 103 mV, respectively, relative to n-Si photoanodes protected by a columnar NOx film. Hence, although an n-Si photoanode with an ALD CoOx layer exhibited higher solar-to-O2 conversion efficiency than an n-Si/SiOx,RCA/NiOx photoanode,29 the interfacial-energetics control of the photovoltage provided by n-Si/SiOx,RCA/CoOx heterojunctions, in combination with the active OER electrocatalysis and protection provided by the NiOx overlayer, provides a superior combination of properties for a Si heterojunction photoanode effecting water oxidation in alkaline electrolytes.
Prior work involving atomic-layer deposition of CoOx films (∼4–5 nm thick) onto planar np+-Si substrates concluded: (i) that such devices are characterized by significant corrosion and a decline in J–E response after 30 min of operation in contact with 1.0 M NaOH(aq) while biased at 1.6 V versus RHE and under 1 Sun illumination; and (ii) that nanoscale texturing of the substrate surface prior to deposition of the CoOx film improved both the performance and the stability of devices.19 For comparison, a CoOx film was deposited herein onto a planar n-Si/SiOx,RCA photoanode using 100 ALD cycles (n-Si/SiOx,RCA/100C-CoOx, CoOx film 4–5 nm thick). When operated in contact with 1.0 M KOH(aq) under 1 Sun of simulated illumination, the n-Si/SiOx,RCA/100C-CoOx device yielded a photocurrent density of 20.0 mA cm−2 at E0′(O2/H2O) and a light-limited current density of 27.3 mA cm−2 (Fig. S7, ESI†). The performance of the n-Si/SiOx,RCA/100C-CoOx compared favorably to that reported for the planar and nanotextured np+-Si/CoOx photoanodes, which exhibited photocurrent densities of 12 mA cm−2 and 17 mA cm−2 at E0′(O2/H2O) and light-limited current densities of 27 mA cm−2 and 30 mA cm−2 under the same conditions, respectively.19 An n-Si/SiOx,RCA substrate was additionally coated with a thinner layer of CoOx (∼2–3 nm thick) that was deposited using 60 ALD cycles (n-Si/SiOx,RCA/60C-CoOx). Fig. S8B (ESI†) shows the initial J–E behavior of the n-Si/SiOx,RCA/60C-CoOx, as well as the behavior after the one-hour stability test. No significant corrosion nor decline in J–E response was observed, in contrast to prior work on planar np+-Si/60C-CoOx devices, which exhibited a decay in performance after 30 min of operation under similar conditions.19
Several factors may contribute to the improved performance and stability observed in this work for n-Si/SiOx,RCA substrates coated by ALD CoOx as compared to prior observations of the behavior of np+-Si homojunctions coated by ALD CoOx. The n-Si/SiOx,RCA/CoOx heterojunctions exhibit improved spectral response in the short-wavelength region of the solar spectrum (<500 nm) relative to devices with np+-Si homojunctions, because the heterojunctions do not suffer from the parasitic absorption inherent to a p+-Si emitter layer. In addition, several differences in the methods for ALD of the CoOx layer may be important contributors to the differences in observations, particularly with respect to the initiation and adhesion of the ALD film, the uniformity of the resulting CoOx layer, and the nature of the SiOx layer at the interface. These factors may include: (i) different oxidants (ozone versus oxygen plasma); (ii) different deposition temperatures (150 °C versus 300 °C); and, (iii) different preparation of substrates prior to ALD (RCA cleaning leaves a thin SiOx layer on the surface, relative to HF etching which leaves a hydride-terminated Si surface). In combination with the previously reported behavior of n-Si/SiOx,RCA/CoOx/NiOx photoanodes, the present work clearly demonstrates that CoOx films deposited using ALD are capable of providing a basis for high photovoltage, stabilized photoanodes on a variety of Si substrates.
We investigated whether the use of 50 nm ALD CoOx films could be extended readily to produce stable, high-performance heterojunctions with other semiconductors that could produce self-passivating oxides under anodic conditions, specifically n-CdTe. The n-CdTe/CoOx photoanode exhibited a photocurrent-onset potential of ∼+220 mV relative to E0′(O2/H2O), and a light-limited current density of ∼14 mA cm−2, but produced no light-induced current at E0′(O2/H2O) in 1.0 M KOH(aq) under 1 Sun of simulated solar illumination. The poor performance of n-CdTe/CoOx devices might be due to lattice mismatch, energy-band misalignment, absence of a passivation layer analogous to the silicon oxide layer on n-Si, or to detrimental effects of the ozone used during the ALD process. Although the n-CdTe/CoOx devices exhibited increased stability under oxygen-evolution conditions relative to unprotected n-CdTe devices (Fig. S9, ESI†), loss of performance over time was significant. For example, the current density from the n-CdTe/CoOx device operated in contact with 1.0 M KOH(aq) and biased to 2.8 V versus RHE while under 1 Sun of simulated solar illumination decreased by ∼10% over a 200 h period, whereas n-CdTe photoanodes that did not have CoOx protection layer showed rapid surface oxidation, as expected (Fig. S4B, ESI†).
In conclusion, stable and efficient photoanodes for the oxidation of water to O2(g) in alkaline aqueous electrolytes can be constructed using heterojunctions between planar n-Si(100) substrates bearing a thin chemically grown oxide layer and a 50 nm thick ALD CoOx film. The Si/SiOx,RCA/CoOx heterojunction simplifies the processing required to produce high-performance stable Si photoanodes by eliminating the need for homogeneous buried np+ junctions, nanotexturing, or intermediate interfacial engineering layers. The CoOx film provides multiple desired functions for fully integrated solar-fuels devices, including a high-energy barrier for charge separation, catalytic activity for the water-oxidation reaction, chemical stability, and structural compactness to enable the long-term operation of Si photoanodes.
Acknowledgements
This work was supported by the Joint Center for Artificial Photosynthesis, a DOE Energy Innovation Hub, supported through the Office of Science of the U.S. Department of Energy under Award Number DE-SC0004993. Atomic-force microscopy and UV-Vis spectroscopy were performed at the Molecular Materials Resource Center (MMRC) of the Beckman Institute at the California Institute of Technology. This work was additionally supported by the Gordon and Betty Moore Foundation under Award No. GBMF1225.References
- J. A. Turner, Science, 2004, 305, 972–974 CrossRef CAS PubMed.
- K. Maeda, K. Teramura, D. Lu, T. Takata, N. Saito, Y. Inoue and K. Domen, Nature, 2006, 440, 295 CrossRef CAS PubMed.
- M. J. Kenney, M. Gong, Y. Li, J. Z. Wu, J. Feng, M. Lanza and H. Dai, Science, 2013, 342, 836–840 CrossRef CAS PubMed.
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed.
- E. A. Hernandez-Pagan, N. M. Vargas-Barbosa, T. H. Wang, Y. X. Zhao, E. S. Smotkin and T. E. Mallouk, Energy Environ. Sci., 2012, 5, 7582–7589 CAS.
- J. Jin, K. Walczak, M. R. Singh, C. Karp, N. S. Lewis and C. X. Xiang, Energy Environ. Sci., 2014, 7, 3371–3380 CAS.
- S. Haussener, C. X. Xiang, J. M. Spurgeon, S. Ardo, N. S. Lewis and A. Z. Weber, Energy Environ. Sci., 2012, 5, 9922–9935 CAS.
- J. R. McKone, N. S. Lewis and H. B. Gray, Chem. Mater., 2014, 26, 407–414 CrossRef CAS.
- K. Sun, S. Shen, Y. Liang, P. E. Burrows, S. S. Mao and D. Wang, Chem. Rev., 2014, 114, 8662–8719 CrossRef CAS PubMed.
- S. Hu, N. S. Lewis, J. W. Ager, J. Yang, J. R. McKone and N. C. Strandwitz, J. Phys. Chem. C, 2015, 119, 24201–24228 CAS.
- S. Hu, M. R. Shaner, J. A. Beardslee, M. Lichterman, B. S. Brunschwig and N. S. Lewis, Science, 2014, 344, 1005–1009 CrossRef CAS PubMed.
- M. F. Lichterman, A. I. Carim, M. T. McDowell, S. Hu, H. B. Gray, B. S. Brunschwig and N. S. Lewis, Energy Environ. Sci., 2014, 7, 3334–3337 CAS.
- M. R. Shaner, S. Hu, K. Sun and N. S. Lewis, Energy Environ. Sci., 2015, 8, 203–207 CAS.
- M. T. McDowell, M. F. Lichterman, A. I. Carim, R. Liu, S. Hu, B. S. Brunschwig and N. S. Lewis, ACS Appl. Mater. Interfaces, 2015, 7, 15189–15199 CAS.
- Y. W. Chen, J. D. Prange, S. Duhnen, Y. Park, M. Gunji, C. E. Chidsey and P. C. McIntyre, Nat. Mater., 2011, 10, 539–544 CrossRef CAS PubMed.
- B. Mei, T. Pedersen, P. Malacrida, D. Bae, R. Frydendal, O. Hansen, P. C. K. Vesborg, B. Seger and I. Chorkendorff, J. Phys. Chem. C, 2015, 119, 15019–15027 CAS.
- K. Sun, F. H. Saadi, M. F. Lichterman, W. G. Hale, H.-P. Wang, X. Zhou, N. T. Plymale, S. T. Omelchenko, J.-H. He, K. M. Papadantonakis, B. S. Brunschwig and N. S. Lewis, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 3612–3617 CAS.
- K. Sun, Y. Kuang, E. A. Verlage, B. S. Brunschwig, C. W. Tu and N. S. Lewis, Adv. Energy Mater., 2015, 5, 1402276 Search PubMed.
- J. Yang, K. Walczak, E. Anzenberg, F. M. Toma, G. Yuan, J. Beeman, A. Schwartzberg, Y. Lin, M. Hettick, A. Javey, J. W. Ager, J. Yano, H. Frei and I. D. Sharp, J. Am. Chem. Soc., 2014, 136, 6191–6194 CrossRef CAS PubMed.
- L. Chen, J. Yang, S. Klaus, L. J. Lee, R. Woods-Robinson, J. Ma, Y. Lum, J. K. Cooper, F. M. Toma, L. W. Wang, I. D. Sharp, A. T. Bell and J. W. Ager, J. Am. Chem. Soc., 2015, 137, 9595–9603 CrossRef CAS PubMed.
- K. Sun, M. T. McDowell, A. C. Nielander, S. Hu, M. R. Shaner, F. Yang, B. S. Brunschwig and N. S. Lewis, J. Phys. Chem. Lett., 2015, 6, 592–598 CrossRef CAS PubMed.
- N. C. Strandwitz, D. J. Comstock, R. L. Grimm, A. C. Nichols-Nielander, J. Elam and N. S. Lewis, J. Phys. Chem. C, 2013, 117, 4931–4936 CAS.
- H. Gerischer, J. Electroanal. Chem., 1977, 82, 133–143 CrossRef CAS.
- E. Verlage, S. Hu, R. Liu, R. J. R. Jones, K. Sun, C. Xiang, N. Lewis and J. H. A. Atwater, Energy Environ. Sci., 2015, 8, 3166–3172 CAS.
- A. Heller, in Photoeffects at Semiconductor-Electrolyte Interfaces, American Chemical Society, 1981, vol. 146, ch. 4, pp. 57–77 Search PubMed.
- G. Grube and O. Feucht, Z. Elektrochem., 1922, 28, 568–579 CAS.
- H. Willems, A. G. C. Kobussen, J. H. W. Dewit and G. H. J. Broers, J. Electroanal. Chem., 1984, 170, 227–242 CrossRef CAS.
- M. F. Lichterman, M. R. Shaner, S. G. Handler, B. S. Brunschwig, H. B. Gray, N. S. Lewis and J. M. Spurgeon, J. Phys. Chem. Lett., 2013, 4, 4188–4191 CrossRef CAS.
- X. Zhou, R. Liu, K. Sun, D. Friedrich, M. T. McDowell, F. Yang, S. T. Omelchenko, F. H. Saadi, A. C. Nielander, S. Yalamanchili, K. M. Papadantonakis, B. S. Brunschwig and N. S. Lewis, Energy Environ. Sci., 2015, 8, 2644–2649 CAS.
- R. H. Coridan, A. C. Nielander, S. A. Francis, M. T. McDowell, V. Dix, S. M. Chatman and N. Lewis, Energy Environ. Sci., 2015, 8, 2886–2901 CAS.
- M. R. Shaner, K. T. Fountaine and H.-J. Lewerenz, Appl. Phys. Lett., 2013, 103, 143905 CrossRef.
- J. C. Hill, A. T. Landers and J. A. Switzer, Nat. Mater., 2015, 14, 1150–1155 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ee03655k |
| This journal is © The Royal Society of Chemistry 2016 |