
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Some of tomorrow's catalysts for processing renewable and non-renewable feedstocks, diminishing anthropogenic carbon dioxide and increasing the production of energy†‡
John Meurig
Thomas
*a and
Kenneth D. M.
Harris
*b
aDepartment of Materials Science, University of Cambridge, Cambridge CB0 3FS, England, UK. E-mail: jmt2@cam.ac.uk
bSchool of Chemistry, Cardiff University, Park Place, Cardiff CF10 3AT, Wales, UK. E-mail: HarrisKDM@cardiff.ac.uk
First published on 6th January 2016
Abstract
This review provides a wide-ranging summary of several aspects of heterogeneous catalysis and its impact on the increasing need to generate more energy, less CO2 and the production of more commodities required by an expanding world population. Particular attention is paid to the options (some of which are already a practical reality) now available for the use of anthropogenic CO2 as a source for the production of platform chemicals required to sustain civilized life. In this connection, Rubisco-inspired methods of utilizing CO2 are discussed, as is the utilization of algae to yield ethanol and O2 from water, CO2 and sunlight. In addition, the increasing use of methanol (derived from CO2) as an energy vector, as well as a source of ethene and propene (which are in growing worldwide demand), is adumbrated. As far as strategies for the design of new solid catalysts are concerned, summarizing accounts are given of the emerging popularity and recent successes of supported “single-atom”, chemo-selective catalysts (of Pt, Pd, Ir and Au), of so-called “single-atom alloy” catalysts for selective hydrogenations, and of monophasic single-site heterogeneous catalysts (SSHCs) for a range of chemical processes, some of which have already been commercialized. SSHCs can, in general, be assembled from earth-abundant elements (C, N, O, Mg, Al, P, Fe), and are effective for shape-selective, regio-selective and enantio-selective catalytic conversions. We also briefly discuss the prospect of converting anthropogenic CO2 into CH4, and touch upon the action needed to reduce atmospheric CO2 so as to fulfil the aims of the recent (December 2015) UN Climate Change Conference in Paris (COP-21).
 John Meurig Thomas | Sir John Meurig Thomas is a pioneer of Solid-State Chemistry, which he initiated in the University of Wales (Bangor and Aberystwyth) and extended after taking up (in 1978) the Headship of the Department of Physical Chemistry, University of Cambridge and the Directorship of the Royal Institution of Great Britain, London (1986). On returning to Cambridge as Master of Peterhouse (1993), he became an Honorary Professor in the Department of Materials Science, Cambridge. Over the years, he has designed and exploited numerous new solid catalysts and devised several novel techniques for their characterization. He has pioneered the use of electron microscopy in chemistry since the early 1960s. The author of several university texts in Heterogeneous Catalysis, and a biography of Michael Faraday, he has been the recipient of a multitude of awards including the Faraday Medal and several other awards of the Royal Society of Chemistry, the Willard Gibbs Gold Medal of the American Chemical Society, the Linus Pauling Gold Medal, the Stokes Gold Medal, the Davy Medal of the Royal Society, and the Zewail Gold Medal (2015), and he was the first recipient of the American Chemical Society award for Creative Research in Homogeneous or Heterogeneous Catalysis in 1999. He was knighted in 1991 for services to chemistry and the popularization of science. A new mineral, meurigite, was named in his honour in 1995. |
 Kenneth D. M. Harris | Kenneth D. M. Harris received a stimulating education in Solid-State Chemistry by carrying out research for his PhD degree under the supervision of Sir John Meurig Thomas FRS at the University of Cambridge and at the Royal Institution, between 1985 and 1988. After holding Lectureships at the University of St. Andrews and then at University College London, he was appointed Professor of Structural Chemistry at the University of Birmingham in 1995. He took up his present appointment as Distinguished Research Professor at Cardiff University in 2003. He has been awarded the Meldola (1991), Marlow (1996), Corday-Morgan (1999), Structural Chemistry (2001) and Tilden (2007/8) Medals of the Royal Society of Chemistry, and has been elected as a Fellow of the Royal Society of Edinburgh (2008), Fellow of the Learned Society of Wales (2011) and Member of Academia Europaea (2013). His research in the Physical Chemistry of Solids has advanced new directions of research focused on understanding fundamental properties of solids, and the development of new aspects of techniques (particularly powder X-ray diffraction, solid-state NMR and X-ray Birefringence Imaging) for interrogating these properties. He has held several appointments as Visiting Professor in Japan, France, USA, Spain and Taiwan. |
Broader contextSolid catalysts, as well as being the backbone of the chemical industry and the major means of manufacturing useful materials for a wide variety of different applications, are also of crucial importance in numerous energy-releasing processes. Small quantities of the correct, judiciously designed (atom-efficient) heterogeneous inorganic catalysts play a vital role in both the conservation and generation of various forms of energy. Many new catalysts are required to cope with the successful utilization of the increasing massive amounts of anthropogenic CO2. Moreover, ways of designing new (e.g. supported single-atom, chemo-selective and single-site heterogeneous) catalysts to effect more efficient, cleaner, environmentally benign chemical processes are outlined. Single-atom catalysts are the most atom-efficient conceivable type of catalyst. In view of the very recently acknowledged assessment1 that natural gas supplies are likely to prove plentiful for the next 230 years (Fig. 1),2 the frequently expressed notion (see Dhakshinamoorthy et al.,3 for example) that there will soon be a shortage of fossil fuels is no longer tenable. We therefore also discuss a selection of important manufacturing processes that are likely to continue to require non-renewable (fossil-derived) feedstocks, but using them in environmentally cleaner and more energetically efficient ways. Ways of stabilizing and ultimately decreasing the content of CO2 in the atmosphere are also critically discussed. |
1. Introduction
As it is always hazardous to look too far into the future, it is with considerable caution, given the title we have chosen, that we embark on this article. Fundamentally new types of catalyst, of which we are currently oblivious, may be discovered and developed in the short-term future; moreover, there may well be existing catalysts of which, again through ignorance, we are currently unaware, and which could well meet some of the needs identified below.It is salutary to recall that, when the Commission set up in 1937 by President F. D. Roosevelt to advise his administration of the likely future scientific and technological developments (over a ten-year horizon) submitted its report, it was seen (when viewed in retrospect many years later) to have been seriously inadequate. Unsurprisingly, it predicted that agricultural science would play an increasingly prominent role in the economy of the nation, which indeed it did and continues to do. It also described how hydrocarbons for various kinds of transport and heating fuels, as well as lubricants, could be produced from coal – again, unsurprisingly, as the Fisher-Tropsch catalytic synthesis of hydrocarbons from a mixture of CO and H2 had been discovered in Germany in 1925.
The Commission could be forgiven for not mentioning nuclear fission and nuclear fusion or magnetic resonance imaging or transistors, masers and lasers: none of these phenomena or devices had been discovered by 1937. But there were other omissions that were less excusable. For example, the fax machine, invented by a Scottish watchmaker in the early 1840s, was not mentioned; neither was there reference to the fuel cell that had been invented by a Welsh lawyer, also in the early 1840s, nor were antibiotics identified even though Fleming had discovered penicillin in 1928 (see Table 1).
| The Physical Sciences and Technology |
|---|
| a For heuristic purposes, we have extended the 10 year horizon requested by President Roosevelt up until the end of the 20th century. |
| Fission, fusion, nuclear energy |
| Radar |
| Laser, maser |
| Transistor, integrated circuit, VLSI |
| Personal computer |
| Laser disk, compact disk, CD-ROM |
| Synchrotron radiation |
| Jet aircraft, rocketry, space travel |
| Stereo-regular polymers (polythene, polypropene) |
| Natural gas to liquid fuels |
| Fax machine, mobile phone |
| Fuel cell |
| Tomography, magnetic resonance imaging (MRI) |
| Positron emission tomography (PET) |
| Charge-coupled device (CCD) |
| Internet, Google, Wikipedia |
| The Biological Sciences and Biotechnology |
|---|
| Antibiotics |
| Immunosuppressive drugs |
| Transplantation medicine |
| Structure of DNA, molecular genetics |
| Genomics, proteomics |
| Protein engineering |
| Genetic fingerprinting and manipulation |
| Monoclonal antibodies |
| Contraceptive pill |
| Pharmaceuticals |
| Flexible endoscope |
These facts remind us that, in seeking to identify the catalysts that are likely to be used in the future, we may well fall far short of satisfactory predictions. Be that as it may, it is incumbent upon us to try and ascertain both the key questions to be asked, and their possible answers, bearing in mind several pressing issues that currently face the human race.
In Section 2, we focus on two key questions pertaining to CO2: first, its continued massive production – the current anthropogenic emission rate hovers around 40 G ton year−1, with little sign of easing off even during the growth of various kinds of renewable energy; second, the urgent need to “recycle” CO2 by, inter alia, utilizing it as a feedstock in the manufacture of materials required for civilized life. (We do not discuss the sequestration of CO2 by such means as the conversion of the minerals cordierite and olivine (magnesium-rich silicates), which occur in abundance, to form the involatile minerals magnesium carbonate and silica; this topic is fully discussed in the article by Fennell and colleagues.4)
In Section 3, we focus on some of the important applied chemical manufacturing processes that are likely to be difficult to operate using renewable, rather than natural gas and other fossil-based, feedstocks. Section 4 deals with aspects of the use of inorganic catalysts in the chemical industry and especially with the need for environmentally-friendly processes. Our environmentally-conscious age also requires either the use of atom-efficient catalysts, composed of relatively rare elements (Rh, Pt, Pd, Ir, Os, Au, etc.) – provided they can be utilized in atomically dispersed forms – or of earth-abundant elements (such as C, N, O, Mg, Al, P and Fe) in appropriate, readily synthesizable forms. Fortunately, there has been significant recent progress in pursuing these two aims in the evolution of new catalysts. And this leads us to ponder the recent spectacular successes of single-atom supported catalysts, and also to elaborate the already useful concept and reality of single-site heterogeneous nanoporous catalysts, which can be utilized in the context of the facile preparation of platform chemicals by the conversion of chemical energy, and in effecting asymmetric syntheses for the production of enantiopure substances.
In looking towards tomorrow's catalysts, we must also bear in mind, as emphasized by Campbell5 a decade or so ago, that computational science is likely to play an increasingly important role in the design and discovery of new catalysts. The growing precision of density functional theoretical methods, as illustrated by the work of Reuter et al.,6 Nørskov, Chorkendorff and co-workers,7–10 van Santen and Neurock11 and others, gives reason to believe that such fundamental properties as activation energies and transition-state vibrational frequencies may now be calculated with good accuracy for all relevant elemental surface reactions, as well as kinetic Monte Carlo simulations of overall reaction processes. To emphasize this point, one need quote only the work by Hinnemann et al.,9 who have made rapid progress in the drive towards replacing platinum by non-precious metal electrocatalysts as the currently preferred catalyst for the hydrogen evolution reaction (HER). This reaction is of cardinal importance in photoelectrochemical and electrolytic applications, as we describe at the end of this article (see Section 5 for further details).
A final introductory comment pertains to future supplies of natural gas. Contrary to the implications of the frequently expressed mantra that fossil fuels are likely soon to be in short supply, we must recognize that this prediction is invalid: gas resources are plentiful, growing and geographically diverse (as recently emphasized by Brown1 – see Fig. 1).
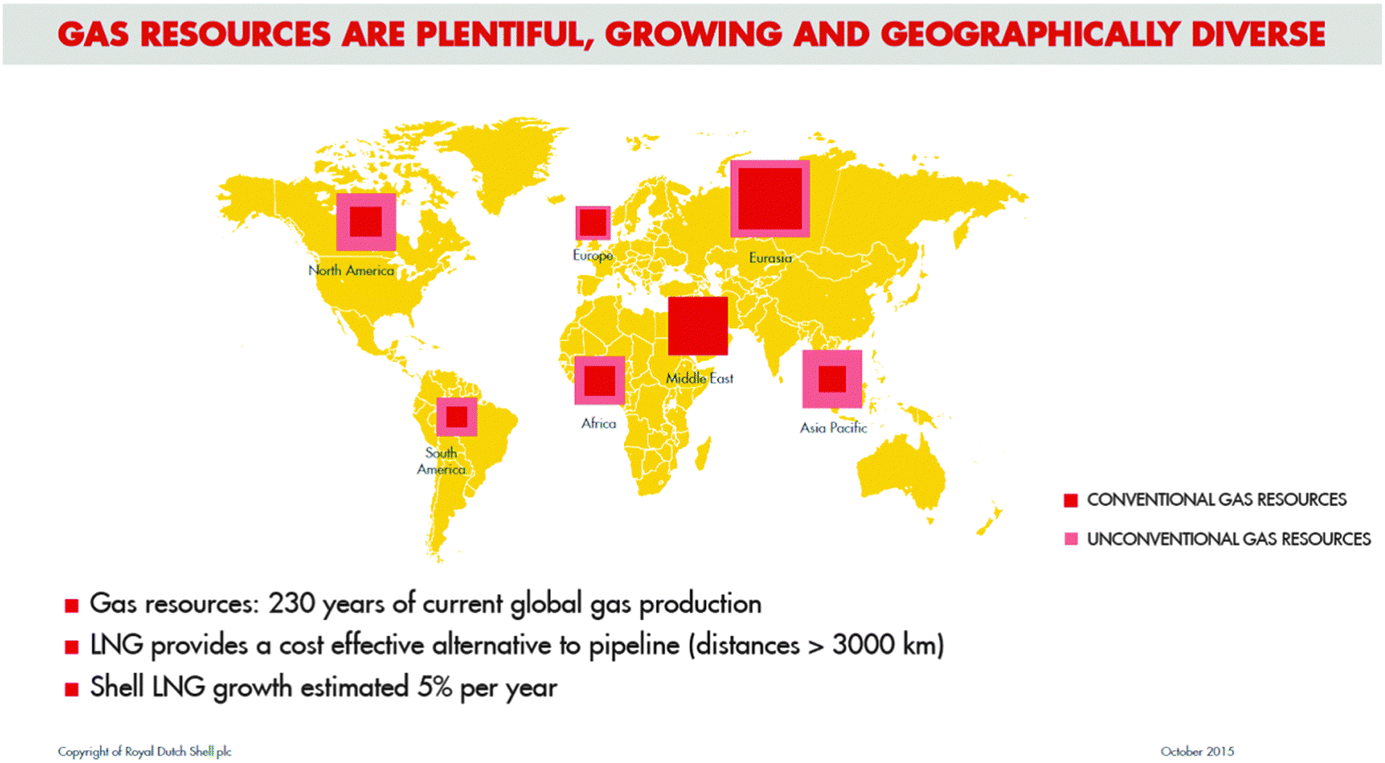 | ||
| Fig. 1 Assessment, by Royal Dutch Shell plc, of the current situation pertaining to gas resources. Note that liquefied natural gas (LNG) is a viable alternative to pipelines as a means of distribution. (By kind permission of A. Brown). | ||
2. The question of CO2
It is almost a platitude to remark that CO2 presents a serious threat to the future of the human race. Far too much of it is liberated into the atmosphere every year. Even though there are now several feasible operations for CO2 storage and sequestration, comparatively little progress has yet been made towards the massive use of CO2 as a chemical feedstock. In addition, there is an urgent need to minimize CO2 production, by seeking alternatives to oil, coal and gas – or improvements in the cleanliness and efficiency of their use – as the primary sources of energy and the chemical materials that sustain civilized life. (In Section 5, we return to the question of converting anthropogenic CO2 into CH4, which, on use, generates CO2, thereby maintaining a carbon neutral situation, which would stabilize the amount of CO2 in the Earth's atmosphere).Much has already been said and written about various aspects of these questions. In 2010, Jiang et al.12 discussed at length the thermodynamic and materials science aspects of turning CO2 into fuel; and many others have addressed the principles and possible practices that involve the use of CO2, not only as a source of fuel, but also as the basis of the manufacture of a wide range of chemicals. In this regard, the comprehensive reviews by Centi et al.13 and by Aresta et al.14 are invaluable as they deal with all the relevant work published up to 2014. Also, the reviews by one of us15,16 and especially the work of Centi and Perathoner et al.17–20 and others,21–23 including a recent missive by Schlögl,24 provide much useful background.
Leaving aside the controversial issue of generating biofuels25 (for transport and other purposes), we discuss first the more attractive and environmentally responsible procedure of using algae as the means of utilizing CO2 to produce ethanol.26 A brief account of potential new biotechnological approaches27 follows, before we make reference to a revolutionary way, pioneered by Kanan,28 of recycling CO2.
2.1 The photo-production of ethanol from CO2
Among the reasons why biofuels are frequently frowned upon are the following facts: their production generally competes with that of foodstuffs, in that land use for the latter has to be sacrificed in favour of the former, and moreover their production requires large quantities of fresh water. Algae, on the other hand, can utilize seawater, and the photo-production of liquids such as ethanol, from CO2 and water by solar radiation, is both environmentally responsible and economically attractive.In the discussion below, we pay particular attention to the work of the company Algenol Biofuels, which produces ethanol and biomass (from CO2, water and sunlight) using their genetically enhanced cyanobacteria (chosen from 2300 strains collected globally as candidates for development), as summarized in Fig. 2. (While we highlight the recent developments of Algenol Biofuels in this article, we note that the first report of the synthesis of ethanol by genetic engineering in cyanobacteria was published29 by Deng and Coleman in 1999, and we emphasize that other commercial companies, such as Joule Co., are also involved in producing fuels from sunlight using cyanobacteria). The metabolic pathway for this process is shown in Fig. 3.
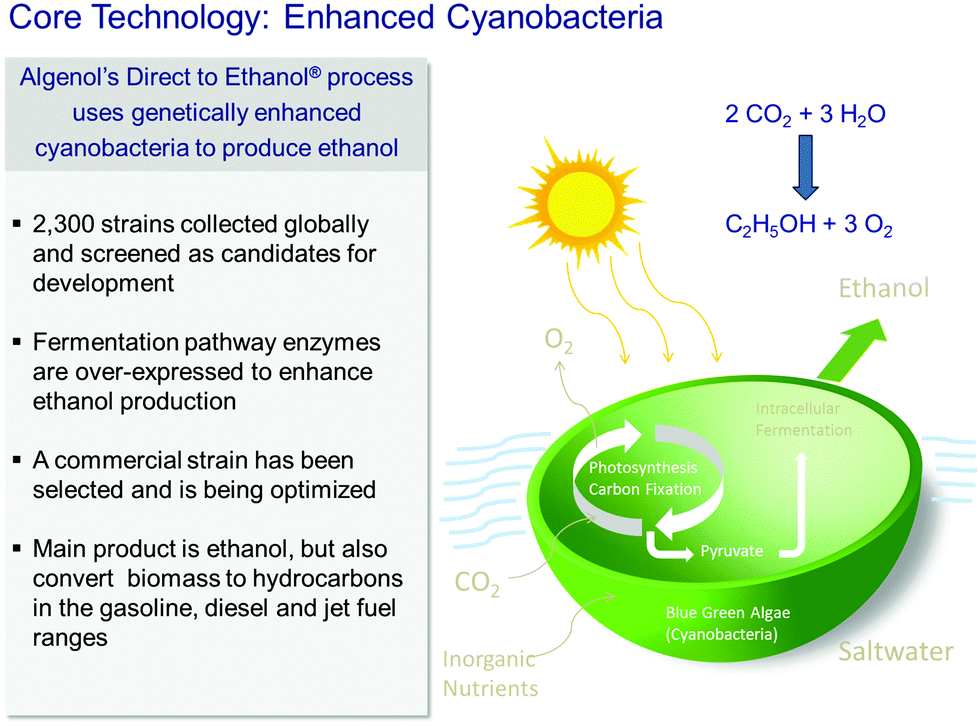 | ||
| Fig. 2 Representation of the use of enhanced cyanobacteria (algae) employed by the Algenol Biofuels Company to convert CO2 and H2O in sunlight to ethanol. (By kind permission of R. R. Chance). | ||
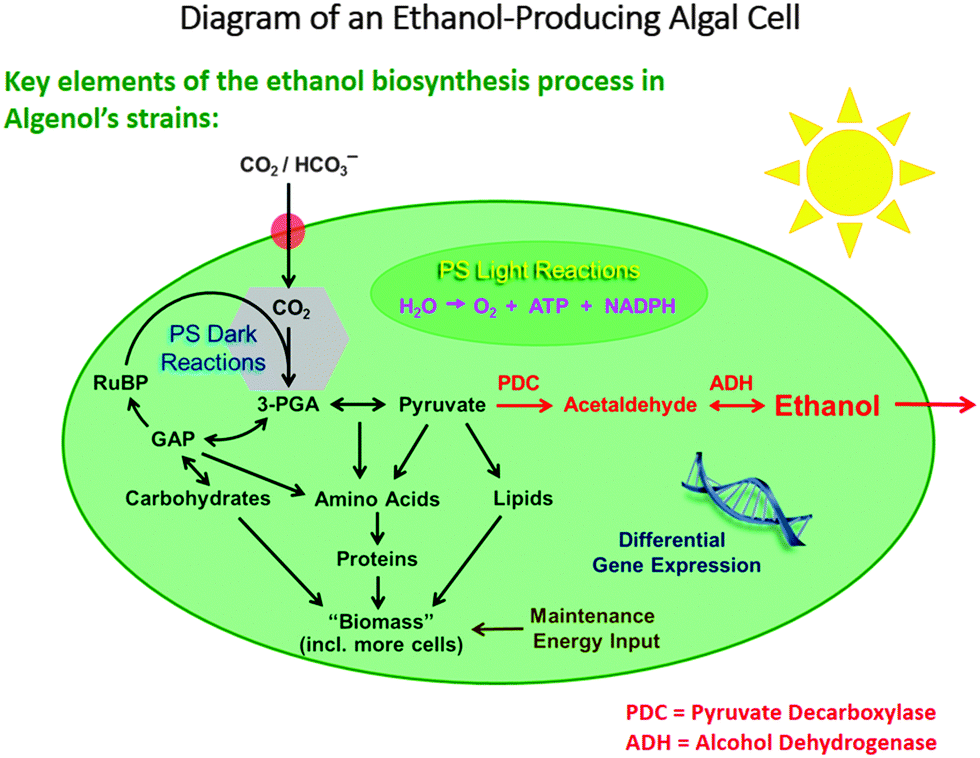 | ||
| Fig. 3 Outline of the metabolic pathway for ethanol production photochemically from CO2 and H2O in sunlight, and the parallel production of biomass. (By kind permission of R. R. Chance and P. Roessler). | ||
In 2013, Algenol Biofuels utilized CO2 gas (liberated from an industrial plant in Florida) to produce 8000 gallons of ethanol per acre per year, and some 2% to 3% solar energy conversion (to ethanol) was possible. When their plant grows to occupy 2000 acres, the Algenol company will produce ca. 14 × 106 gallons of ethanol per annum.26,30 They produce ca. 130 to 140 gallons of biofuel per tonne of CO2, and the energy required to run their facility is ca. 25% to 30% of the heat value of the fuel. We note that O2 is also produced along with ethanol. Several possibilities exist for the further use of this O2, one being energy production for the manufacturing facility. Another possibility is the production of the widely-used chemical, ethylene oxide. It is easy to produce ethylene by catalytic dehydration of the ethanol over a single-site Brønsted acid catalyst31,32 (see Fig. 4.15 on page 70 of ref. 31). The ethylene could, in association with a well-known Ag on α-Al2O3 catalyst for example, be converted readily to ethylene oxide, which in turn can be readily hydrolysed to the valuable platform molecule ethylene glycol.
2.2 Rubisco-inspired methods of utilizing CO2
Rubisco (D-ribulose-1,5-bisphosphate carboxylase/oxygenase), a member of the carboxylase family of enzymes, is one of the most abundant enzymes in the biosphere. Each year it turns over 1011 tons of CO2 in the process of atmospheric CO2 fixation.The active site not only accommodates CO2 but is also capable of accepting O2, which acts as an alternative electrophile during the conversion of the CO2 to glucose.33,34 This causes an oxygenation side reaction, and is a contributor to the rather slow (for an enzyme) turnover frequency (ca. 3 s−1) of Rubisco. Remarkably, the mode of action of Rubisco entails (at least) three distinct mechanistic steps: first, ribulose-1,5-bisphosphate is enolized to form the reactive enolate intermediate; second, this intermediate attacks the CO2 to form the primary carboxylation product, 2-carboxy-3-ketoarabinitol-1,5-bisphosphate; third, this unstable C6-intermediate is hydrolytically cleaved to yield the final reaction products, specifically two molecules of phosphoglycerate.
The multitude of carboxylation reactions that occur in the natural world makes carboxylases interesting targets for applications in organic chemistry, biotechnology and synthetic biology, and these opportunities have been discussed by Erb and colleagues,33,34 who have outlined ways in which synthetic biology could be used to create novel, customized, CO2-fixation pathways to produce biomass and fine chemicals from atmospheric CO2. Up to now, however,34 practical applications of CO2-fixing enzymes in organic chemistry have been limited by the availability of suitable carboxylases, because many of them are highly substrate specific and also oxygen sensitive, or require complex co-factors/co-substrates such as biotin, ATP or ferredoxin for catalysis to ensue.
Nevertheless, Erb has very recently34 elucidated the first step in the process of extracting CO2 by evolving a CO2-fixing enzyme from an ancient purple photosynthetic proteobacteria called Rhodobacter sphaeroides. This enzyme, called crotonyl-CoA carboxylase/reductase (CCR, where CoA stands for coenzyme A), extracts CO2 out of the atmosphere nearly 100 times as fast as the enzyme (Rubisco) used by plants. Erb is currently focused on reducing atmospheric CO2 in this way, and also in producing, from CO2, useful high-value carbon building blocks such as glycerol and aspartic acid, as well as generating biofuels and antibiotics.
Rubisco chemistry has also inspired the recent work of Kanan et al.28 In endeavouring to emulate nature's strategy for C–C bond formation, which is to deprotonate C–H bonds to form carbanions and then to trap these intermediates with CO2 to form C–CO2− (carboxylates), Kanan opted for a purely inorganic approach, which circumvents the need for co-factors and other complications associated with the approach of the synthetic biologists. The challenge that he addressed was how to effect the above deprotonation in a useful way without relying on extremely strong bases. Full details of the experimental work carried out by Kanan et al.28 await publication. Remarkably, they are able to synthesize ethylene glycol and ethanol using only CO2 and H2. Such procedures lead to the ready synthesis of polyethylenefurandicarboxylate (also designated polyethylenefuranoate, PEF), a very viable substitute for polyethyleneterephthalate (PET),35 which is used extensively as a container material for water and mineral drinks.
The H2 required for the preparation of ethylene glycol, as prepared by Kanan,28 may be produced from either wind-powered or solar-powered water-splitting (using, for example, photovoltaics for electrolysis), meaning that the ethylene glycol, an important platform chemical, can be produced cheaply. Moreover, by replacing the entire 15 M tonne year−1 PET market with PEF, it is estimated35 that 20 to 35 M ton year−1 of CO2 would be saved from liberation to the environment. In summary, the methods developed by Kanan rely on the use of simple inorganic bases to generate carbon-centred nucleophiles that are trapped by CO2 to form C–C bonds.
It is relevant to note that, in work only recently submitted for publication by H.-J. Freund et al.,36 an ingenious method has been devised for attaching a neutral CO2 molecule to a radical ion of CO2, thus forming a (CO2)2− species, which may then be transformed into an oxalate species whereby a C–C bond is formed. According to Freund,36 these oxalate species may then be further catalytically transformed with water or ammonia into useful chemicals.
2.3 Synthesizing methanol from CO2
Much has been written about this topic, especially by its leading protagonist Olah,37–39 and about its potential role both in utilizing super-abundant CO2 and in generating a range of fuels and essential chemicals. A cycle illustrating sustainable production of methanol is shown in Fig. 4.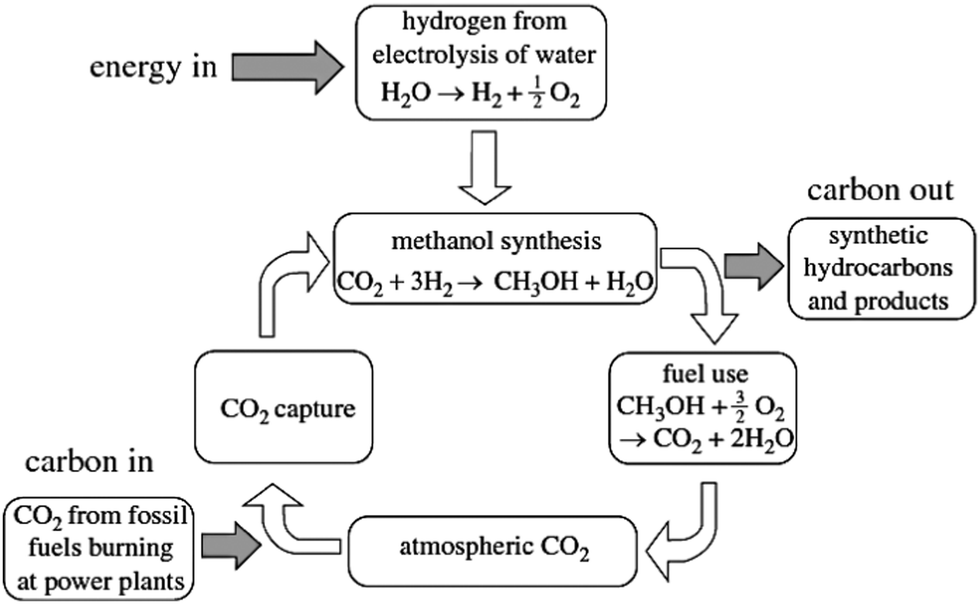 | ||
| Fig. 4 A cycle for sustainable methanol production. | ||
In essence, the sustainable methanol economy requires an abundant source of H2 (which can, as mentioned earlier, readily be produced at an appropriate cost by electrolysis of water using solar, wind or geothermal power) which is combined with CO2 over an appropriate catalyst (usually composed of Cu/ZnO and Al2O3) to yield methanol. (Carbon Recycling International in Iceland utilize geothermal power to produce the H2 that is combined with CO2 in their commercial operation).
As pointed out by Jacobson et al.,40,41 there are, in principle – and increasingly in practice – several ways in which the power of wind, water and sunlight can lead to the abundant generation of H2. It is relevant to mention that numerous concentrated solar power plants of 200 MW to 300 MW are also capable of providing the electricity to electrolyze water for the generation of H2.
As described by Ampelli et al.,17 methanol has many material advantages over other competing substances (such as the hydrocarbon products of Fischer–Tropsch catalysis). For example, it is a raw material for the chemical industry in that it can be readily converted (with known, earth-abundant catalysts) to the building blocks for petrochemical production, such as olefins, or methyl-tertiary-butyl ether (MTBE), which is a high-octane-number component of gasoline.
Ampelli et al.17 have also drawn attention to the fact that methanol turbines have been demonstrated on pilot units to have low NOx emissions. The use of methanol-fuelled gas turbines allows high efficiencies of over 60% to be attained together with recovery of CO2. In this way, it becomes possible to construct a closed-loop system for importing renewable energy from remote areas.
2.4 The growing importance of methanol as an energy vector
Given that the global demand for olefins is about the same as that for liquid natural gas (LNG), it has recently been suggested42 that methanol, derived from CO2, could compete as a useful feedstock. It is now relatively easy to design and construct methanol-to-olefin (MTO) plants in a very short time. (In a joint venture between the Dalian Institute of Chemical Physics and BP, such a plant took less than 18 months to become fully operational from the time of a feasibility study). With a SAPO-34 (single-site heterogeneous) acid catalyst, such a facility has exhibited 100% conversion of methanol with ca. 40% selectivity to C2 olefins and ca. 35% selectivity to C3 olefins over a period of 90 days (see Fig. 5). | ||
| Fig. 5 Photograph of the MTO facility built in less than two years by a Chinese-BP consortium. (By kind permission of M. Atkins). | ||
An illuminating account of the scientific path, taken by workers in the Dalian Institute of Chemical Physics, in proceeding from fundamental studies to commercialization has recently been given by Liu and co-workers.43 Their account briefly summarizes the key issues for the process development, including studies on the reaction mechanism, the synthesis of the molecular sieve (single-site acid catalyst) SAPO-34, and its manufacturing scale-up. As well as the MTO process, these workers also deal with a “dimethyl ether or methanol to olefin” (DMTO) process, the world's first. Their methanol is derived from syngas, which in turn is generated from coal. In fact, China established the world's first coal-to-olefin plant. The world's first DMTO commercial unit (operated from May 2010) produces 0.6 M ton of polyethylene and polypropylene per year. In future, the challenge will be to use coal as a feedstock in an environmentally benign manner.
Fig. 6, taken from the work of Atkins42 and others at BP, shows the ease with which dimethylether (DME) – another important platform molecule – can be generated (again with a single-site acidic, nanoporous catalyst) by dehydration of methanol. It also shows how other products like acetic acid and methyl acetate may be readily made as part of a closed cycle. Another important speciality molecule, dimethylcarbonate (DMC), is formed from methanol (via an H2 carrier) in an evolving economy based on methanol.
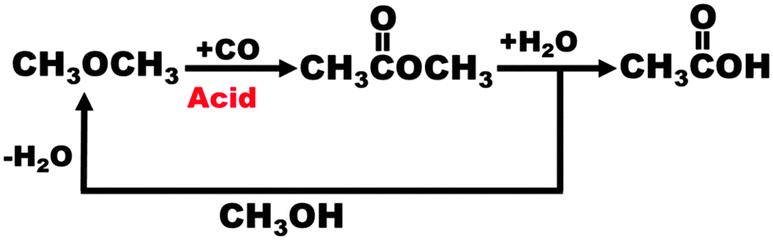 | ||
| Fig. 6 Illustration of the ease of interconversion of important platform molecules using methanol and dimethyl ether. (By kind permission of M. Atkins). | ||
Finally, we note that an increasing prospect, following the recent resolution at the UN Climate Change Conference (COP-21) in December 2015, is that ultimately syngas will have to be generated from biomass, which is both abundant and renewable.
3. A selection of important manufacturing processes that are not easily effected by renewable feedstocks
According to the Technology Roadmap: Energy and Greenhouse Gas Reductions in the Chemical Industry via Catalytic Processes,44 of the several tens of thousands of chemicals that are manufactured, only a small number of products account for 80% of energy demand and are responsible for 75% of greenhouse gas emissions. Table 2 lists these products.| Acrylonitrile | Polypropylene |
| Caprolactam | Propylene oxide |
| Cumene | p-Xylene |
| Ethylene glycol | Styrene |
| Ethylene oxide | Terephthalic acid |
| Phenol | Vinyl chloride |
| Polyethylene |
Some of the sustainable ways in which several of these products may be produced – in place of using non-renewable feedstocks – have been described elsewhere (see ref. 13–17 and 20) and numerous ingenious new sustainable processes are continually being reported. But many crucially important manufacturing processes, such as those listed in Table 3, are likely to be slower to replace using only sustainable feedstocks.
It is rather paradoxical that, with the burgeoning of the fracking industry and the consequential ease of generating methane locally, there are now plans among automobile manufacturers to accelerate the production of fuel-cell vehicles45 because the ease of generating H2 from methane at local (filling) stations is attractive commercially. (It is possible to generate pure H2 from methane using a nickel on silica catalyst46).
The Toyota Company is currently manufacturing a fuel-cell driven car that burns H2. As a consequence, liquid H2 filling stations (where the H2 is produced by steam-reforming of natural gas) are being built in many countries, especially in the USA and Denmark. The Shell Company has recently constructed 200 such filling stations in Germany. And two such filling stations are in London.
Steam reforming and the water-gas shift reaction have long been the corner-stone of the ammonia synthesis industry, since they produce the high-purity H2 necessary for the reaction. Where hydroelectric power is plentiful – as in the Nasser Dam in Upper Egypt – very high purity H2 can be produced (by electrolysis of water) for ammonia synthesis, and hydroelectric power is currently used in that location. (Reliable methods of electrolysis on the industrial scale were evolved long ago by Nørsk Hydro and other companies). However, several workers, notably Gallezot47 and Dumesic,48,49 have convincingly demonstrated that carbohydrate-based feedstocks can be used to generate H2 by aqueous-phase reforming. This can also be carried out with glycerol, which is a plentifully available by-product, as a result of the increased production of biodiesel (fatty acid methyl esters) that is generated from naturally occurring triglyceride, either by transesterification (with methanol) or by alkali saponification.
Acrolein (CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCHO), the precursor to acrylic acid, can now be prepared straightforwardly from glycerol using a single-site Brønsted acid catalyst that effects dehydration (see Fig. 7).50 Caprolactam, the precursor to nylon 6, which unlike nylon 6,6 is recyclable, can also be readily produced in an environmentally benign way (using O2 and NH3 as reactants), but it still requires cyclohexanone as the starting material (Fig. 8) which has not yet been produced from renewable feedstocks.
CHCHO), the precursor to acrylic acid, can now be prepared straightforwardly from glycerol using a single-site Brønsted acid catalyst that effects dehydration (see Fig. 7).50 Caprolactam, the precursor to nylon 6, which unlike nylon 6,6 is recyclable, can also be readily produced in an environmentally benign way (using O2 and NH3 as reactants), but it still requires cyclohexanone as the starting material (Fig. 8) which has not yet been produced from renewable feedstocks.
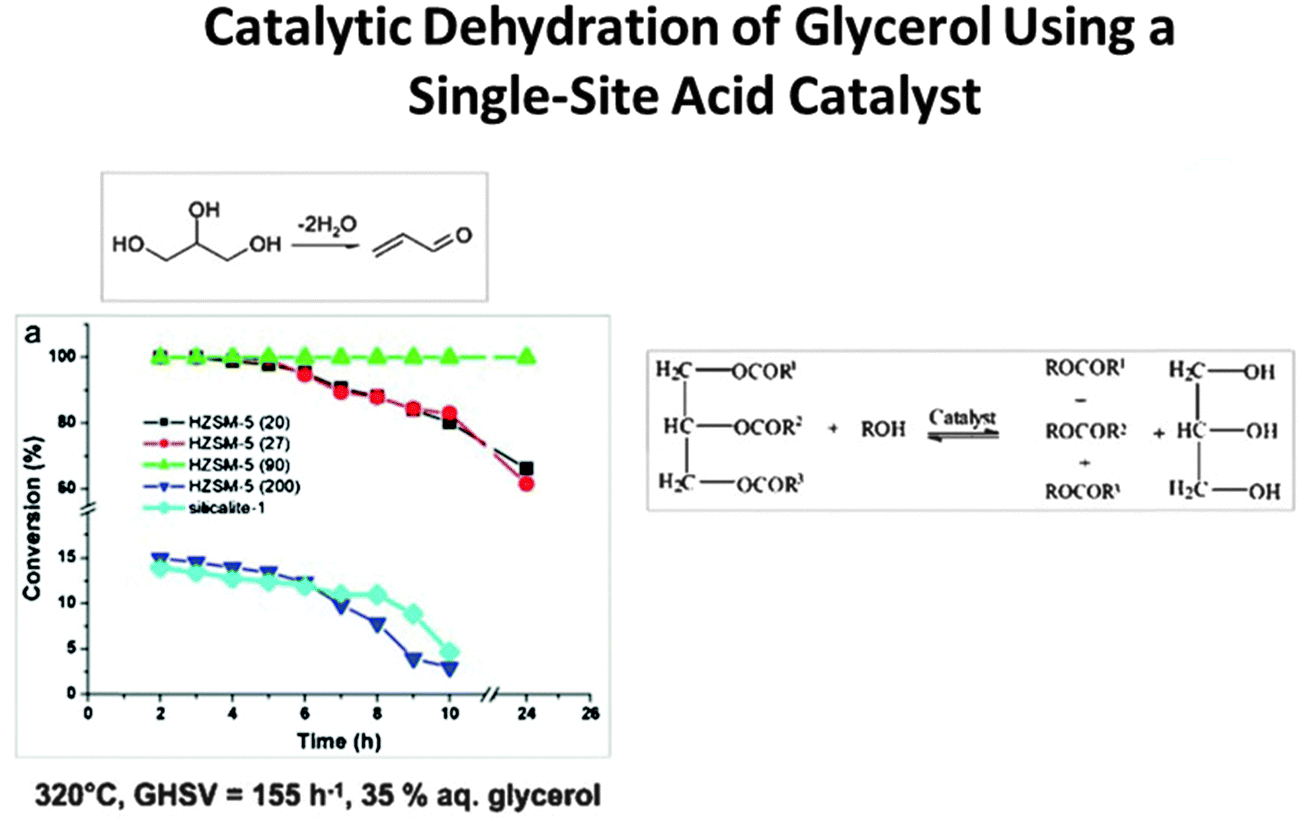 | ||
| Fig. 7 Jia, Schüth and co-workers50 have shown how the single-site H-ZSM-5 catalyst, with appropriate Si/Al ratio, converts glycerol to acrolein with 100% efficiency and possesses long life. | ||
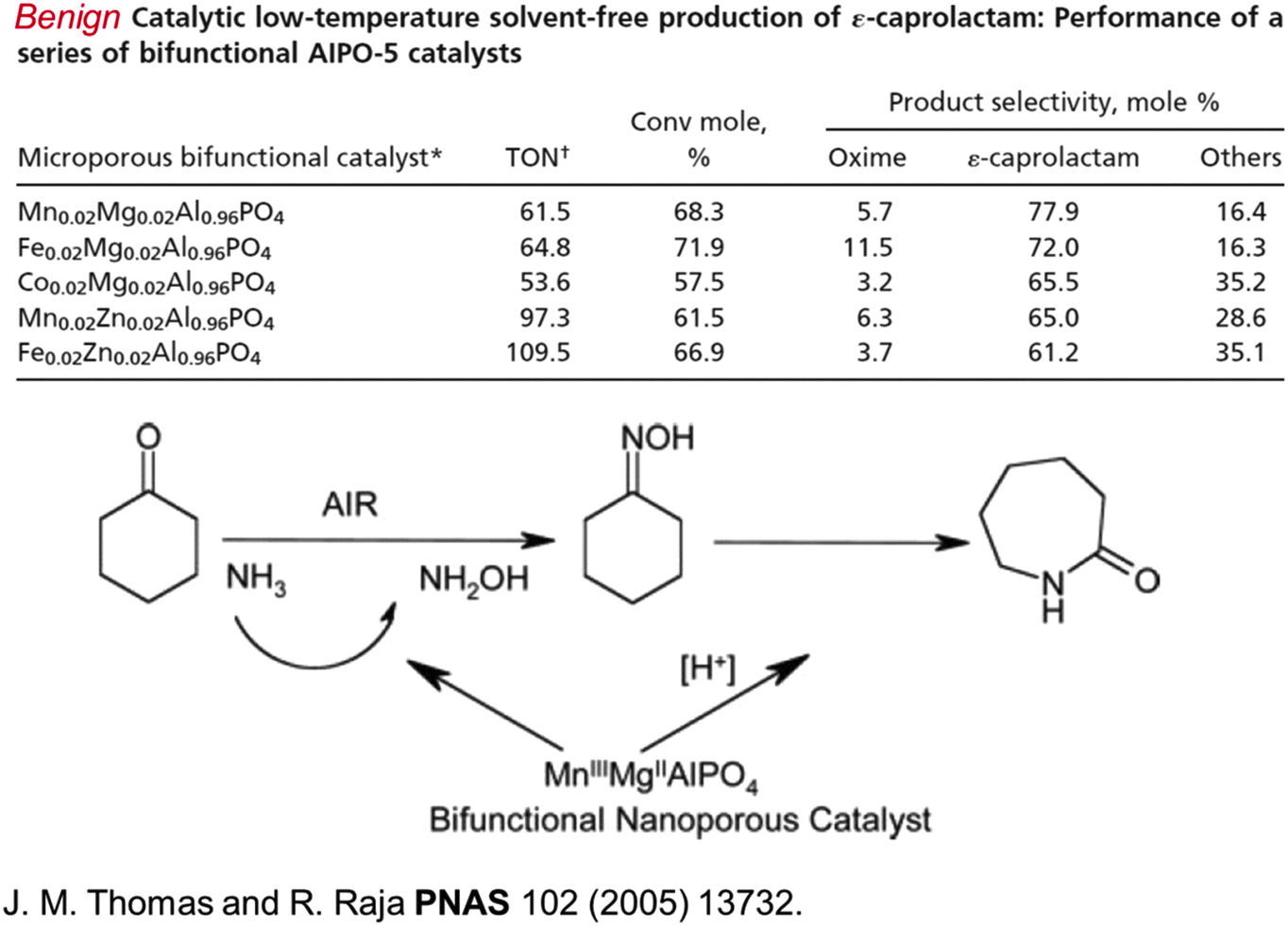 | ||
| Fig. 8 A set of effective nanoporous (see Fig. 18a) catalysts (top part of figure) for the conversion of cyclohexanone, using NH3 and air (or O2) first to its oxime and then to caprolactam, the precursor of nylon 6, as schematized in the bottom part of the figure.51 | ||
This facile production of ε-caprolactam is made possible by the designed, nanoporous, bifunctional aluminophosphate catalyst shown in Fig. 9 (see also Fig. 18), and discussed fully elsewhere.51 The key point about this particular single-site nanoporous catalyst is that it can readily be made bifunctional. When high-oxidation state substituents (like MnIII or CoIII) replace an AlIII (tetrahedral) site, a redox active centre is generated. When a metal ion, such as MgII or ZnII, isomorphously replaces an AlIII ion, a Brønsted acid active centre is generated; a loosely attached proton is formed, and is bound to a structural oxygen atom as an ionic-covalent (polarized covalent) bond.
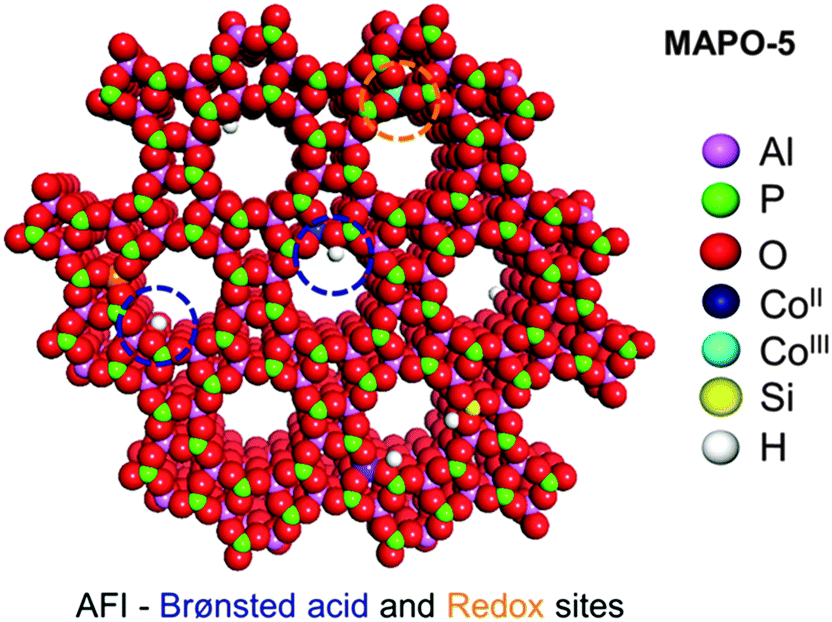 | ||
| Fig. 9 Schematic of a bifunctional single-site heterogeneous catalyst derived from the microporous solid aluminophosphate ALPO-5. The Brønsted acid catalytic centres are well-separated from the redox sites. | ||
Reverting to the discussion in Section 2, many of the polymeric products previously synthesized from non-renewable sources can now be readily prepared in an entirely sustainable manner. The case of polyurethane is a good example: Leitner, Langanke and co-workers52 have shown how CO2 can be used as a feedstock for polyurethane production (see Fig. 10). The Bayer Materials Science (BMS) facility now does this on the scale of a 103 ton year−1 capacity.
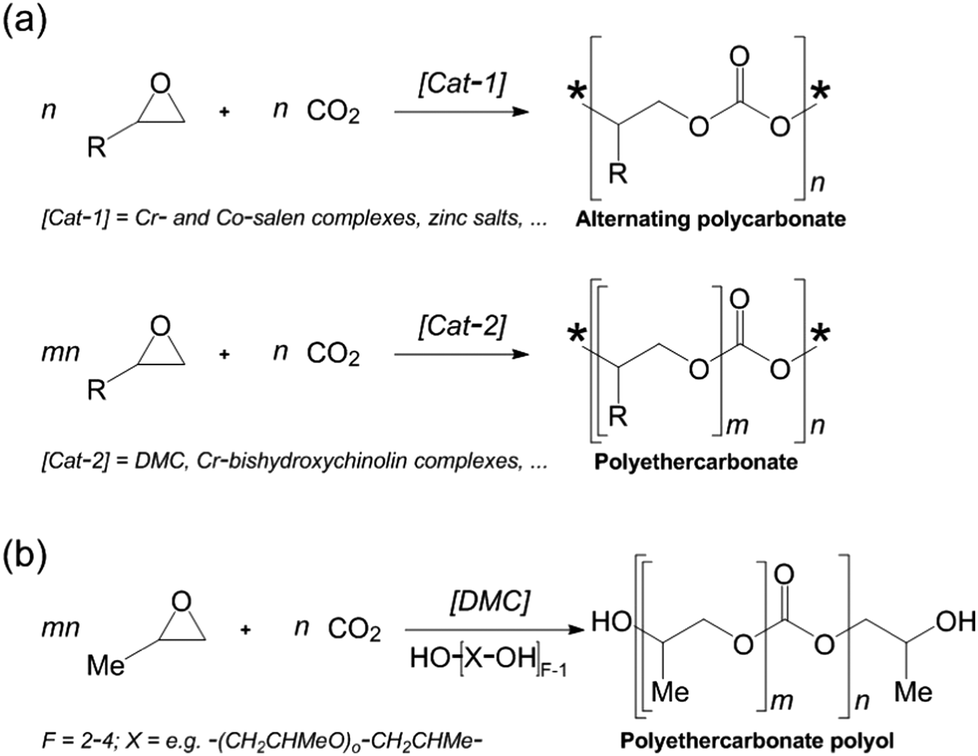 | ||
| Fig. 10 (a) Copolymerization of epoxides and CO2 to alternating polycarbonates (top; * = end group) and polyethercarbonates (bottom). (b) Tailored polyethercarbonate polyols obtained from propylene oxide and CO2 using zinc hexacyanocobaltate (DMC) as a catalyst and a multifunctional alcohol as a starter. (After Langanke et al.52) | ||
So far as the water-gas shift reaction (CO + H2O ⇌ CO2 + H2) is concerned, remarkably powerful so-called “single-atom” catalysts have been developed, as described in Section 4.
4. Inorganic catalysts in the chemical industry. The need for atom efficiency and environmentally benign processes
Fig. 11 summarizes the important role played by catalysis in manufacturing processes. However, this illustration, produced over a decade ago (kindly provided by Gerhard Ertl), does not fairly reflect the growing number of biocatalysts that are now utilized industrially. Although biocatalysts are not yet extensively used, one of the merits of employing enzymes as applied catalysts is that they can be tailored to facilitate cascade processes, in which a given reactant is converted, through multiple steps, to a desired product without the necessity of separating out all the intermediates. Moreover, such is the present, whirlwind pace of advances in chemical biology, epitomized by the work of Keasling53 and Arnold,54 that it is expected that a significant proportion of industrial chemicals will be produced in future using microorganisms by transferring product-specific enzymes, or even whole metabolic pathways, from rare and/or genetically intractable organisms to those that can be readily engineered.55 The data in Fig. 11 are also somewhat biased in favour of heterogeneous catalysts, partly because volume factors loom large, and petrochemical refining and associated processes entail utilization of massive quantities of material.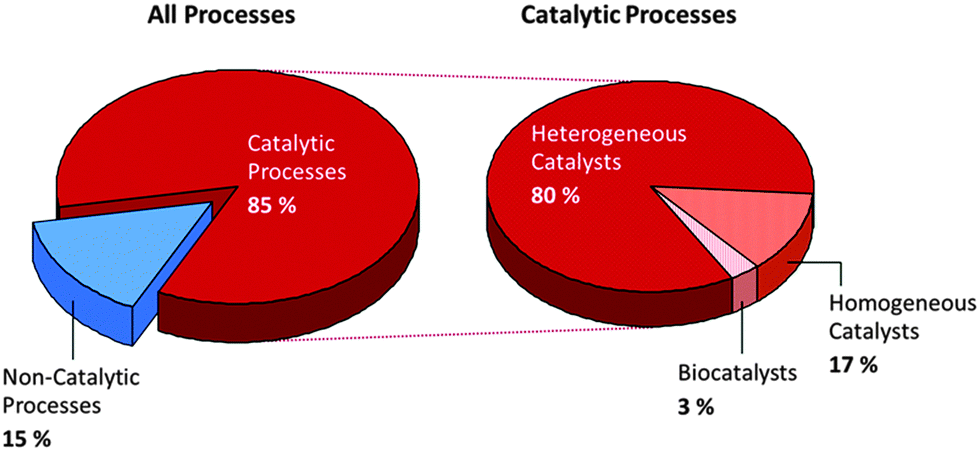 | ||
| Fig. 11 Pie charts showing the percentages of all industrial processes that entail the use of heterogeneous, homogeneous and bio-catalysis. (Source: Winnacker U. Küchler, Chemische Technik, vol. 1, 2004, BASF estimates. By kind permission of BASF). | ||
As pointed out by Leitner,56 there have recently been a number of large-scale plants using homogeneous catalysts, mostly of organometallic origin. Typical examples include Pd compounds for the production of ibuprofen and acetaldehyde (Wacker processes), and Rh compounds for the production of menthol and acetic acid. The latter are used extensively by several companies (BASF, Union Carbide, Exxon, Sasol and Evonik) in hydroformylations.
Nevertheless, one of the supreme and self-evident advantages of heterogeneous catalysts is that they greatly facilitate separation of product from reactants – and are also usually amenable to facile reactivation, as exemplified by the rare-earth-zeolite Y fluidized cracking catalysts.
Insofar as environmental protection is concerned, several informative monographs exist (see Cavani et al.57 and Beller et al.58). And one of us has written extensively on the role of appropriate catalysts to this effect.15,16,31,59
We now proceed to discuss two approaches to the design of atom-efficient catalysts for atom-efficient processes. The first approach which has great promise – single-atom heterogeneous catalysts – is now burgeoning, as described below. The second – single-site heterogeneous catalysts (SSHCs) – has been exploited already for more than a decade, and some industrial processes are operated on its principles. SSHCs have been alluded to already above and are discussed further in Section 4.2.
4.1 Single-atom heterogeneous catalysts
Recent advances in structural characterization have prompted many students of catalysis to argue that the traditional distinctions between heterogeneous and homogeneous catalysts are now becoming increasingly blurred.60 One reason for this state of affairs has arisen because of the results that have emerged from the study of supported metal catalysts by aberration-corrected high-resolution electron microscopy (see Fig. 12).61–65 It has become startlingly apparent that catalytic systems such as nanoclusters or nanoparticles of Au, Pd or Pt on oxides (e.g. TiO2 or MgO) or on elemental carbon, can behave quite differently from one another. Whereas a nanoparticle, of typically 5 nm diameter, contains tens of thousands of atoms, a nanocluster of a pure metal or of a bimetallic entity may contain a very small number of atoms, usually 5 to 15 atoms. | ||
| Fig. 12 Illustrative examples of minute nanoclusters of Au on a titania support (top) and on an activated carbon support (bottom). Sub-nanometre clusters (white circles) of ca. 0.5 nm diameter and containing ca. 10 Au atoms are observed, together with individual Au atoms (black circles). Data from ref. 60. Micrographs kindly provided by P. L. Gai. | ||
Several discussions in the past have pursued the question of whether a single atom attached to a support, or otherwise a constitutional part of the support, may function as an efficient catalyst.66–69 Indeed, Flytzani-Stephanopolous and co-workers70 found no evidence for the participation of nanoparticles of supported Au as active catalysts in the low-temperature water-gas shift reaction. It was deduced by these workers that only single atoms of Au, present as Au–Ox surface species, function as active catalysts. Later work by Flytzani-Stephanopolous and Mavrikakis,71,72 described more fully below, showed how a special kind of single-atom catalyst consisting of an individual Pt atom is stabilized by closely attached Na+ or K+ ions on a SiO2 surface (as depicted72 in Fig. 13).
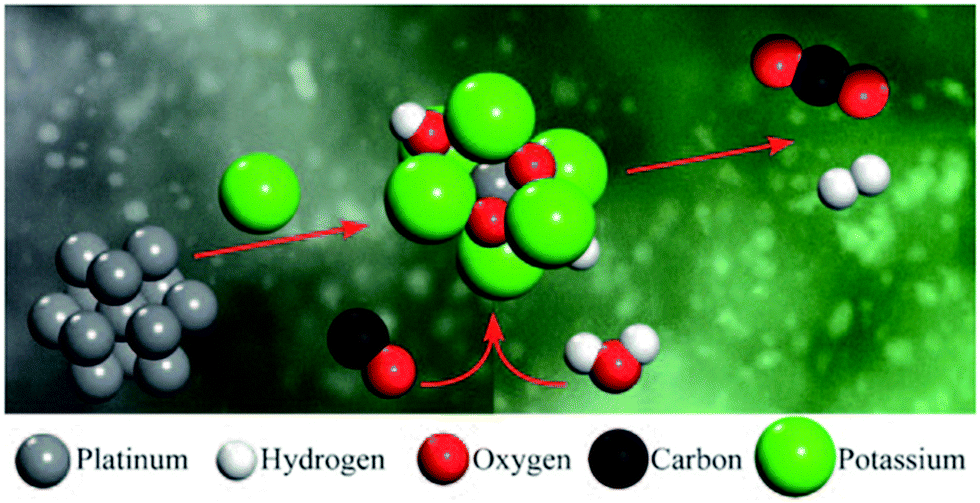 | ||
| Fig. 13 Schematic depiction of the alkali metal-ion-stabilized PtII–O(OH)x species (centre) that catalyses the water-gas shift reaction. (By kind permission of M. Flytzani-Stephanopoulos). | ||
In addition to the work of Flytzani-Stephanopolous and her associates, workers at the Dalian Institute of Chemical Physics73–76 have provided cogent evidence that individual atoms of the precious metals Ir, Pt and Pd are capable of catalysing a number of commercially important reactions. In 2013, they showed73 that single atoms of Ir supported on FeOx exhibit remarkably high performance in catalysing the water gas shift reaction. And in a short review later that year, Yang, Zhang et al.74 and other work by Flytzani-Stephanopolous77,78 reported advances in the preparation, characterization and catalytic performance of single-atom catalysts for a number of oxidations, hydrogenations and the water-gas shift reaction. Also, Wei et al.75 reported on single-atom Pt catalysts (again supported on FeOx) for the chemoselective hydrogenation of nine different functionalized nitroarenes.
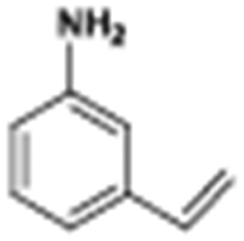
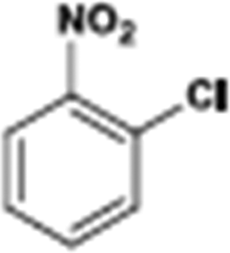
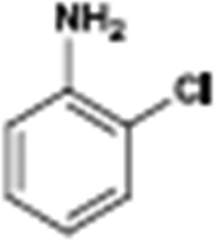
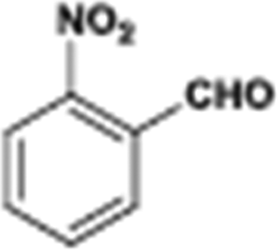
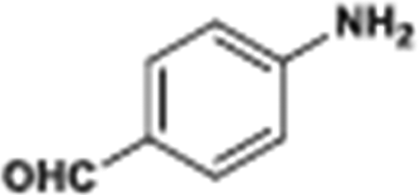
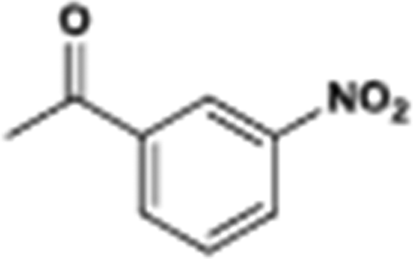
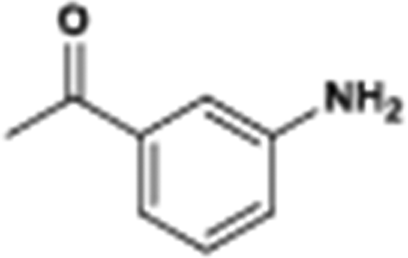
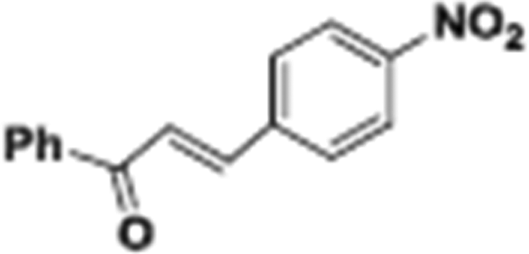
The catalytic hydrogenation of nitroarenes is an environmentally benign technology for the production of anilines that are key intermediates for manufacturing agrochemicals, pharmaceuticals and dyes. These workers point out that most nanoparticle versions of precious metal catalysts suffer from low chemoselectivity when one or more reducible groups is present in a nitroarene molecule. Their single-atom Pt catalysts, however, are highly active, chemoselective and re-usable for the hydrogenation of a variety of substituted nitroarenes. For the hydrogenation of 3-nitrostyrene, for example, the single-atom Pt catalyst of Wei et al.75 yields a turnover frequency of ca. 1500 h−1, which is 20-fold as high as the best result reported in the literature, and has a selectivity to 3-aminostyrene of ca. 99%, which is the highest reported value for a Pt group metal catalyst.79 These authors attribute the exceptional catalytic performance to the presence of positively charged Pt centres and the absence of Pt–Pt bonds. Their reported results are spectacular and superior to those obtained with nanoparticle Pt or Au catalysts (Table 4).
| Entry | Substrate | Product | Time (min) | Conv. (%) | Sel. (%) |
|---|---|---|---|---|---|
| Reaction conditions: T = 40 °C, P = 3 bar, 0.08 wt% Pt/FeOx catalyst, Pt/substrate = 0.08%; 5 ml reaction mixture: 0.5 mmol substrate, toluene as solvent, o-xylene as internal standard.a Pt/substrate = 0.32%, T = 50 °C, p = 6 bar.b Pt/substrate = 0.16%.c Pt/substrate = 0.41%, refer to yields of isolated products. | |||||
| 1 |
|
|
50 | 96.5 | 98.6 |
| 2 |
|
|
60 | 100 | 97.4 |
| 3a |
|
|
44 | 96.9 | 98.0 |
| 4a |
|
|
120 | 90.3 | 97.8 |
| 5 |
|
|
30 | 93.4 | 99.4 |
| 6b |
|
|
122 | 93.7 | 99.3 |
| 7b |
|
|
80 | 99.5 | 92.8 |
| 8 |
|
|
114 | 95.9 | 99.7 |
| 9c |
|
|
10 | 100 | 90 |
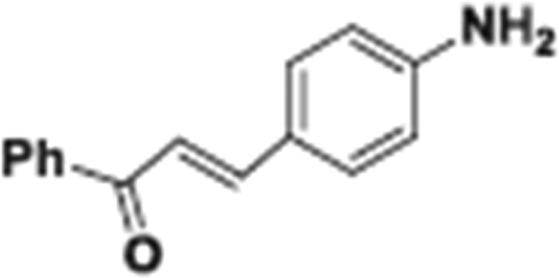
The whole domain of single-atom, precious metal (especially Pt and Au) catalysts has been the subject of several definitive studies by Flytzani-Stephanopoulos and her co-workers.80–82 In the paper by Lucci et al.,78 advantage is taken of a concept introduced earlier by this group (see Kyriakou et al.83) described by them as “single-atom alloys”, in which isolated atoms of Pd on a metallic copper host catalyse carbon deposition. The work of Lucci et al. entailed the design of a new generation of platinum–copper nanoparticle catalysts, at the single-atom limit. And these catalysts selectively hydrogenate 1,3-butadiene to 1-butene (an extremely important industrial reaction, as it is used to eliminate impurities of deleterious butadiene in propylene gas that is to be used in polymerization). Significantly, the isolated Pt atom species in the catalyst allows hydrogen activation and spillover, leading to hydrogenation, but is unable to effect C–C bond scission, which would result in loss of selectivity and catalyst deactivation. A pictorial summary of the mechanistic steps in this selective hydrogenation reaction is given in Fig. 14b.
 | ||
| Fig. 14 (a) STM images showing atomically dispersed Pd atoms in a Cu(111) surface and hydrogen atoms that have dissociated and spilled over onto the Cu surface.83 (A) Pd is alloyed into the Cu(111) surface preferentially above the step edges as evidenced by the rumpled appearance of the upper terrace (scale bar indicates 5 nm). (inset) Atomic resolution of the Pd/Cu alloy on the upper terrace showing individual, isolated Pd atoms in the surface layer appearing as protrusions (scale bar, 2 nm). (B) Schematic showing H2 dissociation and spillover at individual, isolated Pd atom sites in the Cu surface layer. (C) Islands of H atoms imaged after hydrogen uptake appear as depressed regions on the clean Cu(111) lower terrace (scale bar, 5 nm). (inset) High-resolution image of individual hydrogen atoms on Cu(111) (scale bar, 2 nm). Images recorded at 5 K. (b) Mechanistic aspects of the selective hydrogenation of 1,3-butadiene to 1-butene using a platinum–copper nanoparticle catalyst at the single-atom limit.78 (By kind permission of M. Flytzani-Stephanopoulos). | ||
While it is well known that different types of oxide support have different capabilities to anchor a metal, and hence to tailor the catalytic behaviour, it is not always clear whether the support is a mere carrier of the active metal site or whether it participates directly in the overall reaction. Recent work of Flytzani-Stephanopoulos81 sheds light on this question.
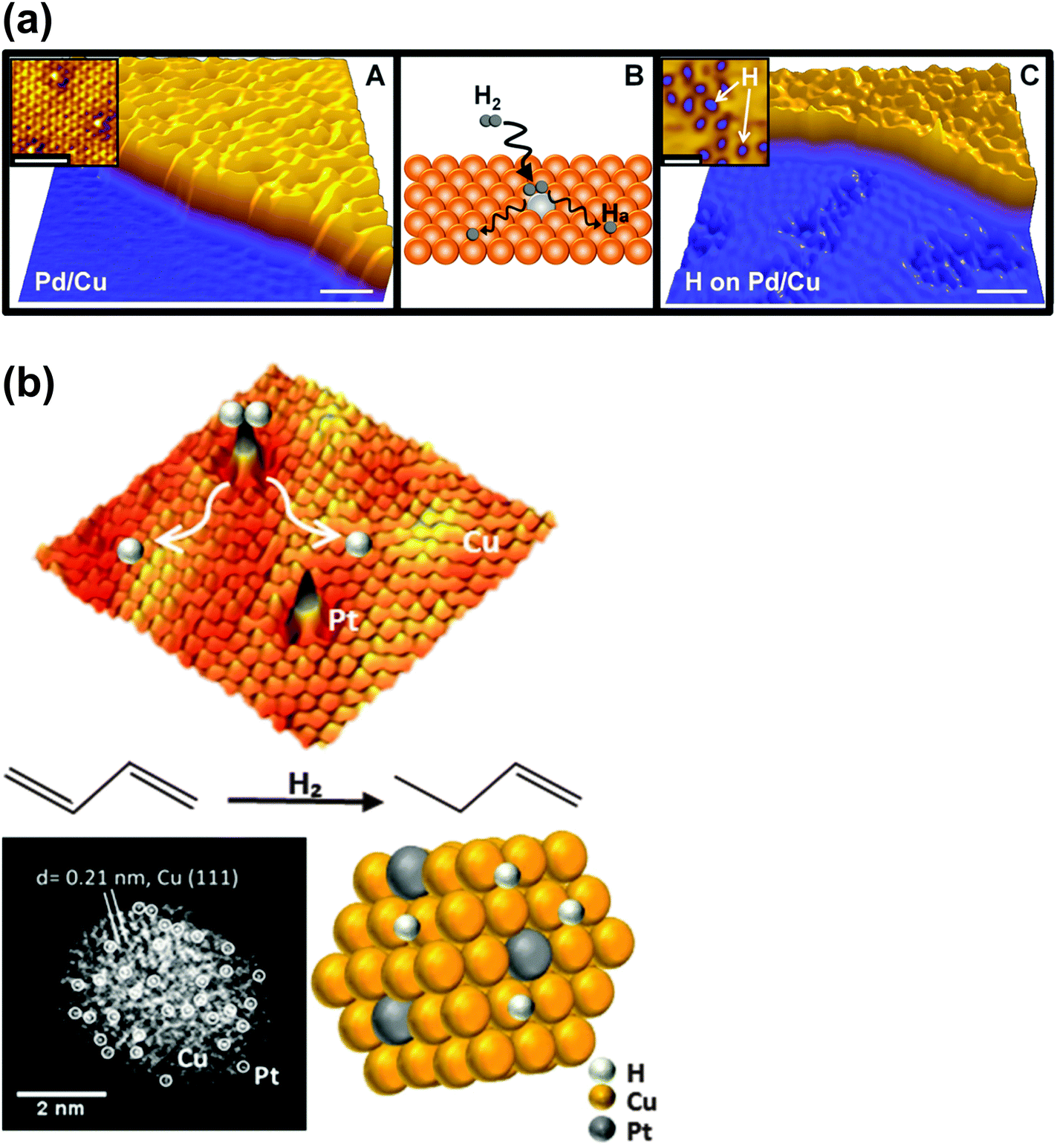
The kind of single-atom site that she envisages (depicted in Fig. 13) is essentially an isolated PtII atom that is covalently bonded to oxygen atoms of the support and shrouded by the alkali metal atoms (either Na or K). Their high-resolution electron micrographs (recorded by scanning transmission electron microscopy) of the single-site PtII–O(OH)x catalytic centres on three distinct supports are shown in Fig. 15. All three catalysts are active in the water-gas shift reaction. These workers determine the oxidation state of the active Pt species by in situ X-ray absorption spectroscopy, leaving little doubt that the active species is in the PtII state.
 | ||
| Fig. 15 Aberration-corrected high-angle annular dark field scanning transmission electron micrographs of atomically-dispersed PtII entities anchored on three different supports. (After Yang et al.81). | ||
An unusual but ingenious experiment utilizing isolated metal atom geometries to improve heterogeneous catalytic hydrogenations has been devised by Sykes and co-workers.83 Starting from the known facts16 that both facile dissociation of H2 and rather weak bonding of reactants at a metal (especially copper) surface are pre-requisites for efficient catalysis, these workers “placed” individual atoms of Pd on a Cu(111) surface. When a molecule of H2 impinges upon a Pd atom, it readily dissociates and “spills over” on to the Cu substratum. Reactant molecules of styrene or acetylene weakly bonded on the latter can, with such so-called “single-atom” alloys, be readily hydrogenated by the mobile H atoms. Fig. 14a represents pictorially the essence of this unusual single-atom catalysis.
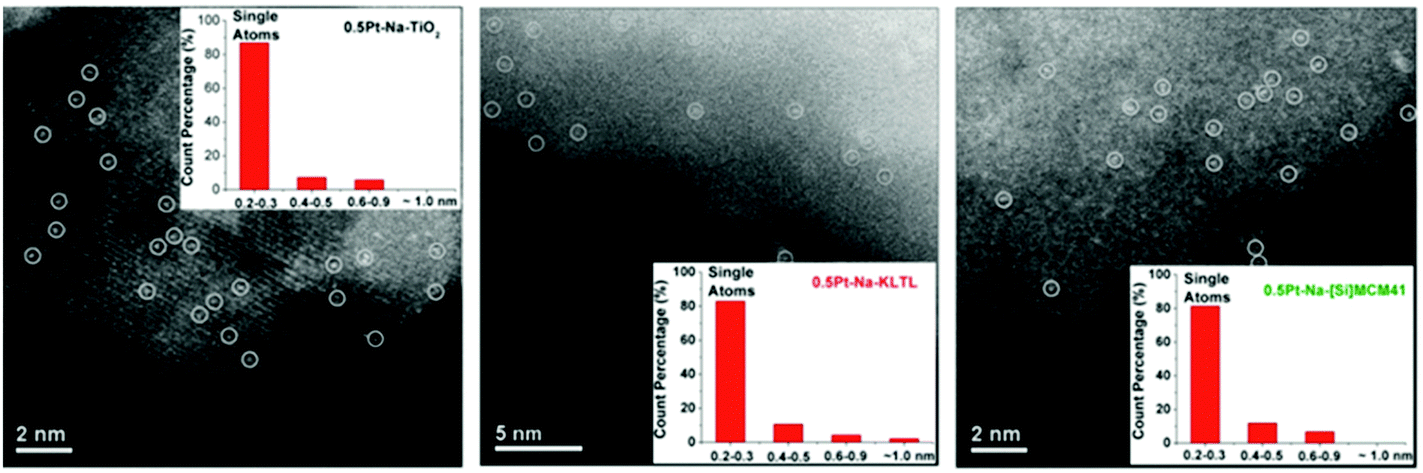
A spectacular example of a stable single-atom Pd catalyst for selective hydrogenation has recently been reported by Lopez and Perez-Ramirez and colleagues.84 Unlike many previous workers in this field, they use a structurally well-defined solid as the support for atomically dispersed Pd, namely nanoporous carbon nitride, C3N4, a material that has been known since the days of Berzelius and Liebig. The great merit of their system (see Fig. 16) is that the individual atoms of Pd are so firmly anchored to the nanoporous walls of the C3N4 that they exhibit no tendency to migrate and coalescence to form nanoclusters or nanoparticles. This catalyst was shown to be active in a three-phase hydrogenation of alkynes in flow mode, and both its activity and selectivity surpassed those of nanoparticulate Pd. The wider significance of the concept of single-atom catalysis is that it augurs well for the more economical (atom-efficient) use of the platinum group metals – Ru, Rh, Pd, Os, Ir and Pt, as well as Au – which are extensively used as catalysts in industries that produce fuels and compounds such as agrochemicals, dyestuffs and pharmaceuticals, and several of them are components of catalytic converters in internal combustion and diesel engines.85
 | ||
| Fig. 16 (a) Schematic of a single-atom Pd catalyst comprising isolated Pd atoms on a solid support of carbon nitride (C3N4; carbon, grey; nitrogen, purple), which acts as a catalyst for hydrogenation reactions. Strong bonds to the nitrogen atoms firmly anchor the Pd atoms in roughly triangular pores in the stacked, two-dimensional layers of the support. There is approximately one, roughly triangular cage per 50 Å2 in each layer, and it is estimated that up to 10% of them are occupied by a single Pd atom. (b) View parallel to the C3N4 plane showing the DFT-optimized position of the Pd atom incorporated in the C3N4 support. (After Vilé et al.84). | ||
4.2 Single-site heterogeneous catalysts (SSHCs)31,86,87
A single-site heterogeneous catalyst does not necessarily have to be made up of a single atom, as in the cases described in Section 4.1. Indeed, the work of Flytzani-Stephanopolous et al.70,78,81,82 shows that the active centre is composed of several other atoms besides the crucial Pt or Au noble metal entities. It has been recognized for well over a decade86 that numerous other single-site catalysts can occur at solid surfaces. In the classic examples of SSHCs,31,86,87 all the single-sites are chemically (and energetically) identical and are spatially well-separated from one another on a high-area support – usually a nanoporous solid.31,87The prime exemplars in this category are zeolites and framework-substituted aluminophosphates (ALPOs), typified by MALPO-5, MALPO-18, MALPO-31 and MALPO-36, where M may be Co, Mn or Fe in one of two oxidation states, usually MII and MIII.86,88 Here, there is little doubt that a single (ionized) atom does, in each case (see below), act as a catalytically active site in such processes as the aerial, selective oxidation of hydrocarbons. A specific example is the oxyfunctionalization of n-hexane to adipic acid with CoIIIALPO-18.89,90
Fig. 17 shows how a reactant (n-hexane) fits snugly inside the chabazitic cage of a CoIIIALPO-18 molecular sieve catalyst. With a high loading (ca. 10 atom percent) of CoIII incorporated into the catalyst in place of AlIII, it follows statistically that a high proportion of the cages in the molecular-sieve catalyst will have two CoIII ions as tenants at sites situated at opposite ends of the cage. Moreover, given the dimensions of n-hexane relative to the cage, these two CoIII active sites would be in close proximity to the two terminal CH3 groups of the n-hexane molecule. The cage and its apertures are large enough to allow free access to the O2 reactant.
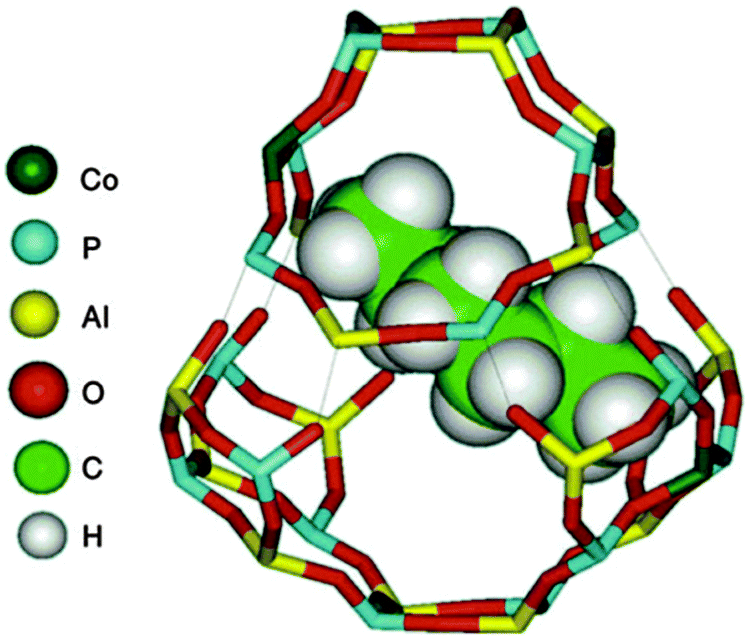 | ||
| Fig. 17 View of the chabazite cage in the ALPO-18 structure, showing two separated CoIII ions in the cage and the energy minimized configuration of an n-hexane molecule. For clarity, the top half of the cage has been separated from the bottom half, and the thin lines indicate the specific atoms in the two separated halves that are linked to each other. The distance between the van der Waals surfaces of the two CoIII ions is ca. 7.6 Å. Figure from ref. 90. | ||
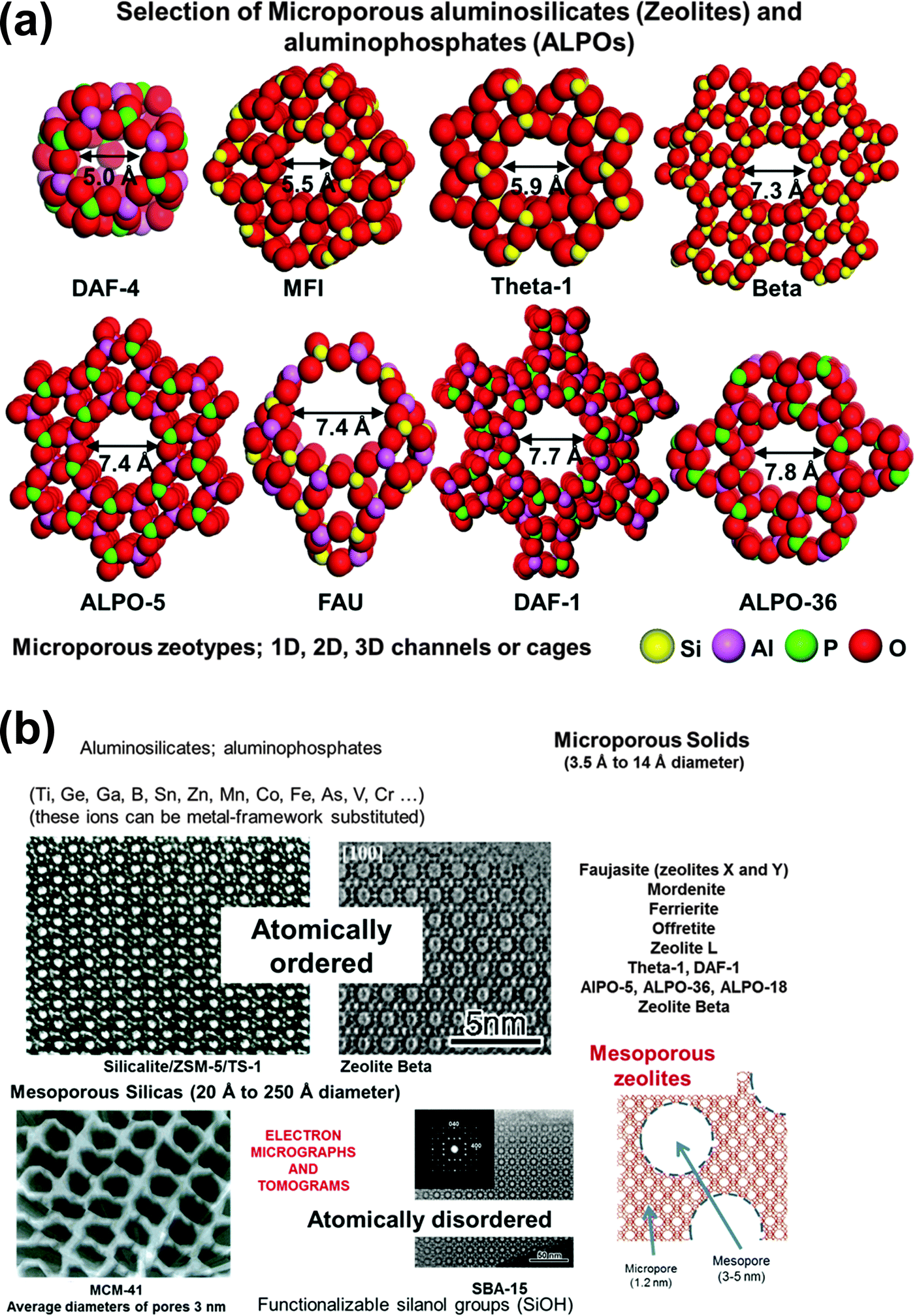 | ||
| Fig. 18 (a) Examples of structural features of microporous zeolites and ALPOs, and (b) a selection of electron micrographs, an electron tomogram and schematic illustration of various kinds of microporous and mesoporous solids used to create SSHCs. | ||
XAFS measurements90 leave little doubt as to the precise ionic states of the catalytically active site: it is CoIII in the framework of the open-structure catalyst. We note in passing that this ionic active site (CoIII) displays multi-functionality in that, in the presence of O2, the terminal –CH3 groups are sequentially converted (as in a cascade reaction) first to –CH2OH, then to –CHO and finally to –COOH. A free-radical reaction is involved in this sequential series of steps.88
Similar arguments pertaining to an ionic active site (in this case FeIII), that also behaves multi-functionally, apply to the FeALPO-31 catalyst that selectively oxidizes cyclohexane to adipic acid in O2.91 In this case, the intermediate, partially oxidized, products are cyclohexyl hydroperoxide, cyclohexanol and cyclohexanone. In all the cases in which MnII/MnIII, CoII/CoIII and FeII/FeIII function as predominantly ionic oxidation catalysts, it has been shown92 that a combination of coordinative unsaturation at the ionic site, and its facility to exhibit redox behaviour, is the essential requirement for its role as the active centre. At the risk of excessive emphasis, the redox sites in the framework-substituted ALPO nanoporous solids, especially CoIII, MnIII and FeIII, are all effective in converting a –CH3 group in the presence of ambient O2 to –CH2OH and on to the aldehyde and the carboxylic acid.
It is interesting to reflect that one of the most successful ever SSHCs is the so-called TS-1 (titanosilicalite) selective oxidation catalyst, developed in the laboratories of Enichem in Italy and discovered by accident.93
In the examples of SSHCs cited above, the key point to note is that, although a single ion (e.g. CoIII, MnIII or TiIV) is the principal locus of the active site, the attached oxygen atoms of the nanoporous matrices, in which the (accessible) active sites are embedded, also participate in the catalytic turnover. The paper by Gomez-Hortiguela et al.92 gives full details of such participation.
Thanks to advances in the synthesis of novel solids, almost all inorganic oxides can now be readily prepared as nanoporous solids possessing very large surface areas – up to and often beyond 1000 m2 g−1 – and with well-defined pore structures of controllable diameters in the range ca. 3.5 to 500 Å (see Fig. 18a). These solids can be fashioned into heterogeneous catalysts with a single type of spatially well-separated, highly selective, active site. These sites can be readily introduced either during synthesis or by post-synthesis modification.31 Moreover, SSHCs of this kind – just like homogeneous catalysts – are readily amenable to delicate design and to in situ characterization by a wide variety of spectroscopic methods and other techniques.31,86,87
Micrographic and related aspects of a selection of the inorganic nanoporous oxides that are used to create SSHCs are shown in Fig. 18b. Recent advances in preparing mesoporous and hierarchical zeolitic structures have added further scope to the range of desired SSHCs that may now be designed. For example, the mesoporous zeolites (shown at the bottom right of Fig. 18) prepared by Garcia-Martinez,94 have substantially improved the performance of catalytic-cracking catalysts (such as LaIII–Y zeolite), in that greater conversion to desired hydrocarbons and less deleterious coking are achieved. (See also Milina et al.95).
Metal–organic frameworks (MOFs), of which there are now estimated to be over 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 different structural types,96 are currently finding increasing use as SSHCs (see Fig. 19). Many MOFs are relatively stable thermally, and are readily amenable to post-synthesis modification so as to enable single-site catalytic centres to be introduced (see Section 4.2.2 below in the context of enantioselective catalysts). Nowadays, MOFs are being used increasingly in contexts that impinge directly on questions pertaining to energy and environmental science. Here we give a few examples.
000 different structural types,96 are currently finding increasing use as SSHCs (see Fig. 19). Many MOFs are relatively stable thermally, and are readily amenable to post-synthesis modification so as to enable single-site catalytic centres to be introduced (see Section 4.2.2 below in the context of enantioselective catalysts). Nowadays, MOFs are being used increasingly in contexts that impinge directly on questions pertaining to energy and environmental science. Here we give a few examples.
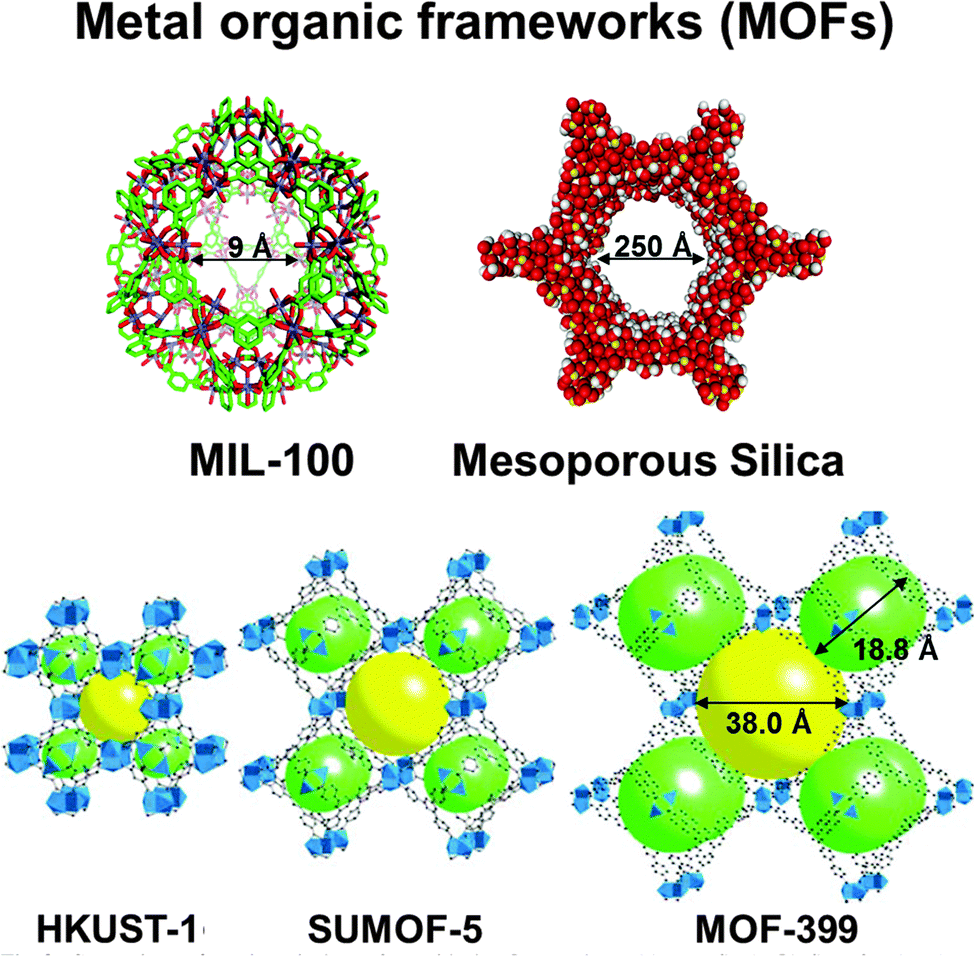 | ||
| Fig. 19 A selection of MOFs (and a mesoporous silica). HKUST ≡ Hong Kong University of Science and Technology; SUMOF ≡ Stockholm University Metal–organic framework; MIL ≡ Institut Lavoisier. | ||
Long and co-workers98,99 designed a MOF – Cr3(1,3,5-benzenetricarboxylate)2 [designated Cr3(BTC)2] – which is highly selective (and reversible) in binding O2 from O2/N2 mixtures. This work promises to replace the industrial (energy intensive) cryogenic distillation process.
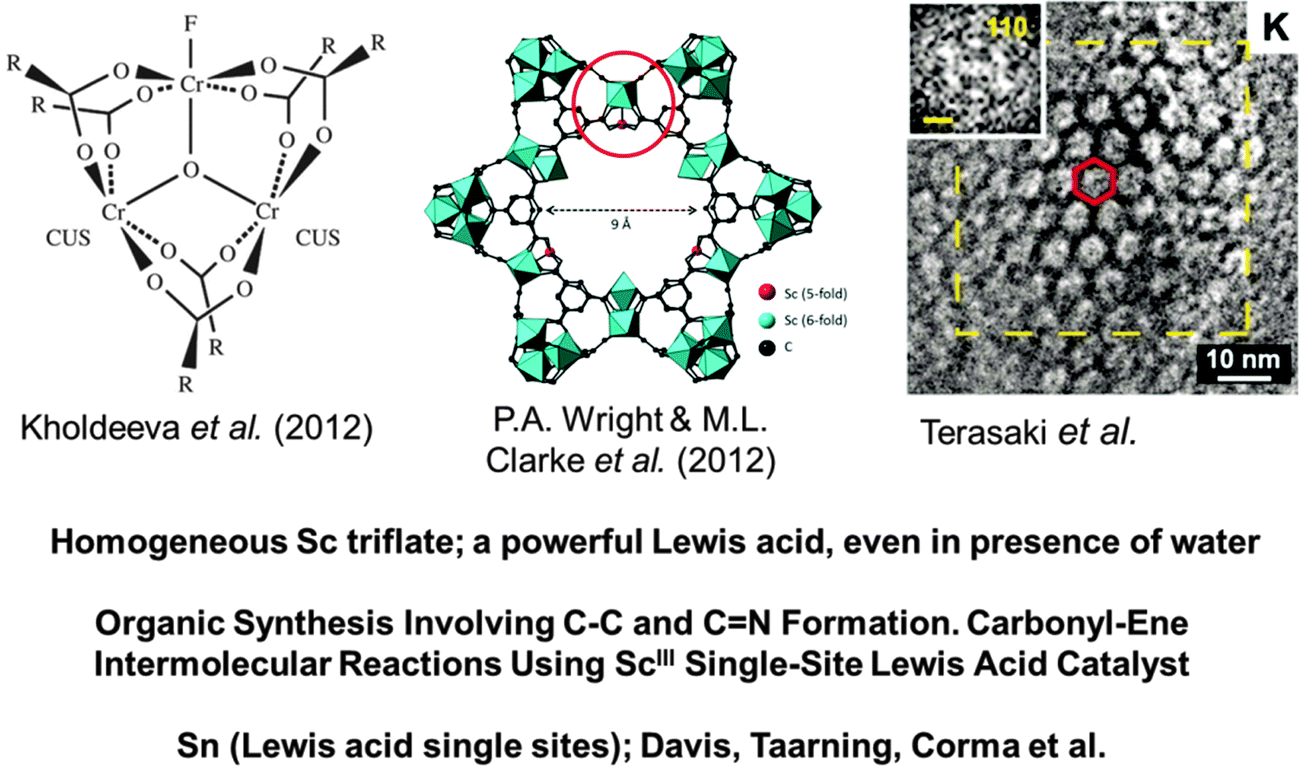 | ||
| Fig. 20 Metal–organic-frameworks, shown schematically here (left and centre), and in an electron micrograph (right), are ideal solids for the creation of SSHCs. In the centre, a coordinatively unsaturated ScIII ion is the single isolated site, which is an ideal Lewis acid catalyst for the formation of a variety of C–C and CN bonds in organic synthetic chemistry. (After Clarke, Wright and co-workers68). The high-resolution electron micrograph on the right, taken by O. Terasaki, illustrates the openness of the MOF structure and the consequential ease with which reactants can reach the active sites in this nanoporous structure. | ||
In the work of Wright, Clarke et al.,68 the performance of a scandium ion-substituted MOF has been investigated in several Lewis acid catalysed reactions that are of great importance in organic synthesis. The circled region of the MOF structure shown in the top of the centre drawing of Fig. 20 constitutes a coordinatively unsaturated ScIII ion that is part of the organic framework. (At this juncture it is relevant to recall that, in homogeneous solution, scandium triflate is a very powerful Lewis acid catalyst, the discovery of which106 led to major advances in the application of Lewis acid catalysis in organic synthesis, even in the presence of water). Wright, Clarke and co-workers68 have convincingly demonstrated that the ScIII single-site catalytic centre in their MIL-type MOF solids are exceptionally good for the following reactions:
• intermolecular carbonyl-ene reactions of nucleophilic alkenes and electron-poor aldehydes;
• Friedel–Crafts type Michael addition between electron-rich heterocycles and electron-deficient alkenes; and
• ketamine and aldimine formation.
In other words, this ScIII-form of the MIL-type MOF is an effective means of catalysing the formation of C–C and CN bonds, which is of vital importance in synthetic organic chemistry. This ScIII substituted MIL-type MOF is a paradigm of SSHCs.
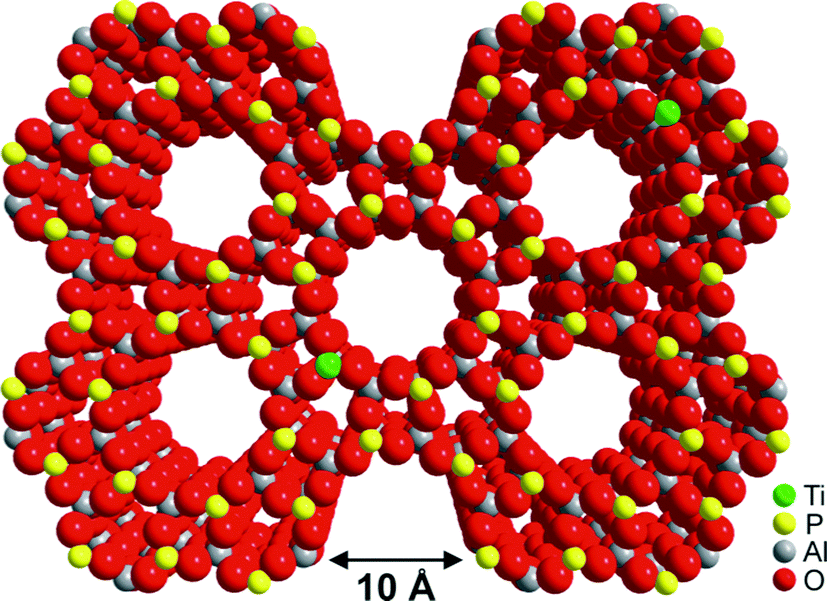 | ||
| Fig. 21 The structure of TAPO-5, which is based on the ALPO-5 framework but with TiIV ions (represented in green) substituted into the framework. | ||
We carried out107 a laboratory-scale set of cascade reactions using TAPO-5 to catalyse the conversion of cyclohexene to adipic acid by H2O2. In this system, several distinct processes are all catalysed at the same active site (a TiIV ion in tetrahedral coordination): epoxide production followed by the formation of cis- and trans-diols, a keto-alcohol and so forth, ultimately producing adipic acid (see Fig. 22). From the results of parallel 13C and 1H NMR studies and GC-MS analysis conducted during the course of the catalytic conversion of cyclohexene (1) to adipic acid (2) in the presence of the catalyst TAPO-5, and from additional analytical measurements (starting from some of the identified intermediates 3 and 4), we concluded that the mechanistic pathway from 1 to 2 is as shown in Fig. 22.
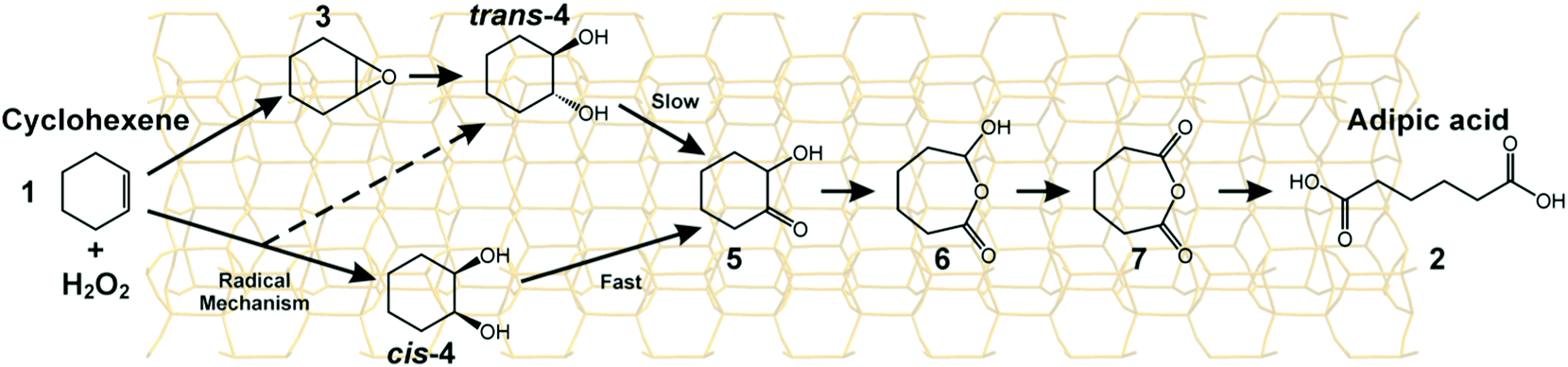 | ||
| Fig. 22 The mechanism of the conversion of cyclohexene to adipic acid using H2O2 and a TAPO-5 catalyst (the structure of the catalyst is depicted in the background).107 | ||
The key point here is that the active site in TAPO-5, just as the catalytically active site in Rubisco (see Section 2.2), functions for several different reactions. This synthesis of adipic acid is solvent-free, but the yield of the acid is not as high as in other methods of benign synthesis in which either n-hexane90 or c-hexane91,108 are converted to adipic acid using O2 as the oxidant and using other transition metal framework-substituted ALPOs as the shape-selective single-site catalysts.
4.3 Other variants and applications of single-site heterogeneous catalysts
As described fully elsewhere31 several other examples of SSHCs are of use both in effecting environmentally benign processes and also enantioselective ones. Here we focus on the former. | ||
| Fig. 23 A series of illustrations taken from an electron tomographic study117 of mesoporous silica in which nanoclusters (shown in red) of Pt10Ru2 are distributed within the mesopores. Each of the slices (1, 2 and 3) is 10 nm thick. (Thomas and Midgley, unpublished work). | ||
In describing the exceptional performance of these nanocluster catalysts – for example, they are capable of effectively hydrogenating muconic acid (from biological sources) to adipic acid (i.e. converting compounds 1 and 3 in Fig. 24 to compound 2) – for the purposes of quantitatively expressing their catalytic activity, each of the clusters is regarded as a single site. If it actually transpires that only one or two of the atoms in the cluster are the locus of the catalytic activity, it simply means that the observed catalytic performance is even greater than originally calculated. Thus, a nanocluster of n atoms will be more active by a factor of n or n/2, respectively.
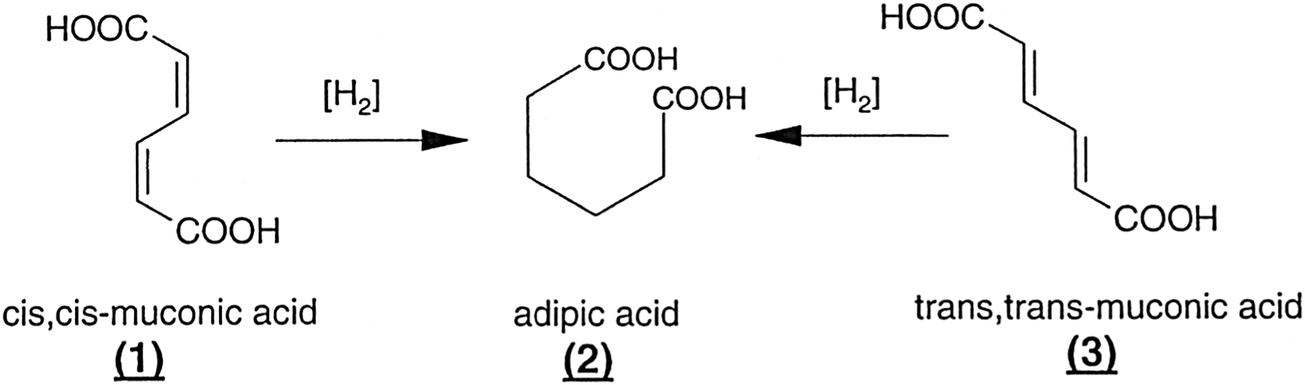 | ||
| Fig. 24 Examples of transformations effected by nanocluster single-site catalysts, as discussed in the text.118 | ||
4.4 Summarizing examples of single-site heterogeneous catalysts (SSHCs)
So far as single-atom heterogeneous catalysts are concerned, their discovery augurs well for the future efficient use of platinum group metals (especially Pt, Pd, Rh and Ir). If, as seems possible from what we have described in Section 4.1, they can be routinely prepared in a thermally stable form, they can be used, in association with supports composed of earth-abundant elements such as C, N, O, Mg, Al, Si, P and Fe, as relatively inexpensive but very powerful catalysts for numerous industrial production and energy-releasing processes. In this regard, nanoporous C3N4 (see Fig. 16) is an ideal support since its structure renders surface diffusion of the single atom improbable.The SSHCs described in Sections 4.2 and 4.3 (and in Table 5 below) are, in the main, also composed of earth-abundant elements. We know that SSHCs are especially useful in the field of biomass conversion, especially for the sugars that can be readily manufactured from biomass through metabolic engineering.86,119 (According to Keasling,53 metabolic engineering is a procedure that rivals, and is potentially capable of eclipsing, synthetic organic chemistry.) For example, sugars derived from biomass can be transformed with one or more species of microorganism to form, not just C3 or C4 alcohols (like iso-butanol, for instance), but also C5, C6 and higher alcohols. These alcohols can be readily dehydrated catalytically, using a SSHC such as H+-ZSM-5 of the appropriate Si/Al ratio (compare Fig. 7, above).
| 1 | Ammoximation of cyclohexanone (active sites are MnIII or CoIII ions) in microporous aluminophosphates (ALPOs) |
| 2* | Beckmann rearrangement of cyclohexanone oxime to ε-caprolactam (ALPOs) |
| 3* | Selective isomerization of (a) butenes to 2-methylpropene and *(b) subsequently its reaction with methanol to yield methyl tertiary butyl ether (MTBE), a gasoline additive, using Theta-1 catalyst |
| 4* | Anchored organometallic complexes (on mesoporous silica) for a range of hydrogenations, chiral allylic aminations and synthesis of pharmaceutically active products |
| 5* | Synthesis of gasoline (and H2) from propane and butane using a Ga framework-substituted zeolite |
| 6 | Terminal selective oxidation (in O2) of dodecane and several other n-alkanes at MnIII or CoIII substitutional sites in ALPOs: (i) n-hexane to n-hexanoic acid, and (ii) n-hexane to adipic acid |
| 7 | Aerobic, shape-selective oxidation of cyclohexane to adipic acid |
| 8 | Bayer–Villiger conversions of cyclic ketones to lactones using MnIII in ALPO or SnIV single sites in silicalite |
| 9 | Conversion of bioethanol to ethene at low temperatures using DAF-4 (a chabazite related microporous acidic catalyst containing CoII active centres) |
| 10 | Conversion of methanol to ethene and propene using MgII or MnII framework-substituted ALPO-18 microporous solids |
| 11 | Formation of niacin (vitamin B3) from nicotine or 3-picoline in the presence of MnIIIALPO-5 catalyst |
| 12 | Benign selective oxidation of toluene to either benzyl alcohol or benzaldehyde or benzoic acid (depending on the reaction conditions) |
| 13 | Epoxidation of renewable fatty acid methyl esters (from natural oils) and terpenes (using TiIV or NbV-centred active sites in mesoporous silica) |
| 14 | Nanocluster bimetallic catalysts, anchored in mesoporous silica, for a wide range of selective hydrogenations and syntheses of key organic compounds (e.g. adipic acid from corn products) |
In turn, the resulting olefins can again, using appropriate SSHCs, be oligomerized or polymerized. And subsequent hydrogenation of these molecules can lead to the clean production of various kinds of transportation fuels that are sulphur-free.
Davis120 has recently shown how several Brønsted acid and Lewis acid SSHCs offer scope for the synthesis of commodity polymers from biomass. The dehydration of lactic acid, for example, opens up new routes to acrylic acid and acrylonitrile, and also to another novel way of generating (from renewable sources) polyethylene furanoate, echoing some of the issues that we discussed in Section 3.
Recently, Davis has also advanced121 the use of tandem catalysis with the aim of converting biomass-derived hexoses and pentoses to industrially important platform chemicals (such as C2, C3 and C4 α-hydroxy carboxylic acids). Specifically, by combining known catalysts (e.g. molybdenum oxide or molybdate species) for retro-aldol reactions with solid Lewis acid materials (e.g. the tin-containing zeotypes Sn-MFI and Sn-Beta) that are known to catalyse 1,2-intramolecular hydride shifts, he has demonstrated that ketohexoses can be converted to lactic acid and alkyl lactates at significantly lower temperature (ca. 100 °C) than the hitherto best reported catalysts (which operate above ca. 160 °C). The advantages of operating at such reduced temperatures include significantly lower catalyst deactivation.
There are many other novel examples of SSHCs of a kind not hitherto elaborated in this review. Thus, the work of Marks and co-workers122 describes single-site d0 heterogeneous arene hydrogenation catalysts, derived from the precatalyst (η5-C5H5)2ZrR2 (R = H, CH3) and adsorbed on Brønsted superacidic sulphated alumina. (It is relevant to note that the well-known Phillips catalyst, CrOx, which has been used over the last half century to produce 50 different types of polyethylene and is the catalyst nowadays used to generate 40% to 50% of all high-density polyethylene,123 is a classic example of a SSHC – see ref. 124). Table 5 enumerates examples, developed mainly in the laboratory of one of us (JMT), that illustrate the industrial and laboratory-scale viability of many different kinds of SSHCs. This Table shows that the concept of SSHCs, as described previously,16,31,86,114 leads to a strategy for the design of many new catalysts of practical importance.
5. Envoi
This perspective article has focused almost exclusively on thermally activated catalytic processes that are of vital importance in the chemical industry, not only in the context of producing new materials, but also in regard to energy releasing conversions. Although we have made many references to renewable sources of energy, comparatively little has been said about the field of solar energy and its relevance to the generation of solar fuels. We have, however, given an analysis of how CO2 may be used as a feedstock, that has led to a significant advance in the solar-assisted algal production of ethanol. It is of interest to note that, very recently,125 engineers have been engaged in the establishment of zero – or even surplus – energy buildings as part of the goal to achieve sustainable architecture (it is estimated that some 40% of all fossil fuels are utilized in heating homes and other buildings). An apartment block in Hamburg in Germany has been built that uses microalgae placed within the façade to generate heat and biomass. We have also hinted (Section 2.2) that, one day, an appropriately genetically modified (Rubisco-like) carboxylase may be able to process anthropogenic CO2 and convert it to useful products.Members of the Solar Fuel Network (SFN) and other individuals and organizations are dedicated to the photocatalytic production of useful chemicals such as CH4, CH3OH and HCOOH by the photoreduction of CO2. A comprehensive review (up to 2012) of the use of various kinds of Ti-containing substances has been given by Dhakshinamoorthy et al.3 They conclude that the current average productivity value of the best TiO2-based photocatalysts is about 100 μmol g−1 h−1 of catalyst with sunlight. They also emphasize the need to increase this productivity by orders of magnitude, a fact also emphasized in a short review by one of us in 2014.126 In the main, the solar fuels community has concentrated most of its efforts on improving the efficiency of the hydrogen evolution reaction (HER), by employing solar light to drive a photoelectrochemical cell typified by the set-up: TiO2–Ti–pn+Si as photocathode. Striking advances have been made recently, as described by Chorkendorff and co-workers,10,127,128 in substituting new materials, such as MoS2 and Ni2P, each of which may well prove nearly as effective as Pt as the “best” HER catalyst. These workers also examined128 the viability of using ultra-low loadings of Pt in photoelectrochemical H2 evolution (with the TiO2–Ti–pn+Si photocathode described above), and estimated that, if 30% of the world's current annual production of Pt could be used in such H2 evolution catalysts (with a loading of 100 ng cm−2 and a current density of 10 mA cm−2), 1 TW of H2 production could be achieved.
At the recent SFN Symposium in London (July 2015), Gray and others urged those working on the generation of solar fuels to concentrate more on using CO2, rather than H2O, as the material for the production of useful fuels. In this connection, the recent work of Ozin and co-workers in Toronto merits attention. They use the non-stoichiometric In2O3−x(OH)y material for the photoconversion of CO2 to CO – see Fig. 25.129 As pointed out by Ozin,130 an attractive feature of the photocatalytic conversion of CO2 to CH4 is that, if successful on a large scale, the current practice of blending H2 with natural gas can be replaced by the much safer and utilitarian blending of sunlight-derived CH4 with the natural gas grid. As mentioned in Section 2, this strategy, if and when successful, would ultimately stabilize the amount of CO2 in the Earth's atmosphere (CO2 → CH4 → CO2 → CH4, etc.).
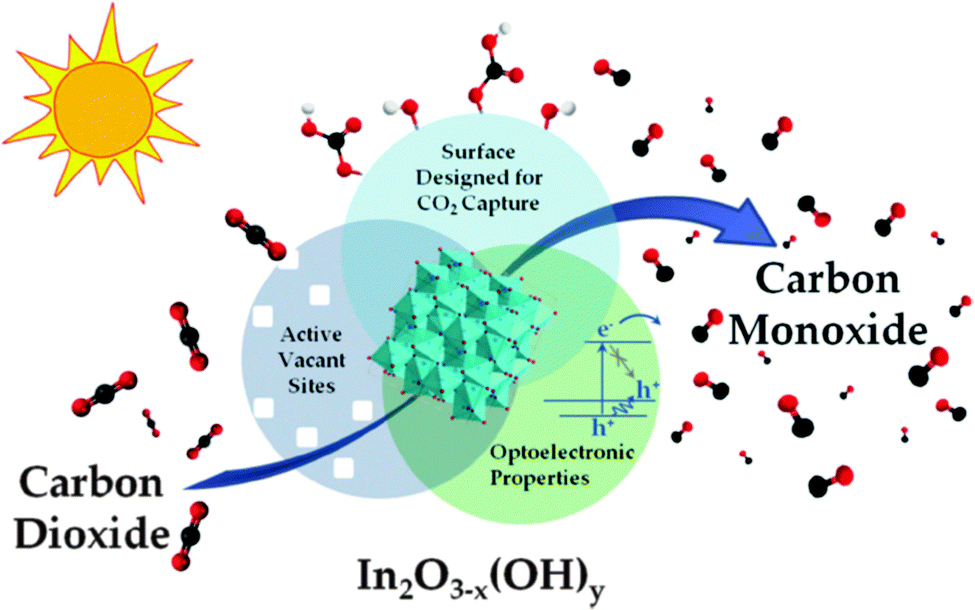 | ||
| Fig. 25 Schematic illustration of key materials attributes of In2O3−x(OH)y nanocrystals that give rise to their activity towards photocatalytic reduction of CO2 to CO in an environment containing H2. (After Hoch et al.129). | ||
In another direction, aimed at artificial photosynthesis, Bell has emphasized the advantages of producing alcohols by photo/electrochemical conversion of CO2 and water, motivated by the advantages of such products in terms of high energy density and high market value per unit energy input. Nevertheless, a number of major challenges must be overcome, including high membrane fuel-crossover losses, low product selectivity and high cost of product separation. Very recently, however, he has proposed131 a viable scheme for direct synthesis of almost pure ethanol by artificial photosynthesis, which is associated with minimal product crossover (phase separation of the ethanol, which is produced in a saturated salt electrolyte, is readily achieved using a microemulsion). It is argued that the annual rate of production of ethanol using this artificial photosynthesis system under reasonable operating conditions would correspond to 7% of the industrial ethanol capacity of California. A detailed analysis of the thermodynamics of such systems for solar-driven electrochemical conversion of CO2 to transportation fuels, and an assessment of their realistic efficiency limits, has been reported.132
In none of the examples of new or improved catalysts mentioned above have we made reference to the importance of Life Cycle Analysis (LCA). We surmise that, in the future, LCA will be mandatory in assessing the merits of a new catalyst to look, not only at its chemical performance, but also at its impact on the environment such as its carbon footprint. An early example of the application of LCA is contained in the work of Luo et al.133 on greenhouse gas emissions for an ethanol production process based on blue-green algae, as described in Section 2.1 above. Very recently, Edwards and co-workers134 have used the so-called Catalyst Selectivity Index (CSI) as a metric to assess the impact of catalyst efficiency enhancement upon energy and CO2 footprints, and we anticipate that the concept of CSI will also assume an increasingly significant role in the future. It is also relevant here to emphasize the importance, from both fundamental and applied perspectives, of fully optimizing catalytic processes, both via detailed thermodynamic analysis (see, for example, the work of Xiao and co-workers135) and via the application of multi-technique studies in the development of optimized catalyst materials (see, for example, the recent work of Edwards and co-workers136). However, increasingly, such optimization must also take into consideration the implications with regard to environmental issues, of the type facilitated by LCA and CSI.
The general picture constructed in the mind of non-specialists looking in at the present, confusing scene in renewable energy is a rather enigmatic one. Whilst, on the one hand, there are some encouraging signs that the human race will cope satisfactorily with the energy needs of the future, by utilizing currently available know-how, from wind, water and sunshine – see Jacobson et al.41 and Herron et al.137 – there are numerous detailed, current reports in which it is implied that, even as late as 2030, the reliance that humans will be placing on non-renewable sources of energy, such as coal, oil and natural gas, will still be a high percentage of all possible, utilizable energy. Reference to the energy outlooks published recently by Exxon Mobil, by BP and by Shell, reveals that over 80% of all transport fuel in 2030 will, it is estimated, be derived from oil. Yet, many prominent experts and members of the public, as well as the 2015 UN Climate Change Conference in Paris (COP-21), are calling for an ultimate ban on the use of fossil fuels. As stated earlier, however, real prospects exist for processing non-renewable feedstocks such as natural gas in a cleaner and more efficient fashion. But, if the UN target of holding the increase in Earth's temperature to less than 2 °C is to be achieved, more drastic action may be required.138 Stabilizing the total amount of CO2 in the atmosphere (by burning CH4 generated by photoreduction of anthropogenic CO2) may not be sufficient, and to reduce the total amount of CO2 will require a “negative carbon” policy. Not only may there be a need to abandon the use of fossil fuels, but strict political action involving imposition of carbon prices may need to be implemented.
In 2009, former U.S. Vice-President Al Gore called for the progressive abandonment of fossil fuel sources. In early August 2015, President Obama announced his Clean Power Plan, an audacious political step to wean the USA off coal. In 2014, the USA's consumption of coal was 453 million metric tons of oil equivalent, in comparison to those of the UK, Russia and Germany of 29, 85 and 77 million metric tons of oil equivalent, respectively. For China, however, the usage was 1962 million metric tons of oil equivalent.139 In line with these aims and aspirations, it is gratifying to see the significant progress recently made using concentrated solar furnaces. Soon there will be solar plants generating 200 MW to 300 MW of power.140 It has recently been reported141 that the world's largest concentrated power plant, powered by the Saharan sun, is in the process of completion outside the Moroccan city of Quarzazate.
Clearly, world agreement is needed (exemplified by the agreement of over 180 countries achieved at the 2015 Paris UN Climate Change Conference) if, indeed, coal (and other fossil fuels) are to be abandoned as sources of energy and materials. As described in Section 2.4 above, China's highly successful MTO and DMTO plants – with a total capacity of 15 M ton year−1 of ethylene and propylene – all use coal to generate the syngas for the formation of the methanol feedstock. Expert judges surmise that, by 2030, China may well have in operation some 200 such coal-based MTO and DMTO plants, unless political action is taken to switch to other sources of carbon, such as biomass.142,143
As we have described in Section 2.2 above, there are several promising options already available, or on the horizon, for the massive conversion of anthropogenic CO2, and one can only hope that these, and others yet to be discovered, will hold sway. Reducing the amount of atmospheric CO2, while also increasing the production of energy, are the key drivers of future action. And catalysts, new and old, will figure eminently in this endeavour.
Acknowledgements
We thank the Kohn Foundation for supporting the work of JMT and EPSRC for supporting the work of KDMH. We also appreciate helpful discussions with M. Atkins, A. T. Bell, A. Brown, R. R. Chance, L. Di Marco, M. Flytzani-Stephanopoulos, M. Kanan and G. A. Ozin. We thank Andrew Williams and Yating Zhou for help in preparing some of the figures.References
- A. Brown, Upstream International Director, Royal Dutch Shell plc. Remarks made at a lecture in the University of Cambridge, 29 October 2015.
- This view is also held by other multinational oil companies, and is implied in several of their energy outlooks – see BP Energy Outlook (2014) and “Technology Roadmap”, International Energy Agency, Dechema (2013).
- A. Dhakshinamoorthy, S. Navalon, C. Corma and H. Garcia, Energy Environ. Sci., 2012, 5, 9217–9233 CAS.
- M. E. Boot-Handford, J. C. Abanades, E. J. Anthony, M. J. Blunt, S. Brandani, N. MacDowell, J. R. Fernández, M.-C. Ferrari, R. Gross, J. P. Hallett, R. S. Haszeldine, P. Heptonstall, A. Lyngfelt, Z. Makuch, E. Mangano, R. T. J. Porter, M. Pourkashanian, G. T. Rochelle, N. Shah, J. G. Yao and P. S. Fennell, Energy Environ. Sci., 2014, 7, 130–189 CAS.
- C. T. Campbell, Nature, 2004, 432, 282–283 CrossRef CAS PubMed.
- K. Reuter, D. Frenkel and M. Scheffler, Phys. Rev. Lett., 2004, 93, 116105 CrossRef PubMed.
- J. K. Nørskov, T. Bligaard and J. Kleis, Science, 2009, 324, 1655–1656 CrossRef PubMed.
- J. K. Nørskov, T. Bligaard, J. Rossmeisl and C. H. Christensen, Nat. Chem., 2009, 1, 37–46 CrossRef PubMed.
- B. Hinnemann, P. G. Moses, J. Bonde, K. P. Jørgensen, J. H. Nielsen, S. Horch, I. Chorkendorff and J. K. Nørskov, J. Am. Chem. Soc., 2005, 127, 5308–5309 CrossRef CAS PubMed.
- P. C. K. Vesborg, B. Seger and I. Chorkendorff, J. Phys. Chem. Lett., 2015, 6, 951–957 CrossRef CAS PubMed.
- R. A. van Santen and M. Neurock, Molecular Heterogeneous Catalysis: A Conceptual and Computational Approach, Wiley-VCH, 2006 Search PubMed.
- Z. Jiang, T. Xiao, V. L. Kuznetsov and P. P. Edwards, Philos. Trans. R. Soc., A, 2010, 368, 3343–3364 CrossRef CAS PubMed.
- G. Centi, E. A. Quadrelli and S. Perathoner, Energy Environ. Sci., 2013, 6, 1711–1731 CAS.
- M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709–1742 CrossRef CAS PubMed.
- J. M. Thomas, ChemSusChem, 2014, 7, 1802–1831 CrossRef PubMed.
- J. M. Thomas and W. J. Thomas, Principles and Practice of Heterogeneous Catalysis, Wiley-VCH, Weinheim, 2nd edn, 2015, ch. 9 Search PubMed.
- C. Ampelli, S. Perathoner and G. Centi, Philos. Trans. R. Soc., A, 2015, 373, 20140177 CrossRef PubMed.
- G. Centi and S. Perathoner, Catal. Today, 2009, 148, 191–205 CrossRef CAS.
- E. A. Quadrelli, G. Centi, J.-L. Duplan and S. Perathoner, ChemSusChem, 2011, 4, 1194–1215 CrossRef CAS PubMed.
- G. Centi and S. Perathoner, in The Chemical Element: Chemistry's Contribution to our Global Future, ed. J. Garcia-Martinez and E. Serrano Torregrosa, Wiley-VCH, Weinheim, 2011, pp. 269–307 Search PubMed.
- M. Aresta and A. Dibenedetto, Dalton Trans., 2007, 2975–2992 RSC.
- M. Mikkelsen, M. Jorgensen and F. C. Krebs, Energy Environ. Sci., 2010, 3, 43–81 CAS.
- M. Aresta, CO2 as a Chemical Feedstock, Wiley-VCH, Weinheim, 2010 Search PubMed.
- R. Schlögl, Angew. Chem., Int. Ed., 2015, 54, 4436–4439 CrossRef PubMed.
- H. Michel, Angew. Chem., Int. Ed., 2012, 51, 2516–2518 CrossRef CAS PubMed.
- R. R. Chance, private communication to JMT, June 2015.
- L. S. von Borzyskowski, R. G. Rosenthal and T. J. Erb, J. Biotechnol., 2013, 168, 243–251 CrossRef PubMed.
- M. Kanan, private communication to JMT, 25 June 2015 and 8 July 2015.
- M.-D. Deng and J. R. Coleman, Appl. Environ. Microbiol., 1999, 65, 523–528 CAS; See also: R. P. Woods, J. R. Coleman and M. D. Deng, US Pat., 6,306,639, 2001 Search PubMed; R. P. Woods, J. R. Coleman and M. D. Deng, US Pat., 6,699,696, 2004 Search PubMed.
- R. P. Lively, P. Sharma, B. A. McCool, J. Beaudry-Losique, D. Luo, V. M. Thomas, M. Realff and R. R. Chance, Biofuels, Bioprod. Biorefin., 2015, 9, 72–81 CrossRef CAS.
- J. M. Thomas, Design and Application of Single-Site Heterogeneous Catalysts: Contributions to Green Chemistry, Clean Technology and Sustainability, Imperial College Press, London & Singapore, 2010 Search PubMed.
- M. E. Potter, M. E. Cholerton, J. Kezina, R. Bounds, M. Carravetta, M. Manzoli, E. Gianotti, M. Lefenfeld and R. Raja, ACS Catal., 2014, 4, 4161–4169 CrossRef CAS.
- T. J. Erb, Appl. Environ. Microbiol., 2011, 77, 8466–8477 CrossRef CAS PubMed.
- T. J. Erb, B. S. Evans, K. Cho, B. P. Warlick, J. Sriram, B. M. Wood, H. J. Imker, J. V. Sweedler, F. R. Tabita and J. A. Gerlt, Nature Chem. Biol., 2012, 8, 926–932 CrossRef CAS PubMed . See also S. Everts, C&E News, 6 July 2015, p. 13.
- A. J. J. E. Eerhart, A. P. C. Faaij and M. K. Patel, Energy Environ. Sci., 2012, 5, 6407–6422 CAS.
- H.-J. Freund, private communication to JMT, 11 August 2015.
- G. A. Olah, A. Goeppert and G. K. S. Prakash, J. Org. Chem., 2009, 74, 487–498 CrossRef CAS PubMed.
- G. A. Olah, A. Goeppert and G. K. S. Prakash, Beyond Oil and Gas: The Methanol Economy, Wiley-VCH, 2nd edn, 2011 Search PubMed.
- G. A. Olah, Angew. Chem., Int. Ed., 2013, 52, 104–107 CrossRef CAS PubMed.
- M. Z. Jacobson and M. A. Delucchi, Sci. Am., 2009, 301, 58–65 CrossRef CAS PubMed.
- M. Z. Jacobson, M. A. Delucchi, G. Bazouin, Z. A. F. Bauer, C. C. Heavey, E. Fisher, S. B. Morris, D. J. Y. Piekutowski, T. A. Vencill and T. W. Yeskoo, Energy Environ. Sci., 2015, 8, 2093–2117 CAS.
- M. Atkins (of BP), presentation at Satellite Meeting on Catalysis, Royal Society Discussion, June 2015.
- P. Tian, Y. Wei, M. Ye and Z. Liu, ACS Catal., 2015, 5, 1922–1938 CrossRef CAS.
- Energy Technology Perspective produced by the International Energy Agency, Dechema and the International Council of Chemical Associations, 2013.
- Remarks by D. G. Nocera at the Solar Fuels Network meeting in the Royal Society, London, 8 July 2015.
- K. Otsuka, Y. Shigeta and S. Takenaka, Int. J. Hydrogen Energy, 2002, 27, 11–18 CrossRef CAS.
- P. Gallezot, Top. Catal., 2010, 53, 1209–1213 CrossRef CAS.
- R. D. Cortright, R. R. Davda and J. A. Dumesic, Nature, 2002, 418, 964–967 CrossRef CAS PubMed.
- J. N. Chheda, G. W. Huber and J. A. Dumesic, Angew. Chem., Int. Ed., 2007, 46, 7164–7183 CrossRef CAS PubMed.
- C.-J. Jia, Y. Liu, W. Schmidt, A.-H. Lu and F. Schüth, J. Catal., 2010, 269, 71–79 CrossRef CAS.
- J. M. Thomas and R. Raja, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 13732–13736 CrossRef CAS PubMed.
- J. Langanke, A. Wolf, J. Hofmann, K. Böhm, M. A. Subhani, T. E. Müller, W. Leitner and C. Gürtler, Green Chem., 2014, 16, 1865–1870 RSC.
- J. D. Keasling, Nature, 2012, 492, 188 CrossRef CAS PubMed.
- S. Bastian, X. Liu, J. T. Meyerowitz, C. D. Snow, M. M. Y. Chen and F. H. Arnold, Metab. Eng., 2011, 13, 345–352 CrossRef CAS PubMed.
- F. Malpartida and D. A. Hopwood, Nature, 1984, 309, 462–464 CrossRef CAS PubMed.
- W. Leitner, private communication to JMT, 22 July 2015.
- Sustainable Industrial Chemistry, ed. F. Cavani, G. Centi, S. Perathoner and F. Trifiro, Wiley-VCH, Weinheim, 2009 Search PubMed.
- Catalysis from Principles to Application, ed. M. Beller, A. Renken and R. A. van Santen, Wiley-VCH, Weinheim, 2012 Search PubMed.
- Materials Design and Chemistry of Environmentally Acceptable Catalysts, ed. J. M. Thomas and P. L. Gai, Ann. Rev. Mater. Sci., 2005, vol. 35, pp. 1–465. See especially the articles by Schüth, Hutchings, Iwasawa and Marchese Search PubMed.
- J. M. Thomas and R. Raja, Top. Catal., 2010, 53, 848–858 CrossRef CAS.
- E. P. Ward, I. Arslan, P. A. Midgley, A. Bleloch and J. M. Thomas, Chem. Commun., 2005, 5805–5807 RSC.
- A. A. Herzing, C. J. Kiely, A. F. Carley, P. Landon and G. J. Hutchings, Science, 2008, 321, 1331–1335 CrossRef CAS PubMed.
- K. T. Rim, D. Eom, L. Liu, E. Stolyarova, J. M. Raitano, S. W. Chan, M. Flytzani-Stephanopolous and G. W. Flynn, J. Phys. Chem. C, 2009, 113, 10198–10205 CAS.
- J. H. Kwak, J. Z. Hu, D. Mei, C. W. Yi, D. H. Kim, C. H. F. Peden, L. F. Allard and J. Szanyi, Science, 2009, 325, 1670–1673 CrossRef CAS PubMed.
- J. M. Thomas, R. Raja, P. L. Gai, H. Grönbeck and J. C. Hernandez-Garrido, ChemCatChem, 2010, 2, 402–406 CrossRef CAS.
- J. M. Thomas, Z. Saghi and P. L. Gai, Top. Catal., 2011, 54, 588–594 CrossRef CAS.
- S. L. Wegener, T. J. Marks and P. C. Stair, Acc. Chem. Res., 2012, 45, 206–214 CrossRef CAS PubMed.
- L. Mitchell, B. Gonzalez-Santiago, J. P. S. Mowat, M. E. Gunn, P. Williamson, N. Acerbi, M. L. Clarke and P. A. Wright, Catal. Sci. Technol., 2013, 3, 606–617 CAS.
- A. Bruix, Y. Lykhach, I. Matolinova, A. Neitzel, T. Skala, N. Tsud, M. Vorokhta, V. Stetsovych, K. Sevcikova, J. Myslivecek, R. Fiala, M. Vaclavu, K. C. Prince, S. Bruyere, V. Potin, F. Illas, V. Matolin, J. Libuda and K. M. Neyman, Angew. Chem., Int. Ed., 2014, 53, 10525–10530 CrossRef CAS PubMed.
- Q. Fu, H. Saltsburg and M. Flytzani-Stephanopolous, Science, 2003, 301, 935–938 CrossRef CAS PubMed.
- J. M. Thomas, Angew. Chem., Int. Ed., 2011, 50, 49–50 CrossRef CAS PubMed.
- Y. Zhai, D. Pierre, R. Si, W. Deng, P. Ferrin, A. U. Nilekar, G. Peng, J. A. Herron, D. C. Bell, H. Saltsburg, M. Mavrikakis and M. Flytzani-Stephanopoulos, Science, 2010, 329, 1633–1636 CrossRef CAS PubMed.
- J. Lin, A. Wang, B. Qiao, X. Liu, X. Yang, X. Wang, J. Liang, J. Li, J. Liu and T. Zhang, J. Am. Chem. Soc., 2013, 135, 15314–15317 CrossRef CAS PubMed.
- X. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef CAS PubMed.
- H. Wei, X. Liu, A. Wang, L. Zhang, B. Qiao, X. Yang, Y. Huang, S. Miao, J. Liu and T. Zhang, Nat. Commun., 2014, 5, 5634 CrossRef CAS PubMed.
- G. X. Pei, X. Y. Liu, A. Wang, A. F. Lee, M. A. Isaacs, L. Li, X. Pan, X. Yang, X. Wang, Z. Tai, K. Wilson and T. Zhang, ACS Catal., 2015, 5, 3717–3725 CrossRef CAS.
- M. Flytzani-Stephanopoulos, Acc. Chem. Res., 2014, 47, 783–792 CrossRef CAS PubMed.
- F. R. Lucci, J. Liu, M. D. Marcinkowski, M. Yang, L. F. Allard, M. Flytzani-Stephanopoulos and E. C. H. Sykes, Nat. Commun., 2015, 6, 8550 CrossRef CAS PubMed and references therein.
- P. Serna, M. Boronat and A. Corma, Top. Catal., 2011, 54, 439–446 CrossRef CAS.
- Q. Fu, W. Deng, H. Saltsburg and M. Flytzani-Stephanopoulos, Appl. Catal., B, 2005, 56, 57–68 CrossRef CAS.
- M. Yang, J. Liu, S. Lee, B. Zugic, J. Huang, L. F. Allard and M. Flytzani-Stephanopoulos, J. Am. Chem. Soc., 2015, 137, 3470–3473 CrossRef CAS PubMed and references therein.
- M. Yang, S. Liu, Y. Wang, J. A. Herron, Y. Xu, L. F. Allard, S. Lee, J. Huang, M. Mavrikakis and M. Flytzani-Stephanopoulos, Science, 2014, 346, 1498–1501 CrossRef CAS PubMed.
- G. Kyriakou, M. B. Boucher, A. D. Jewell, E. A. Lewis, T. J. Lawton, A. E. Baber, H. L. Tierney, M. Flytzani-Stephanopoulos and E. C. H Sykes, Science, 2012, 335, 1209–1212 CrossRef CAS PubMed.
- G. Vilé, D. Albani, M. Nachtegaal, Z. Chen, D. Dontsova, M. Antonietti, N. López and J. Perez-Ramírez, Angew. Chem., Int. Ed., 2015, 54, 11265–11269 CrossRef PubMed.
- J. M. Thomas, Nature, 2015, 525, 325–326 CrossRef CAS PubMed.
- J. M. Thomas, R. Raja and D. W. Lewis, Angew. Chem., Int. Ed., 2005, 44, 6456–6482 CrossRef CAS PubMed.
- J. M. Thomas, Phys. Chem. Chem. Phys., 2014, 16, 7647–7661 RSC.
- J. M. Thomas, R. Raja, G. Sankar and R. G. Bell, Nature, 1999, 398, 227–230 CrossRef CAS.
- R. Raja, G. Sankar and J. M. Thomas, Angew. Chem., Int. Ed., 2000, 39, 2313–2316 CrossRef CAS.
- J. M. Thomas, R. Raja, G. Sankar and R. G. Bell, Acc. Chem. Res., 2001, 34, 191–200 CrossRef CAS PubMed.
- M. Dugal, G. Sankar, R. Raja and J. M. Thomas, Angew. Chem., Int. Ed., 2000, 39, 2310–2313 CrossRef CAS.
- L. Gomez-Hortiguela, F. Cora, G. Sankar, C. M. Zicovich-Wilson and C. R. A. Catlow, Chem. – Eur. J., 2010, 16, 13638–13645 CrossRef CAS PubMed.
- G. Bellussi, A. Carati, M. G. Clerici, G. Maddinelli and R. Millini, J. Catal., 1992, 133, 220–230 CrossRef CAS.
- N. Linares, A. M. Silvestre-Albero, E. Serrano, J. Silvestre-Albero and J. Garcia-Martinez, Chem. Soc. Rev., 2014, 43, 7681–7717 RSC.
- M. Milina, S. Mitchell, P. Crivelli, D. Cooke and J. Perez-Ramirez, Nat. Commun., 2014, 5, 3922 Search PubMed.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 123044 CrossRef PubMed.
- M. Kanniche, R. Gros-Bonnivard, P. Jaud, J. Valle-Marcos, J.-M. Amann and C. Bouallou, Appl. Therm. Eng., 2010, 30, 53–62 CrossRef CAS.
- L. J. Murray, M. Dinca, J. Yano, S. Chavan, S. Bordiga, C. M. Brown and J. R. Long, J. Am. Chem. Soc., 2010, 132, 7856–7857 CrossRef CAS PubMed.
- E. D. Bloch, L. J. Murray, W. L. Queen, S. Chavan, S. N. Maximoff, J. P. Bigi, R. Krishna, V. K. Peterson, F. Grandjean, G. J. Long, B. Smit, S. Bordiga, C. M. Brown and J. R. Long, J. Am. Chem. Soc., 2011, 133, 14814–14822 CrossRef CAS PubMed.
- K. D. M. Harris and J. M. Thomas, ChemCatChem, 2009, 1, 223–231 CrossRef CAS.
- J. S. Seo, D. Whang, H. Lee, S. I. Jun, J. Oh, Y. J. Jeon and K. Kim, Nature, 2000, 404, 982–986 CrossRef CAS PubMed.
- L. Ma, C. Abney and W. Lin, Chem. Soc. Rev., 2009, 38, 1248–1256 RSC.
- Z.-X. Xu, Y.-X. Tan, H.-R. Fu, J. Liu and J. Zhang, Inorg. Chem., 2014, 53, 12199–12204 CrossRef CAS PubMed.
- K. Mo, Y. Yang and Y. Cui, J. Am. Chem. Soc., 2014, 136, 1746–1749 CrossRef CAS PubMed.
- N. V. Maksimchuk, O. V. Zalomaeva, I. Y. Skobelev, K. A. Kovalenko, V. P. Fedin and O. A. Kholdeeva, Proc. R. Soc. A, 2012, 468, 2017–2034 CrossRef CAS , and references therein.
- D. A. Evans and J. Wu, J. Am. Chem. Soc., 2005, 127, 8006–8007 CrossRef CAS PubMed.
- S.-O. Lee, R. Raja, K. D. M. Harris, J. M. Thomas, B. F. G. Johnson and G. Sankar, Angew. Chem., Int. Ed., 2003, 42, 1520–1523 CrossRef CAS PubMed.
- J. M. Thomas, R. Raja and B. F. G. Johnson, Chem. – Eur. J., 2001, 7, 2973–2978 CrossRef CAS.
- J. H. Sinfelt, Bimetallic Catalysts, Exxon Publication, 1980 Search PubMed.
- M. Haruta, Faraday Discuss., 2011, 152, 11–32 RSC.
- M. Valden, X. Lai and D. W. Goodman, Science, 1998, 281, 1647–1650 CrossRef CAS PubMed.
- M. D. Hughes, Y.-J. Xu, P. Jenkins, P. McMorn, P. Landon, D. I. Enache, A. F. Carley, G. A. Attard, G. J. Hutchings, F. King, E. H. Stitt, P. Johnston, K. Griffin and C. J. Kiely, Nature, 2005, 437, 1132–1135 CrossRef CAS PubMed.
- J. M. Thomas, B. F. G. Johnson, R. Raja, G. Sankar and P. A. Midgley, Acc. Chem. Res., 2003, 36, 20–30 CrossRef CAS PubMed.
- J. M. Thomas, J. Chem. Phys., 2008, 128, 182502 CrossRef PubMed.
- E. C. Tyo and S. Vajda, Nat. Nanotechnol., 2015, 10, 577–588 CrossRef CAS PubMed , and references therein.
- J. M. Thomas, R. Raja, B. F. G. Johnson, S. Hermans, M. D. Jones and T. Khimyak, Ind. Eng. Chem. Res., 2003, 42, 1563–1570 CrossRef CAS.
- R. Leary, P. A. Midgley and J. M. Thomas, Acc. Chem. Res., 2012, 45, 1782–1791 CrossRef CAS PubMed.
- J. M. Thomas, R. Raja, B. F. G. Johnson, T. J. O'Connell, G. Sankar and T. Khimyak, Chem. Commun., 2003, 1126–1127 RSC . See also editor's choice in Science, 2003, 300, 867.
- J. D. Keasling, Science, 2010, 330, 1355–1358 CrossRef CAS PubMed.
- M. E. Davis, Top. Catal., 2015, 58, 405–409 CrossRef CAS.
- M. Oratov and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 11777–11782 CrossRef PubMed.
- L. A. Williams, N. Guo, A. Motta, M. Delferro, I. L. Fragalà, J. T. Miller and T. J. Marks, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 413–418 CrossRef CAS PubMed.
- A. Chakrabarti and I. E. Wachs, Catal. Lett., 2015, 145, 985 CrossRef CAS.
- A. Zecchina and E. Groppo, Proc. R. Soc. A, 2012, 468, 2087–2098 CrossRef CAS.
- J. Wurm and J. Entwhistle, Ingenia, 2015, 64, 30 Search PubMed.
- J. M. Thomas, Energy Environ. Sci., 2014, 7, 19–20 CAS.
- P. C. K. Vesborg and T. F. Jaramillo, RSC Adv., 2012, 2, 7933–7947 RSC.
- E. Kemppainen, A. Bodin, B. Sebok, T. Pedersen, B. Seger, B. Mei, D. Bae, P. C. K. Vesborg, J. Halme, O. Hansen, P. D. Lund and I. Chorkendorff, Energy Environ. Sci., 2015, 8, 2991–2999 CAS.
- L. B. Hoch, T. E. Wood, P. G. O'Brien, K. Liao, L. M. Reyes, C. A. Mims and G. A. Ozin, Adv. Sci., 2014, 1, 1400013 Search PubMed.
- G. A. Ozin, Adv. Mater., 2015, 27, 1957–1963 CrossRef CAS PubMed.
- M. R. Singh and A. T. Bell, Energy Environ. Sci., 2016, 9, 193–199 CAS.
- M. R. Singh, E. L. Clark and A. T. Bell, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E6111–E6118 CrossRef CAS PubMed.
- D. Luo, Z. Hu, D. G. Choi, V. M. Thomas, M. J. Realff and R. R. Chance, Environ. Sci. Technol., 2010, 44, 8670–8677 CrossRef CAS PubMed.
- T. Xiao, T. Shirvani, O. Inderwildi, S. Gonzalez-Cortes, H. AlMegren, D. King and P. P. Edwards, Top. Catal., 2015, 58, 682–695 CrossRef CAS.
- N. Luo, F. Cao, X. Zhao, T. Xiao and D. Fang, Fuel, 2007, 86, 1727–1736 CrossRef CAS.
- B. Liu, L. France, C. Wu, Z. Jiang, V. L. Kuznetsov, H. A. Al-Megren, M. Al-Kinany, S. A. Aldrees, T. Xiao and P. P. Edwards, Chem. Sci., 2015, 6, 5152–5163 RSC.
- J. A. Herron, J. Kim, A. A. Upadhye, G. W. Huber and C. T. Maravelias, Energy Environ. Sci., 2015, 8, 126–157 CAS.
- See the data and discussion presented in the following: http://climateparis.org/COP21.
- From BP and the US Energy Information Administration, quoted in The Independent newspaper, London, 4 August 2015, p. 22.
- See: brightsourceenergy.com and the Solar Power Alliance http://www.csp-alliance.org, 2014.
- See: http://www.theguardian.com/environment/2015/oct/26/morocco-poised-to-become-a-solar-superpower-with-launch-of-desert-mega-project.
- M. Saidi, F. Samimi, D. Karimipourfard, T. Nimmanwudipong, B. C. Gates and M. R. Rahimpour, Energy Environ. Sci., 2014, 7, 103–129 CAS.
- V. K. Venkatakrishnan, W. N. Delgass, F. H. Ribeiroa and R. Agrawal, Green Chem., 2015, 17, 178–183 RSC.
Footnotes |
| † Based in part on a lecture given by JMT at the Nobel Workshop, Gothenburg, 5 May 2015. |
| ‡ Dedicated to Professor Bengt Nordén on the occasion of his seventieth birthday. |
| This journal is © The Royal Society of Chemistry 2016 |