
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Boronic acid and boronic ester containing polyoxometalates†
Hedi
Karoui
and
Chris
Ritchie
*
School of Chemistry, The University of Melbourne, Parkville, Melbourne, Victoria 3010, Australia. E-mail: critchie@unimelb.edu.au
First published on 11th November 2016
Abstract
Three organoboron functionalized polyoxometalates have been synthesized using Schiff base chemistry including a boronic acid, its methyl ester and its trimethylene glycol ester. The compounds have been characterized in the solid state by single crystal diffraction (XRD), FT-IR and elemental analysis and in solution using 1H NMR spectroscopy.
Boronic acids have attracted the attention of chemists since the mid-19th century, largely due to their relatively straightforward preparation and chemical versatility. As precursors, boronic acids are particularly useful for the formation of (C–C) bonds via Suzuki Miyaura, Petasis, and Liebeskind–Srogl couplings, as well as the Chan–Lam coupling for the formation of (C–N and C–O bonds).1–4 Further to these attractive cross coupling reactions, boronic acids can also be relatively easily condensed with a large range of substrates, as well as acting as strong Lewis acids.5 Consequently, an extensive library of molecules has been prepared from boronic acid precursors including pharmaceuticals,6,7 molecular cage compounds, crystalline covalent organic frameworks,8–12 polymers13–15 and sensors.16–18 Despite this large and growing collection of molecules and materials, very few boronic acid containing inorganic–organic hybrids are known.19
Polyoxometalates (POMs) are a class of molecular metal oxides that possess notable structural and compositional versatility. The major structural components of POMs are typically assembled from the condensation of early transition metal oxyanions with the metal in high oxidation states, although this is by no means exclusive. One striking feature of these molecules is their ability to incorporate almost any element within their skeletal frameworks as heteroatoms, thus enabling further functionalization of the molecules, including the grafting of organic ligands. In POM chemistry, only a few molecular platforms exist that can be reliably utilized to yield predesigned molecules. For example, the routine and predictable surface modification of Lindqvist polyanions to yield organo-imido containing compounds,20–22 the insertion of organo-phosphate and silicate species into lacunary polyoxotungstate ligands23–27 and the elaboration of the trisalkoxo functionalized Anderson–Evans POM by formation of imine and amide linkages.28–30
Herein, we report the first examples of organoboron containing Anderson–Evans POMs meshing the rich chemistries of these two important chemical classes. This initial report details the structures and characterization of the functionalized Mn(III) Anderson–Evans POM with the general formula [Mn(III)Mo6(VI)O18((OCH2)3)CN![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHC6H4(B(OR)2)2]3−, where R = H, 1a; Me, 1b and [Mn(III)Mo6(VI)O18((OCH2)3)CNCHC6H4(BO2(CH2)3)2]3−, 1c. Bond valence sum calculations (BVS), confirm the assigned metal oxidation states with no evidence for protonation of the polyanion (Tables 1–3 in ESI†). The three novel compounds are obtained through one pot synthesis. Mixing of 2-formylphenyl boronic acid with (TBA)3[Mn(III)Mo6(VI)O18{(OCH2)3CNH2}2] at room temperature in dimethylformamide (DMF) results in the crystallisation of 1a through slow evaporation, with the addition of 1,3 propanediol to the same reaction mixture resulting in the crystallization of 1c on vapour diffusion of diethyl ether. A pure micro-crystalline sample of 1b is obtained when the reaction is conducted in methanol, with recrystallization from DMF yielding crystals suitable for single crystal X-ray diffraction. All crude products were thoroughly washed with cold methanol then diethyl ether to remove residual starting materials.
CHC6H4(B(OR)2)2]3−, where R = H, 1a; Me, 1b and [Mn(III)Mo6(VI)O18((OCH2)3)CNCHC6H4(BO2(CH2)3)2]3−, 1c. Bond valence sum calculations (BVS), confirm the assigned metal oxidation states with no evidence for protonation of the polyanion (Tables 1–3 in ESI†). The three novel compounds are obtained through one pot synthesis. Mixing of 2-formylphenyl boronic acid with (TBA)3[Mn(III)Mo6(VI)O18{(OCH2)3CNH2}2] at room temperature in dimethylformamide (DMF) results in the crystallisation of 1a through slow evaporation, with the addition of 1,3 propanediol to the same reaction mixture resulting in the crystallization of 1c on vapour diffusion of diethyl ether. A pure micro-crystalline sample of 1b is obtained when the reaction is conducted in methanol, with recrystallization from DMF yielding crystals suitable for single crystal X-ray diffraction. All crude products were thoroughly washed with cold methanol then diethyl ether to remove residual starting materials.
Compound 1a (Fig. 1a, left) crystallizes in the monoclinic space group I2/a with cell axes a = 24.0713(6), b = 14.3578(5), c = 28.5472(15) where β = 94.407(3)° and V = 9837.1(7) Å with half a polyanion in the asymmetric unit. The molecular structure of 1a shows the expected coordination of the tripodal ligand to the surface of the polyanion as observed in the starting material (TBA)3[Mn(III)Mo6(VI)O18{(OCH2)3CNH2}2]. Condensation of the di-amino terminated precursor with 2-formylphenylboronic acid results in the formation of the anticipated secondary aldimine as confirmed by single-crystal X-ray diffraction, IR (S1) and 1H NMR studies (S4). Unfortunately, significant crystallographic disorder of the grafted ligand is observed which prevented accurate determination of bond lengths and angles, with a significant number of crystallographic restraints being imposed to satisfactorily refine the disorder. The aldimine nitrogen refines well over two positions as does the remainder of the ligand with thermal parameters suggesting 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 occupancy of the two crystallographic positions. Refinement of the aromatic portion of the ligand indicates rotational disorder, with two positions for the boronic acid functional group being identified with approximately equal contributions using the FVAR command. Subsequently the two disordered positions were fixed with equal occupancies. Intramolecular interactions include N–H–O–B hydrogen bonding between the aldimine nitrogen and the protons of the boronic acid, with intermolecular hydrogen bonding between the boronic acid functional groups of neighbouring molecules resulting in supramolecular ribbons in the solid state. Disorder of the TBA cations and solvent required the use of a solvent mask, with the composition and bulk purity of 1a verified by combustion. The solution state structure of 1a was also probed using 1H in d6-DMSO and is discussed in detail later.
:1 occupancy of the two crystallographic positions. Refinement of the aromatic portion of the ligand indicates rotational disorder, with two positions for the boronic acid functional group being identified with approximately equal contributions using the FVAR command. Subsequently the two disordered positions were fixed with equal occupancies. Intramolecular interactions include N–H–O–B hydrogen bonding between the aldimine nitrogen and the protons of the boronic acid, with intermolecular hydrogen bonding between the boronic acid functional groups of neighbouring molecules resulting in supramolecular ribbons in the solid state. Disorder of the TBA cations and solvent required the use of a solvent mask, with the composition and bulk purity of 1a verified by combustion. The solution state structure of 1a was also probed using 1H in d6-DMSO and is discussed in detail later.
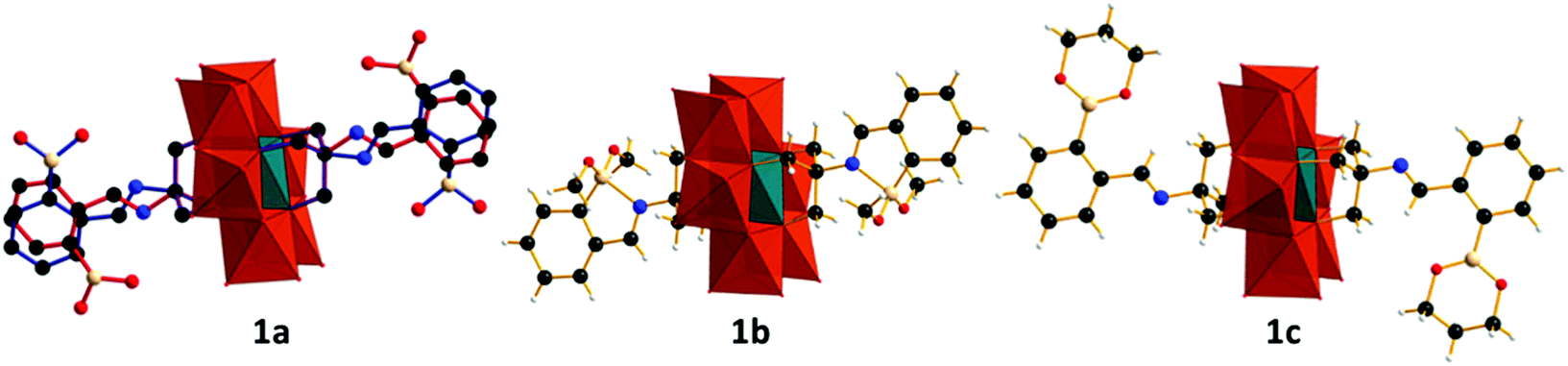 | ||
| Fig. 1 Representation of compounds 1a–c from left to right. Crystallographic disorder of 1a is clarified by the use of red and blue bonds. MoO6, orange polyhedra; MnO6, teal polyhedra; C, black spheres; O, red spheres; N, blue spheres; B, beige spheres; H, white spheres. | ||
Compound 1b (Fig. 1, middle) crystallizes in the monoclinic space group P21/c with cell axes a = 22.8863(8), b = 24.7348(7) and c = 18.1665(6) where β = 93.009(3)° and V = 10269.7(6) with two crystallographically independent halves of the functionalised cluster residing in the asymmetric unit. Reaction of (TBA)3[Mn(III)Mo6(VI)O18{(OCH2)3CNH2}2] with 2-formylphenylboronic acid in methanol yields the boronate methyl ester functionalized polyanion 1b (Fig. 1, middle). Crystallographic analysis of 1b shows an intramolecular B–N dative bond between the aldimine nitrogen and the boron of the boronate methyl ester with a bond length of 1.830(4) Å. The formation of which is facilitated by the conversion of the boronic acid to its methyl ester, negating the possibility of intramolecular hydrogen bonding between the aldimine nitrogen lone pair and the protons of the boronic acid as observed in 1a (Fig. 2). As a result of the B–N dative bond, the boron bonding is distorted from idealized C3 symmetry expected for sp2 hybridization, to that lying between trigonal and tetrahedral. Bond lengths and angles are summarized in Table 4 in ESI.† The extended structure of 1b includes intermolecular C–H⋯π hydrogen bonds between the methyl protons of the {B(OMe)2} moiety and the aromatic system of neighbouring functionalized molecules, resulting in the formation of 1D supramolecular chain structures. A more extensive discussion regarding the crystallographic disorder of the polyanions can be found in the ESI.†
 | ||
| Fig. 2 Ball and stick representation of the different boron nitrogen environments in the boronic acid and boronic ester moieties of 1a, 1b and 1c. C, black spheres; O, red spheres; N, blue spheres; B, beige spheres; H, white spheres. | ||
The ability of boron to form reversible covalent complexes with vicinal 1,2 and 1,3 diols prompted us to probe this chemistry. Stoichiometric addition of 1,3 propane-diol to the reaction mixture which would otherwise yield 1a, results in the almost quantitative crystallization of 1c where the boronic acid has been converted to its trimethylene glycol ester. 1c crystallizes in the monoclinic space group C2/c with cell axes a = 35.796(3), b = 13.2361(3) and c = 25.8822(19) where β = 128.966(12)° and V = 9534.7(19) with one-half of the functionalized cluster residing in the asymmetric unit. As with 1a, no intramolecular interactions between the aldimine nitrogen and boron are observed in the solid state. This is due to unfavourable steric interactions between the glycol ester and the POM surface. Crystallographic data for compounds 1a, 1b and 1c are summarized in Table 5 in ESI.† The IR spectra of 1a–c (S1, S2 and S3) feature characteristic vibrational bands between 890–950 cm−1 for the terminal MoO units and between 660 and 710 cm−1 for the bridging Mo–O–Mo groups of the tris functionalized Mn-Anderson. Additional boronate stretches are observed between 1350–1380 and 1430–1460 cm−1 with the vibrational band located between 2800–3000 cm−1 assigned to the aldimine functionality.
Attempts to collect 13C and 11B NMR spectra were unsuccessful due to the presence of the paramagnetic MnIII ion located at the centre of the polyanion, meanwhile all 1H resonances show significant broadening (Fig. 3).
 | ||
| Fig. 3 Representation of the intra and intermolecular interactions for 1a – top, 1b – middle and 1c – bottom. MoO6, orange polyhedra; MnO6, teal polyhedra; C, black spheres; O, red spheres; N, blue spheres; B, beige. | ||
Spectroscopic analysis of 1a–b using 1H NMR (S4 and S5) supports the crystallographic observations. The 1H NMR of 1a shows broad signals at δ = 7.20, 7.48, 7.63, 8.33 and 8.48 ppm with the anticipated integrations of 1:1:2:2:1. The strongly deshielded (OCH2) protons are also observed at δ = 62.96 ppm due to their proximity to the MnIII center. Residual signals are observed in the aromatic region between 7.8–8 ppm due to 2-formylphenyl boronic acid starting material, meanwhile no changes were observed over time suggesting stability in solution.
On dissolution of a freshly prepared sample of 1b a very similar 1H NMR spectrum is obtained indicating that the solid state B–N interaction is not maintained in solution. Furthermore, the methyl ester protons are not observed, with additional resonances at 3.14 ppm indicating partial hydrolysis of the boronic methyl ester releasing 1 equivalent of methanol. The study of B–N dative bond dynamics and the calculation of activation parameters for nitrogen inversion are typically investigated using variable temperature VT-NMR and ab initio calculations with correlation to 11B NMR and crystallographic data.31 Using the same techniques it is also possible to probe the effect of solvents on the boron environment due to the applicability of boronic acids in molecular sensors.32 Unfortunately, the presence of the paramagnetic Mn(III) ion in 1a–1c precluded the possibility of these experiments on this particular system. We are currently developing a series of diamagnetic POM based molecules that will be investigated using these approaches.
1H NMR of compound 1c shows overlapping of diol-boronic acid functionalized POMs and boronic acid functionalized POMs peaks in the aromatic region suggesting partial hydrolysis of the diol-boronic acid complex in d6-DMSO. This was confirmed by the observation of two sets of peaks, one broad peak at 3.99 ppm corresponding to the boronic acid bonded methylene glycol in the functionalized POM and a triplet at 4.32 ppm corresponding to the free propanediol in solution. Hydrolysis of the imine linkage was also observed with a supplementary aldehyde singlet observed at 10.33 ppm and diol-boronic acid triplet at 4.11 ppm. To quantify the hydrolysis of imine linkage, titration experiments with propanediol were undertaken. Upon addition of 0.5 eq. of propanediol to 1a, two sets of (OCH2) peaks at 61.68 ppm and 63.59 ppm were observed corresponding respectively to a mixture of 1c and [Mn(III)Mo6(VI)O18{(OCH2)3CNH2}2]3− in the ratio 2.5:1 (S8). This ratio decreases to reach 1:6 upon addition of 2 eq. propanediol (S9) and 1:13 upon addition of 4 eq. propanediol (S10). The methylene protons of the boronate ester 1c appear at 4.03 ppm on addition of 0.5 eq. propanediol to a solution of 1a. At this ratio, the methylene protons integrate as 2:1 with respect to the aromatic resonances indicating 50% of 1a has been converted to 1c. On adding sequential aliquots of propanediol, the resonance at 4.03 ppm diminishes, finally disappearing after addition of 4 eq. indicating complete imine hydrolysis (S8–S10).
We have successfully coupled 2-formylphenyl boronic acid to the Mn-Anderson platform using Schiff base chemistry, and additionally investigated the conversion of the boronic acid functionality to the corresponding methyl and trimethylene glycol esters. As anticipated, these species display a degree of hydrolytic instability in solution, yet they can be readily obtained as solid state structures. Transfer of these species into non-polar solvents is underway as are investigations into their structural elaboration using the boronic acid functional group. Furthermore, hydrolytically stable and diamagnetic derivatives are currently in preparation and will be reported in due course.
Notes and references
- N. Miyaura and A. Suzuki, Chem. Soc. Rev., 1995, 95(7), 2457–2483 CrossRef CAS.
- N.-A. Petasis and I. Akritopoulou, Tetrahedron Lett., 1993, 34, 583–586 CrossRef CAS.
- L. S. Liebeskind and J. Srogl, J. Am. Chem. Soc., 2000, 122, 11260–11261 CrossRef CAS.
- D. Chan, K. Monaco, R. Wang and M. Winter, Tetrahedron Lett., 1998, 39, 2933–2936 CrossRef CAS.
- D. Salazar-Mendoza, J. Cruz-Huerta, H. Höpfl, I. F. Hernández-Ahuactzi and M. Sanchez, Cryst. Growth Des., 2013, 13(6), 2441–2454 CAS.
- N. Iwasawa, H. Takahagi, K. Ono, K. Fujii and H. Uekusa, Chem. Commun., 2012, 48, 7477–7479 RSC.
- N. Christinat, R. Scopelliti and K. Severin, Chem. Commun., 2008, 3660–3662 RSC.
- N. Christinat, R. Scopelliti and K. Severin, Angew. Chem., Int. Ed., 2008, 47, 1848–1852 CrossRef CAS PubMed.
- J. Hunt, C. Doonan, J. LeVangie, A. Côté and O. Yaghi, J. Am. Chem. Soc., 2008, 130, 11872–11883 CrossRef CAS PubMed.
- N. Christinat, R. Scopelliti and K. Severin, Chem. Commun., 2008, 31, 3660–3662 RSC.
- N. Christinat, R. Scopelliti and K. Severin, J. Org. Chem., 2007, 72, 2192–2200 CrossRef CAS PubMed.
- A. Côté, H. El-Kaderi, H. Furukawa, J. Hunt and O. Yaghi, J. Am. Chem. Soc., 2007, 129, 12914–12915 CrossRef PubMed.
- T. D. James, K. R. A. S. Sandanayake and S. Shinkai, Angew. Chem., Int. Ed. Engl., 1996, 35(17), 1910–1922 CrossRef.
- Z. Guo, I. Shin and J. Yoon, Chem. Commun., 2012, 48, 5956–5967 RSC.
- G. Mirri, S. Bull, P. Horton, T. James, L. Male and J. Tucker, J. Am. Chem. Soc., 2010, 132, 8903–8905 CrossRef CAS PubMed.
- Y. Zhu, Z. Xiao, N. Ge, N. Wang, Y. Wei and Y. Wang, Cryst. Growth Des., 2006, 6, 1620–1625 CAS.
- J. Zhang, F. Xiao, J. Hao and Y. Wei, Dalton Trans., 2012, 41, 3599–3615 RSC.
- G. Sima, Q. Li, Y. Zhu, C. Lu, R. N. Khan, J. Hao, J. Zhang and Y. Wei, Inorg. Chem., 2013, 52, 6551–6558 CrossRef CAS PubMed.
- J. Cambre, D. Roy, S. Gondi and B. Sumerlin, J. Am. Chem. Soc., 2007, 129, 10348–10349 CrossRef CAS PubMed.
- M. Healey, S. Best, L. Goerigk and C. Ritchie, Inorganics, 2015, 3(2), 82–100 CrossRef CAS.
- N. Joo, S. Renaudineau, G. Delapierre, G. Bidan, L.-M. Chamoreau, R. Thouvenot, P. Gouzerh and A. Proust, Chem. – Eur. J., 2010, 16(17), 5043–5051 CrossRef CAS PubMed.
- A. Proust, R. Thouvenot and P. Gouzerh, Chem. Commun., 2008, 1837–1852 RSC.
- C. Dablemont, A. Proust, R. Thouvenot, C. Afonso, F. Fournier and J.-C. Tabet, Inorg. Chem., 2004, 43, 3514–3520 CrossRef CAS PubMed.
- S. Bareyt, S. Piligkos, B. Hasenknopf, P. Gouzerh, E. Lacôte, S. Thorimbert and M. Malacria, J. Am. Chem. Soc., 2005, 127, 6788–6794 CrossRef CAS PubMed.
- C. Boglio, K. Micoine, É. Derat, R. Thouvenot, B. Hasenknopf, S. Thorimbert, E. Lacôte and M. Malacria, J. Am. Chem. Soc., 2008, 130, 4553–4561 CrossRef CAS PubMed.
- B. Matt, S. Renaudineau, L.-M. Chamoreau, C. Afonso, G. Izzet and A. Proust, J. Org. Chem., 2011, 76, 3107–3112 CrossRef CAS PubMed.
- J. Li, I. Huth, L.-M. Chamoreau, B. Hasenknopf, E. Lacôte, S. Thorimbert and M. Malacria, Angew. Chem., Int. Ed., 2009, 48, 2035–2038 CrossRef CAS PubMed.
- C. Allain, S. Favette, L. Chamoreau, J. Vaissermann, L. Ruhlmann and B. Hasenknopf, Eur. J. Inorg. Chem., 2008, 22, 3433–3441 CrossRef.
- D. Schaming, C. Allain, R. Farha, M. Goldmann, S. Lobstein, A. Giraudeau, B. Hasenknopf and L. Ruhlmann, Langmuir, 2010, 26, 5101–5109 CrossRef CAS PubMed.
- C. Yvon, A. Surman, M. Hutin, J. Alex, B. O. Smith, D.-L. Long and L. Cronin, Angew. Chem., Int. Ed., 2014, 53, 3336–3341 CrossRef CAS PubMed.
- C. Hoang, I. Prokes, G. Clarkson, M. Rowland, J. Tucker, M. Shipman and T. Walsh, Chem. Commun., 2013, 49, 2509–2525 RSC.
- (a) L. Zhu, S. H. Shabbir, M. Gray, V. M. Lynch, S. Sorey and E. V. Anslyn, J. Am. Chem. Soc., 2006, 128, 1222 CrossRef CAS PubMed; (b) B. E. Collins, S. Sorey, A. E. Horgrove, S. H. Shabbir, V. M. Lynch and E. V. Anslyn, J. Org. Chem., 2009, 74, 4055 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1496433–1496435. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt04197c |
| This journal is © The Royal Society of Chemistry 2016 |