
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rational serendipity: “undirected” synthesis of a large {MnIII10CuII5} complex from pre-formed MnII building blocks†
Jamie M.
Frost
 ,
Fraser J.
Kettles
,
Claire
Wilson
and
Mark
Murrie
*
,
Fraser J.
Kettles
,
Claire
Wilson
and
Mark
Murrie
*
WestCHEM, School of Chemistry, University of Glasgow, University Avenue, Glasgow, UK G12 8QQ. E-mail: Mark.Murrie@glasgow.ac.uk
First published on 28th October 2016
Abstract
Use of an aminopolyalcohol-based MnII complex in solvothermal CuII chemistry leads to a rare example of a high nuclearity heterometallic {MnIII10CuII5} system, in which four CuII(H1Edte) units trap an inner {MnIII10CuII} oxide core.
High-symmetry, high-nuclearity transition metal complexes have always been a source of fascination for coordination chemists. This is not simply because of the interesting chemical and physical properties that such molecules can often display, applications range from catalysis1 to magnetism,2 but also because of the synthetic challenges involved in isolating such systems.3 One well-established method for preparing high nuclearity complexes is to employ flexible multidentate organic ligands (or combinations thereof) which can simultaneously chelate and bridge, in simple one-pot reactions with appropriate metal ions.4 Polyalkoxide ligands have yielded beautiful examples of homometallic systems including; a {Mn32} truncated cube,5 a {Mn19} complex6a (which until recently possessed the largest spin ground state known; S = 83/2),6b {Fe16} square-wheels7 and {Ni10} supertetrahedra,8 to name but a few.
The main issue with this approach however is that it is difficult, if not impossible, to predict the structure of the resulting products a priori. Indeed, this method is frequently referred to in the literature as “serendipitous self-assembly”.9 In an effort to transcend this serendipity and gain some degree of control over product formation, one can react pre-formed complexes which feature multiple (and vacant) ligand binding sites capable of coordinating additional metal ions. This is particularly useful for assembling heterometallic complexes and constitutes a kind of synthetic “half-way house”, between serendipitous self-assembly at one extreme and the more rigid building-block approach exemplified by cyanometallate chemistry at the other (so-called “rational design”).10 Aminopolyalcohol ligands are ideally suited to constructing systems in this manner, since they can form stable [M(HnL)] monometallic complexes in which one or more of the alkoxide arms remain protonated and thus amenable to later coordination, thereby facilitating cluster nucleation.
To this end, we have previously investigated the coordination chemistry of the ligand bis–tris propane (H6L = {2,2′-(propane-1,3-diyldiimino)bis[2-(hydroxymethyl)-propane-1,3-diol]}), demonstrating that a pre-formed monomeric CuII complex of H6L can be used to trap a cubooctahedral manganese oxide core, thereby directing the synthesis of a large {MnIII12MnII6CuII6} complex (Scheme 1).11 Here, we demonstrate that the coordination chemistry of H6L's smaller cousin H4Edte (2,2′,2′′,2′′′-(1,2-ethanediyldinitrilo)tetraethanol, Scheme 1 right) is much less predictable (though no less interesting), by presenting the synthesis, structure and magnetic characterisation of the {CuII(H1Edte)}-capped {MnIII10CuII} complex; [MnIII10CuII5O8(O2CPh)8(HEdte)4(H2O)4][NO3]4·7MeOH·13H2O (2) – the unexpected product of a reaction initially intended to produce large {Mnx(HnEdte)}-capped CuII complexes (Scheme 1) as part of our investigation into core–shell molecular interfaces.
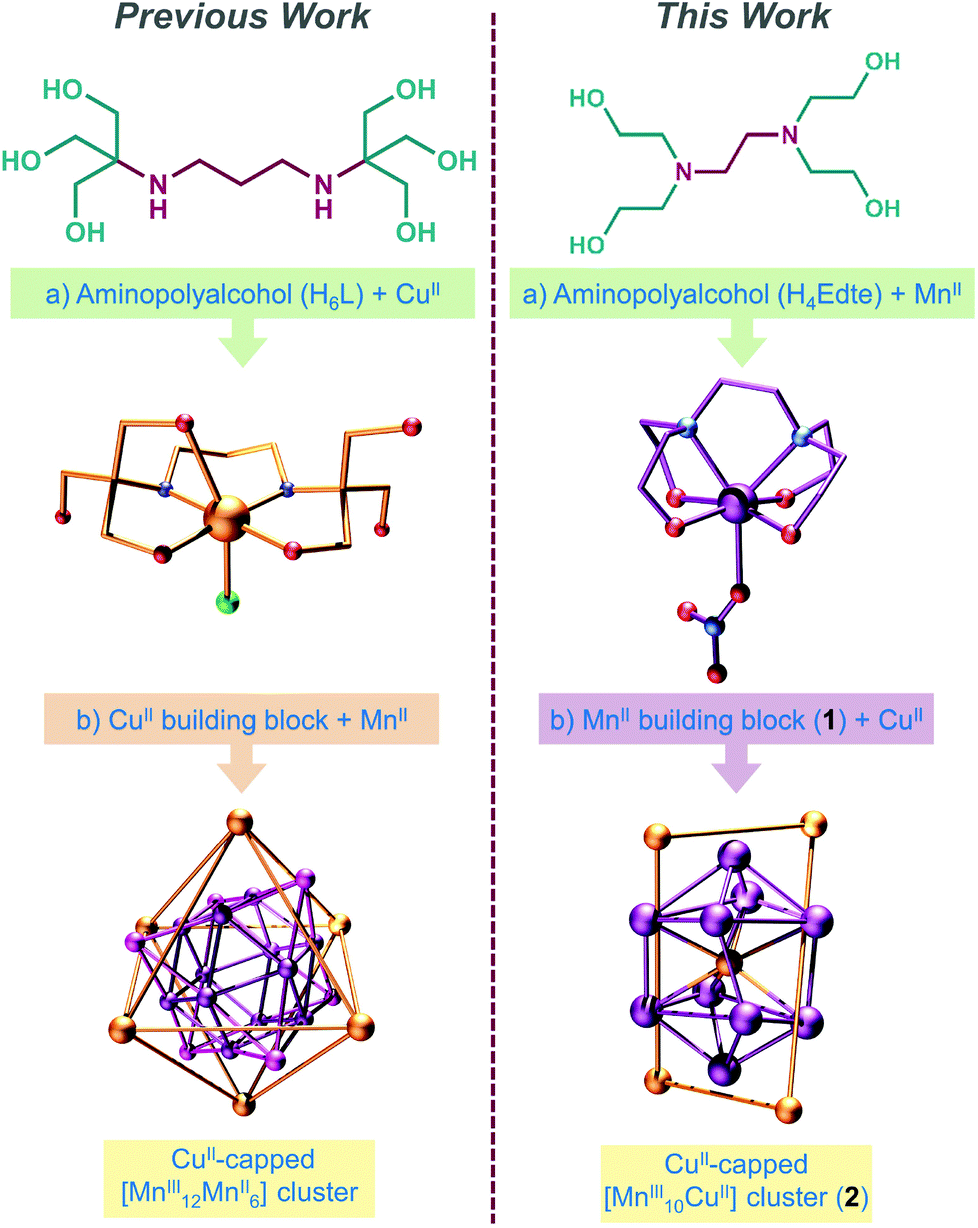 | ||
| Scheme 1 Highlighting a modular approach to the design of high nuclearity heterometallic complexes (left) and its attempted application herein with H4Edte (right). | ||
Reaction of Mn(NO3)2·4H2O with H4Edte in MeCN yields a light-pink precipitate which can be recrystallised from EtOH via Et2O vapour diffusion to yield pink needle-like crystals of [Mn(H4Edte)(NO3)][NO3] (1, see ESI† for details). Complex 1 (Fig. S1†) crystallises in the orthorhombic space group Pbca, with its structure describing a single H4Edte ligand coordinated to a single MnII ion via its two N and four O donor atoms (the latter of which all remain protonated). The MnII ion is heptacoordinate and in distorted capped octahedral geometry (C3v, ChSM = 0.932),12 with a single terminal monodentate NO3− ligand, which along with a uncoordinated NO3− anion maintain charge balance (see cif file and Tables S1 and S2† for full details).
Heating of 1 in a methanolic solution of Cu(OAc)2·2H2O and NaO2CPh under solvothermal conditions, yields small dark brown block-like crystals of 2 after slow evaporation of the filtered mother liquor over a period of 6 weeks (see ESI† for details). The use of 1 under these conditions was essential to isolate 2. Compound 2 crystallises in the tetragonal space-group I41/a (Fig. S2, Table S3†). Bond lengths and angles pertinent to the discussion herein are given in Table S4.† The structure of 2 is complex and unusual (Fig. 1a). We suggest that it is best thought of as comprising; two pairs of symmetry equivalent {CuII(H1Edte)}-capped corner-sharing oxo-centered pseudo-{MnIII3} triangles, that form two heavily distorted square-based {MnIII5} pyramids which are stacked and off-set with respect to one another, and in-turn linked at their square faces by a single CuII ion (Fig. 1b). All Mn ions are hexacoordinate, exhibit distorted octahedral coordination geometries and are in the +3 oxidation state, as confirmed by a combination of bond length considerations, BVS calculations (Table S5†) and charge balance. Two η1:η2:μ3-benzoate ligands link two pairs of MnIII ions (Mn1–Mn2 and Mn1–Mn3), creating two edges of each {MnIII3} sub-unit, with the η2-O arm facilitating corner sharing between triangles (via Mn1) to create each “{MnIII5} pyramid” (see Fig. 1). The remaining edge of each triangular sub-unit is completed by a single μ-bridging H2O ligand. Although a rare occurrence in Mn chemistry, there are previous reports of polymetallic complexes which feature Mn ions bridged by H2O ligands.13 The Jahn–Teller (JT) axes of the MnIII ions lie along the edges of the {MnIII3O} triangle (again something of an unusual occurrence, see Fig. 1d), with the central μ3-O2− anion within each deviating ∼0.16 Å from the mean {MnIII3} planes. Each {MnIII3O} triangle is capped on its face by a single {CuII(H1Edte)} unit, which employs three of its deprotonated alkoxide arms to coordinate to the triangular face (one to each MnIII ion), with the resulting CuII–O–MnIII angles all falling within an extremely narrow range (∠Cu–O–Mn = 120.34(13)–121.03(13)°). The remaining R–O− arm of each Edte ligand remains protonated and unbound, exhibiting significant crystallographic disorder. Each CuII ion within the {CuII(H1Edte)} units is pentacoordinate and in an {O3N2} coordination environment, with continuous shape measurements revealing a heavily distorted coordination geometry which is best described as square pyramidal (C4V, ChSM = 2.298).12a,14
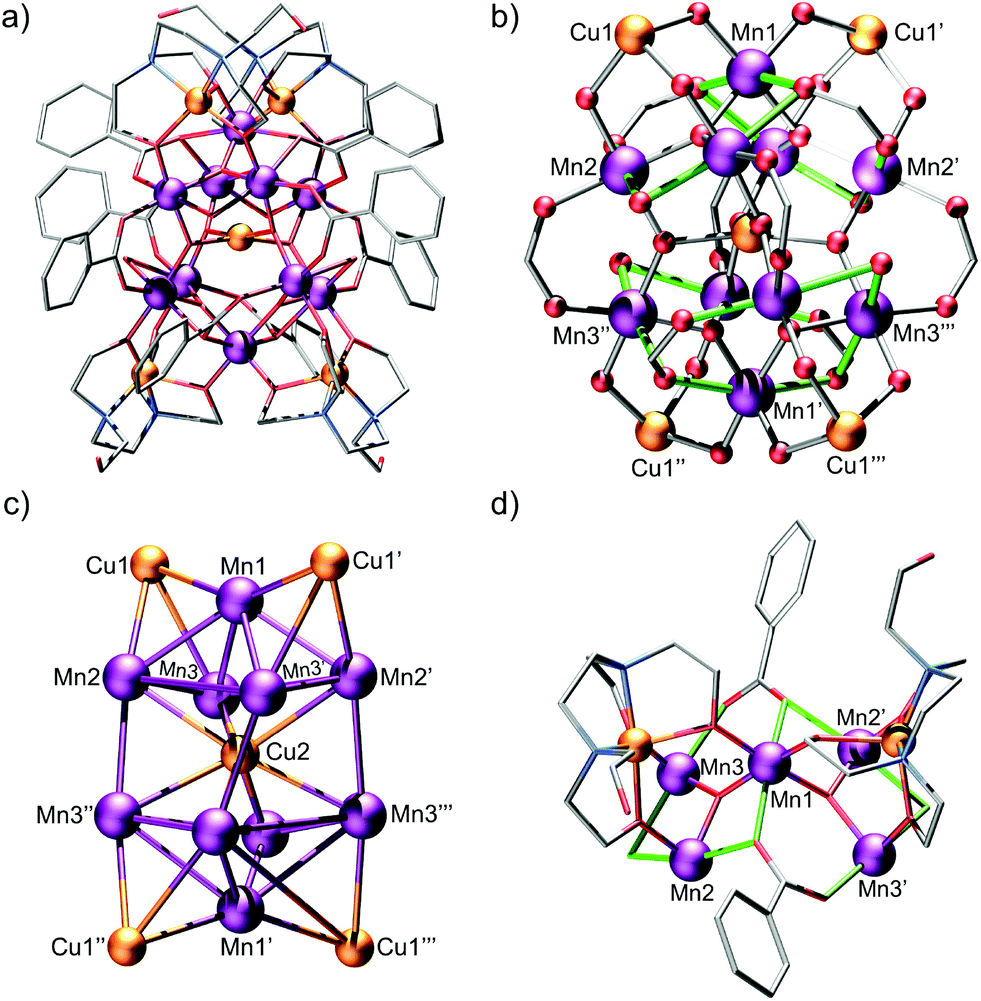 | ||
| Fig. 1 (a) Molecular structure of the cation in 2 with H-atoms, MeOH solvent molecules and NO3− anions omitted for clarity. (b) Magnetic core of 2 with the JT axes of the MnIII ions highlighted in green. (c) The metallic skeleton of 2 highlighting the CuII-capped {MnIII3} triangular sub units and the {MnIII5} square-based pyramids, which the system can be thought of as comprising. (d) Highlighting the role of the η1:η2:μ3-benzoate ligands in linking {MnIIIO3} triangular sub-units together to form an {MnIII5} square pyramid, and, the capping of [MnIIII3] triangular faces by {CuII(H1Edte)} units. The JT axes of the MnIII ions are highlighted in green for clarity. Colour code: Mn = purple, Cu = orange, O = red, N = blue, C = silver. | ||
Four η1:η1:μ-benzoate ligands link the square faces of the two {MnIII5} pyramids at their corners (Mn2/Mn3 and symmetry equivalents), with a single CuII ion (Cu2) lying at the centre of the “{MnIII8} cubane” created by the joining of the square faces of the two {MnIII5} sub-units. This CuII ion is ligated by four μ3-O2− ligands, which link the CuII ion to the eight MnIII ions of the {MnIII8} cubane, and is in distorted square-planar geometry (D4h, ChSM = 1.172).12a,15 The distance between Cu2 and the mean MnIII4 planes created by the square faces of the two {MnIII5} pyramids is just 1.6 Å, with the corresponding distance between Cu2 and Mn1/Mn1′ being ∼3.1 Å. The entire cluster is ∼18.5 Å at its widest point with the largest intramolecular metal–metal separation being ∼10.5 Å (Cu1–Cu1′′′/Cu1′–Cu1′′). The charge on the complex is balanced by four lattice NO3− anions.
There are intramolecular H-bonding interactions between the η1-O atoms of η1:η2:μ3-benzoate ligands and the H-atoms of coordinated H2O ligands (O6⋯O9 = 2.808(4) Å). These same H2O ligands also form intermolecular H-bonds with the O atoms of disordered MeOH solvent molecules (O9⋯O1M = 2.58(9) Å). The protonated arms of Edte ligands also form an extensive network of intermolecular H-bonding contacts, with the O-atoms of NO3− counter ions (O12⋯O42 = 3.16(2) Å), which in-turn, are H-bonded to disordered MeOH solvent molecules (O13⋯O1M = 2.756(17) Å). The closest intercluster contacts are between the aromatic rings of the η1:η2:μ3-benzoate ligands of neighbouring clusters (∼3.56 Å), with the shortest intercluster metal–metal separation being between the CuII ions of {CuII(H1Edte)} units (∼9.24 Å, see Fig. S3† for packing diagram).
To the best of our knowledge 2 represents just the fifth reported example of a discrete heterometallic Mn/Cu complex of nuclearity greater than ten.11,16 It is also an extremely rare example of a high nuclearity Mn/Cu system featuring Mn exclusively in the +3 oxidation state – the only other example being the {MnIII6CuII10} system of Oshio and co-workers.16a
It is interesting to note that despite the use of the {MnII(H4Edte)} unit as a starting material, the Edte ligands in 2 encapsulate Cu and not Mn. This was unexpected, and our initial intention had been to assemble Mn-capped oxo-bridged CuII complexes using {MnII(H4Edte)}, in a similar fashion to our previous work using CuII-bis-tris-propane building blocks (see Scheme 1).17 One can only assume that under solvothermal conditions the {CuII(HnEdte)} unit is more thermodynamically stable than the corresponding {Mnz(HnEdte)} one. Studies are currently underway to examine the structural integrity of {MII(HnEdte)} building blocks under a variety of reaction conditions, in an effort to better understand, and hence exploit, this ligand for the controlled assembly of heterometallic structures.
The magnetic susceptibility of 2 (χ) was measured from 290 K down to 1.8 K in an applied field of 0.1 T, with the results plotted in Fig. S5† as the χMT vs. T product (see Fig. S6† for the corresponding M vs. H plot). The χMT value of 30.64 cm3 K mol−1 at 290 K, is close to the theoretical value of 31.47 cm3 K mol−1 expected for ten MnIII and five CuII non-interacting ions; MnIII (S = 2, g = 1.98) and CuII (S = 1/2, g = 2.1). This value decreases steadily until around 100 K, before decreasing more sharply to reach a minimum value of 3.1 cm3 K mol−1 at 1.8 K. This behaviour is indicative of dominant antiferromagnetic exchange interactions between the constituent spin carriers. Unfortunately, despite the high symmetry of the cluster the sheer number of distinct M–L–M exchange pathways precludes any meaningful quantitative analysis of the susceptibility data. AC susceptibility studies in the 10–1.8 K temperature range in a 3.5 G field at oscillating at frequencies up to 1300 Hz, do not reveal any frequency dependence in the out-of-phase component of the AC susceptibility (χ′′), indicating that 2 does not exhibit slow relaxation of the magnetisation in the temperature regime investigated. The lack of slow relaxation behaviour is perhaps unsurprising given the presence of dominant antiferromagnetic exchange interactions and the relative orientation of the JT axes of the MnIII ions in the structure.
Use of the pre-formed building block {MnII(H4Edte)} in solvothermal CuII chemistry has led to isolation of a fascinating {MnIII10CuII5} complex, an extremely rare example of a heterometallic Mn/Cu system featuring Mn ions exclusively in the +3 oxidation state. Despite our initial intention to “direct” the assembly of {Mnn(HnEdte)}-capped CuII complexes, as part of an investigation into core–shell molecular interfaces and magnetic behaviour, the resulting molecule was a consequence of serendipity with the Edte ligands in the structure of 2 found to encapsulate Cu and not Mn. These results suggest that the structural integrity of molecular building blocks derived from flexible multidentate chelates must be carefully considered if any degree of synthetic control is to be maintained, but also that “undirected” synthesis can nevertheless produce fascinating new molecules.
We thank the University of Glasgow and the UK Engineering and Physical Sciences Research Council for financial support (grant ref. EP/IO27203/1 & EP/K033662/1). The data which underpin this work are available at http://dx.doi.org/10.5525/gla.researchdata.369.
Notes and references
- T. M. Powers and T. A. Betley, J. Am. Chem. Soc., 2013, 135, 12289 CrossRef CAS PubMed.
- C. J. Milios and R. E. P. Winpenny, Struct. Bonding, 2015, 164, 1 CrossRef CAS.
- G. Aromí and E. K. Brechin, Struct. Bonding, 2006, 122, 1–69 CrossRef.
- E. K. Brechin, Chem. Commun., 2005, 5141–5153 RSC.
- R. T. W. Scott, S. Parsons, M. Murugesu, W. Wernsdorfer, G. Christou and E. K. Brechin, Angew. Chem., Int. Ed., 2005, 44, 6540–6543 CrossRef CAS PubMed.
- (a) M. Ako, I. J. Hewitt, V. Mereacre, R. Clérac, W. Wernsdorfer, C. E. Anson and A. K. Powell, Angew. Chem., Int. Ed., 2006, 45, 4926 CrossRef PubMed; (b) S. Kang, H. Zheng, K. Hamachi, S. Kanegawa, K. Sugimoto, Y. Shiota, S. Hayami, M. Mito, T. Nakamura, M. Nakano, M. L. Baker, H. Nojiri, K. Yoshizawa, C. Duan and O. Sato, Nat. Commun., 2015, 6, 5955 CrossRef PubMed.
- (a) L. F. Jones, A. Batsanov, E. K. Brechin, D. Collison, M. Helliwell, T. Mallah, E. J. L. McInnes and S. Pilikgos, Angew. Chem., Int. Ed., 2002, 41, 4318 CrossRef CAS; (b) R. Carrasco, J. Cano, T. Mallah, L. F. Jones, D. Collison and E. K. Brechin, Inorg. Chem., 2004, 43, 5410 CrossRef CAS PubMed.
- R. Shaw, I. S. Tidmarsh, R. H. Laye, B. Breeze, M. Helliwell, E. K. Brechin, S. L. Heath, M. Murrie, S. Ochsenbein, H.-U. Güdel and E. J. L. McInnes, Chem. Commun., 2004, 1418 RSC.
- R. E. P. Winpenny, J. Chem. Soc., Dalton Trans., 2002, 1–10 RSC.
- See for example: (a) V. Mervaud, C. Dercroix, A. Scullier, C. Guyard-Duhayon, J. Vaissermann, F. Gonnet and M. Verdauger, Chem. – Eur. J., 2003, 9, 8 Search PubMed; (b) K. S. Pedersen, J. Bendix and R. Clérac, Chem. Commun., 2014, 50, 4396 RSC.
- V. A. Milway, F. Tuna, A. R. Farrell, L. E. Sharp, S. Parsons and M. Murrie, Angew. Chem., Int. Ed., 2013, 52, 1949–1952 CrossRef CAS PubMed.
- (a) M. Pinsky and D. Avnir, Inorg. Chem., 1998, 37, 5575 CrossRef CAS PubMed; (b) S. Alvarez, D. Avnir, M. Llunell and M. Pinsky, New J. Chem., 2002, 26, 996 RSC.
- (a) J.-L. Liu, F.-S. Gao, Z.-S. Meng, Y.-Z. Zheng, J.-D. Leng, M.-L. Tong, L. Ungur, L. F. Chibotaru, K. J. Heroux and D. N. Hendrickson, Chem. Sci., 2012, 2, 1268 RSC; (b) V. Chandrasekhar, A. Dey, S. Das, M. Rouxieres and R. Clérac, Inorg. Chem., 2013, 52, 2588 CrossRef CAS PubMed; (c) I. A. Kühne, N. Magnani, V. Mereacre, W. Wernsdorder, C. E. Anson and A. K. Powell, Chem. Commun., 2014, 50, 1882 RSC.
- S. Alvarez and M. Llunell, J. Chem. Soc., Dalton Trans., 2000, 3288 RSC.
- J. Cirera, P. Alemany and S. Alvarez, Chem. – Eur. J., 2004, 10, 190 CrossRef CAS PubMed.
- (a) S. Yamashita, T. Shiga, M. Kurashina, M. Nihei, H. Nojiri, H. Sawa, T. Kakiuchi and H. Oshio, Inorg. Chem., 2007, 46, 3810 CrossRef CAS PubMed; (b) W.-G. Wang, A.-J. Zhou, W.-X. Zhang, M.-L. Tong, X.-M. Chen, M. Nakano, C. C. Beedle and D. N. Hendrickson, J. Am. Chem. Soc., 2007, 129, 1014 CrossRef CAS PubMed.
- (a) M. Heras Ojea, V. A. Milway, G. Velmurugan, L. H. Thomas, S. J. Coles, C. Wilson, W. Wernsdorfer, G. Rajaraman and M. Murrie, Chem. – Eur. J., 2016, 22, 12839 CrossRef CAS PubMed; (b) M. Heras Ojea, C. Wilson, S. J. Coles, F. Tuna and M. Murrie, Dalton Trans., 2015, 44, 19275 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, single crystal X-ray crystallography experimental data tables, packing diagrams, FT-IR and magnetic data. CCDC 1501411 and 1501412. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt03914f |
| This journal is © The Royal Society of Chemistry 2016 |