
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
DFT calculations in the assignment of solid-state NMR and crystal structure elucidation of a lanthanum(III) complex with dithiocarbamate and phenanthroline†
Vasantha
Gowda
 *ab,
Risto S.
Laitinen
c,
Ville-Veikko
Telkki
a,
Anna-Carin
Larsson
b,
Oleg N.
Antzutkin
b and
Perttu
Lantto
*a
*ab,
Risto S.
Laitinen
c,
Ville-Veikko
Telkki
a,
Anna-Carin
Larsson
b,
Oleg N.
Antzutkin
b and
Perttu
Lantto
*a
aNMR Research Unit, University of Oulu, P.O. Box 3000, FI-90014 Oulu, Finland. E-mail: gowda.vasantha.k@gmail.com; perttu.lantto@oulu.fi
bChemistry of Interfaces, Luleå University of Technology, SE-97187, Luleå, Sweden
cLaboratory of Inorganic Chemistry, University of Oulu, P.O. Box 3000, FI-90014, Finland
First published on 16th November 2016
Abstract
The molecular, crystal, and electronic structures as well as spectroscopic properties of a mononuclear heteroleptic lanthanum(III) complex with diethyldithiocarbamate and 1,10-phenanthroline ligands (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) were studied by solid-state 13C and 15N cross-polarisation (CP) magic-angle-spinning (MAS) NMR, X-ray diffraction (XRD), and first principles density functional theory (DFT) calculations. A substantially different powder XRD pattern and 13C and 15N CP-MAS NMR spectra indicated that the title compound is not isostructural to the previously reported analogous rare earth complexes with the space group P21/n. Both 13C and 15N CP-MAS NMR revealed the presence of six structurally different dithiocarbamate groups in the asymmetric unit cell, implying a non-centrosymmetric packing arrangement of molecules. This was supported by single-crystal X-ray crystallography showing that the title compound crystallised in the triclinic space group P
:1) were studied by solid-state 13C and 15N cross-polarisation (CP) magic-angle-spinning (MAS) NMR, X-ray diffraction (XRD), and first principles density functional theory (DFT) calculations. A substantially different powder XRD pattern and 13C and 15N CP-MAS NMR spectra indicated that the title compound is not isostructural to the previously reported analogous rare earth complexes with the space group P21/n. Both 13C and 15N CP-MAS NMR revealed the presence of six structurally different dithiocarbamate groups in the asymmetric unit cell, implying a non-centrosymmetric packing arrangement of molecules. This was supported by single-crystal X-ray crystallography showing that the title compound crystallised in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . In addition, the crystal structure also revealed that one of the dithiocarbamate ligands has a conformational disorder. NMR chemical shift calculations employing the periodic gauge including projector augmented wave (GIPAW) approach supported the assignment of the experimental 13C and 15N NMR spectra. However, the best correspondences were obtained with the structure where the atomic positions in the X-ray unit cell were optimised at the DFT level. The roles of the scalar and spin–orbit relativistic effects on NMR shielding were investigated using the zeroth-order regular approximation (ZORA) method with the outcome that already the scalar relativistic level qualitatively reproduces the experimental chemical shifts. The electronic properties of the complex were evaluated based on the results of the natural bond orbital (NBO) and topology of the electron density analyses. Overall, we apply a multidisciplinary approach acquiring comprehensive information about the solid-state structure and the metal–ligand bonding of the heteroleptic lanthanum complex.
. In addition, the crystal structure also revealed that one of the dithiocarbamate ligands has a conformational disorder. NMR chemical shift calculations employing the periodic gauge including projector augmented wave (GIPAW) approach supported the assignment of the experimental 13C and 15N NMR spectra. However, the best correspondences were obtained with the structure where the atomic positions in the X-ray unit cell were optimised at the DFT level. The roles of the scalar and spin–orbit relativistic effects on NMR shielding were investigated using the zeroth-order regular approximation (ZORA) method with the outcome that already the scalar relativistic level qualitatively reproduces the experimental chemical shifts. The electronic properties of the complex were evaluated based on the results of the natural bond orbital (NBO) and topology of the electron density analyses. Overall, we apply a multidisciplinary approach acquiring comprehensive information about the solid-state structure and the metal–ligand bonding of the heteroleptic lanthanum complex.
Introduction
Rare earth element (REE) dialkyldithiocarbamate complexes have found wide practical applications as photo-luminescent materials,1 in heteroleptic compounds as precursors for REE sulphide nanoparticles and thin films,2–4 as high pressure lubricating additives,5,6 for the solvent extraction of REEs,7etc. For a given set of ligands, most REEs are known to form complexes of similar crystal structure types.2 In general, due to similar chemical properties (oxidation state and ionic radii), REEs can readily substitute one another that often renders their refinement to pure metals a challenging task.8 Moreover, their thermal stability and physiochemical properties such as solubility, sorption, etc. are highly dependent on the structure of the material in the solid state. In this context, being able to understand subtle structural differences in the complexation of REEs to a particular type of ligand is highly important. Also, the coordination chemistries of lanthanum and yttrium have received much less attention than that of most d-block metals or even other REEs, in part because the only accessible oxidation state of Y(III) and La(III) ions in solution (d0 and f0, respectively) lacks both magnetic and UV-vis spectroscopic information regarding the electronic d–d and charge transfer transitions. On the other hand, for NMR studies, complexes of La and Y are desirable since they are diamagnetic, while most of the REE complexes are paramagnetic, which makes NMR analysis difficult, although recent developments in paramagnetic NMR theories and their novel implementations facilitate the analysis of complicated paramagnetic NMR spectra significantly.9–12 In any case, studies on diamagnetic REE complexes can be used to probe the chemistry of complexes analogous to those containing paramagnetic REEs.Solid-state NMR is often used as an analytical tool to acquire information regarding local and long-range electronic and molecular structures.13,14 Obtaining high-quality single crystals, which is essential for X-ray crystallography, can be troublesome. In this regard, interpretations of the unique information, which can be gained from solid-state 13C and 15N NMR, particularly from the nuclear shielding interaction, require a systematic investigation of NMR parameters of simple REE complexes. The knowledge gained from such exercises may then be applied to NMR investigations of more complex materials in the solid state. Although solid-state 139La-NMR (I = 7/2) has been performed on many materials for structural analyses,15,16 the technique has not been used routinely because of a moderately high quadrupole moment (Q = 20.0 fm2) and a moderate magnetogyric ratio, both leading to a low sensitivity particularly for lanthanum complexes with a low symmetry.17 Our attempts to obtain any useful data for the title complex from static solid-state 139La NMR using either the WURST-QCPMG18 sequences or echo-based experiments19 were unsuccessful. However, in such difficult situations, the coordination behaviour and local structure of the complexes may still be explored by studying other NMR active nuclei such as the 13C and 15N present in the molecule.
In addition, quantum chemical calculations could also play an important role in obtaining the electronic and molecular properties of heavy metal atoms and their coordination complexes. They will also assist in analysing the experimental results and studying the relationship between the structure and properties. To account for factors such as crystal lattice effects and long-range intermolecular dispersion forces which largely affect the properties of periodic or extended materials and to obtain highly accurate results at the same time, computationally expensive ab initio electron correlation methods are required.20 However, in the last two decades several schemes have been developed within the framework of density functional theory (DFT) methods, which not only offer an alternative use of inexpensive computational methods but can also handle relatively large molecules efficiently.21 DFT plane-wave methods22–25 have been found to be useful for periodic systems to compare the theoretical NMR shielding parameter calculations, chemical shift tensors, and electric field gradients (EFG) with the experimental solid-state NMR results. Nevertheless, combining a number of methods, such as experimental solid-state NMR and X-ray diffraction techniques with theoretical calculations using the gauge including projector augmented wave (GIPAW) approach, often referred to as the “SMARTER Crystallography” approach,26–29 can provide much deeper insight compared to any individual approach alone.
In this work, we report on the solid-state structure elucidation and electronic and spectroscopic property evaluation of a heteroleptic tris(N,N-diethyldithiocarbamato)(PHEN)lanthanum(III) complex with the formula [La{S2CN(C2H5)2}3PHEN], where PHEN = 1,10-phenanthroline, by combining solid-state NMR spectroscopy, X-ray diffraction, and first principles DFT calculations. Since REEs form hard ions and sulphur is considered as a soft ligand, the rare-earth metal sulphur coordination is regarded as a less favourable combination. However, the inclusion of PHEN as a strong N-donor ligand contributes to the stabilisation of the heteroleptic 8-coordinate (with a central core REE-S6N2) complex.2,30 To the best of our knowledge, only mononuclear and centrosymmetric structures have been reported in the literature for other tris(N,N-diethyldithiocarbamato)(PHEN)REE(III) complexes, where REE = (Nd, Sm, Gd, Ho and Er),2 Eu,31 Pr,1 Yb,32 and Y.30 Their crystal structures are all monoclinic with the space group P21/n. Herein, a new single-crystal X-ray structure, crystallised in the triclinic P space group, is reported for the title complex. Even though the implications of this new crystal structure on the properties of the material are worth studying, it is beyond the scope of this paper. However, in general, a different crystal packing indicates differences in metal–ligand interactions, stability, and thermal and other physicochemical properties. In addition, different crystal structures for different REE complexes may have implications in the selection or/and design of novel more specific ligands for separating rare earth metals, which have very similar affinities towards a large range of currently used collectors.
After geometry optimisation, we have calculated the NMR shielding values of 13C and 15N nuclei using the GIPAW method in CASTEP.22 The results were compared with the experimental data in order to support the NMR spectral assignments and to follow the evolution of calculated NMR parameters in the course of structure optimisation. To quantify the role of relativistic effects on the chemical shifts of light atoms (LA) because of the neighbouring heavy atoms (HA), HALA effects, both scalar relativistic (SR-ZORA) as well as relativistic zeroth-order regular approximation including both scalar and spin–orbit relativity (SO-ZORA) calculations were performed for one molecular unit of the complex using the ADF code.33 In addition, the nature of the metal–ligand bonding as well as the ground state stabilisation interactions were studied by the natural bond orbital (NBO)34 and the quantum theory of atoms in molecules (QTAIM)35,36 method. Finally, we showed that the quantum-chemical calculations reproduced the observed NMR shifts in both liquid (in CDCl3 solutions) and solid state, supported the assignments, and provided more precise structural information.
Results and discussion
Powder X-ray diffraction (PXRD) patterns
Based on the fact that most crystalline materials have unique PXRD patterns, the technique can be used to identify the fingerprints of crystalline phases leading to a qualitative characterisation of materials. The PXRD pattern for the newly prepared lanthanum dialkyldithiocarbamate (hereafter Dtc) complex [La{S2CN(C2H5)2}3PHEN] was primarily compared with the PXRD patterns for a previously reported yttrium [Y{S2CN(C2H5)2}3PHEN] complex (Fig. 1).30 The comparison based on a visual assessment of the PXRD patterns revealed that they are crystallographically different. For example, the highest intensity peak was observed at 2θ° 9.70 (d-spacing 4.89 Å) and 10.43 (d-spacing 8.51 Å) for the lanthanum and yttrium Dtc complexes, respectively. On the other hand, a good matching in terms of the peak positions and the relative peak intensities between the experimental and the calculated PXRD patterns for the title compound (see Fig. 1) ensures that the obtained single crystal structure (vide infra) corresponds to the structure of the powdered material studied in the solid-state NMR experiments.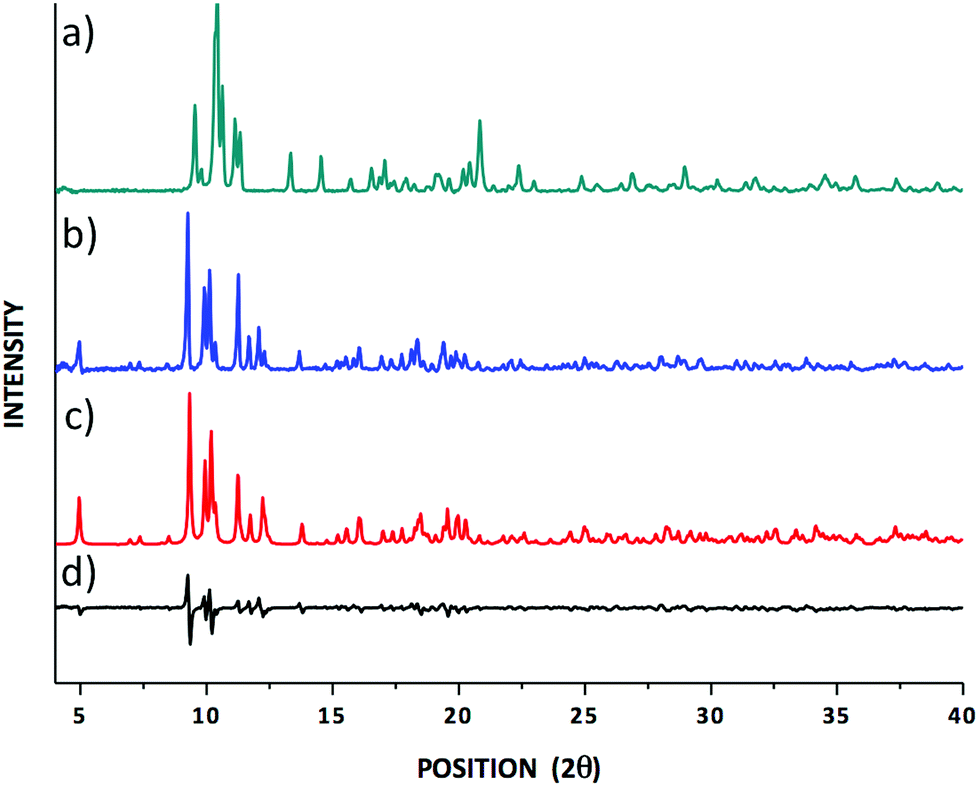 | ||
| Fig. 1 (a) An experimental PXRD pattern for tris(N,N-diethyldithiocarbamato)(PHEN)yttrium(III);30 (b) experimental and (c) calculated PXRD patterns for the tris(N,N-diethyldithiocarbamato)(PHEN)lanthanum(III) complexes; (d) the difference spectrum comparing (b) and (c). | ||
Solution-state NMR
Baba et al. have reported on the isotropic 1H NMR spectrum for a tris(N,N-di-n-propyldithiocarbamato)(PHEN)lanthanum(III) complex.37 However, in that spectrum the fine spectral features of PHEN protons were missing and no J-couplings were reported due to the broad signals, especially for H1, H2, and H3 (for labelling see the molecular structure of the complex in Fig. S1 in the ESI†). It is noteworthy that there have been discrepancies in the assignment of the PHEN protons (and carbons) in different metal complexes where PHEN is involved as one of the ligating moieties.38 In our case, we found the following chemical shift order of signals: H1 > H3 > H6 > H2 and C1 > C5 > C3 > C4 > C6 > C2 for PHEN as a ligand, which follow the same order as the chemical shifts of free PHEN molecules (see Fig. S1 and S2 in the ESI†). The above order in chemical shifts is also supported by our SO-ZORA DFT calculations, where the solvation effect on the complex geometry was taken into account using the Conductor-like Screening Model (COSMO)39 in geometry optimisation (see details in Table S1 in the ESI†). Interestingly, the aromatic protons next to the nitrogen atoms become deshielded by 0.6 ppm (known as the coordination shift) upon the bonding of nitrogen to a metal centre. Similar nuclear deshielding or shielding trends were observed for other protons and carbons in the complex but to a lesser extent (see Table S1 in the ESI†).The analysis of J-couplings was also performed in order to assess whether the coordination of PHEN to lanthanum(III) had any effect on the coupling pattern. We have measured 3JHH and 1JCH in both the PHEN and the Dtc ligands. In the case of PHEN, two of the measured ortho H–H couplings for H2 are different; 3JH2–H1 is 4.8 Hz, whereas 3JH2–H3 is 8.4 Hz. Similarly, the measured 1JCH values for PHEN show large variations, e.g., 1JC1–H1 = 182.4 Hz, whereas 1JC6–H6 = 163.7 Hz. However, when compared with PHEN as a free ligand, the 1H–1H or 1H–13C J-coupling constants remained almost similar before and after the coordination. In other words, in liquids, La-PHEN coordination deshields certain nuclei but it neither changes the order of the resonance lines (of 1H and 13C) nor the 1H–1H/1H–13C J-coupling patterns. As can be seen from the 1H decoupled 13C spectrum (Fig. S2a in the ESI†), coupling to quadrupolar 14N (I = 1) only slightly broadens the Dtc signal (half width ca. 5.5 Hz). The observed triplet for the –NC(S)S carbon sites in the 1H coupled 13C spectrum (gated decoupling) (see Fig. S2b in the ESI†) can be attributed to the coupling of Dtc to the methylene protons (3JCH = 4.5 Hz). A cross-peak between the Dtc carbons and methylene protons in the 2D 1H–13C HMBC (nJCH optimised for 4.0 Hz) experiment additionally supports this conclusion (see Fig. S3a in the ESI†). Similarly, in the 1H–15N HMBC experiment, a signal at 175.4 ppm is assigned to the nitrogen atoms in the –S2CN = groups which shows two-bond as well as four-bond correlations to the methylene and methyl protons, respectively (see Fig. S3b in the ESI†).
Solid-state NMR
Cross-polarisation magic-angle-spinning (CP-MAS) NMR is an efficient method for determining the local structure by means of a comparison of the number of observed resonance lines with the number of non-equivalent sites present in the molecule. The experimental 13C and 15N chemical shifts along with the theoretically calculated shifts are listed in Table 1. The molecular structure of the complex and the labelling of nuclei are depicted in Fig. 2. The structure assignment was partially done by combining information about 1H, 13C, and 15N isotropic chemical shifts in liquid (solution in CDCl3) and 13C/15N CP-MAS NMR in the solid state. In the solid-state NMR spectra, we have observed five distinct 13C resonance lines (with integral intensities 1:2:1:1:1) and three 15N signals (with integral intensities 2:3:1) in the range of 202.1–207.8 ppm and 172.3–179.4 ppm, respectively (see Fig. 3 and 4). These NMR patterns for 13C and 15N are a clear indication of the fact that the title complex has non-centrosymmetric molecular arrangements in the unit cell with at least six non-equivalent Dtc ligands. The aromatic carbons can be characterised by fifteen resonance lines in the 13C CP-MAS NMR spectra, covering a chemical shift range of 19.9 ppm (122.3–152.2 ppm).
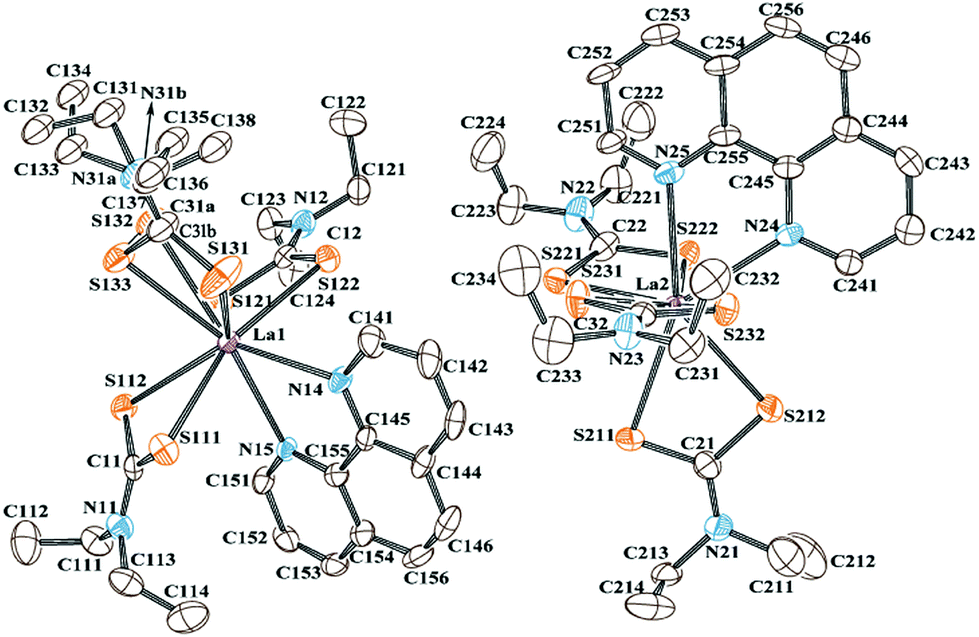 | ||
| Fig. 2 The ORTEP diagram of the title complex, showing two mononuclear [La{S2CN(C2H5)2}3C12N2H8] molecules with 50% thermal ellipsoids and the crystallographic atom labelling scheme. H atoms are omitted for clarity. | ||
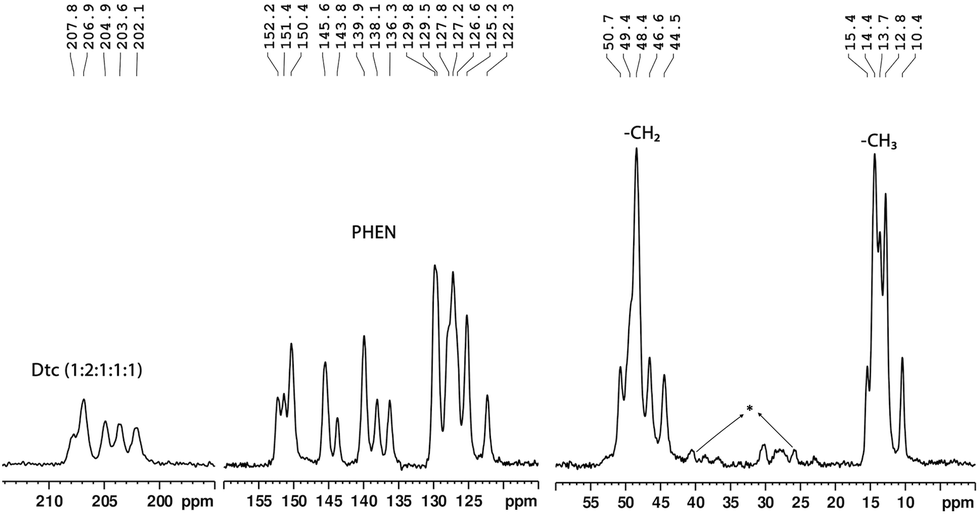 | ||
| Fig. 3 A solid-state 13C CP-MAS NMR spectrum (90.52 MHz) of the tris(N,N-diethyldithiocarbamato)(PHEN)lanthanum(III) complex (*spinning side bands). | ||
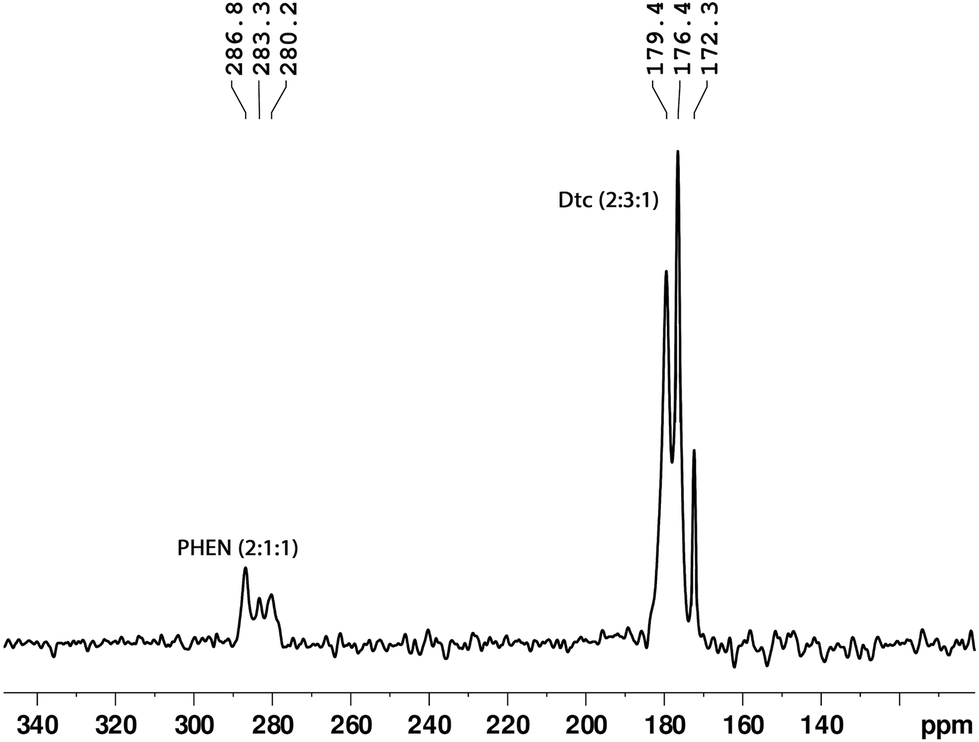 | ||
| Fig. 4 A solid-state 15N CP-MAS NMR spectrum (40.0 MHz) of the tris(N,N-diethyldithiocarbamato)(PHEN)lanthanum(III) complex. | ||
| Label | 1A/1Bb | 2A/2Bb | SR-ZORAc | SO-ZORAd | 3A/3Bb | Expt. |
|---|---|---|---|---|---|---|
| a All values are averages of results for A and B isomers. Expt. = experimental solid-state NMR results. b Periodic GIPAW/PBE NMR shifts with CASTEP optimised structures: 1A/1B = all protons relaxed; 2A/2B = partial geometry optimisation (all H, S131–S133, N31A/N31B, C31A/C31B, C131–C138); 3A/3B = all atoms relaxed. c ADF SR-ZORA/PBE/TZ2P result in the 3A/3B structure. d ADF SO-ZORA/PBE/TZ2P result in the 3A/3B structure. e Averages over all methylene and methyl groups, respectively. f The standard deviations (SD) and the mean absolute errors (MAE, in parenthesis) between the calculated and experimental chemical shifts. | ||||||
| 13 C Chemical shifts (ppm) | ||||||
| C31 | 228.1 | 211.9 | 213.7 | 208.2 | 208.5 | 207.8 |
| C32 | 204.8 | 205.3 | 212.8 | 207.3 | 206.8 | 206.9 |
| C12 | 203.5 | 203.5 | 212.8 | 207.3 | 206.1 | 206.9 |
| C22 | 202.0 | 202.2 | 211.5 | 206.0 | 204.5 | 204.9 |
| C21 | 198.7 | 198.8 | 210.5 | 205.0 | 203.8 | 203.6 |
| C11 | 197.6 | 198.2 | 210.3 | 204.8 | 201.1 | 202.1 |
| C241 | 155.2 | 155.4 | 159.1 | 158.4 | 154.7 | 152.2 |
| C251 | 153.0 | 153.5 | 156.8 | 156.1 | 154.2 | 151.4 |
| C141 | 152.7 | 152.0 | 156.7 | 156.0 | 153.2 | 150.4 |
| C251 | 151.5 | 151.7 | 155.9 | 155.2 | 152.0 | 150.4 |
| C145 | 148.8 | 149.2 | 153.4 | 152.7 | 148.6 | 145.6 |
| C245 | 148.4 | 148.2 | 153.3 | 152.6 | 148.2 | 145.6 |
| C255 | 147.8 | 147.9 | 152.7 | 152.0 | 147.7 | 145.6 |
| C155 | 146.0 | 146.2 | 152.5 | 151.8 | 146.3 | 143.8 |
| C243 | 141.8 | 142.3 | 146.5 | 145.8 | 144.0 | 143.8 |
| C143 | 141.7 | 141.3 | 142.0 | 141.3 | 143.6 | 139.9 |
| C155 | 138.2 | 138.3 | 141.9 | 141.2 | 142.1 | 139.9 |
| C253 | 138.1 | 138.1 | 141.8 | 141.1 | 139.3 | 138.1 |
| C154 | 131.5 | 131.6 | 136.9 | 136.2 | 135.7 | 136.3 |
| C246 | 131.3 | 131.5 | 136.9 | 136.2 | 131.9 | 129.9 |
| C254 | 131.2 | 131.2 | 135.9 | 135.2 | 131.8 | 129.9 |
| C144 | 130.7 | 130.6 | 135.9 | 135.2 | 131.6 | 129.5 |
| C256 | 130.2 | 129.8 | 135.2 | 134.5 | 131.4 | 129.5 |
| C244 | 129.4 | 129.3 | 133.9 | 133.2 | 131.4 | 127.8 |
| C156 | 128.9 | 129.0 | 133.0 | 132.3 | 130.3 | 127.8 |
| C242 | 128.1 | 128.8 | 132.8 | 132.1 | 130.1 | 127.2 |
| C156 | 127.5 | 127.5 | 131.2 | 130.5 | 129.0 | 127.2 |
| C152 | 125.2 | 125.2 | 129.3 | 128.6 | 127.8 | 126.6 |
| C142 | 124.7 | 125.1 | 128.5 | 127.8 | 127.6 | 125.2 |
| C252 | 124.2 | 124.0 | 128.0 | 127.3 | 124.1 | 122.3 |
| –CH2e |
50.2 | 48.9 | 51.8 | 51.1 | 47.7 | 48.3 |
| –CH3e |
10.8 | 10.2 | 14.2 | 13.5 | 11.0 | 13.4 |
| SD (MAE)f | 4.3 (2.6) | 2.4 (2.0) | 2.1 (5.2) | 2.4 (3.7) | 1.5 (1.8) | — |
| 15 N Chemical shifts (ppm) | ||||||
| N24 | 304.8 | 304.9 | 308.2 | 307.4 | 306.8 | 286.8 |
| N25 | 301.7 | 303.3 | 305.8 | 304.9 | 306.9 | 286.8 |
| N14 | 299.1 | 300.2 | 303.2 | 302.2 | 301.6 | 283.3 |
| N15 | 297.0 | 297.2 | 301.0 | 300.2 | 295.9 | 280.2 |
| N11 | 190.1 | 189.6 | 188.7 | 189.4 | 190.7 | 179.4 |
| N21 | 194.3 | 193.9 | 184.8 | 185.5 | 189.5 | 179.4 |
| N12 | 186.3 | 185.6 | 182.6 | 183.3 | 187.4 | 176.4 |
| N22 | 186.1 | 186.8 | 179.7 | 180.5 | 187.4 | 176.4 |
| N23 | 189.8 | 190.2 | 179.7 | 180.4 | 185.9 | 176.4 |
| N31 | 175.0 | 180.7 | 179.4 | 180.1 | 181.2 | 172.3 |
| SD (MAE)f | 4.5 (12.8) | 3.6 (13.6) | 6.9 (12.7) | 6.9 (11.8) | 4.4 (13.7) | — |
On the other hand, three 15N resonance lines (with integral intensities 2:1:1), in the range of 280.1–286.7 ppm, were observed for PHEN nitrogen atoms (see Fig. 4). The 13C resonance lines corresponding to methylene and methyl carbons appeared in the range of 44.5–50.7 ppm and 10.4–15.4 ppm, respectively. For atom wise assignments of the solid-state 13C and 15N NMR spectra, we used the relative chemical shift differences from the first principles DFT (GIPAW) calculated values (see Table 1).
X-ray crystallography
The details of the molecular and crystal structure determination of the title complex are explained in the Experimental section, crystal structure data are given in Table 2, and the molecular structure of the complex with a crystallographic labelling scheme is depicted in Fig. 2. La(III) has a coordination number of 8, and crystallised in the triclinic system with the space group P. The unit cells of the title complex comprise four orientationally non-equivalent mononuclear molecules where a centre of symmetry relates two pairs. In other words, there are two structurally non-equivalent molecules in the asymmetric unit. Overall, single-crystal X-ray diffraction results confirm the conclusions of 13C and 15N CP-MAS NMR that the complex under consideration contains in total six structurally non-equivalent Dtc carbon ligands. The distances and angles for some selected bonds in the experimental structure are compared with the corresponding calculated values from the DFT-CASTEP in Tables S2 and S3 in the ESI.† The La(III) coordination displays a distorted square antiprismatic geometry, where each lanthanum ion is coordinated in a S,S′-aniso-bidentate manner to three non-equivalent Dtc ligands, with the neutral PHEN ligand filling the coordination sphere forming two basal planes.
| a R 1 = ∑||Fo| − |Fc||/∑|Fo|, wR2 = [∑w(Fo2 − Fc2)2/∑wFo4]1/2. | |
|---|---|
| Empirical formula | C27H38N5S6La |
| Formula weight | 763.89 |
| Temperature (K) | 150 |
| Crystal system | Triclinic |
| Space group |
P |
| a (Å) | 11.082(2) |
| b (Å) | 17.106(3) |
| c (Å) | 18.304(4) |
| α (°) | 91.96(3) |
| β (°) | 103.92(3) |
| γ (°) | 94.31(3) |
| V (Å3) | 3353.4(12) |
| Z | 4 |
| F(000) | 1552 |
| D c (g cm−3) | 1.513 |
| μ (Mo Kα) (mm−1) | 1.673 |
| Crystal size (mm) | 0.15 × 0.12 × 0.10 |
| Independent/observed reflectionsa | 12908/10676 |
| R INT | 0.0894 |
| R 1/wR2 [I ≥ 2σ(I)]a | 0.0687/0.1680 |
| R 1/wR2 (all data)a | 0.0870/0.1822 |
| GOF | 1.123 |
Among the three Dtc ligands coordinating around La2, S221, S222, S231, and S232 form the first basal plane while the other plane is formed by S211, S212, N24, and N25. The La–S, C–S, and N–C(S)S bond lengths vary in the ranges 2.926(2)–3.012(4) Å, 1.692(9)–1.789(9) Å and 1.324(11)–1.365(2) Å, respectively. The C–N bond lengths reflect a partial double bond character. One of the three (Et2NCS2) ligands, coordinated around La1 (see Fig. 2), is disordered assuming two different orientations (denoted as A and B) with different occupation parameters. The disordered Dtc ligand is randomly distributed in the crystal with the probabilities 0.53 and 0.47, respectively, for the A and B conformers. This kind of orientational disorder of a ligand is a rather common phenomenon in crystals, especially when there are alkyl groups in the structure of the ligands. Their flexibility can bring dynamics and/or conformational disorder to the structure of the complexes.
For the disordered Dtc ligand (orientations A and B), we can see a difference not only in the orientation but also in certain bond distances and bond angles (Tables S2 and S5 in the ESI†). In addition, the average bond parameters for the disordered ligand show many differences compared to all other Dtc ligands in the complex. For example, for the conformer A, the C–S bond lengths are equal within the experimental error bar (1.69 Å) whereas one of the C–S bond lengths for the conformer B (C31B–S133) is much larger (1.79 Å) compared to all other bonds (1.72 Å). On the other hand, the N31A–C131/C135 and N31B–C133/C137 bond lengths are much shorter (1.37 Å) compared to all other N–C(Et2) bonds (1.48 Å). The S–C–S bond angle is 113.1(5)° for orientation B, whereas for the other Dtc ligands it is in the range of 117.3(5)°–120.3(5)°. Similarly, significant differences were also observed in the disordered Dtc ligands in terms of C–C bond distances as well as in bond angles such as N–C–S and C–N–C (see Table S3 in the ESI†). The effects of these discrepancies of the bond parameters on the NMR chemical shift calculations are explained in the following sections.
Geometry optimisation and GIPAW NMR chemical shift calculations
The high sensitivity of nuclear shielding to the electronic structure, observed as NMR chemical shifts, makes it extremely useful to study subtle changes in the chemical environment of a nucleus. Knowing the fact that it is often difficult to determine hydrogen atom positions accurately from X-ray diffraction, the initial XRD structure was subjected to a geometry optimisation of hydrogen positions prior to NMR calculations.25 The NMR shielding constants were then computed using the coordinates of the modified structure (denoted as 1A/1B). The bond angles and bond lengths obtained after the geometry optimisation of the atomic positions are given in Tables S2 and S3 (see the ESI†). The calculated NMR chemical shifts for structures obtained at different geometry optimisation levels are given in Table 1 along with the experimental results. The absolute nuclear shielding values are also given in Table S4 (see the ESI†). It must be noted that in the experimental 13C and 15N NMR spectra (Fig. 3 and 4), due to signal broadening, the resonance lines for the Dtc carbon and nitrogen nuclei do not allow us to distinguish between different conformers. Therefore, in all our cases, although the geometry optimisations were carried out for the conformers A and B separately, an average value for the chemical shift was reported. The H-optimised structure produced insubstantial agreement with the experimental NMR data and gave a poor fit of the computed 13C and 15N chemical shift values especially for atoms in the disordered ligands. It is surprising to see such an unreasonably large difference in the shielding values for both 13C and 15N nuclei (Table S4†) in the disordered ligands when the X-ray diffraction structures are optimised just for protons (1A and 1B). For example, there is a 19.9 ppm difference between the 13C shielding values of C31A and C31B and a 32.8 ppm difference between N31A and N31B shielding (15N) values. These large discrepancies in solid-state NMR results indicate that further atomic coordinate refinements are essential. This was evident from the large remaining forces (>3 eV Å−1) on some of the atoms in the 1A/1B structures. The standard deviation (SD) between the calculated and experimental 13C and 15N chemical shifts was 4.3 ppm and 4.5 ppm, respectively (see Fig. S4a and S4c in the ESI†). In the next step, to get an idea on how much these (large) forces affect the chemical shifts, only the positions of the above mentioned atoms on the disordered ligands were relaxed (2A/2B structures). This leads to a significant improvement since the NMR shift calculation of these partially optimised structures shows a difference of only 1.9 ppm and 3.6 ppm, respectively, for the C31A–C31B and N23A–N24B atoms. This is also seen in reduced SD values between the calculated and experimental 13C (2.4 ppm) and 15N (3.6 ppm) chemical shifts for the 2A/2B structures (see Fig. S4b and S4d in the ESI†). The partial geometry optimisation did not result in any notable changes in the subsequent nuclear shielding calculations for the other unrelaxed atoms.A significant improvement in the agreement between experimental and computational NMR chemical shifts was obtained in the final refinement by adjusting the positions of all atoms while holding the lattice parameters constant at the XRD values (3A/3B structures). As seen in Table 1, the computed NMR results for 3A/3B structures agree with the experimental results better than for any other structure in terms of both the chemical shift range and the spectral pattern. Moreover, the differences between the CASTEP calculated nuclear shielding values for the Dtc carbon (C31A/C31B) and nitrogen (N31A/N31B) atoms in the disordered ligands of conformers 3A and 3B are within the experimental error limits (see Table S4 in the ESI†) and hence support the notion that they are not distinguishable using the current experimental set up. In all the above cases, the 15N calculations show a systematic offset in the isotropic chemical shift values (Table 1).
There clearly is a good agreement between the experimental and calculated (for structures 3A/3B) isotropic chemical shift values, as observed from the close to unity slope of their linear regression plots (Fig. 5a and b). The SD between the experimental and calculated 13C chemical shifts is further reduced to 1.5 ppm while for 15N the SD remained almost similar (4.4 ppm) compared to the 1A/1B structures (see Fig. 5a and b) because of the full optimisation (3A/3B structure). The RMSD of the heavy atom displacements with respect to the proton optimised X-ray geometry (3A/3B vs. 1A/1B) is 0.12 Å. It may, therefore, be concluded that the most significant part of the improvement between the calculated and experimental chemical shifts obtained from solid-state NMR comes from the refinement of non-hydrogen atoms.
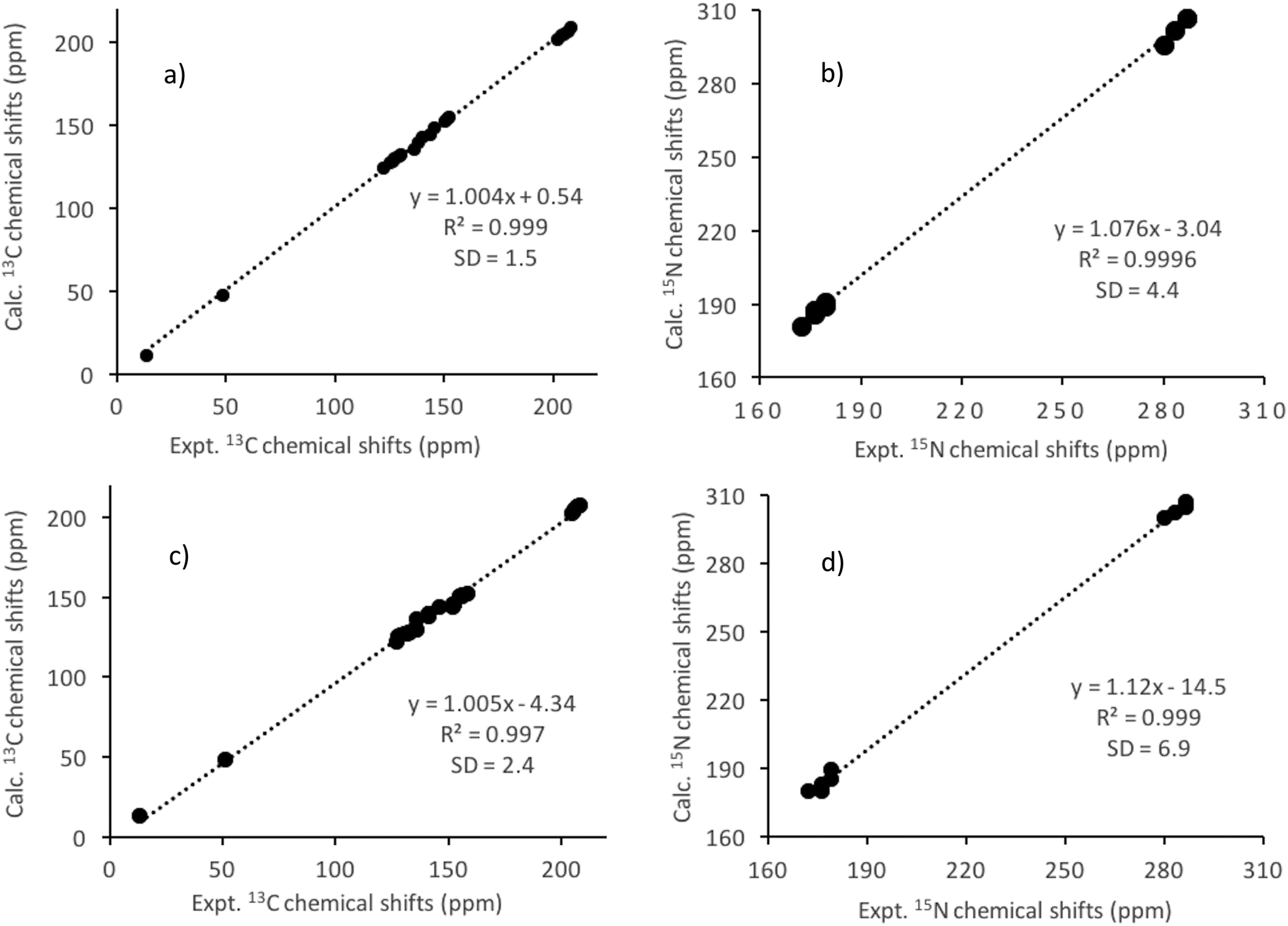 | ||
| Fig. 5 Computed (GIPAW/PBE) vs. experimental 13C (a) and 15N (b) chemical shift plots for the title lanthanum complex. Average values for the isomers 3A and 3B were used. The shielding values were converted to chemical shifts using a reference value (σref) of 170.9 ppm and 234.7 ppm for 13C (TMS) and 15N (NH3), respectively. Computed (ADF/PBE/SO-ZORA) vs. experimental 13C (c) and 15N (d) chemical shift plots for the title lanthanum complex. Average values for the isomers A and B were used. The shielding values were converted to chemical shifts using a reference value (σref) of 185.6 ppm and 251.3 ppm for 13C (TMS) and 15N (NH3), respectively. The standard deviations (SD) of the mean errors between the calculated and experimental chemical shifts are also shown. | ||
Explanation for the anomaly in chemical shifts
The NMR calculations on the partially and fully optimised structures mentioned above demonstrate the exquisite sensitivity of nuclear shieldings to local atomic configurations, i.e., bond structures (bond lengths and bond angles) and arrangements of non-bonded neighbours, which means that any motional change in these configurations is accompanied by changes in the nuclear shieldings. We have obtained the best fit with experimental NMR results for structures 3A/3B, where all atoms in the complex were relaxed (forces on each atom were <0.01 eV Å−1). The bond angles and distances of the disordered ligands before (experimental) and after geometry optimisation are compared in Tables S2 and S3 (see the ESI†). As explained earlier, significant changes were revealed in the structures of the disordered Dtc ligand, which has two different orientations, A and B, both showing many discrepancies in terms of bond lengths and bond angles compared to all the other Dtc sites in the unit cell. On the other hand, the CASTEP/PBE optimised bond lengths and bond angles for the two orientations, A and B, are very similar to all other sites and well within the expected range of values for the present theoretical level calculations.40 The optimised structures (3A and 3B) have S(S)C–N bond lengths in the range of 1.35–1.36 Å and C–S bond lengths in the range of 1.71–1.73 Å. At the same time, the alkyl C–C and N–CH2– bond lengths were within the range of experimental results.The following sources of errors in the computed 13C and 15N chemical shifts can be considered: (i) the level of theory used (scalar relativistic); (ii) the limitations of the density functional (PBE);40 (iii) the effects of the thermal rovibrational motions of atoms,41–43etc. Also, the uncertainties in the atomic positions of the disordered ligands as determined from X-ray diffraction measurements may cause significant chemical shift variations.25,44–46 The accuracy of the X-ray diffraction data depends on the quality of the single crystals available, which is also correlated with the narrowness of the NMR resonance lines and thus the resolution of the NMR spectra. In addition, dynamic averaging (vibrations, conformational averaging, molecular aggregation, etc.) may worsen the quality of the X-ray diffraction structure, but these effects are here neglected. However, based on the convergence of calculated NMR shifts (compare data for structures 2A/2B and 3A/3B with the experimental isotropic chemical shifts as well as the reduced SD values), it can be concluded that the low-quality of the original single crystal X-ray diffraction structure (R-factor 6.8%) is probably the main reason for the above observed irregularity in the calculated chemical shifts.
ZORA-DFT chemical shift calculations
It is an established fact that the NMR shielding calculations of light atoms such as hydrogen, carbon, nitrogen, etc. in molecular systems with heavy elements have to take into account an appropriate treatment of the relativistic effects because of the Heavy Atom (HA) on the neighbouring Light-Atoms (LA), referred to as the HALA effects.47–52 We have carried out the calculations at the optimised geometries, 3A and 3B, of both conformers A and B. The average chemical shifts were calculated from the results of both A and B conformers and are given in Table 1. The calculated spin–orbit (SO) contributions to the NMR shielding constants of the Dtc carbon atoms are about 5.5 ppm while they are less than 0.5 ppm for the other carbon atoms (Table 1). The SO contribution to the shieldings of the PHEN and Dtc nitrogen nuclei is <1.0 ppm, although the PHEN nitrogen atoms are directly bonded to lanthanum. This is in accord with our previous report on a heteroleptic tris(N,N-diethyldithiocarbamato)(PHEN) yttrium(III) complex, in which the SO contribution to the calculated NMR shielding was found to be important only for carbon atoms directly bonded to sulphur atoms and was negligibly small for other 13C, 1H and 15N nuclei in the complex.30 Interestingly, additional NMR calculations performed on a model [S2CNEt2]− fragment have revealed that the SO contribution to the shielding of the S2CN carbon nucleus is actually of a similar size (6.6–7.7 ppm) because of the highly delocalised orbitals of the sulphur atoms.30 Thus, it was concluded that the SO contribution from the heavy atom (Y) to two-bond distant carbon sites in the Dtc ligands is relatively small.30 Similarly, for the title tris(N,N-diethyldithiocarbamato)(PHEN)lanthanum(III) complex the calculated chemical shifts for the S2CN carbons at the SR-ZORA level differ ca. 5.5 ppm from those calculated at the SO-ZORA level; however, SO has practically no effect on the chemical shifts of the other carbon atoms. Overall, the SO contributions to the Dtc carbons resulted in chemical shifts, which do fit better with the experimental values.The lack of a ‘crystal lattice effect’ in ADF calculations seems to decrease the chemical shift difference between the Dtc carbons when compared to those observed in the solid-state NMR spectrum. For example, the experimental chemical shift range (6 ppm) is almost reproduced by the periodic GIPAW calculations (7 ppm) naturally incorporating the crystal lattice effects, whereas the chemical shifts of a single molecular unit in A and B conformations by ADF are spread over a rather small range (ca. 3 ppm both at SR- and SO-ZORA levels). Nonetheless, the ADF calculations reproduce the same spectral pattern in terms of shift ordering as the GIPAW calculations. However, as indicated by the lower SD value (see Fig. 5a–d and Table 1), the correlation between the calculated and experimental chemical shifts is much better with GIPAW. In other words, GIPAW calculations clearly show that the long-range structural organisation influences the chemical shielding of nuclei in REE(Dtc)3PHEN complexes.
Topological analysis of the electron density
For a qualitative understanding of the nature of metal–ligand bonding interactions of the title complex, we performed a topological analysis of the electron density (ρ) according to the quantum theory of atoms in molecules (QTAIM) in the ground state.35,53 In this theory, the topology of an electron-density distribution is described by a set of characteristic critical points (CrPs), viz. bond critical point (BCrP), ring critical point (RCrP), etc. (for definitions see Note 1 in the ESI†). The gradient of electron density (∇ρ) is zero at any critical point. Generally, ρ < 0.10 a.u. and positive values for the Laplacian of the electron density (∇ρ2) and energy density [H(r)] indicate ionic interactions, while ρ > 0.20 a.u. and negative values for both ∇ρ2 and H(r) correspond to a covalent bond. The pictorial representation of different types of critical points for the title compound is depicted in Fig. S5 (see the ESI†). As shown in Table 3, ∇ρ2 is positive at all BCrPs between La–N and La–S indicating, with the small values of ρ < 0.10 a.u., that both bond types are predominantly ionic.| Type | ρ | ∇ ρ | ∇ ρ 2 | H(r) |
|---|---|---|---|---|
| BCrP La–N | 0.034 | 0.32 × 10−16 | 0.094 | −0.0013 |
| BCrP La–S1 | 0.034 | 0.28 × 10−15 | 0.060 | −0.0036 |
| BCrP La–S2 | 0.034 | 0.10 × 10−16 | 0.061 | −0.0038 |
| RCrP N–La–N | 0.014 | 0.13 × 10−16 | 0.054 | 0.0016 |
| RCrP S–La–S | 0.017 | 0.24 × 10−16 | 0.057 | 0.0014 |
However, the slightly negative H(r) values of the above bonds suggest that these metal–ligand bonds may have some covalent character. Furthermore, the larger ∇ρ2 for La–N than La–S BCrP suggests that the former is more ionic than the latter. The contour plot representations of the Laplacian ∇ρ2 in the plane spanned by the atoms that involve some selected metal–ligand bonds, depicted in ESI Fig. S6a (La–S) and Fig. S6b (La–N),† both indicate a predominantly ionic bonding.
Natural bond orbital (NBO) analysis
The NBO analysis method has been widely used for a quantitative description of the electronic structure and bonding properties of molecules. Within the NBO method, the interpretation of the bonding situation is performed in terms of Lewis structures, which are popular bonding models in chemistry. In our case, the NBO search program (see Note 2 and Table S5 in the ESI†) yielded the best Lewis structure which described about 97.98% of the total electron density. In Table S6 (see the ESI†), we have listed some donor–acceptor (bond–antibond) interactions between “filled” (donor, L) Lewis-type NBOs and “unfilled” (acceptor, NL) non-Lewis-type NBOs, which are important from the description point of view of metal–ligand bonding. The energetic importance of the NBOs was estimated by the 2nd-order perturbation theory. Some of the most intensive donor–acceptor energy transfers include: donation of an electronic charge from sulphur lone pairs to antibonding (BD*) NBOs: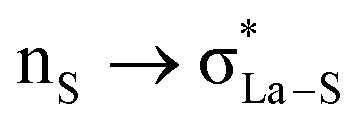 ,
, 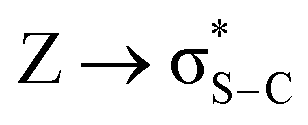 ,
, 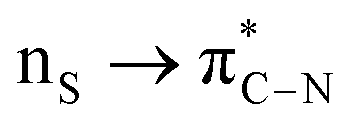 ; from a “lone spn pair” of PHEN nitrogen atoms to BD*, i.e.,
; from a “lone spn pair” of PHEN nitrogen atoms to BD*, i.e., 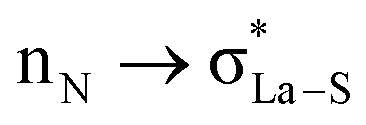 ; interaction between bonding La–S NBOs to antibonding La–S, i.e.,
; interaction between bonding La–S NBOs to antibonding La–S, i.e., 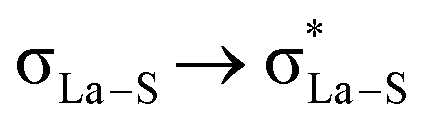 , etc. Among donor–acceptor interactions (ground state), the strongest stabilisation was found for
, etc. Among donor–acceptor interactions (ground state), the strongest stabilisation was found for  of energy 58.06 kcal mol−1. In Table 4, we have listed the natural atomic orbitals of which the above NBOs are composed, giving the percentage s- and p-characters of the NBO on each hybrid. For example, the σLa-S NBO is formed from the sd4.23f0.54 hybrid (72.9% d- and 9.4% f-character) on La interacting with an sp3.03 hybrid (75.1% p-character) on sulphur,
of energy 58.06 kcal mol−1. In Table 4, we have listed the natural atomic orbitals of which the above NBOs are composed, giving the percentage s- and p-characters of the NBO on each hybrid. For example, the σLa-S NBO is formed from the sd4.23f0.54 hybrid (72.9% d- and 9.4% f-character) on La interacting with an sp3.03 hybrid (75.1% p-character) on sulphur,| σLa-S = 0.3(sd4.23f0.54)La + 0.96(sp3.03)S |
| NBO-type | Occ | % Occ | Hybrids |
|---|---|---|---|
| LP(1) N | 1.846 | N(100) | s(30.0%) p 2.3 (69.9%) |
| LP(1) S | 1.928 | S(100) | s(59.0%) p 0.7 (41.0%) |
| LP(2) S | 1.596 | S(100) | s(0.1%) p 1.0 (99.9%) |
| BD(1) La–S | 1.936 | L(8.1) | 0.3 × La s(17.2%) p 0.02 (0.4%) d 4.2 (72.9%) f 0.5 (9.4%) |
| S(91.9) | 0.96 × S s(24.9%) p 3.0 (75.1%) | ||
| BD(1) S–C | 1.971 | S(46.0) | 0.7 × S s(20.0%) p 3.9 (79.1%) |
| C(54.0) | 0.7 × C s(32.5%) p 2.1 (67.4%) | ||
| BD(1) C–N | 1.990 | C(38.4) | 0.6 × C s(35.3%) p 1.8 (64.6%) |
| N(61.6) | 0.8 × N s(38.06%) p 1.6 (61.8%) | ||
| BD(2) C–N | 1.947 | C(28.3) | 0.5 × C s(0.1%) p 1.0 (99.8%) |
| N(71.7) | 0.8 × N s(0.0%) p 1.0 (99.9%) | ||
| BD*(1) La–S | 0.102 | La(91.9) | 0.96 × La s(17.2%) p 0.02 (0.4%) d 4.2(73.0%) f 0.5 (9.4%) |
| S2(8.1) | −0.28 × S s(24.8%) p 3.0 (75.1%) | ||
| BD*(1) S–C | 0.051 | S(54.0) | −0.7 × S s(20.0%) p 3.95 (79.1%) d 0.04 (0.8%) |
| C(46.0) | 0.7 × C s(32.5%) p 2.0 (67.4%) | ||
| BD*(1) C–N | 0.089 | C(62.1) | 0.8 × C s(35.5%) p 1.82 (64.4%) |
| N(37.9) | −0.6 × N s(38.0%) p 1.63 (61.9%) | ||
| BD*(2) C–N | 0.604 | C(71.5) | 0.8 × C s(0.13%) p 1.00 (99.7%) |
| N(28.5) | −0.5 × N s(0.02%) p 1.00 (99.9%) |
Fig. S7 in the ESI† shows surface diagrams for the overlapping hybrids and bond (σLa-S) and anti-bond (π*) NBOs of one of the La–S bonds. In addition, based on the natural population analysis (NPA), the natural charge on the La atom is considerably smaller than that on the free La3+ ion (see Table S7 in the ESI†), suggesting that a significant ligand-to-metal charge transfer occurs in the molecule. Besides, the net charges of the PHEN nitrogen atoms are more negative than those of the Dtc sulphur atoms, indicating the stronger electron-donating abilities of the nitrogen atoms. Fig. S8 in the ESI† shows the lowest unoccupied molecular orbital (LUMO) and the highest occupied molecular orbital (HOMO) of the title complexes. The calculated HOMO–LUMO gap of the molecule at the SR-ZORA/B3LYP/TZ2P theory level is ΔE = EHOMO − ELUMO = 2.431 eV.
Experimental
Materials
Lanthanum nitrate hexahydrate (La(NO3)3·6H2O), PHEN, diethylamine, carbon disulphide (99%), acetonitrile (99.8%), ethanol (99.8%), acetone, p-ether, diethyl ether, and chloroform were of analytical grade (Sigma-Aldrich) and used without further purification.Synthesis of the diethylammonium diethyldithiocarbamate ligand
The preparation of N,N-dialkyldithiocarbamate ligands is reported elsewhere.54 Briefly, carbon disulphide was added in excess to a 25% (volume/volume) solution of diethylamine (100 mmol) in acetone. The reaction mixture was stirred at 273 K for about 2 hours. The solvent and unreacted reagents were evaporated in a rotary evaporator at a low pressure. The crude product was purified by washing several times with petroleum ether. The resulting pale yellow product was recrystallised from diethyl ether. The yield was 80%.Synthesis of the [La(S2CNEt2)3PHEN] complex
The complex was prepared in the same way as the other REE(Dtc)3PHEN complexes reported.2 A solution of diethylammonium diethyldithiocarbamate (0.67 g, 3 mmol) and PHEN (0.18 g, 1 mmol) in acetonitrile (8 mL) and an ethanol mixture (4 mL) were added to a solution of lanthanum nitrate hexahydrate (La(NO3)3·6H2O) (0.43 g, 1 mmol) in acetonitrile (3 mL). A light yellow precipitate appeared upon stirring the reaction mixture. The solution was stirred for 15 minutes. The precipitate was washed with acetonitrile/ethanol several times and dried at 40 °C. Crystals suitable for X-ray crystallography were obtained from a saturated solution of the complex in a chloroform/ethanol mixture (20:1 ratio) by a slow evaporation of the solvents at room temperature over a few days. The yield was 75%.
Powder X-ray diffraction (PXRD)
The PXRD patterns were collected using a PANalytical Empyrean X-ray diffraction spectrometer (45 kV and 40 mA) and Cu Kα radiation with λ = 1.54 Å. Data collection was performed over the range 2θ = 5–50° with a scan interval of θ = 0.013°.X-ray crystallography
Unit cell parameters of a single crystal of the title complex (0.15 × 0.12 × 0.1 mm3) were collected at 150 K on a Bruker Nonius Kappa CCD diffractometer using graphite monochromatic Mo Kα radiation (λ = 0.71073 Å). The structures were solved using SHELS-2013 and refined using SHELXL-2013.55,56 After the full-matrix least-squares refinement of the non-hydrogen atoms with anisotropic thermal parameters, the hydrogen atoms were placed at the calculated positions in the aromatic ring (0.95 Å), methyl groups (0.98 Å) and methylene groups (0.99 Å). In the final refinement, hydrogen atoms were treated using a riding model in which the isotropic thermal parameters of the hydrogen atoms are proportional to those of the carbon atoms to which they are bonded. The isotropic thermal parameters of the aromatic and methylene hydrogen atoms were fixed at 1.2 times, and the methyl hydrogen atoms were fixed at 1.5 times the corresponding carbon atoms. The scattering factors for the neutral atoms were those incorporated with the programs. The site occupation factors were refined along with the positional and displacement parameters. Because of the correlation between the thermal parameters and the occupation factors, the anisotropic displacement parameters of the disordered pair of atoms were constrained to be equal. The final site occupancy factors were 0.528(4) and 0.472(4) for the two disordered orientations. Further information in the form of CIF has been deposited as depository CCDC 1453517 for C27H38La1N5S6.Solution-state NMR
One-dimensional 1H and 13C and two-dimensional 1H–13C and 1H–15N heteronuclear multiple bond correlation (HMBC) NMR spectra were measured on a Bruker Avance III spectrometer operating at 600.13, 150.92 and 60.80 MHz for 1H, 13C, and 15N, respectively. The spectral parameters for 1H–13C and 1H–15N HMBC experiments were as follows: π/2 pulse lengths 10.8 μs (1H), 11.3 μs (13C), and 17.3 μs (15N); 2048 data points in the 1H dimension and 256 increments (incremental delay 78.0 μs and 54.8 μs, respectively for 13C and 15N); the 1H–13C and 1H–15N HMBC experiments were optimised for nJ(C, H) = 5 Hz and nJ(N, H) = 8 Hz, respectively. The 1H and 13C spectra were referenced to TMS (0.0 ppm), while the 15N spectrum was referenced to liquid NH3 (0.0 ppm) using neat CH3NO2 at 380.2 ppm.571H NMR (CDCl3, Fig. S1 in ESI†). 9.84 ppm (dd, 3JHH = 4.8 Hz, 4JHH = 1.6 Hz, 2H), 8.38 ppm (dd, 3JHH = 8.4 Hz, 4JHH = 1.6 Hz, 2H), 7.86 ppm (s, 2H), 7.72 ppm (dd, 3JHH = 8.0 Hz, 4JHH = 4.8 Hz, 2H), 3.89 ppm (q, 3JHH = 7.2 Hz, 12H), 1.15 ppm (t, 3JHH = 7.2 Hz, 18H).
13C NMR (CDCl3, Fig. S2a and S2b in the ESI†). 206.05 ppm (–S2CN![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) , t, 3JCH = 4.6 Hz), 151.20 ppm (CH–, d, 1JCH = 182.4 Hz), 145.76 ppm (C–, 2JCH = 9.9 Hz), 138.32 (CH–, d, 1JCH = 164.23 Hz), 129.73 ppm (C–), 127.13 ppm (CH–, d, 1JCH = 163.7 Hz), 123.84 ppm (CH–, dd, 1JCH = 166.9 Hz, 2JCH = 7.3 Hz), 46.40 ppm (–CH2–, qt, 1JCH = 138.8 Hz, 2JCH = 3.7 Hz), 12.51 ppm (–CH3, tq, 1JCH = 127.4 Hz, 2JCH = 3.0 Hz).
, t, 3JCH = 4.6 Hz), 151.20 ppm (CH–, d, 1JCH = 182.4 Hz), 145.76 ppm (C–, 2JCH = 9.9 Hz), 138.32 (CH–, d, 1JCH = 164.23 Hz), 129.73 ppm (C–), 127.13 ppm (CH–, d, 1JCH = 163.7 Hz), 123.84 ppm (CH–, dd, 1JCH = 166.9 Hz, 2JCH = 7.3 Hz), 46.40 ppm (–CH2–, qt, 1JCH = 138.8 Hz, 2JCH = 3.7 Hz), 12.51 ppm (–CH3, tq, 1JCH = 127.4 Hz, 2JCH = 3.0 Hz).
15N NMR (CDCl3, Fig. S3b in the ESI†). 174.5 ppm (–CNEt2).
Solid-state NMR
13C and 15N CP-MAS NMR spectra were recorded on a Bruker Avance III spectrometer operating at 100.64 and 40.56 MHz, respectively. For both 13C and 15N CP-MAS experiments about 60 mg of the polycrystalline lanthanum(III) complex was pressed in a Zirconia rotor of 4.0 mm diameter. The following spectral parameters were used for 13C/15N CP-MAS acquisitions: 1H π/2 pulse length 2.7/2.5 μs, 1H–13C/1H–15N CP contact time 3.0/2.0 ms, 5 s recycle time for CP, and broadband spinal64 decoupling58 at 1H field strength 92.6/100.0 kHz. The 13C NMR chemical shifts were externally referenced to the most deshielded 13C signal of powder adamantane (38.48 ppm with respect to TMS)59 whereas 15N chemical shifts were referenced to ammonium chloride (98%) (39.2 ppm with respect to liquid NH3).60Computational details of periodic calculation
Geometry optimisations and NMR shielding calculations were carried out by the DFT based CASTEP code22,24,61 using the Perdew–Burke–Ernzerhof generalised gradient approximation (PBE-GGA).62 The structure was described as an extended solid using periodic boundary conditions. In all calculations, the so-called “ultra-soft” pseudopotentials (USPP)61 were used to describe the interaction of the valence electrons with the nuclei and core electrons. Within the periodic boundary conditions, the positions of all hydrogen atoms were allowed to relax because X-ray diffraction often has difficulty in resolving the accurate proton positions. Moreover, since one of the ligands in the isomeric molecule [La{S2CN(C2H5)2}3PHEN] has two possible orientations (A and B) it was needed to optimise the positions of the protons of these two isomers separately (hereafter, 1A and 1B). We also did a partial geometry optimisation by relaxing the positions of selected atoms (A: C64A, N23A, S14, C65, C66, C67, C68; B: C64B, N24B, S15, C69, C70, C71, C72) of the disordered ligand (La2) for both A and B conformers (2A and 2B). Finally, we performed a geometry optimisation by relaxing all atoms in the unit cell (3A and 3B), while keeping the unit cell parameters fixed. For the atomic relaxation, a force tolerance parameter of 0.01 eV Å−1 was used.The all-electron information, needed for the calculation of NMR shielding constants, was reconstructed using the GIPAW22 method as implemented in the NMR CASTEP 7.0 code.24 The wavefunctions were expanded using a plane wave (PW) basis set with a kinetic energy cut-off of 500 eV that produces converged results for both the geometry optimisation and the calculation of NMR parameters. The self-consistent field (SCF) calculations were considered to be converged when the total energy of the system was stable within 10−5 eV. The integrals were calculated over the Brillouin zone with k-point spacing <0.03 Å−1 on the 4 × 2 × 2 Monkhorst–Pack (MP) mesh. This grid contains 8 k-points in the irreducible Brillouin zone for the P space group and was tested to provide a well-converged geometry as well as the NMR shielding tensors. In order to compare the GIPAW calculated 13C shielding values directly with the experimentally measured isotropic chemical shift values the following expression was used:
| δiso, calc = σref − σiso | (1) |
![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) space group (TMS) and the P212121 space group (CH3NO2), respectively.
space group (TMS) and the P212121 space group (CH3NO2), respectively.
A linear regression between the isotropic calculated chemical shift (δiso, calc) and the experimental isotropic chemical shift (δiso, expt) values represents the amount of agreement between the calculation and the experiment.
Computational details of molecular calculations
Calculations of a one-molecular unit of the CASTEP optimised structures (3A/3B) were performed with the ADF2014 program package.63 For the NMR shielding calculations, the Slater-type orbital (STO) basis, a triple-ζ basis set with two polarisation functions (TZ2P), from the standard ADF basis set library was employed. The calculations were performed at the PBE theory level using both scalar-relativistic (SR) only and scalar plus spin–orbit relativistic (SO) ZORA formalisms.64,65 The 13C chemical shifts were calculated as the difference between the absolute shielding constant for the reference compounds and the shielding constant of each nucleus on the sample molecule using eqn (1), where the reference is TMS for 1H and 13C and NH3 for 15N. The reference shielding values were calculated at the same level of theory as the complexes (geometry optimisations were done at the PBE/TZ2P level, σref values for ADF calculations: 1H = 31.4 ppm, 13C = 185.6 ppm, and 15N = 251.3 ppm).The CASTEP optimised geometry (3A) was used in NBO 6.034 analysis, and implemented in ADF 2013, at the SR-ZORA/PBE theory level with the DZ basis set. The NBO method utilises the second order perturbation theory (PT2) analysis of the Fock matrix in the NBO basis set. According to PT2, for each donor NBO (i) and acceptor NBO (j), the stabilisation energy E(2) due to the delocalisation of an electron pair from (i) to (j) can be estimated as
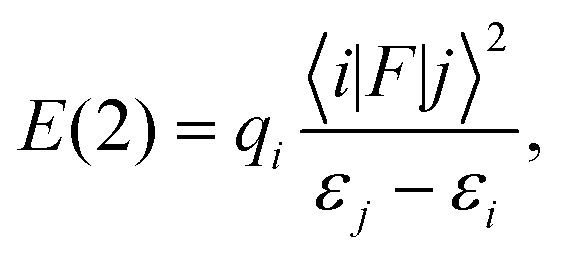 | (2) |
Conclusions
We have successfully used a multidisciplinary approach to establish the solid-state structure and to explore the electronic and spectroscopic properties of a heteroleptic tris(N,N-diethyldithiocarbamato)(PHEN)lanthanum(III) complex. Compared to all previously reported REE(Dtc)3PHEN complexes, a substantially different powder XRD pattern and 13C and 15N solid-state NMR spectra of the title compound, which indicated the presence of at least six Dtc sites in the asymmetric part of the unit cell, were obtained. The experimental NMR results were substantiated by single crystal XRD studies showing two structurally non-equivalent molecules in the unit cell. The DFT geometry optimisation of the crystal structure removed anomalies in the modelled NMR chemical shifts for the experimental X-ray structure. While the inclusion of spin–orbit (SO-ZORA) relativistic effects improved the correspondence with experiments, especially for the –S2CN– carbons, the periodic GIPAW modelling at the SR-ZORA level was sufficient for the full assignment of both 13C and 15N experimental spectra. A topological analysis of the electron density reveals the metal–ligand interactions to be largely ionic. Overall, our work demonstrates how different experimental and theoretical methods can be successfully combined affording insights into solid-state structures and bonding environments of heavy-metal complexes.Acknowledgements
Computations were carried out at CSC - the Finnish IT Centre for Science (Espoo, Finland). The current project is financed by the Centre for Advanced Mining and Metallurgy (CAMM) at LTU, Nordic Mining School and Svenska Kulturfonden (Oulu), and the Academy of Finland (#289649, #294027, #125316, #218191, #255641, and #285666). The CMX-360 spectrometer was purchased with a grant from the Swedish Council for Planning and Coordination of Research (FRN). We thank the foundation in the memory of J. C. and Seth M. Kempe for a grant from which a part of the NMR equipment has been purchased.Notes and references
- M. D. Regulacio, M. H. Pablico, J. A. Vasquez, P. N. Myers, S. Gentry, M. Prushan, S. Tam-Chang and S. L. Stoll, Inorg. Chem., 2008, 47, 1512–1523 CrossRef CAS PubMed.
- M. D. Regulacio, N. Tomson and S. L. Stoll, Chem. Mater., 2005, 17, 3114–3121 CrossRef CAS.
- M. D. Regulacio, K. Bussmann, B. Lewis and S. L. Stoll, J. Am. Chem. Soc., 2006, 128, 11173–11179 CrossRef CAS PubMed.
- L. Peisen, L. Huanyong and J. Wanqi, J. Rare Earths, 2011, 29, 317–320 CrossRef.
- C. Boshui, D. Junxiu and Y. Yi, J. Chin. Rare Earth Soc., 1998, 16, 358–360 Search PubMed.
- R. B. Rastogi, J. L. Maurya, V. Jaiswal and D. Tiwary, Int. J. Ind. Chem., 2012, 3, 1–10 CrossRef.
- S. A. Afrillya, Y. Mulyasih and I. Hastiawan, in Proceedings of the International Seminar on Chemistry, Jatinagor, 2008, pp. 186–190 Search PubMed.
- S. B. Castor and J. B. Hedrick, Industrial Minerals volume, Society for Mining, Metallurgy, and Exploration, Littleton, Colorado, 7th edn, 2006, pp. 769–792 Search PubMed.
- T. O. Pennanen and J. Vaara, Phys. Rev. Lett., 2008, 100, 133002 CrossRef PubMed.
- W. Van den Heuvel and A. Soncini, J. Chem. Phys., 2013, 138, 054113 CrossRef PubMed.
- J. Vaara, S. A. Rouf and J. Mareš, J. Chem. Theory Comput., 2015, 11, 4840–4849 CrossRef CAS PubMed.
- F. Gendron, K. Sharkas and J. Autschbach, J. Phys. Chem. Lett., 2015, 6, 2183–2188 CrossRef CAS PubMed.
- H. C. Hoffmann, M. Debowski, P. Müller, S. Paasch, I. Senkovska, S. Kaskel and E. Brunner, Materials, 2012, 5, 2537–2572 CrossRef CAS.
- D. D. Laws, H. L. Bitter and A. Jerschow, Angew. Chem., Int. Ed., 2002, 41, 3096–3129 CrossRef CAS.
- M. J. Willans, K. W. Feindel, K. J. Ooms and R. E. Wasylishen, Chem. – Eur. J., 2006, 12, 159–168 CrossRef CAS PubMed.
- L. Spencer, E. Coomes, E. Ye, V. Terskikh, A. Ramzy, V. Thangadurai and G. R. Goward, Can. J. Chem., 2011, 89, 1105–1117 CrossRef CAS.
- R. K. Harris, E. D. Becker, C. de Menezes, M. Sonia, R. Goodfellow and P. Granger, Pure Appl. Chem., 2001, 73, 1795–1818 CrossRef CAS.
- L. A. O'Dell and R. W. Schurko, Chem. Phys. Lett., 2008, 464, 97–102 CrossRef.
- R. Bhattacharyya and L. Frydman, J. Chem. Phys., 2007, 127, 194503 CrossRef PubMed.
- C. Müller and B. Paulus, Phys. Chem. Chem. Phys., 2012, 14, 7605–7614 RSC.
- J. Vaara, Phys. Chem. Chem. Phys., 2007, 9, 5399–5418 RSC.
- C. J. Pickard and F. Mauri, Phys. Rev. B: Condens. Matter, 2001, 63, 245101 CrossRef.
- M. Segall, P. J. Lindan, M. A. Probert, C. Pickard, P. Hasnip, S. Clark and M. Payne, J. Phys.: Condens. Matter, 2002, 14, 2717 CrossRef CAS.
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. I. Probert, K. Refson and M. C. Payne, Z. Kristallogr. - New Cryst. Struct., 2005, 220, 567–570 CAS.
- R. K. Harris, P. Hodgkinson, C. J. Pickard, J. R. Yates and V. Zorin, Magn. Reson. Chem., 2007, 45, S174–S186 CrossRef CAS PubMed.
- S. E. Ashbrook and D. McKay, Chem. Commun., 2016, 52, 7186–7204 RSC.
- C. Martineau, A. Cadiau, B. Bouchevreau, J. Senker, F. Taulelle and K. Adil, Dalton Trans., 2012, 41, 6232–6241 RSC.
- C. Martineau, Solid State Nucl. Magn. Reson., 2014, 63, 1–12 CrossRef PubMed.
- R. K. Harris, R. E. Wasylishen and M. J. Duer, NMR Crystallography, Wiley, New York, 2009 Search PubMed.
- V. Gowda, B. Sarma, S. Öberg, V.-V. Telkki, A.-C. Larsson, P. Lantto and O. N. Antzutkin, Eur. J. Inorg. Chem., 2016, 20, 3278–3291 CrossRef.
- V. Varand, L. Glinskaya, R. Klevtsova and S. Larionov, J. Struct. Chem., 1998, 39, 244–252 CrossRef CAS.
- N. Kuz'mina, R. Ivanov, A. Ilyukhin and S. Paramonov, Koord. Khim., 1999, 25, 635–638 Search PubMed [Russ. J. Coord. Chem. (Engl. Transl.), 1999, 25, 593–596].
- G. Te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS.
- E. D. Glendening, C. R. Landis and F. Weinhold, J. Comput. Chem., 2013, 34, 1429–1437 CrossRef CAS PubMed.
- J. I. Rodríguez, J. Comput. Chem., 2013, 34, 681–686 CrossRef PubMed.
- J. I. Rodríguez, R. F. Bader, P. W. Ayers, C. Michel, A. W. Götz and C. Bo, Chem. Phys. Lett., 2009, 472, 149–152 CrossRef.
- I. Baba, I. Raya and B. M. Yamin, Sains Malays., 2009, 38, 185–190 CAS.
- L. Pazderski, T. Pawlak, J. Sitkowski, L. Kozerski and E. Szłyk, Magn. Reson. Chem., 2010, 48, 450–457 CrossRef CAS PubMed.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 5, 799–805 RSC.
- R. Laskowski, P. Blaha and F. Tran, Phys. Rev. B: Condens. Matter, 2013, 87, 195130 CrossRef.
- J. Vaara, J. Lounila, K. Ruud and T. Helgaker, J. Chem. Phys., 1998, 109, 8388–8397 CrossRef CAS.
- J. Dumez and C. J. Pickard, J. Chem. Phys., 2009, 130, 104701 CrossRef PubMed.
- M. Dracinsky, P. Bouř and P. Hodgkinson, J. Chem. Theory Comput., 2016, 12, 968–973 CrossRef CAS PubMed.
- R. J. Iuliucci, J. C. Facelli, D. Alderman and D. M. Grant, J. Am. Chem. Soc., 1995, 117, 2336–2343 CrossRef CAS.
- A. C. de Dios and C. J. Jameson, Annu. Rep. NMR Spectrosc., 2012, 77, 1–69 CrossRef.
- S. Cadars, A. Lesage, C. J. Pickard, P. Sautet and L. Emsley, J. Phys. Chem. A, 2009, 113, 902–911 CrossRef CAS PubMed.
- M. Kaupp, O. L. Malkina, V. G. Malkin and P. Pyykkö, Chem. – Eur. J., 1998, 4, 118–126 CrossRef CAS.
- J. Vaara, K. Ruud, O. Vahtras, H. Ågren and J. Jokisaari, J. Chem. Phys., 1998, 109, 1212–1222 CrossRef.
- A. M. Kantola, P. Lantto, J. Vaara and J. Jokisaari, Phys. Chem. Chem. Phys., 2010, 12, 2679–2692 RSC.
- P. Lantto, S. Standara, S. Riedel, J. Vaara and M. Straka, Phys. Chem. Chem. Phys., 2012, 14, 10944–10952 RSC.
- J. Vícha, M. Straka, M. L. Munzarová and R. Marek, J. Chem. Theory Comput., 2014, 10, 1489–1499 CrossRef PubMed.
- J. Vícha, C. Foroutan-Nejad, T. Pawlak, M. L. Munzarová, M. Straka and R. Marek, J. Chem. Theory Comput., 2015, 11, 1509–1517 CrossRef PubMed.
- K. Kiewisch, G. Eickerling, M. Reiher and J. Neugebauer, J. Chem. Phys., 2008, 128, 044114 CrossRef PubMed.
- K. Cavell, J. Hill and R. Magee, J. Inorg. Nucl. Chem., 1979, 41, 1277–1280 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 1990, 46, 467–473 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2007, 64, 112–122 CrossRef PubMed.
- D. S. Wishart, C. G. Bigam, J. Yao, F. Abildgaard, H. J. Dyson, E. Oldfield, J. L. Markley and B. D. Sykes, J. Biomol. NMR, 1995, 6, 135–140 CrossRef CAS PubMed.
- B. Fung, A. Khitrin and K. Ermolaev, J. Magn. Reson., 2000, 142, 97–101 CrossRef CAS PubMed.
- C. R. Morcombe and K. W. Zilm, J. Magn. Reson., 2003, 162, 479–486 CrossRef CAS PubMed.
- S. Paramasivam, A. Balakrishnan, O. Dmitrenko, A. Godert, T. P. Begley, F. Jordan and T. Polenova, J. Phys. Chem. B, 2010, 115, 730–736 CrossRef PubMed.
- J. R. Yates, C. J. Pickard and F. Mauri, Phys. Rev. B: Condens. Matter, 2007, 76, 024401 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- G. Te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS.
- E. van Lenthe, A. Ehlers and E. Baerends, J. Chem. Phys., 1999, 110, 8943 CrossRef CAS.
- E. Van Lenthe and E. J. Baerends, J. Comput. Chem., 2003, 24, 1142–1156 CrossRef CAS PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: ADF/SO-ZORA calculated 1H and 13C chemical shifts; experimental and CASTEP optimised structural parameters; CASTEP/GIPAW calculated 13C and 15N nuclear shielding values; Cartesian coordinates of the CASTEP optimised geometries; additional experimental NMR spectra in solution; frontier MOs, NBO, NPA, and QTAIM analysis. CCDC 1453517. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt03705d |
| This journal is © The Royal Society of Chemistry 2016 |